
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From curiosity to applications. A personal perspective on inorganic photochemistry
Peter C.
Ford
Department of Chemistry and Biochemistry, University of California, Santa Barbara, CA 93110-9510, USA. E-mail: ford@chem.ucsb.edu
First published on 12th February 2016
Abstract
Over the past several decades, the photochemistry and photophysics of transition metal compounds has blossomed from a relatively niche topic to a major research theme. Applications arising from the elucidation of the fundamental principles defining this field now range from probing the rates and mechanisms of small molecules with metalloproteins to light activated molecular machines. Offered here is a personal perspective of metal complex photochemistry drawn from this author's long involvement with this field. Several examples are described. Topics include characterizing key excited states and tuning these to modify chemical reactivity and/or photoluminescence properties, as well as using photoreactions as an entry to reactive intermediates relevant to homogeneous catalysts. This is followed by discussions of applying these concepts to developing precursors and precursor–antenna conjugates for the photochemical delivery of small molecule bioregulators to physiological targets.
 Peter C. Ford | Professor Ford joined the UCSB faculty after graduate study with Ken Wiberg at Yale and a postdoctoral fellowship with Henry Taube at Stanford. He has been a Visiting Fellow at the Australian National U, Guest Professor at U Copenhagen, Humboldt Fellow at Regensberg and Muenster and Guest Investigator at the US National Cancer Institute. Honors include a Humboldt Senior US Scientist Award, Inter-American Photochemical Society 2008 Award in Photochemistry, ACS 2013 Award for Distinguished Service to Inorganic Chemistry and RSC 2015 Award in Inorganic Mechanisms. He is an AAAS Fellow and a RSC Fellow. His current research focuses on photochemical delivery of bioactive small molecules to physiological targets and biomass conversion to chemicals and fuels. He has advised 65 Ph.D. graduates. |
Introduction
The interaction of light and matter has long intrigued humans of all ages. Indeed this author's personal interest in the photochemistry of transition metal complexes can be traced to the qualitative observation that certain metal compounds left on the benchtop for several days looked very different than those kept in the dark for a comparable period. This was during a postdoctoral fellowship in Henry Taube's Stanford U. laboratory, where as a new convert to inorganic chemistry, I became fascinated by the bright colors of transition metal complexes. The compounds of interest were the pentaammineruthenium(II) complexes Ru(NH3)5L2+ of various aromatic nitrogen heterocycles L. The absorption bands that dominate the visible absorption spectra of these Ru(NH3)5L2+, e.g., Fig. 1, are markedly dependent on the nature of L. For example, if L is a substituted pyridine py-X, this band shifts markedly to the red when X is an electron accepting substituent and to the blue when X is an electron donor.1 This and related observations drove me to study C. K. Jørgensen's book on metal complex spectroscopy2 and to learn sufficient group and ligand field theory to interpret these absorption bands as metal-to-ligand charge transfer (MLCT) transitions.1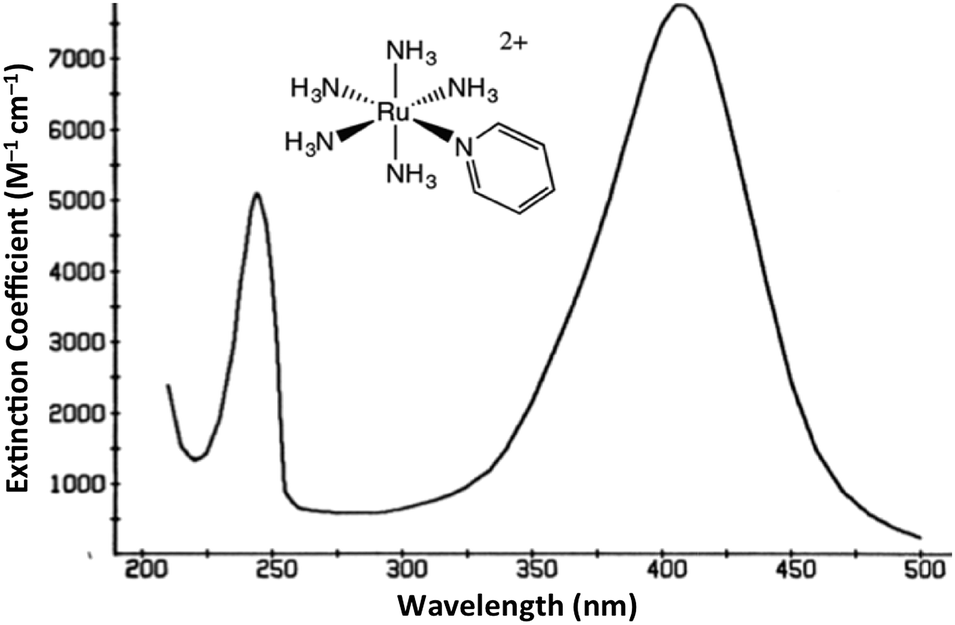 | ||
| Fig. 1 Absorption spectrum of Ru(NH3)5(py)2+ ion in aqueous solution. | ||
It was with these materials that I first noted light induced color changes and learned to store my samples in the dark. Furthermore, curiosity about the underlying causes led me to begin investigating the photoreactions of the Ru(II) ammine complexes, once I joined the chemistry faculty of the University of California, Santa Barbara (UCSB). I was fortunate that several UCSB colleagues were studying gas phase photoreactions and, moreover, were willing to teach this novice some nuances of photochemical science as it was then practiced. To set the context, I should emphasize that there had already been significant activity investigating photoreactions of metal complexes. Much of this work has been summarized in books by Balzani and Carassiti (1970)3 and by Adamson and Fleischauer (1974),4 both of which remain valuable resources. In addition, it would be remise not to draw special attention to the iron(III) oxalate-based actinometry developed by Hatchard and Parker in the UK (1956).5 This provided a convenient and sensitive method for measuring the intensities of the light at the location where solution phase photochemical reactions occur. It is a tool that continues to be in use many decades later.
Transition metal photochemistry continues to evolve, especially as new tools or potential new applications are realized. Offered here is a personal and not very comprehensive perspective of this evolution organized around the author's focus on the reactions and mechanisms that are triggered by light.
Fundamental mechanistic photochemistry
Excited states
The first question one might ask when considering the solution photochemistry of a complex ion like Ru(NH3)5(py-X)2+ is “what excited states (ES) are involved?” From the orbital parentage of these ES, one might develop a qualitative view of expected reaction trajectories. Given the power of density functional theory (DFT) computations now accessible, such visualization might seem primitive, but it does provide intuition of the type that has made the practice of chemistry a very human activity. Stepping back to our earliest photochemical investigations, neither DFT nor laser flash photolysis were readily available. As a consequence, mechanistic conclusions were largely built on determining photoreaction products and quantum yields and on comparing these data to the electronic spectroscopic properties.Fig. 2 illustrates the types of one-electron transitions that are commonly used to assign absorption bands and excited states in mononuclear metal complexes. Those most characteristic of transition metal complexes with partially filled d-orbitals are absorption bands representing excitation between “d” orbitals that are split by mixing with σ-donor and π-donor and acceptor orbitals of the ligands. These “d–d” ligand field (LF) transitions involve molecular orbitals (MOs) that are largely metal in character, hence the resulting ES are often referred to as metal-centered excited states (MC*). In a centrosymmetric complex, LF bands are Laporte forbidden and typically have relatively low extinction coefficients. In contrast, metal-to-ligand charge transfer (MLCT) and ligand-to-metal charge transfer (LMCT) bands are much more intense owing to the dipole moment change between the ground and excited states. The fourth type denoted in this figure is a ligand centered or intraligand (IL) transition, represented here as πL → π*L excitation. While these one-electron designations provide a convenient framework for addressing the natures of the relevant ES, theoretical calculations have long shown that there is much more mixing of the excited state characters than implied by Fig. 2.
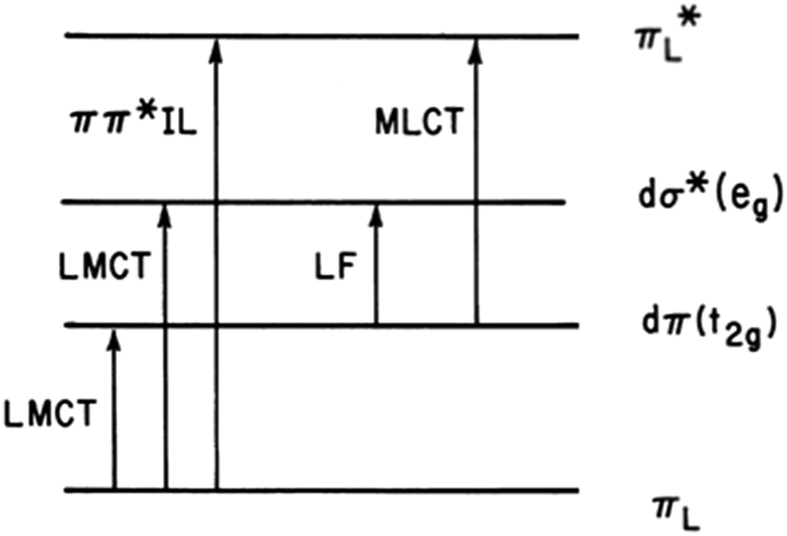 | ||
| Fig. 2 Representation of one-electron transitions between the MOs of a mononuclear metal complex ML6 in Oh symmetry. LF: ligand field transition; LMCT: ligand to metal charge transfer; MLCT: metal to ligand charge transfer; IL: intraligand or ligand centered. | ||
For Ru(NH3)5(py-X)2+ and related complexes such as Ru(NH3)5(pz)2+ (pz = pyrazine), the strong absorption bands (ε ∼ 104 M−1 cm−1) seen in the visible spectra were assigned as spin-allowed (singlet to singlet) MLCT transitions, while those in the ultraviolet region (UV) as IL (πL → π*L) in character. Where would one expect to find the LF absorptions for these low spin d6 complexes? As an empirical model, one might consider the spectrum of Ru(NH3)62+, which does not include any π-unsaturated ligands. The electronic spectrum of this octahedral complex displays two relatively weak absorption bands at 390 nm (ε = 35 M−1 cm−1) and 275 nm (640 M−1 cm−1) as predicted by group theory (1A1g → 1T1g and 1A1g → 1T2g)7 (note: the higher intensity of the second band has been attributed to mixing with a charge transfer to solvent transition, CTTS).7 Analogous LF bands might be expected at comparable energies for the Ru(NH3)5(py-X)2+ ions, but they are obviously obscured by the much stronger MLCT bands.
Quantum yields and ES dynamics
The quantum yield is basically an efficiency measurement and can be defined for a specific photoprocess i as shown in eqn (1). Note that Φi is unit-less. An efficient process has a quantum yield near 1 (or higher if a chain reaction results); however, as discussed below, there are many reasons why ΦA might be much smaller. A key point here is the nature of the denominator, since photoreactions will not occur unless light is absorbed. Quantum yields are typically measured at a specific wavelength of irradiation (λirr), so this concept is less precise when a broadband excitation source such as sunlight is used.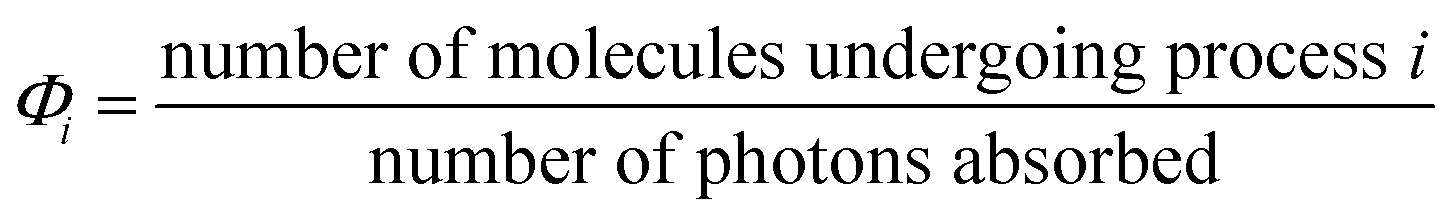 | (1) |
With certain applications, for example, the uncaging of a drug at a physiological target, the overall rate of the photochemical reaction is of key interest. The photoreaction rate equals Φi × Ia, where Ia is the intensity of the light absorbed, typically in units of einsteins per unit time (1 einstein = 1 mole of photons) or in einsteins per unit volume per unit time. For a solution where the photoreactant is the only species absorbing the incident light, Ia = I0(1 − 10−Abs(λ)), where Abs(λ) is the solution absorbance at λirr and equals the product of the molar concentration of the photoactive species (c), the molar extinction coefficient (ελ, in L mol−1 cm−1) at that λ and the path-length of the cell (in cm). There is no unimolecular rate constant associated with this process.
Taking for example Ru(NH3)5(py)2+, continuous photolysis of this ion with 405 nm light in ambient temperature, aqueous solution resulted in the photosubstitution reactions depicted in eqn (2).8 The respective quantum yields for pyridine and ammonia substitution Φpy and ΦNH3 were measured as 0.045 and 0.063 for λirr = 405 nm. This excitation wavelength is close to the λmax of the spin-allowed MLCT band (407 nm), so one can be assured that the vast majority of the photons absorbed lead to MLCT excitation.
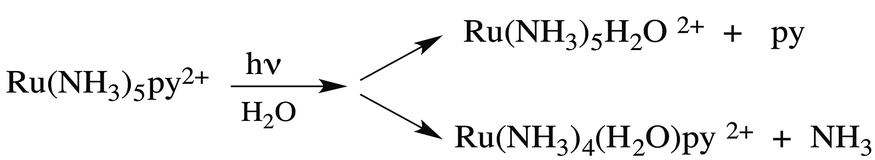 | (2) |
Are the observed photosubstitution reactions consistent with that expected for an MLCT excited state, which can simplistically be represented as having an oxidized metal center and a reduced pyridine, [RuIII(py−)]2+? Given that low spin d5 Ru(III) complexes are not very thermally labile, this behavior is not what one might expect for the MLCT* state. In contrast, the MC* states, which involve promoting an electron from a non-bonding or even π-bonding orbital to one which is σ* relative to the metal–ligand bonds would appear to be logical precursors of ligand photosubstitutions. Consistent with this argument, the hexaammine complex Ru(NH3)62+ is comparably labile toward photodissociation of NH3 (ΦNH3 = 0.27 for λirr = 405 nm).7 Drawing such a conclusion does, however, presume that the mechanism for ES ligand labilization is predominantly dissociative in character. In support of this view, a recent TDDFT calculation on the metal centered 3MC* state of Ru(NH3)5(py)2+ confirms that this state is nearly dissociative in character.9
This reasoning led to our proposal that the ES responsible for the reactions noted in eqn (2) was the result of rapid internal conversion/intersystem crossing from MLCT states populated by excitation to spectrally unobserved MC* states as illustrated in Fig. 3.10 The observation that photolabilization of aq. Ru(NH3)5(py)2+ is largely independent of the excitation wavelength suggested that a common reactive excited state, presumably the lowest energy excited state(s) (LEES), is responsible for these photoreactions. Another consequence of this model is the prediction that we can use ligand substituents to tune the excited state energies to where the 3MLCT is the LEES. In that case, there should be marked decreases in the observed photolability.
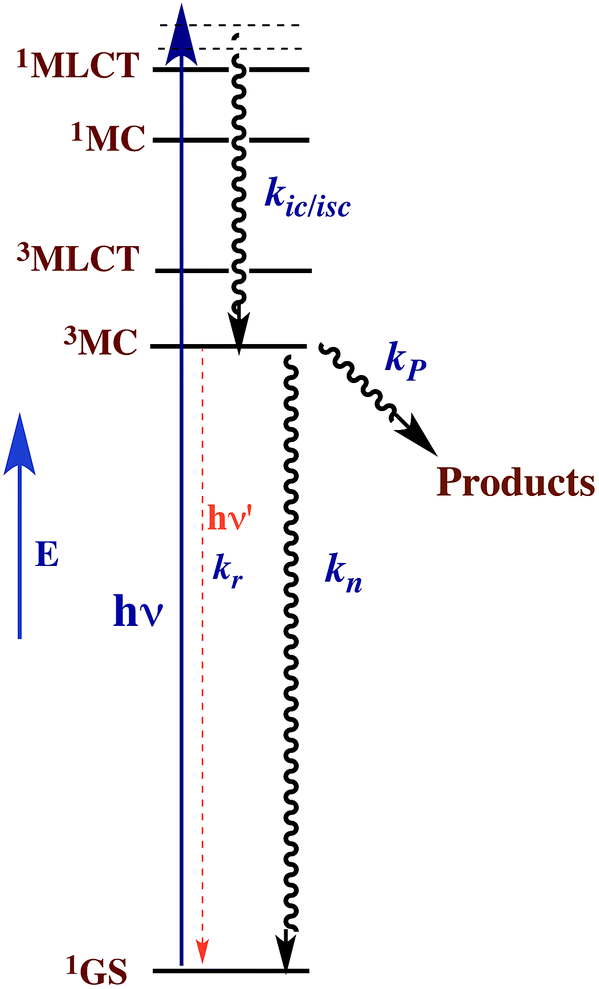 | ||
| Fig. 3 Proposed ES diagram to explain photoaquation of aq. Ru(NH3)5(py)2+ upon irradiation of MLCT bands. kP is the rate constant for reactions to photoproducts. kic/isc, kn and kr are rate constants for internal conversion/intersystem crossing to a lowest energy metal centered excited state, nonradiative deactivation to the ground state 1GS and radiative deactivation (emission), respectively. | ||
Our ES “tuning model” was thus demonstrated by examining the photoreactions of a series of Ru(NH3)5(py-X)2+ complexes.11 Electron-withdrawing substituents that shift the MLCT band to λmax values > 470 nm led to dramatically less photolability than those with λmax values at shorter wavelengths. Similar MLCT/MC state tuning approaches have since been used with a number of systems including those based on the Ru(II) polypyridyl complexes of interest with regard to solar energy conversion devices12 and similar Ir(III) phenylpyridyl complexes of interest for organic light emitting diodes (OLED) applications.13
The observation that photoreaction quantum yields ΦP for aq. Ru(NH3)62+ and Ru(NH3)5(py)2+ are considerably less than unity is explained by the Jablonski diagram shown in Fig. 3. Radiative and nonradiative pathways for ES deactivation to the ground state (GS) compete with the chemical pathways leading to products. In this simplified diagram, all three pathways are represented as occurring from the lowest energy ES of metal complex. However, one should keep in mind that with organic molecules and other compounds of the lighter elements, intersystem crossing (ISC) rates are often not competitive with direct radiative (fluorescence) and non-radiative deactivation to the GS. Furthermore, Franck–Condon states formed directly by excitation from the GS may undergo processes, including ISC, faster even than vibrational relaxation.14 Thus, the ES kinetics can be quite complex owing to the numerous states and deactivation pathways potentially involved (note: for compounds of lighter elements, e.g., organic dyes, internal conversion to the lowest energy ES of a particular spin state is typically much faster than the forbidden ISC to an ES of a different spin state. However, the much greater spin orbit coupling with complexes of heavier elements tends to break down this pattern).
For illustrative purposes, we will continue discussion of quantum yields based on the simple model described by Fig. 3, where a single state (or collection of ES) is populated by rapid and highly efficient internal conversion and ISC from states initially formed by excitation. For such a system, where decay occurs only by first order ES processes, Φi can also be described (eqn (3)), where kP is the rate constant for photoproduct formation, while kn and kr represent those for nonradiative deactivation to the ground state and radiative deactivation (emission), respectively. In the case of aq. Ru(NH3)5(py)2+ emission is at best very weak, and the photoproduct quantum yield ΦP is less than 0.5, so the dominant LEES pathway must be nonradiative deactivation.
| Φi = ki/(kP + kn + kr) | (3) |
Excited state lifetimes can be measured either by transient absorption, transient bleaching or luminescence methods. If the decay pathways from the LEES are unimolecular as implied by Fig. 3, then the observed lifetime τ equals (∑ki)−1, where in this case τ = (kP + kn + kr)−1. Thus eqn (3) can be rewritten eqn (4), and excited state rate constants for individual pathways can be calculated from ki = Φiτ−1. However, it must be emphasized that, while this approach often applies to complexes of the heavier transition metals, the kinetics are more complicated when more than one ES is responsible for the chemical or photoluminescence behavior.
| Φi = kiτ | (4) |
It was long thought that the Ru(NH3)5(py-X)2+ complexes did not display any emission either in solution or as solid salts. This conclusion is a matter of detection sensitivity given the einstein principle that every excited state undergoes spontaneous emission.15 The inherent intensities of such luminescence are determined by the competing processes that deplete that ES. Ultrafast flash photolysis studies by Winkler et al.16 and others17 did not detect emission, but with transient bleaching measurements did demonstrate ES lifetimes from <20 ps to ∼200 ps in ambient temperature solution. The longer values of τ were seen for those complexes for which the LEES are MLCT in character. Recently, Endicott and coworkers9 detected very weak emissions (Φr ∼ 5 × 10−5) from several of these complexes in 77 K organic glasses. Under these conditions, lifetimes were found to be on the 0.1–3 μs timescale, so the weak emissions were attributed to very low values of kr rather than to short lifetimes under these conditions.
The reaction dynamics implied by Fig. 3 and eqn (3) assumes that the lowest energy ES lies on potential surfaces with clearly defined minima. Such states are “bound” ES, for which there is an energy barrier larger than kBT, (kB being the Boltzmann constant) to decay along any relevant reactive coordinate (Fig. 4). It is also possible to have a “dissociative” state, where the barrier is less than kBT. The latter would have exceedingly short lifetimes, and it is not correct to treat their dynamics by classical kinetics methods.
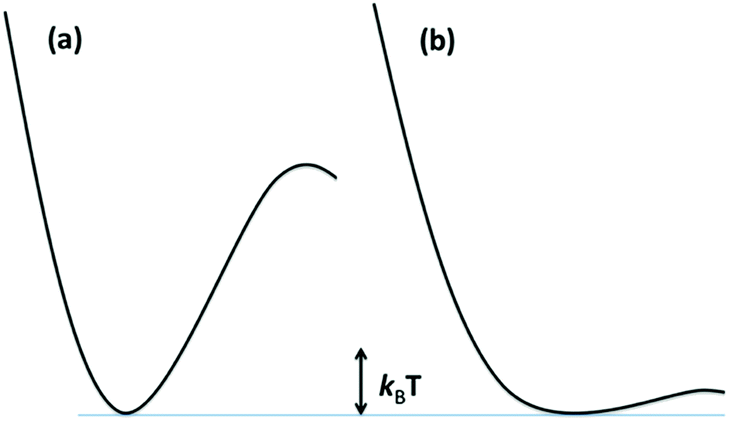 | ||
| Fig. 4 Potential surfaces for (a) a bound excited state where the barrier is >kBT and (b) a dissociative ES where the barrier is <kBT. Since kBT is temperature dependent, if any barrier is present, an ES that is dissociative at a high T, may behave as a bound state at low T. | ||
Coordination compounds
There have been a number of quantitative photochemical studies of coordination compounds, although the earlier studies primarily focused on those that were thermally stable in solution. Consequently, d3 (primarily Cr(III) complexes), low spin d6 (Fe(II), Ru(II) Os(II), Co(III), Rh(III), Ir(III) and some Pt(IV) complexes) and low spin d8 complexes (e.g., Rh(I), Ir(I) and Pt(II)) received the most attention, although this list is not comprehensive. Described here are several Rh(III) ammine complexes for which the detailed ligand photosubstitution dynamics and mechanisms were studied in this laboratory. These provide a valuable case study on the reactions of the metal-centered excited states of low spin d6 complexes, an electronic configuration of metal systems often found in applications such as dye sensitized solar cells (DSSCs) and OLEDs.Rhodium(III) amine complexes
The importance of MC* states in defining the photolability of the Ru(II) species even when initial excitation is MLCT led us to focus attention on the analogous photochemistry of the isoelectronic Rh(III) ammine complexes such as the Rh(NH3)63+ and Rh(NH3)5Cl2+ ions.18 The absorption spectra of these rhodium ammine complexes are dominated by the spin-allowed LF bands (1A1 → 1T2, 1A1 → 1T1 for the Oh complex Rh(NH3)63+, Fig. 5) predicted by group theory, while the emission spectra are characteristic of that expected from the lowest energy 3MC* states (3T1 → 1A1 for Rh(NH3)63+). Our entry into these studies also corresponded to our collaborations with R. J. Watts and D. S. Magde, with whom we were able to conduct ns and ps pulsed laser experiments that allowed determining for the first time the emission and transient absorption lifetimes of the lowest energy MC* states in fluid solutions.19–22 In this manner we were also able to probe the 3MC* state ligand substitution and nonradiative decay mechanisms with hydrostatic pressure effects in collaboration with R. van Eldik.23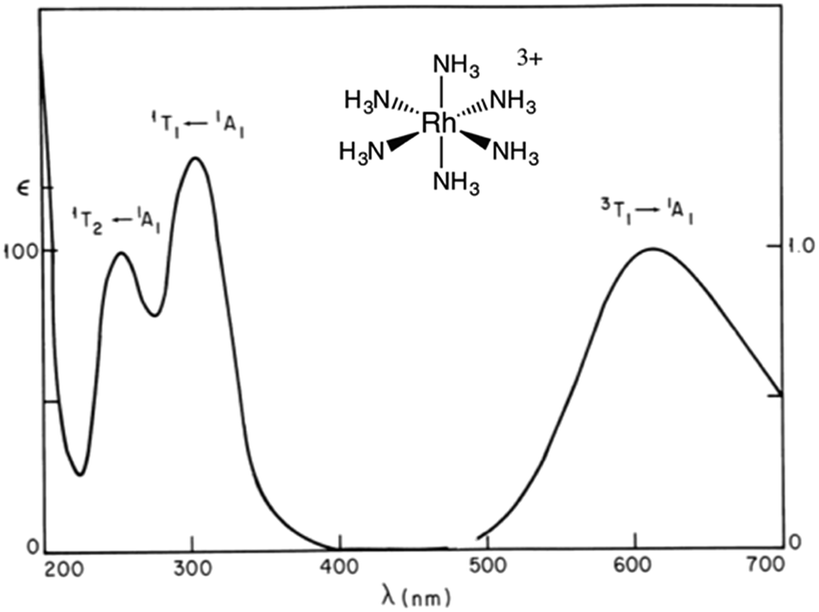 | ||
| Fig. 5 Absorption (left) and emission (right) spectra of Rh(NH3)63+ in aqueous solution. | ||
For Rh(NH3)63+, the photosubstitution chemistry (eqn (5), ΦA = 0.075 in 296 K aq. solution, A = NH3) and the luminescence properties proved to be largely independent of the excitation wavelengths.18 These observations, combined with early sensitization experiments,24 led to the conclusion that initial LF excitation to form 1MC* states was followed by efficient internal conversion/intersystem crossing to the lowest energy 3MC* state (i.e. the 3T1)25 from which emission or reaction largely occurred (note: kISC values > 5 × 109 s−1 have been measured for Rh(III) ammine complexes).25 This progression is envisioned by the Jablonski diagram shown in Fig. 6. Accordingly, one can treat the photoreaction and emission quantum yields in terms of the excited state rate constants as in eqn (3). Thus, for example, ΦA = kA/(kA + kn + kr). As seen for the ruthenium complexes, the modest quantum yield for eqn (5) and the very weak emission clearly indicate that the dominant pathway from the 3T1 is nonradiative deactivation, thus kn > kA ≫ kr. If that were the case, then inhibiting kn would have the potential to increase ΦA.
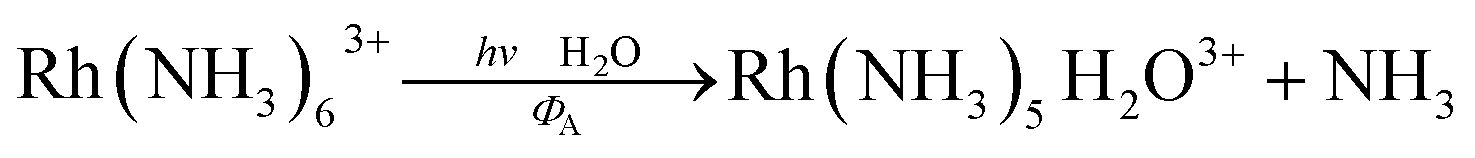 | (5) |
 | ||
| Fig. 6 Jablonski excited state diagram for Rh(NH3)63+ MC states. | ||
The question was: how to test this idea by perturbing kn? To do so, we drew upon studies by Glen Crosby and coworkers, who had shown that the perdeuterated complex Rh(ND3)63+ had dramatically longer luminescence lifetimes in frozen solutions at 77 K than did the perprotio analog.26 Since no photochemistry occurred under those conditions and the emission was a minor deactivation pathway, this meant that exchanging the N–H's for N–D's significantly decreased the rate of nonradiative deactivation. This, now a well recognized phenomena, is the result of a weak-coupling mechanism27 for nonradiative deactivation that occurs through intramolecular excitation of the highest frequency molecular vibrations (νNH in the case of Rh(NH3)63+). The question remained whether this effect on kn would carry over to studies in fluid solutions. Indeed, we were able to show that exchanging the amines with D2O to give Rh(ND3)63+, led to roughly doubled values of ΦA in ambient T solutions, consistent with this premise.19
However, given that eqn (3) interprets the quantum yield as a ratio of rate constants, the increased ΦA is only indirect evidence in support of the conclusion that we were suppressing kn partially by perdeuterating the complex. One could instead draw the seemingly less likely conclusion that the amine perdeuteration leads to an increase in the photoreaction rate constant kA. This conundrum led us to increase efforts to measure excited state lifetimes under conditions directly relevant to the photoreaction pathways. Initially these studies were with Doug Magde at UC San Diego, but this quest was reinforced by access to one of the first ns pulsed Nd/YAG lasers commercially available. We were thus able to measure lifetimes of the faint phosphorescence from the 3MC* states of various perprotio and perdeuterio Rh(III) ammine complexes in fluid aqueous solutions.21,28
By using eqn (4) and the measured ΦA and τ values for Rh(NH3)63+ (0.08 and 21 ns) and Rh(ND3)63+ (0.15 and 40 ns) in 298 K aq. solution, we calculated ES kA values of 3.8 × 106 s−1 and 3.3 × 106 s−1, respectively, for these two ions.28 Obviously, ammine deuteration had only a small effect, and in the wrong direction, on the kA values. Similarly, the respective kn values were calculated as 4.4 × 107 s−1 and 1.9 × 107 s−1. These results clearly confirm that the deuterium effect on ΦA is entirely due to suppression of the weak-coupling component of the nonradiative deactivation (kn), not to the acceleration of the excited state rate of ligand dissociation (kA).
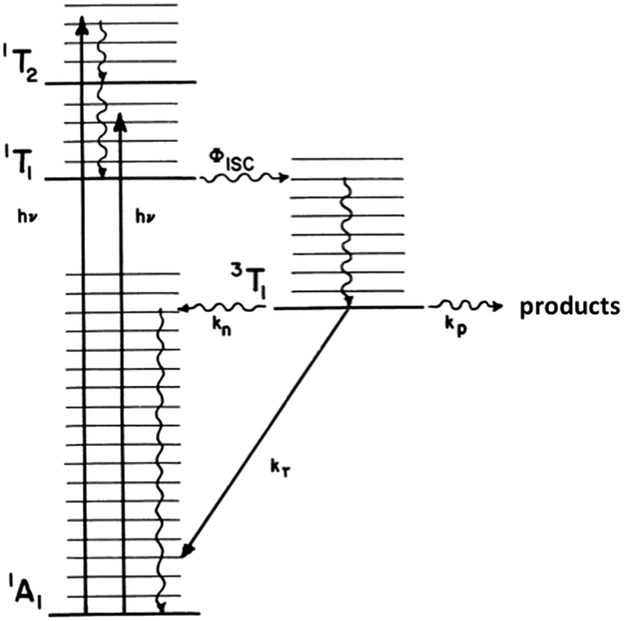
The related chlorido complex Rh(NH3)5Cl2+ provided an opportunity to probe the actual mechanism by which the metal-centered excited state undergoes ligand substitution. Excitation of its ligand field bands leads both to competitive release of an ammine and of a chloride in aq. solution (eqn (6)).21 Of the three likely Rh-containing photoproducts, cis- and trans-Rh(NH3)5(H2O)Cl2+ and Rh(NH3)5H2O3+, only the latter two were observed. However, isotopic labeling studies by Skibsted demonstrated that the labilized NH3 originates roughly equally from axial and equatorial sites, so equatorial NH3 labilization must be accompanied by coordination sphere isomerization.29
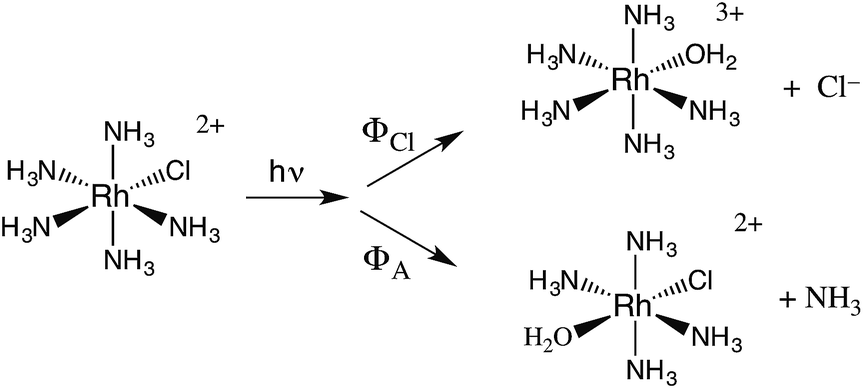 | (6) |
One can rationalize these products from the one-electron excited states predicted by group theory. The lowest energy 3T1 excited state shown for the octahedral complex in Fig. 6 is split in the C4v symmetry of a complex such as Rh(NH3)5X2+ to give 3E and 3A2 states. If X is a weaker σ-donor ligand than NH3, then the 3E is the LEES. The one electron configuration of that ES places the excited electron in the σ-antibonding (dz2) orbital directed along the unique axis, suggesting that trans NH3 or Cl− will be labilized. Consistent with this view, Cl− photoaquation is the principal photoreaction. Furthermore, from a statistical perspective, it appears that the axial ammonia is labilized four times as readily as any single equatorial ammonia. However, the occurrence of significant equatorial labilization suggests that the 3A state may also play a role.
If X− is a stronger σ-donor than is NH3, then the 3A2 state is the LEES, and the excited electron is primarily in the equatorial σ-antibonding (dx2−y2) orbital. Labilization would then be expected along the axes perpendicular to the principal axis. Consistent with this argument, the cyanido complex undergoes NH3 labilization of the equatorial ammine to give cis-Rh(NH3)4(H2O)CN2+ with a ΦNH3 of 0.09 in ambient temperature aqueous solution (eqn (7)).30 Furthermore, selective isotopic labeling demonstrated that the labilized NH3 originates from the equatorial sites as predicted.31
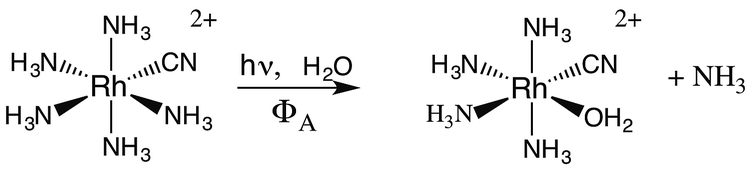 | (7) |
The quantum yields for eqn (6) are ΦA = 0.02 and ΦCl = 0.18 in 298 K aq. solution.21 We measured the emission quantum yield Φr for the 3MC* state of Rh(NH3)5Cl2+ as 3 × 10−5 under these conditions,21 so this compound also represents another case where nonradiative decay is the principal deactivation pathway from the 3MC* state. Accordingly, the emission lifetime (35 ns) of the perdeuterio complex Rh(ND3)5Cl2+ is significantly longer than that (14 ns) of Rh(NH3)5Cl2+. By using eqn (4), the respective excited state rate constants for the latter ion calculated from the quantum yields and lifetimes are kCl = 1.4 × 107 s−1, kA = 0.14 × 107 s−1, kr = 2.3 × 103 s−1 and kn = 5.6 × 107 s−1. By measuring the quantum yields and lifetimes at several temperatures, we calculated an activation energy (Ea) of 25 kJ mol−1 for kCl. This result clearly indicates that the potential well for the reactive 3MC* state of Rh(NH3)5Cl2+ is well defined. In other words, it is a “bound” state as described in Fig. 4.
For comparison, heating an acidic, aqueous solution of Rh(NH3)5Cl2+ leads to Cl− aquation only.32 This displays a first-order rate law, and extrapolation to ambient temperature gives k′Cl = 5.4 × 10−8 s−1 for the ground state reaction. Thus, the 3E triplet ES is >14 orders of magnitude more reactive toward chloride labilization than is the ground state. Furthermore, the Ea (120 kJ mol−1) for k′Cl is much higher than that for the analogous photoreaction.
These comparisons do not differentiate the possible “unimolecular” mechanisms by which the 3MC* state might undergo cleavage of the metal–ligand bonds. For example, is this reaction the result of a dissociative substitution mechanism, where M–L bond breaking is the predominant contribution to the activation energy of that step, as might be implied by the LEES (t2g)5(eg)1 electronic configuration for a hexaammine complex? Alternatively, the t2g vacancy (relative to the (t2g)6 ground state) might enhance a more associative pathway. We explored this by determining the ES reaction rate constants kCl and kA in different solvents.33 The key result was that changing the solvent polarity (and nucleophilic character) had a modest impact on kA but major influence on kCl, reducing the latter dramatically in the less polar solvents. This argues against a major associative component to the exchange of the coordinated NH3 with the solvent molecule. While it could suggest such a component to the Cl− labilization, a more likely explanation is that the barrier to dissociation is strongly affected by the solvation of the developing charge separation as the (NH3)5Rh3+⋯Cl− bond dissociates.
In this context, a collaborative investigation with Rudi van Eldik23 measured the photoreaction quantum yields and lifetimes for aq. Rh(NH3)5Cl2+ as a function of hydrostatic pressure up to 200 MPa, and activation volumes ΔV‡i were determined. These results are self-consistent with the solvent effects on the analogous reactions in the context that the solvation of the developing charge separation as Cl− dissociates from the Rh(III) center has a major impact on the barrier for the kCl pathway. Although no experiment proves a mechanism, these results are clearly consistent dissociative labilization mechanisms from the 3MC* state.
Bimolecular processes
Our discussion has so far focused on unimolecular processes that occur subsequent to excitation. For transition metal complexes these processes will be dominated by the competing nonradiative deactivation, emission and “unimolecular” events such as ligand solvolysis, isomerization or redox reactions like eqn (8) (TPP2− = tetraphenyl-porphyrinato dianion).34 However, excited state complexes may also participate in bimolecular events such as energy or electron transfer to another chromophore. | (8) |
The simplest case is one in which there is a single state or set of states in thermal equilibrium that is largely responsible for the photochemical and photophysical behavior of the metal complex. Such is the case for the extensively investigated Ru(bpy)32+ ion.12 For this species, the situation described in Fig. 3 is reversed, i.e., the lowest energy ES is the 3MLCT ES, which is relatively unreactive toward ligand substitution. As a consequence, Ru(bpy)32+ salts are strongly luminescent even in ambient temperature solutions (Φem = 0.063 in 298 K deaerated H2O, 0.040 in aerated H2O),35 although the major decay pathway remains the non-radiative deactivation which is largely attributed to the back population from 3MLCT state to the lowest energy 3MC state. The roughly microsecond lifetimes and the sensitivity of luminescence detection make such a species ideal for probing bimolecular quenching mechanisms either by energy transfer or by electron transfer. Notably, excited states are both stronger oxidants and stronger reductants than the respective ground state, and this feature has been extensively exploited in applications such as solar energy conversion,36,37 organic synthesis,38 and new photo-initiated cancer drugs.39
From a kinetics perspective, quenching may have a major impact on the luminescent lifetime τ. The quenching rate constants kq in solution can be determined by measuring τ as a function of quencher concentrations [Q] using Stern–Volmer plots of τ0/τ vs. [Q] according to eqn (9) where τ0 is the lifetime in the absence of quencher. The value of kq will depend on the viscosity of the solvent (hence the rate of diffusion) and the operating quenching mechanism(s). If the latter involves energy transfer, the rate will depend on the spectral overlap between the donor and acceptor pair as well as the energies of the relevant excited states. If it involves electron transfer, then the key issue will be the relative redox potentials of the excited state species and of the quencher. We illustrate these points with photo-luminescent Cu(I) complexes.
| τ0/τ = 1 + kqτ0[Q] | (9) |
The Cu(I) cluster Cu4I4py4
Luminescence in first row transition metal complexes is frequently bypassed by the presence of lowest energy MC states that are too short-lived to have measurable emissions. Thus, filled shell d10 metal systems offer an opportunity to observe other excited states.40 Excellent examples are the Cu(I) complexes of the type Cu(DMP)2+ ion (DMP = 2,9-dimethyl-1,10-phenanthroline) described by McMillin et al.40a,b In the absence of MC* states these moieties display MLCT emissions. Here we shall focus on the photophysical/chemical behavior of a different type of Cu(I) complexes, tetranuclear clusters such as Cu4I4py4 that consist of Cu4 tetrahedra with short Cu–Cu distances. Bridging iodides and terminal pyridine ligands complete the unit. Related polynuclear copper(I) clusters have drawn recent interest for potential applications in OLEDs.41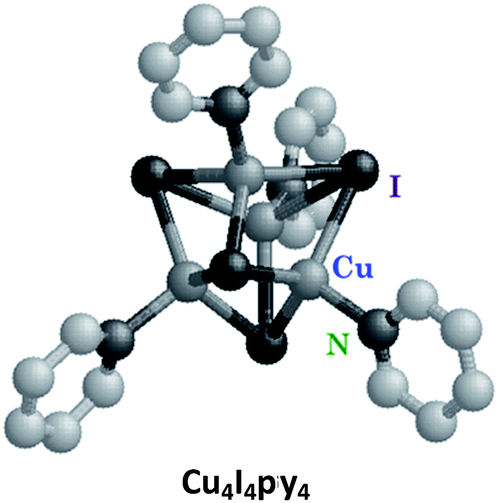
The photophysical properties of Cu4I4py4 demonstrate marked environmental sensitivity, i.e., the luminescence spectra change reversibly by varying the temperature and the rigidity of the medium42 owing to the presence of multiple emitting excited states. In ambient solutions, Cu4I4py4 displays two distinct emission bands that also demonstrate different lifetimes (Fig. 7). This behavior is in obvious contrast to the patterns seen for the Rh(NH3)63+ and Ru(bpy)32+ ions described above, where single emissions from lowest energy MC and MLCT states were respectively recorded.
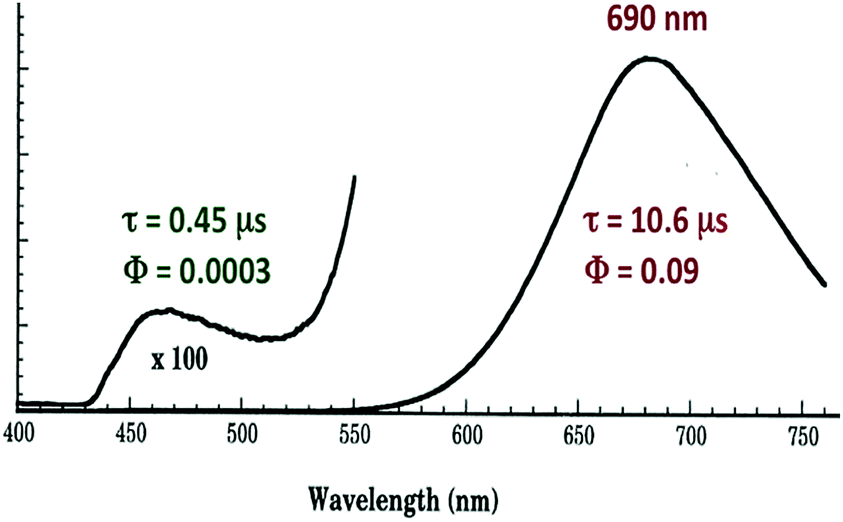 | ||
| Fig. 7 Emission spectrum from Cu4I4py4 in 296 K toluene solution. The excitation wavelength was 350 nm. | ||
The strong, longer-lived (τ = 10.6 μs) lower energy (LE) emission (λmax = 690 nm) from Cu4I4py4 in 296 K toluene solution was assigned to a triplet cluster-centered (3CC) ES, resulting from a combination of delocalized iodide-to-copper charge transfer and d–s transitions.42 The weaker, shorter-lived (τ = 0.45 μs) higher energy (HE) band (λmax = 460 nm) was assigned to a triplet halide-to-ligand (pyridine) charge transfer (3XLCT) ES.
Notably, when the pyridines are replaced by the aliphatic amine piperidine, the LE band is still present, but the HE band is not.43 Relative to the GS, the 3CC and 3XLCT ES of Cu4I4py4 are distorted along different trajectories, since the former ES is formed by promoting an electron from filled orbitals that are non-bonding (or even anti-bonding) to orbitals that are bonding with respect to the Cu4 cluster. As a result the surfaces of the 3CC and 3XLCT states overlap poorly, and there is an energy barrier for internal conversion from the 3XLCT state to the lower energy 3CC excited state (Fig. 8). This is especially apparent at lower temperatures where the 3XLCT emission is much stronger. Computational studies clearly support these assignments.44
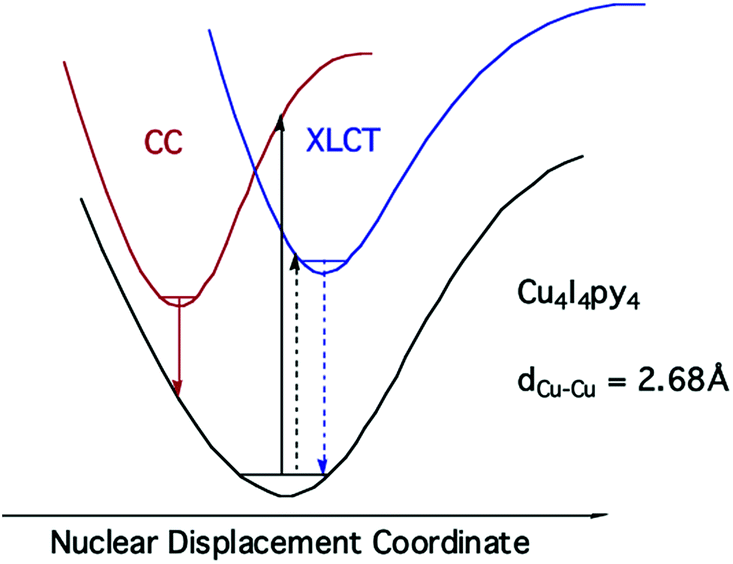 | ||
| Fig. 8 ES diagram to explain multiple emissions from Cu4I4py4. | ||
The kinetics of biomolecular quenching of the Cu4I4py43CC emission were explored in dichloromethane solution using nitrobenzenes and ferricenium cations as well as a series of tris((β-dionato))chromium(III) derivatives CrL3.45 The CrL3 complexes quench by competitive energy transfer and electron transfer mechanisms (Scheme 1). By varying L, one is able to markedly change the reduction potential of the CrL3 species, but the energy (∼1.3 μm−1) of the lowest ES of this species, a Cr(III) centered 2MC state, is essentially unchanged.46
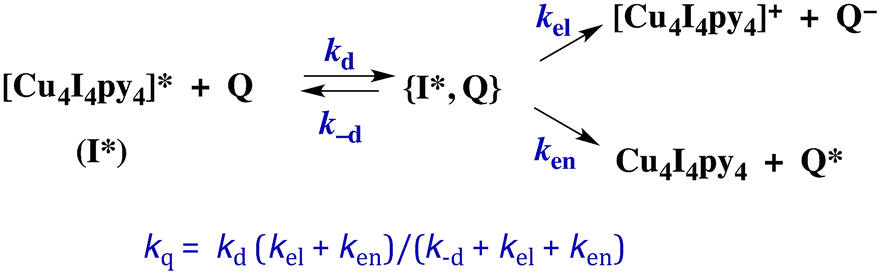 | ||
| Scheme 1 Competitive energy transfer and electron transfer quenching of the 3CC excited state of Cu4I4py4. | ||
The energy of the 3CC state is ∼1.66 μm−1 (2.06 V) and the excited state reduction potential E1/2([Cu4I4py4]+/[Cu4I4py4]*) is ∼−1.78 V (vs. the ferricenium/ferrocene couple). Thus the 3CC state is a very powerful reducing agent, but it also is capable of undergoing energy transfer quenching by the CrL3 centers. The bimolecular rate constant for the latter process was shown to be ∼107.9 M−1 s−1, while contributions to the quenching by an electron-transfer mechanism were evident for those Q with reduction potentials E1/2(Q/Q−) less than 1.4 V. This behavior is demonstrated by the plot of log10(kq) vs. the ΔGo of the electron transfer quenching step calculated according to eqn (10) (Fig. 9). The curve represents the fit to the Marcus model47,48 for the quenching rate constants from Q for which electron transfer processes are expected. The substantial over-potential required before electron transfer quenching becomes dominant can be attributed to a large contribution to the inner-sphere reorganization energy owing to the distortion of the cluster centered excited state.
| ΔGoel = −{E1/2(Q/Q−) − E1/2([Cu4I4py4]+/[Cu4I4py4]*)} | (10) |
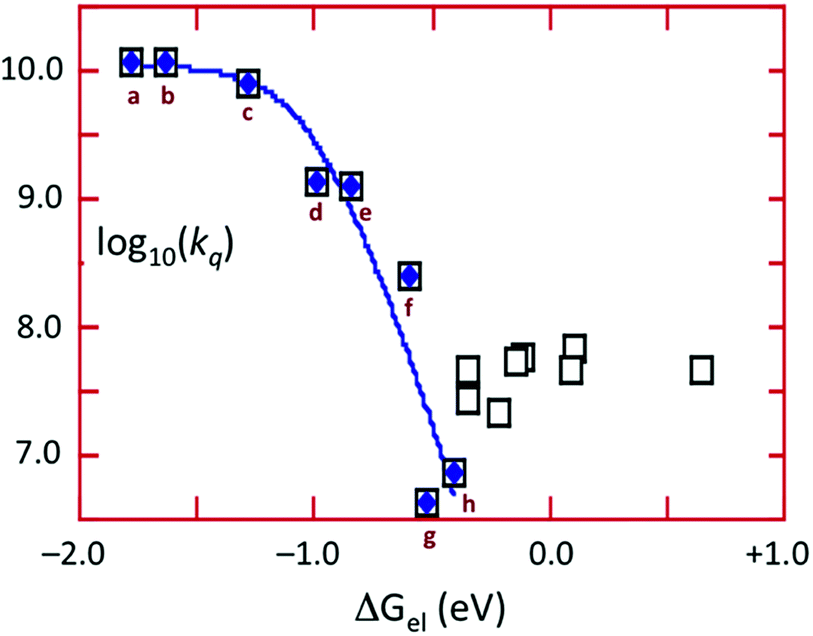 | ||
Fig. 9 Plot of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) kqvs. ΔGoel for the quenching of emission from Cu4I4py4* by oxidants. The curve represents the best fit according to the Marcus model for electron transfer quenching by (a) Fc+, (b) Me2Fc+, (c) Me10Fc+, (d) Cr(hfac)3 (hfac = hexafluoroacetyl-acetonato), (e) 1,4-benzoquinone, (f) p-dinitrobenzene, (g) o-dinitrobenzene, (h) m-dinitrobenzene. Open squares represent CrL3 complexes considered to quench largely by energy transfer. kqvs. ΔGoel for the quenching of emission from Cu4I4py4* by oxidants. The curve represents the best fit according to the Marcus model for electron transfer quenching by (a) Fc+, (b) Me2Fc+, (c) Me10Fc+, (d) Cr(hfac)3 (hfac = hexafluoroacetyl-acetonato), (e) 1,4-benzoquinone, (f) p-dinitrobenzene, (g) o-dinitrobenzene, (h) m-dinitrobenzene. Open squares represent CrL3 complexes considered to quench largely by energy transfer. | ||
Flash photolysis probes of reactive intermediates
Within the anatomy of a photoreaction, there are potentially a number of steps between the initial ground state and the final product(s) as qualitatively illustrated in Fig. 10. The first is the initial absorption of light that prepares a Franck–Condon state (FC*) that is both electronically and vibrationally excited. The FC* may undergo reaction if oriented along a particular trajectory, or it may undergo vibronic and electronic decay via internal conversion to lower energy states of the same spin multiplicity and/or intersystem crossing to lower energy ES of different multiplicity(ies). From either type of ES, luminescence (fluorescence or phosphorescence, respectively), non-radiative deactivation to the GS and/or chemical reaction can occur. The focus here will be on the reactive pathway, since primary photoproduct(s) formed, while no longer electronically excited, may yet be quite reactive under the experimental conditions. Thus, the final products observed may result from secondary pathways of the intermediate(s) I.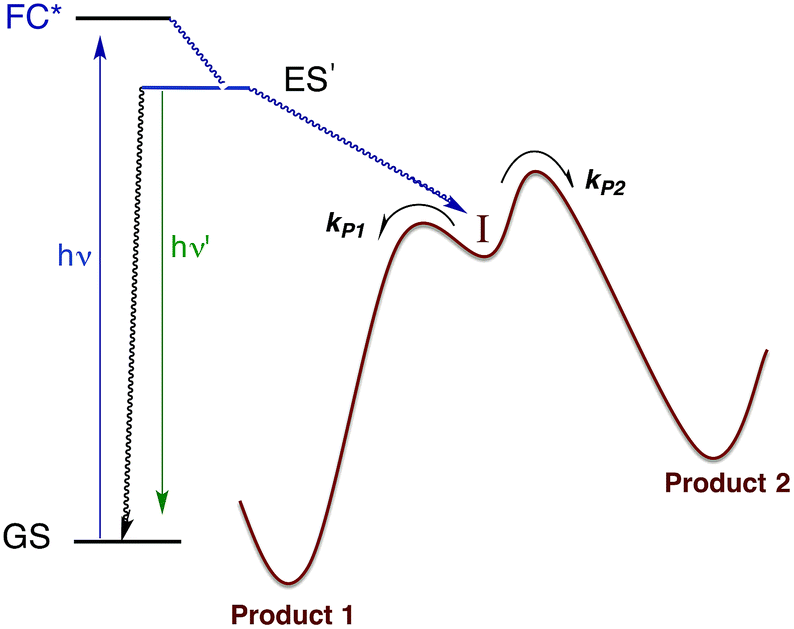 | ||
| Fig. 10 Illustration of reactive intermediate formation as the primary photoproduct from excitation into the Franck–Condon state. I can react by competitive pathways to form more than one product. | ||
There are several ways to probe such intermediates. For example, in low temperature matrices, the activation energies for surmounting the barriers illustrated in Fig. 10 may be sufficient to inhibit further reaction. In that case, photolysis can be used to generate I, which then can be characterized by conventional spectroscopy. An alternative (and complementary) approach is to use flash photolysis to generate such species and fast detection techniques to characterize their spectroscopy and reaction dynamics as they evolve to the final products.
Our first flash photolysis apparatus was a “conventional” flash lamp system with a design not altogether that different from that first built by Norrish and Porter, who were awarded the 1966 Nobel Prize in Chemistry for their contributions to fast reaction kinetics. This system operated by discharging hundreds of Joules of electrical energy from a capacitor bank through 10–20 cm long cylindrical quartz flashlamps filled with xenon gas. The white light flash lasted about 10 μs and irradiated a sample cell with a path of similar length. If one were sufficiently careful to avoid saturating the PMT detector with scattered light, it was possible to record flash-induced temporal changes in the transient spectra.
Flash photolysis of Ru(NH3)5(py)2+
The first system that we used this flash photolysis apparatus to study was the highly colored Ru(NH3)5(py)2+ ion that had first stirred our interest in photochemistry. Our early flash studies indicated the formation of a transient species that partially returned to the starting material on a millisecond time scale.49 In acidic solution, the recovery was slower and less of the starting ion reformed. These observations paralleled our finding that pyridine photoaquation quantum yields are higher at lower pH.8 On these bases, we proposed that pyridine aquation occurs via formation of a transient species by which the Ru–py bonding has isomerized from monodentate coordination to a π-complex such as the η2-py species illustrated in Scheme 2. It is notable that a recent study9b using laser flash photolysis techniques and DFT computations confirmed the formation of such an intermediate, although Ru(II) coordination at a pyridine C–C bond was concluded to be the most stable η2-pyridine complex.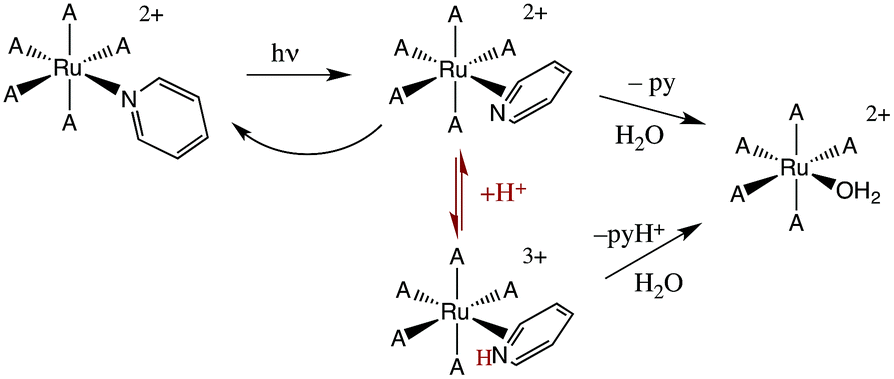 | ||
| Scheme 2 Possible scenario for flash photolysis dynamics of Ru(NH3)5(py)2+ (A = NH3). | ||
Flash photolysis also provides a valuable tool for probing the mechanisms of thermal reactions where the presence of a key reactive intermediate is suspected. We will give several examples to illustrate this approach.
Wilkinson's catalyst
One such study was concerned with the reactivity of intermediates proposed in the homogeneous hydrogenation of olefins by the rhodium(I) species Rh(PPh3)3Cl, first reported by Wilkinson and coworkers.50 Those workers and Halpern51 had suggested that the key intermediate in the catalytic cycle was the “three coordinate” species Rh(PPh3)2Cl formed by spontaneous dissociation of PPh3 (Scheme 3). One way to access this intermediate is by flash photolysis induced CO dissociation from the carbonyl analog trans-Rh(PPh3)2(CO)Cl (Scheme 4). We studied this reaction first with the conventional flash system,52 then later using laser flash photolysis.53 In benzene solution, this substrate undergoes reversible photo-dissociation of CO (Scheme 4, kCO = 7.8 × 108 M−1 s−1). By carrying out the experiment in the presence of reactants such as PPh3, H2 and ethylene, it was possible to determine other second order rate constants for trapping the Rh(PPh3)2Cl intermediate.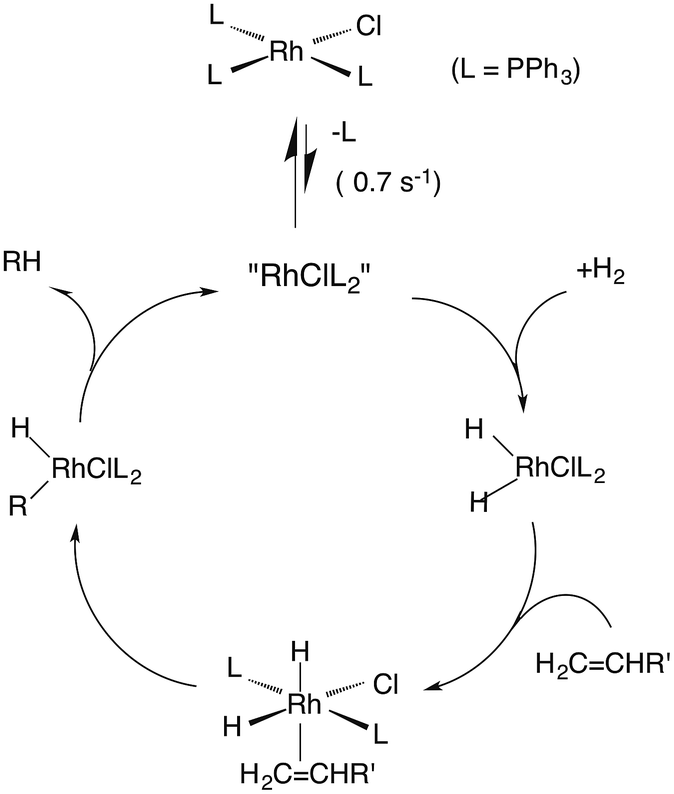 | ||
| Scheme 3 Proposed mechanism for hydrogenation of alkenes by Wilkinson's catalyst (L = PPh3). | ||
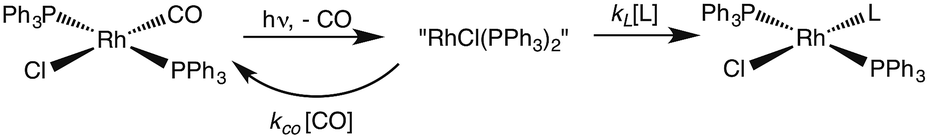 | ||
| Scheme 4 Photodissociation of CO from trans-Rh(PPh3)2(CO)Cl to give the “tricoordinate” intermediate of Wilkinson's catalyst. | ||
For example, the kL for the reaction with PPh3 was determined to be ∼6 × 107 M−1 s−1 in 296 K benzene while that with H2 was nearly a factor of 30 slower. In addition, kCO values in cyclohexane and dichloromethane were ∼2-fold and 5-fold lower, respectively, suggesting that the intermediate is not three-coordinate, but is weakly bound by solvent in the position vacated by CO.53
Metal carbonyls and the migratory insertion reaction
While flash induced changes in the optical spectra are often particularly valuable for following the dynamics of excited states and reactive intermediates, the interpretable structural information from the transient spectra is often limited by the broadening of the absorption and emission bands in solution. In this regard, other time-resolved spectroscopic techniques can provide new dimensions. Time-resolved infrared (TRIR) spectroscopy is especially effective in probing fast reactions of ES and intermediates generated by the photo excitation of metal carbonyls.54–58 These compounds display strong IR absorption bands, the positions of which provide insight into the electronic nature of the metal center in the transient species. In order to access such information we constructed a TRIR apparatus that (initially) used a XeCl Excimer laser as the pulsed excitation source, tunable lead salt IR diodes as the detection source and a Hg/Cd/Te solid state fast response IR detector.59 This allowed pump-probe experiments on the ns to μs timescale with detection in the νCO region of the IR spectrum.An example is shown in Fig. 11, which describes the temporal IR difference spectra in the νCO region upon 308 nm flash photolysis of CH3Mn(CO)5 in cyclohexane solution.60 The IR spectrum can be interpreted in terms of CO photodissociation from this entity to give primarily cis-CH3Mn(CO)4(Sol) (eqn (11), Sol being the solvent). Homolytic fragmentation of the Mn–CH3 bond was a minor (<10%) photochemical pathway as well.
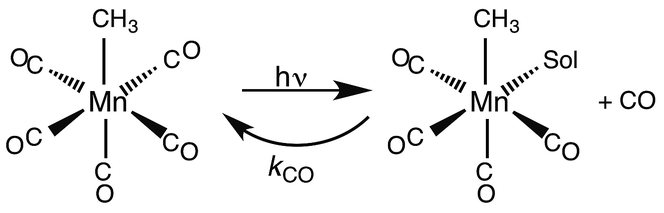 | (11) |
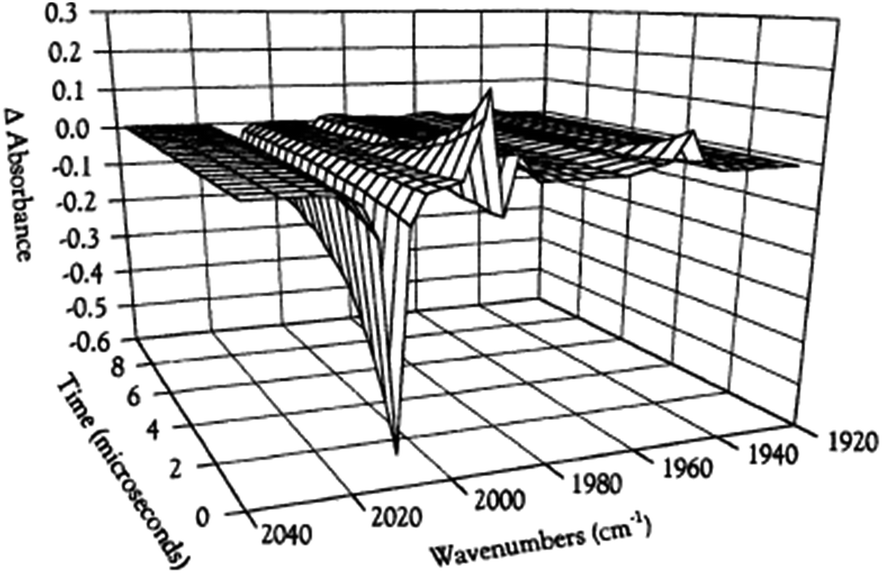 | ||
| Fig. 11 TRIR spectral data resulting form the 308 nm flash photolysis of CH3Mn(CO)5 in 295 K cyclohexane under 0.1 atm CO. | ||
As other studies have shown, the empty site generated by CO photodissociation is often filled by coordination to a solvent molecule, even ones as weakly binding as a saturated hydrocarbon.61,62 In the presence of excess CO this transient undergoes exponential decay back to CH3Mn(CO)5 at rates linearly dependent on [CO] and strongly affected by the nature of the solvent. In cyclohexane, the second order rate constant kCO (295 K) was 4.5 × 108 M−1 s−1, while the value in THF (1.4 × 102 M−1 s−1) was more than 6 orders of magnitude smaller. For reactions in THF, the temperature dependence of kCO was investigated over the range 232–293 K, and an Eyring plot gave ΔH* = 76 kJ mol−l and ΔS* = 59 J mol−l K−l.60 Although these data do not clearly differentiate between associative and dissociative pathways, the positive ΔS* value suggests that considerable Mn–Sol bond breaking is occurring in the rate-limiting step of this substitution pathway.
Another reaction of interest from the catalysis perspective is the “migratory insertion” of carbon monoxide into metal alkyl bonds (eqn (12)). This is the key carbon–carbon bond formation pathway in catalytic carbonylations such as acetic acid synthesis from methanol and alkene hydroformylation.63 Alkyl manganese carbonyl complexes such as CH3Mn(CO)5 were extensively probed as mechanistic models for this important class of organometallic reactions.64 Those studies suggest that alkyl migration to a cis carbonyl leads to a reactive intermediate in a step promoted by more polar solvents.
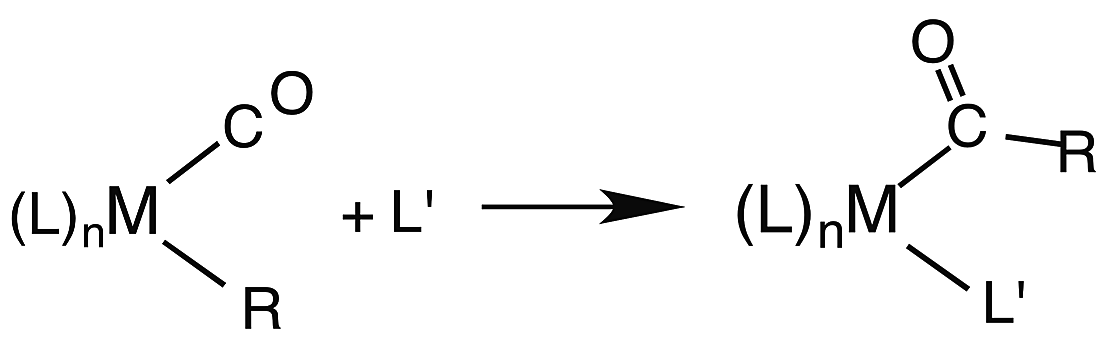 | (12) |
Our strategy for characterizing the structure and reactivity of such intermediates started with the acyl complex. Photodissociation of a ligand L′ from the acyl complex M-C(O)R prepares a reactive species I (eqn (13)) with the same composition as the intermediate proposed for migratory insertion. Time-resolved optical and IR spectral studies were then used to interrogate I and the dynamics of the reactions with various L′ to give the stable acyl products and of reverse alkyl migration to give the metal alkyl complex M–R.
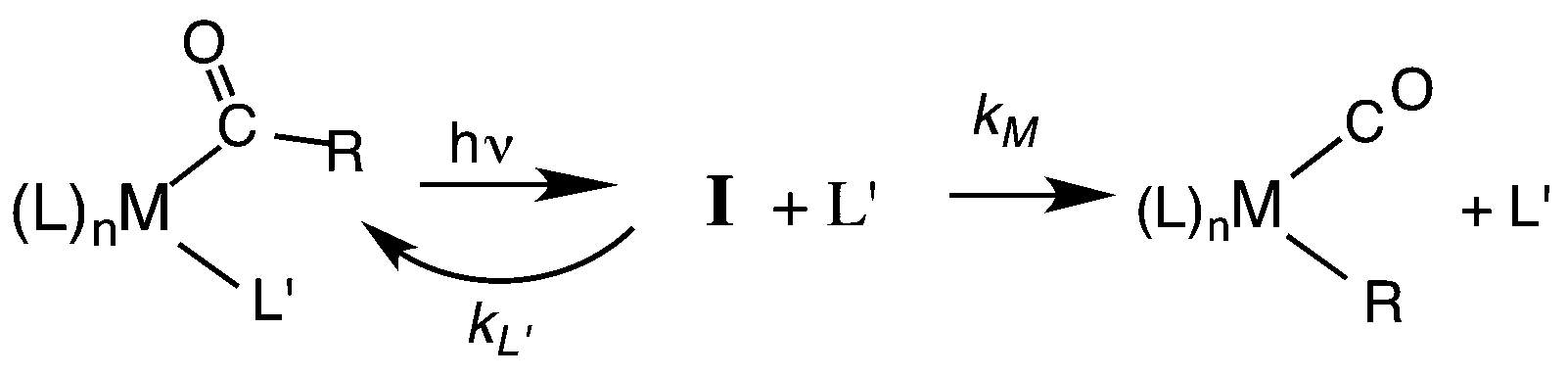 | (13) |
Comparisons of rates measured for I in this manner to the competitive reactivities deduced for intermediates in the thermal migratory insertion kinetics provide guidance relevant to mechanisms of these reactions. We studied such migratory insertion pathways by starting with Mn(CO)5(C(O)R),65,66 (η5-C5H5)Fe(CO)(C(O)R),67 CH3C(O)Co(CO)3PPh3 (ref. 68–70) and [CH3Ir(CO)2I3]−.71 Notably, the latter two systems are relevant to industrial catalysts for the hydroformylation of higher molecular weight alkenes (Shell catalyst)64 and for methanol carbonylation to acetic acid (Cativa catalyst).72 For the latter two studies, it was necessary to build a high pressure/variable temperature (HP/VT) TRIR sample flow cell (Fig. 12)70 so that conditions approaching those of the industrial catalysts could be used. A benefit of this system was precise control of reaction temperatures in order to determine activation parameters.
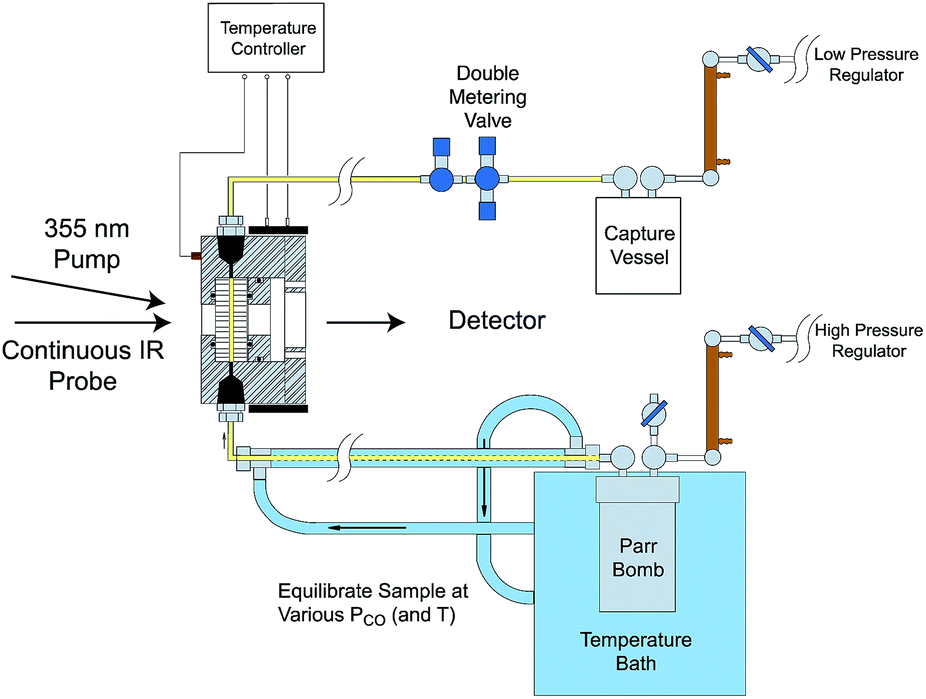 | ||
| Fig. 12 HP/VT flow cell for TRIR studies at higher pressures (up to 100 atm) and temperature (up to 150 °C). Note that the transit of solution through the cell was sufficiently fast that under the repetitive pulsing of the 355 nm pump (3rd harmonic of a Nd:YAG ns laser) solutions are refreshed between pulses (figure adapted from ref. 70). | ||
Homogeneous cobalt carbonyl catalysts for alkene hydroformylation have been in use since their discovery in 1938 by Otto Roelen at Ruhrchemie AG. The original catalyst was based on simple cobalt carbonyl precursors; however, phosphine modified catalysts provide more favorable linear-to-branched selectivity and greater stability at higher temperatures. A key intermediate relevant to such catalysts is the “unsaturated” acyl complex RC(O)Co(CO)2L (ICo).73 This intermediate may be stabilized by as a η2-acyl structure,74 a view that has been supported by DFT calculations.75
We used flash photolysis of the phosphine modified cobalt acyl complex CH3C(O)Co(CO)3PPh3 (ACo) to probe the TRIR spectra and reactivity of the unsaturated intermediate ICo.68–70 The IR spectrum of ACo shows the weak A1 and strong E νCO bands expected for the local C3V symmetry. Events following 355 nm photolysis of ACo are indicated by the temporal TRIR spectra. Readily apparent changes are the prompt bleach of the νCO bands characteristic of ACo, and the prompt formation and decay of a transient species ICo with strong absorbances at 1915 and 1947 cm−1 followed by growth of a photoproduct that was shown to be CH3Co(CO)3PPh3 (MCo).
Regeneration of ACo is linearly dependent on [CO] while formation of MCo was independent of [CO] (kobs = kCO[CO] + kM; kCO = 1.14 × 107 M−1 s−1; kM = 6.2 × 104 s−1 in 298 K benzene-d6) in accord with the scenario illustrated in Scheme 5. Although we will not detail the arguments here, solvent and deuterium isotope effects on kCO and kM indicate that the η2-acyl is indeed the most likely structure for ICo.68Scheme 6 offers proposed mechanisms for the steps depleting ICo. The solvent insensitive kCO has a very small ΔH‡ (6 kJ mol−1) but a sizable, negative ΔS‡ (−92 J mol−1 K−1), and these suggest an associative mechanism to regenerate ACo. The kM rates to give MCo are accelerated by donor solvents and reflect more intimate involvement of solvent in the methyl migration. This may be necessary owing to the orientation of the alkyl group in ICo that is not well suited for migration to the metal center.
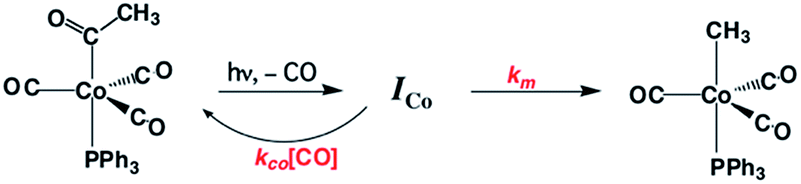 | ||
| Scheme 5 Photolysis of CH3C(O)Co(CO)3(PPh3). | ||
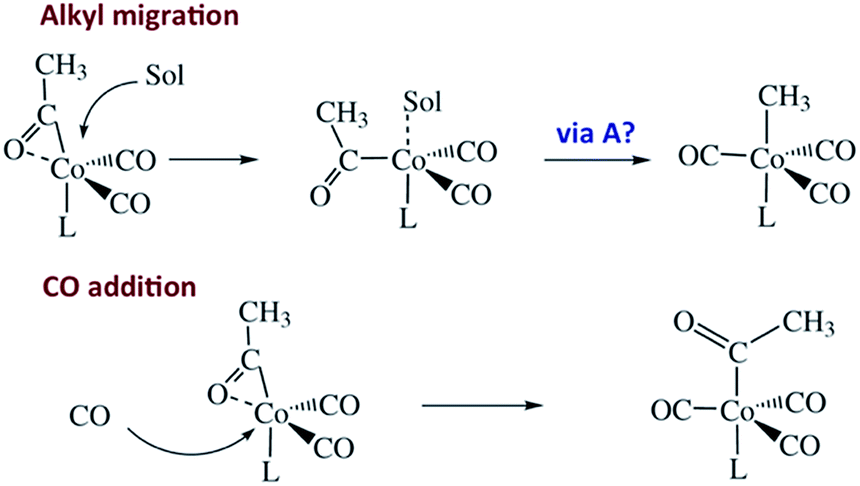 | ||
| Scheme 6 Potential mechanisms for the decay of ICo to MCo and ACo. | ||
Reaction of the bioregulatory molecule NO with heme iron
Nitric oxide plays major roles in cardiovascular regulation, and one key bioregulatory target is the ferroheme protein soluble guanylyl cyclase (sGC), which displays a remarkable specificity for NO.76 In this context we used flash photolysis to study the rates and mechanisms of NO reactions with biologically relevant metal centers.77,78 This approach was not unique to us, but was used by Hoffman and Gibson,79 Magde,80 Olson,81 and others.82 to investigate the kinetics of various heme centers with NO. However, until our studies,83 there had been few systematic mechanistic investigations of metal–NO bond formation.One precedent is the report by Taube and coworkers85 of the nitrosylation kinetics for the Ru(III) complex Ru(NH3)63+ in acidic aqueous solution (eqn (14)). They measured a rate constant (kNO = 0.2 M−1 s−1 at 298 K), which was much larger than that for NH3 substitution by other ligands. They concluded that the reaction with NO must be associative, whereby the low spin d5 Ru(III) center engages the odd electron of NO to give a 7-coordinate Ru(NH3)6(NO)3+ transient. Subsequent activation parameter measurements gave a relatively small ΔH‡ (41 kJ mol−1) and a large and negative ΔS‡ (−114 J K−1 mol−1)86 as well as a negative activation volume ΔV‡ (−14 cm3 mol−1)87 consistent with this view.
| Ru(NH3)63+ + NO + H+ → Ru(NH3)5(NO)3+ + NH4+ | (14) |
Should a similar mechanism be valid for the high spin d5 Fe(III) porphyrinato complexes? To probe this question, we investigated the flash photolysis kinetics of aqueous solutions containing FeIII(TPPS) (TPPS = tetra(4-sulfonato-phenyl)porphyrinato) and NO, which are in labile thermal equilibrium with k11 = 103 M−1 (298 K).77,83 Flash photolysis leads to NO photolabilization from FeIII(TPPS)(H2O)(NO), followed by a relaxation of the system back to equilibrium (eqn (15)). Under excess NO, the latter process is exponential. Linear plots of the resulting kobs values vs. [NO] follow the relationship kobs = koff + kon[NO] from which the values of koff = 0.5 × 103 s−1 and kon = 4.5 × 105 M−1 s−1 (298 K) were determined.
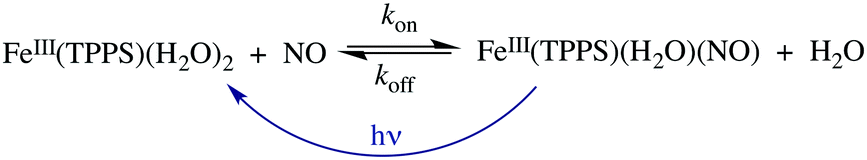 | (15) |
The temperature dependence of these rate constants over the range 298–318 K, gave the ΔH‡ and ΔS‡ values for the NO “on” and “off” pathways of FeIII(TPPS) as 69 kJ mol−1 and +95 J mol−1 K−1 for kon and 76 kJ mol−1 and 60 J mol−1 K−1 for koff, respectively.83
Another parameter is the activation volume ΔV‡, which reflects the sensitivity of the rate constants to pressure (ΔV‡ = −RT(d(lnk))/dPhyd)T, where Phyd is the applied hydrostatic pressure. Pressure effects on laser flash photolysis kinetics were measured in these laboratories using the laser flash photolysis cell illustrated in Fig. 13, which was designed to function from 0.1 to 400 MPa. The experiment involved measuring kobs over a range of [NO] to determine kon and koff for each hydrostatic pressure investigated. Plots of ln(kon) and of ln(koff) vs. P were linear in this pressure region and from these ΔV‡on = +9 cm3 mol−1 and ΔV‡off = +18 cm3 mol−1 were determined.83
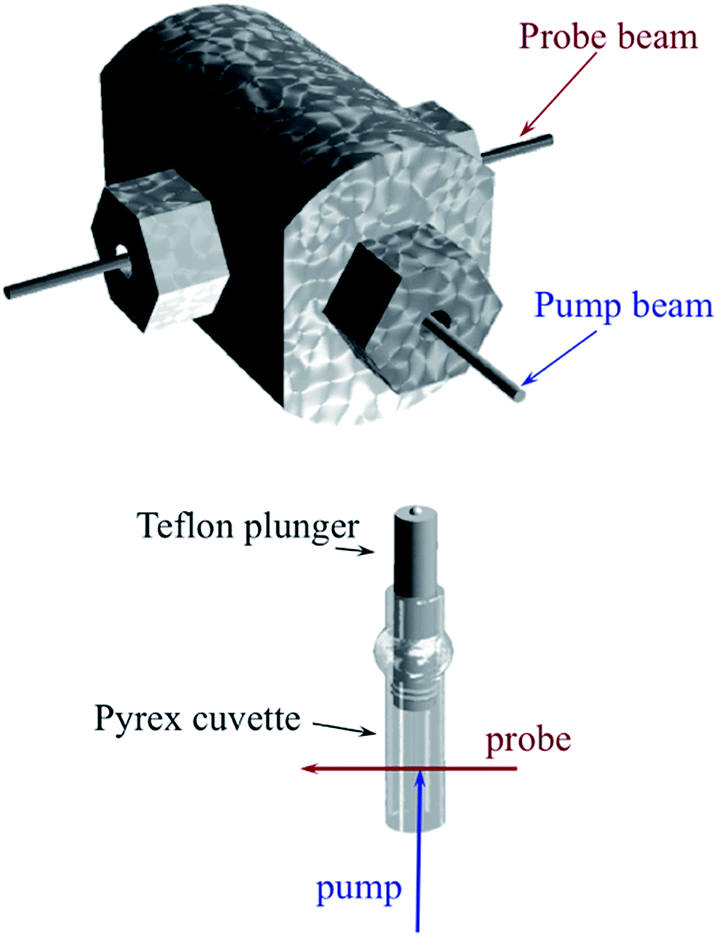 | ||
| Fig. 13 Illustration of cell for measuring the effects of hydrostatic pressures on the dynamics of excited states or intermediates generated by laser flash photolysis (pump) as detected using UV or visible light (probe). The Pyrex or quartz cuvette that is inserted into the HP cell has a sealed Teflon plunger that transmits the applied pressure to the solution. The pump and probe light are transmitted into (and out of) the HP cell through sapphire windows strong enough to withstand the applied pressure. | ||
These activation parameters suggest the reaction pathways envisioned in eqn (16) and (17). This pathway also predicts that FeIII(TPPS)(H2O)2 will exchange its coordinated waters with solvent much faster than the reaction with NO, which was present at mM concentrations. This indeed is the case; H2O exchange with FeIII(TPPS)(H2O)2 occurs with a reported first order rate constant (kex = 1.4 × 107 s−1 in 298 K water)88 far exceeding the kobs values determined at any [NO].
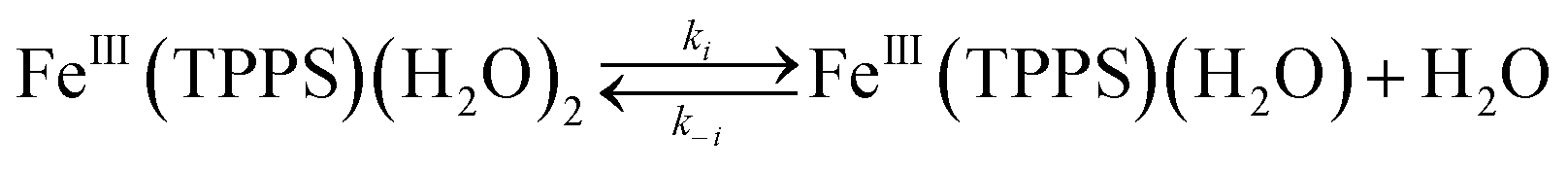 | (16) |
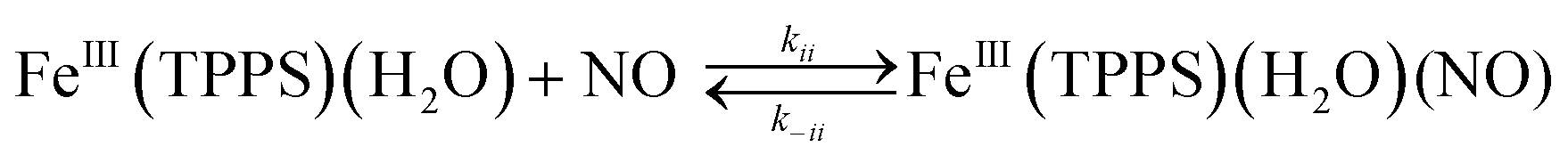 | (17) |
A more recent reexamination of the exchange reaction89 using variable temperature/pressure NMR techniques reported the values ΔH‡ex = 67 kJ mol−1, ΔS‡ex = +99 J mol−1 K−1 and ΔV‡ex = +7.9 cm3 mol−1, which are in remarkable agreement with the activation parameters measured by flash photolysis for the kon pathway with NO.83 The same factors that determine the NO reaction with FeIII(TPPS)(H2O)2 therefore dominate solvent exchange in agreement with a dissociative mechanism, the limiting step being eqn (16). Not surprisingly, the substitution mechanism for the labile, high spin d5 FeIII(TPPS)(H2O)2 complex is markedly different from the associative mechanism described above for the low spin d5 Ru(NH3)63+ ion.
Studies with met-myoglobin came to similar mechanistic conclusions for the reactions of this ferri-heme protein with NO.84 In contrast, a flash photolysis study of the NO reaction with the ferrous analog FeII(TPPS) gave the much larger value of kon = 1.5 × 109 M−1 s−1 (298 K), and a value of koff so small (6.4 × 10−4 s−1) that it had to be measured by a different technique.83 Furthermore, the kon value for the ferrous complex is much less temperature sensitive (ΔH‡on = 24 kJ mol−1) consistent with a reaction pathway having a rate approaching the diffusion limit in aqueous solution. Analogously large kon values have also been reported for the ferrous heme protein sGC.90 Since activating this protein is a key process in the endogenous control of blood pressure by NO, it is obvious that the reaction must be very fast, as endogenous NO concentrations involved are very low.91
Applying what we know and learning new tricks: photochemical uncaging of nitric oxide and other bioactive molecules
In many fundamental investigations, there is often later recognition that the knowledge gained can be utilized for practical applications not perceived at the onset. This is especially true in chemistry, a discipline that lies at the interface between truly fundamental science and the chemical, materials and pharmaceutical industries. Indeed, many practitioners of transition metal photochemistry have for some time been interested in developing methods for converting solar radiation to chemical potential energy, for designing new photo-optical materials and for improving the efficiency of lighting with new types of OLEDs. Biomedical applications include photodynamic therapy, new photoactivated anti-cancer drugs and the photochemical delivery (photo-uncaging) of bioactive substances to physiological targets.Our focus in this regard has been on the photo-uncaging of the small molecule bioregulators (SMBs) nitric oxide and carbon monoxide. This technique has the potential to provide unprecedented control, not only of the location and timing, but also of the dosage for SMB delivery. Thus, photo-uncaging has value both as an investigative tool in physiology and for its ability to affect the progression of specific disease states.
NO, formed endogenously by several isoforms of the enzyme nitric oxide synthase (NOS), has numerous roles in mammalian biology including cardiovascular regulation and immune response. Among potential therapeutic applications of controlled NO delivery are cardiovascular treatment, antibacterial control of biofilms and toxic effects on cancer cells.78,92 With regard to tumors, precise spatiotemporal control is essential, since high levels of NO can kill tissue by inducing apoptosis, but low levels may instead be proliferative.93 Our initial interest in NO delivery derived from its role as a radiation senistizer.94–96 Although gamma-radiation is a common treatment of malignant tumors, the hypoxic regions of tumors are more radiation-resistant than normoxic tissues.95 Therefore, increasing the radiation-sensitivity of a targeted site will reduce collateral damage to healthy tissue. NO is also a potent vasodilator,91 so release at a specific site should enhance oxygenation and, correspondingly, sensitivity of the targeted tissue.
Two themes have guided our photochemical studies of metal nitrosyls. One, described above, used flash photolysis to investigate the mechanisms of ferri- and ferro-heme reactions with NO (e.g., eqn (15)).81 The other was to develop new photochemical precursors for NO delivery to biological targets. Early collaborative experiments with researchers at the Radiation Biology Branch of the US National Cancer Institute (NCI)94 involved probing γ-radiation effects on hypoxic Chinese hamster lung (V79) cells treated with the compound Na2[Fe2S2(NO)4], also know as Roussin's red salt (RRS).97 In the dark, RRS treatment had little effect on radiation survival rates; however, simultaneous white light irradiation led to markedly enhanced cell death attributed to radiation-sensitization by photochemically released NO (eqn (18)). Stimulated by these results, we have since explored NO photo-uncaging from other Fe/S/NO clusters,98 several ruthenium nitrosyls99 and chromium(III) nitrite complexes.100 With the exception of a qualitative application of Roussin's black salt (RBS) as a light-activated NO donor for vascular relaxation,101 these were the first photochemical studies to focus on NO uncaging.
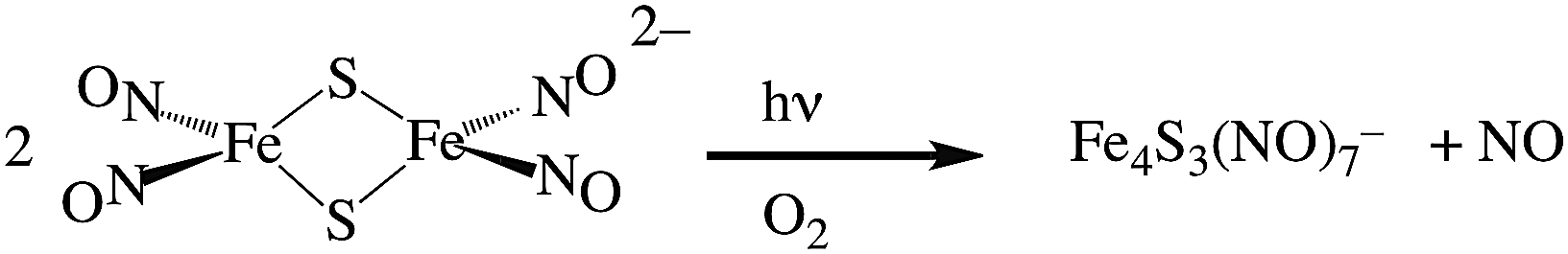 | (18) |
With regard to carbon monoxide, it has long been known that CO is formed endogenously during heme catabolism by the enzyme heme oxygenase.102 More recently, it was shown that exogenously introduced CO is cytoprotective during inflammation and promotes wound healing.103 Moreover, it is anti-bacterial and disrupts biofilms.104 In addition, CO donors called “CORMs” (CO releasing moieties) were shown in animal studies to be effective in alleviating ischemia/reperfusion (I/R) injury with various organs and tissues.105
Thus, for both NO and CO, there is considerable interest in developing methodologies for controlled release at targeted sites. Successful application of photo-uncaging requires elucidating both the fundamental photochemistry and photophysics of the SMB precursors and the mechanisms for transporting these species to the sites of interest. One must also recognize that mammalian tissue is a poor transmitter of shorter visible and UV wavelengths. Tissue penetration improves for longer visible wavelengths and reaches its deepest values in the near infrared (NIR) spectral region (∼700–1100 nm).106
There are several approaches to the problem of limited tissue transmission at certain wavelengths. For example, one might design a small polymeric device attached to an implantable optical fiber.107 Excitation through the fiber could then provide a mechanism for delivering the caged compound upon demand. However, a less invasive approach would be to develop precursors or precursor–antenna conjugates that are responsive to single- or multi-photon excitation by tissue-transmitting light (Scheme 7).
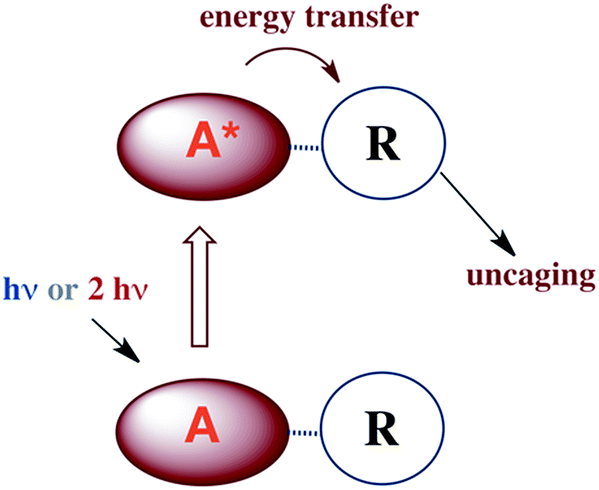 | ||
| Scheme 7 A is the antenna, R is a precursor of a SMB that is uncaged once R is photosensitized. | ||
In order to simplify terminology we will use “photoCORM”, a term we coined several years ago,107 as shorthand for photo-activated CO releasing moiety, and analogously, “photoNORM” for photo-activated NO releasing moiety. In designing new photoCORM and photoNORM systems, certain design guidelines are particularly attractive. Among these are (i) reasonable stability under physiological conditions (e.g. aerated, aqueous media at 37 °C) and (ii) the absence of undesirable toxicity of the photochemical precursor or of the residual photoproduct once the SMB is released. Also desirable are (iii) the ability to activate SMB release using longer wavelength, esp. NIR, excitation, and (iv) to track the location of these species in the organism and to determine whether the system has indeed undergone photo-activated release. In this regard, photoluminescence (PL) is a particularly sensitive imaging method. Lastly, (v) spatio-temporal control of SMB release is essential, and while this is partially achieved by using light as the trigger, developing delivery mechanisms that can target specific sites will enhance efficiency and reduce undesirable side effects.
PhotoNORMs
Following the earlier studies referenced above, we and others developed a number of transition metal containing complexes and materials that release NO when activated by light. These studies have been summarized in various reviews,109 so the discussion here will be confined to systems studied in our laboratory that illustrate key aspects of the design guidelines given.CrONO
The chromium(III) O-nitrito complex trans-Cr(cyclam)(ONO)2+ (“CrONO”, cyclam = 1,4,8,11-tetrazacyclo-tetradecane) meets several of the desired criteria. It is photoactive with quantum yields measured by spectral changes of 0.25–0.30 moles per einstein over the λirr range 365–536 nm and NO is a photoproduct.100,110 Furthermore, CrONO is stable under physiologically relevant conditions (normoxic pH 7.4 aq. buffer at 37 °C), and cell culture studies indicate no acute toxicity. Direct measurement of NO release using a Sievers Nitric Oxide Analyzer (NOA) gave ΦNO = 0.25 when the photoreaction was carried out in the presence of added glutathione (GSH). Photolysis of low concentration CrONO solutions released sufficient NO to activate the key enzyme soluble-guanylate cyclase (s-GC) and, in separate experiments, to contract relaxed endothelium-free porcine arteries with an IC50 concentration ∼320 nM.110a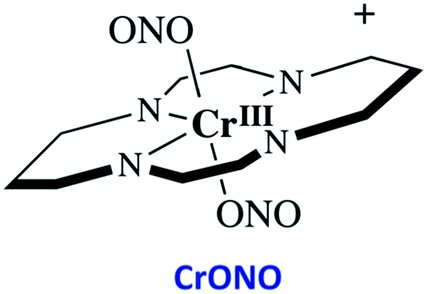
The discovery of CrONO's photochemistry was not accidental. There were previous indications that a Cr(III) nitrito complex might undergo CrO–NO photofragmentation to give NO plus a CrIV(O) species.111 However, photolysis of a simple complex such as Cr(NH3)5(ONO)2+, while showing some NO release, primarily led to ligand photolabilization.112 This is where knowing the photochemical literature proved valuable, since earlier studies by Kutal and Adamson had demonstrated that trans-Cr(cyclam)Cl2+ is relatively inert to photosubstitution, although other chlorido complexes such as Cr(NH3)5Cl2+ are not.113 These differences have been interpreted in terms of the ligand substitution from Cr(III) MC excited states requiring substantial distortion along reaction channels involving the equatorial ligands.6 The relatively rigid coordination of the tetradentate cyclam ligand prevents this distortion, thereby in the case of CrONO, allows the competitive NO fragmentation pathway to dominate. DFT studies suggest that the latter occurs from the lowest energy 2MC state of the Cr(III) center.110b
The CrIV oxo intermediate is also quite reactive. For example, flash photolysis studies showed that it reacts with NO via second order kinetics (kNO = 3.1 × 106 M−1 s−1 in 298 K aq. solution) to regenerate CrONO. Permanent photochemistry is only observed when the NO is removed by entraining the solution with air or helium, or if a trapping agent intercepts the CrIV(O), thereby preventing the back reaction. O2 serves this purpose. Thus photolysis of CrONO in aerated aq. solution leads to permanent photochemistry, presumably via oxidation of Cr(IV) to Cr(V). However, the net NO production is inefficient owing to scavenging by the superoxide presumably also generated in this step. On the other hand, when mM concentrations of the common cellular reductant GSH are present, reduction back to a Cr(III) product dominates (Scheme 8).110
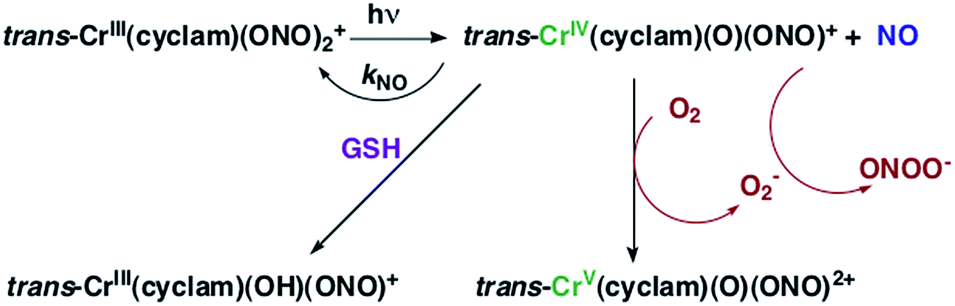 | ||
| Scheme 8 The reversible labilization of NO from CrONO. The net production of NO is effected by the trapping of the Cr(IV) intermediate by oxygen in aerobic media or by GSH in a more reducing environments. | ||
Antennas
A key problem with using CrONO as a photoNORM is that its longer wavelength absorptions are ligand field bands with very low extinction coefficients (ε); they are Laporte-forbidden transitions for this centro-symmetric complex. Thus, the rate of NO generation is relatively slow unless the CrONO concentration and/or excitation light intensity is high. To address this issue, we decided to attach various organic chromophores to the CrONO-type platform to serve as antennas to enhance light absorption and, correspondingly, NO production (Scheme 9).114 | ||
| Scheme 9 Pendant anthracene antenna collects light and photosensitize NO release from a CrONO center. | ||
While these new constructs are proof-of-principle examples of the antenna effect, they do not extend light absorption to the longer visible wavelengths needed for biological systems. For this reason we turned to semiconductor quantum dots (QDs) as photosensitizers, since QD absorption and emission energies can be tuned through the visible spectrum by varying the nanoparticle diameters.115 Accordingly, we prepared water-soluble CdSe:ZnS core:shell QDs by decorating the QD surface with dihydrolipoic acid (Scheme 10). Adding CrONO salts to solutions of such nanoparticles with 3.8 nm core diameters led to progressive quenching of the QD PL centered at 570 nm with increasing CrONO concentrations.116 More importantly, substantially more NO was photogenerated than from solutions containing only CrONO. Subsequent studies confirmed that the PL quenching and the sensitized NO production are due to Förster resonance energy transfer (FRET) from the excited QDs to CrONO.117 However, these electrostatic CrONO:QDs assemblies are unlikely to be stable in physiological fluids, so we are developing protocols to attach CrONO and other photoNORMS to QD surfaces via covalent bonds.
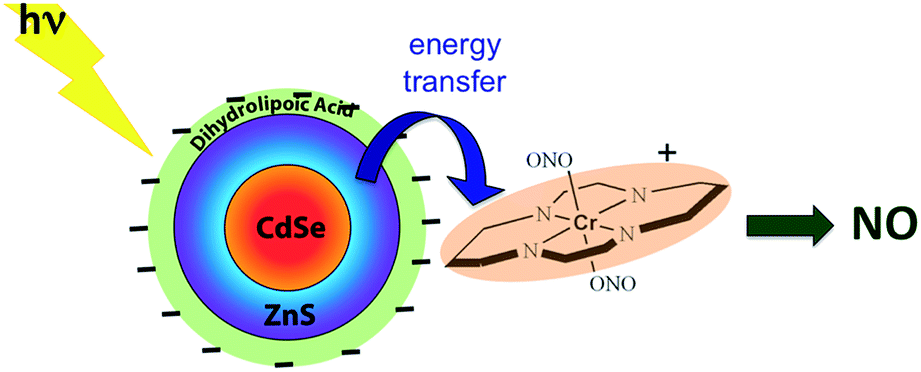 | ||
| Scheme 10 Analogous photosensitization of NO release from CrONO using a water-soluble CdSe:ZnS core:shell semiconductor quantum dot as the antenna. | ||
In parallel studies, we investigated the antenna effect with Roussin's red salt esters (RSE, Fe2(μ-RS)2(NO)4), prepared from the RRS anion by the reaction shown in eqn (19). These RSE are more stable than is RRS and upon photolysis in aerated solution release all 4 NO (eqn (20)). Variations of this synthesis were used to build RSE's with strongly absorbing chromophores, an example being PPIX-RSE (Fig. 14).118 Excitation of the pendant protoporphyrin IX antenna (PPIX) photosensitizes NO release. The enhanced rate of NO release from PPIX-RSE versus that from a simpler RSE such as Fe2(μ-SEt)2(NO)4 (Et-RSE) is largely due to more efficient light absorption (Ia) by the tethered antenna. Energy transfer from PPIX* to the cluster is also evidenced by the quenching of the characteristic PPIX PL (Fig. 14). Similar behavior was seen with Fluor-RSE, where the pendant antennas are fluorescein derivatives.119 Also recent study120 described using carbon quantum dots to photosensitize NO release from the iron sulfur nitrosyl cluster Na[Fe4S3(NO)7] in a nanocarrier made from carboxymethyl chitosan; however, the excitation wavelength was the near UV.
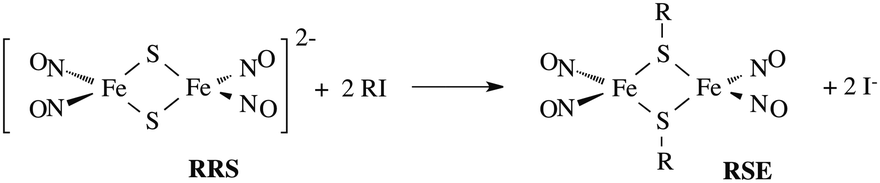 | (19) |
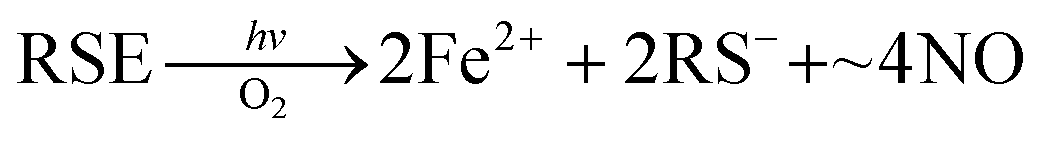 | (20) |
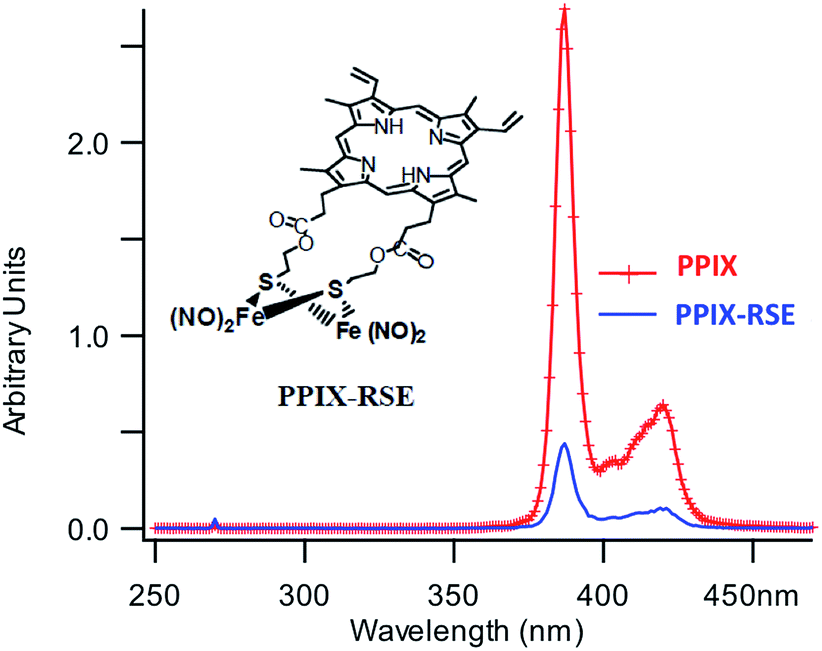 | ||
| Fig. 14 Fluorescence spectra of free PPIX and PPIX-RSE under the same conditions showing that the PPIX fluorescence is about 85% quenched by the Fe/S/NO cluster. PL lifetime measurements confirm that this quenching is due to energy transfer to the cluster (figure adapted from ref. 118). | ||
Multi-photon excitation using NIR light
None of the photoNORMs described above displayed significant absorbance at the NIR wavelengths where tissue transmission is optimized. In principle, this limitation can be addressed by using multi-photon excitation, where the summed energy of several NIR quanta is sufficient to populate the higher energy excited states from which the desired uncaging occurs. See, e.g., the report by Castellano et al.121 that NIR excitation from a fs laser triggers visible emission from Ru(II) bipyridyl complexes. In this laboratory, we are investigating two multi-photon approaches: simultaneous two-photon excitation (TPE) and energy transfer upconversion (ETU), which functions by sequential photon absorptions. Unlike the single photon excitation (SPE) we have been discussing, the rates of TPE and ETU processes have a non-linear dependence on the incident light intensity I0. For example, the probability of TPE is proportional to I02, if a single source is utilized. Thus, TPE induced photoreaction occurs mostly at the focal point of the excitation source, offering three-dimensional spatial resolution, a property widely exploited in imaging. Such resolution would provide unprecedented definition of the uncaging location.122 However, the high intensities needed for TPE require light sources such as pulsed lasers where very high peak powers (photons/unit time) are readily achieved. This may change with the recent description of a lower intensity mechanism for NIR to visible upconversion.123The selection rules for TPE are different from those for SPE. The former is allowed only between two states that have the same parity; the latter requires a parity change.122a Chromophores with high TPE cross sections in the NIR include π-conjugated molecules with electron-donor and -acceptor units arranged symmetrically with respect to the center.124 Semi-conductor QDs are also TPE chromophores with two photon absorption cross-sections (δ) as large as 104 GM (Goeppert-Mayer units, 1 GM = 10−50 cm4 s per photon) seen for CdSe nanoparticles.125
Notably, aqueous solutions of PPIX-RSE that show no absorbance at NIR wavelengths, display a weak PL (λmax 632 nm) when subjected to 810 nm excitation with 100 fs pulses from a Ti/sapphire laser.126 This PL is accompanied by NO generation. These observations thus provide clear evidence that the higher energy states responsible were being accessed by multi-photon excitation, even though the δ value for PPIX is quite low (∼2 GM).
Fluorescein has a higher δ value, which was one rationale for preparing Fluor-RSE. NIR excitation of this species using an ultrafast laser gave the same PL as seen upon SPE at 436 nm, suggesting that analogous emissive excited states are formed by both methods.127 Like PPIX-RSE, the PL from Fluor-RSE was much less than that from the chromophores free of the Fe/S/NO cluster owing to energy transfer to the latter. The log/log plot of the NO generated vs. laser intensity at 800 nm light gave a slope of 1.8 ± 0.2 consistent with the (I0)2 dependence predicted for a photochemical process initiated by TPE.
Subsequent studies have confirmed TPE with pulsed NIR lasers for other RSE derivatives with chromophores having large two photon cross-sections.128 Several other molecular platforms for TPE-induced NO release have also been recently described.129
Multi-photon NIR excitation of caged NO and other bioactive molecules can also be achieved by energy transfer upconversion. The advantage of ETU is that it involves sequential, rather than simultaneous excitation, so lower I0 values are needed. Instead of pulsed lasers, such NIR to visible/UV upconversion can be achieved with relatively inexpensive continuous wave (CW) NIR diode lasers. If the desired upconversion involves the net of two photons, the intensity dependence can vary from second order at low I0 to first order at very high I0 (where the initial excitation step becomes rate limiting). This depends upon the rates of the competing energy transfer and deactivation pathways. The typical situation is between these limits, and as a consequence, the potential for 3-D resolution using light source focusing remains.
In order to exploit ETU, we collaborated with Fan Zhang and Dongyuan Zhao from Fudan U. (China) and UCSB colleague Galen Stucky to test lanthanide ion-doped upconverting nanoparticles (UCNPs) as NIR antennas for NO photo-uncaging. With such UCNPs, a LnIII ion such as Yb3+ is the sensitizer that absorbs the NIR light. This energy is transferred to an activator such as Er3+ as shown in Fig. 15.130,131 Further excitation of the Yb3+ and energy transfer to the long-lived excited state of the activator populates higher energy states from which visible/UV emission occurs. If an appropriate solid matrix is used and the core is coated with a shell of host lattice, silica or polymer, deactivation pathways are suppressed and stronger upconverted emissions result.
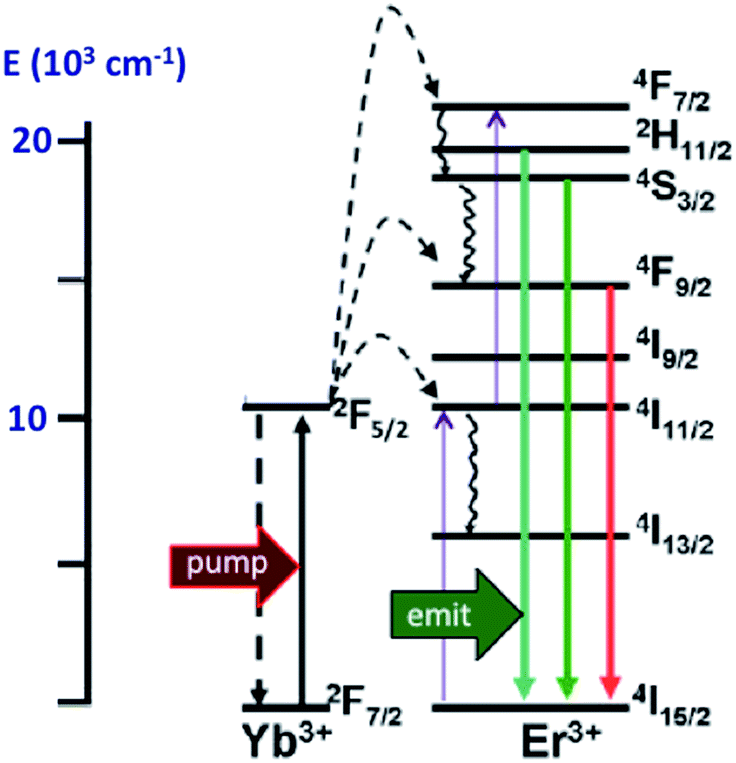 | ||
| Fig. 15 ETU mechanism for NIR of VIS upconversion using a Yb3+/Er3+ doped UCNP with a β-phase NaYF4 host lattice. Pumping the Yb3+ sensitizer with 980 nm light gives an ES that undergoes energy transfers to the Er3+ activator. Continued excitation of Yb3+ and energy transfer to Er3+ gives higher energy states from which several visible range emissions occur. Figure adapted from ref. 131. | ||
There was limited precedent for using such UCNPs for uncaging bioactive substances with NIR excitation. In 2010, Carling et al.132 used 980 nm photolysis of NaYF4:Yb/Tm UCNPs to uncage acetate from 3′,5′-(carboxymethoxy)benzoin acetate loaded on the surface, and in 2012 Y. Yang et al.133 utilized similar UCNPs to uncage D-luciferin in C6 glioma cells and in living mice. Notably, the latter studies showed that NaYF4 (Yb3+/Er3+) UCNPs are non-toxic, their components are eventually excreted, and their location in the organism can be imaged by PL.
Our first UCNP uncaging study, also reported in 2012,134 showed that 980 nm excitation with a CW diode laser of NaYF4:Yb/Er:NaYF4 core:shell UCNPs triggers NO release from the anion of Roussin's black salt Na[Fe4S3(NO)7] (RBS). Solutions of RBS are photoactive under visible excitation (eqn (21))98 but show no such activity when irradiated in the NIR. This first example used UCNPs coated with a silica layer (∼10 nm) and surface modified with cationic –NH3+ groups to render the nanoparticle water-soluble and to facilitate ion pairing with the RBS anions. As shown in Fig. 16, the strong absorptions of RBS overlay the visible emissions from the NaYF4:Yb/Er UCNPs, so the emitted light can be reabsorbed by RBS to facilitate the photoreaction. The NO output as measured with the NOA was linear with the irradiation time at constant power but non-linear in response to systematic increases in excitation intensity (∼I01.5).
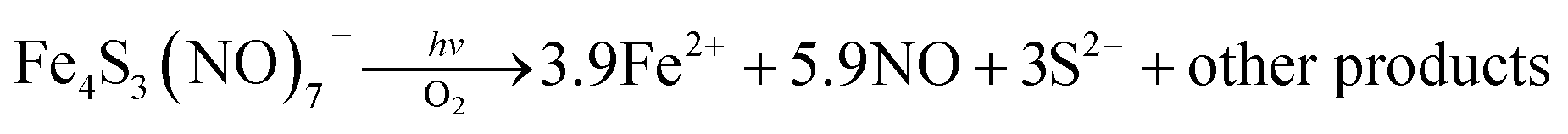 | (21) |
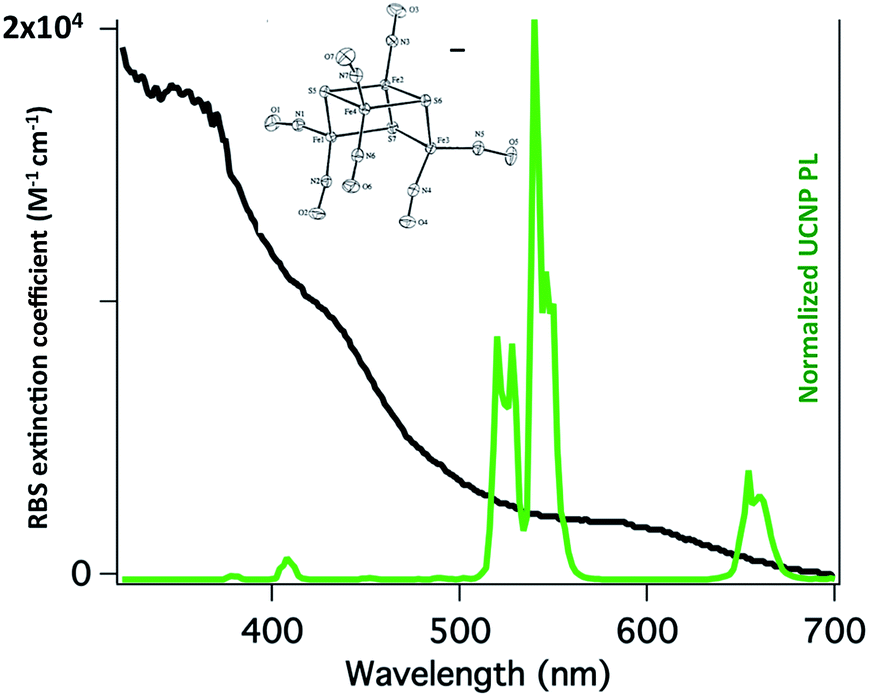 | ||
| Fig. 16 Overlap of RBS absorption spectrum and the upconverted emission spectrum from NaYF4:Yb/Er (20/2%):NaYF4 core:shell UCNPs excited at 980 nm. | ||
A more robust nanocarrier is illustrated in Scheme 11.134 The UCNP core was coated with silica then with a mesoporous silica shell to give a porous layer that was subsequently impregnated with RBS. Coating with poly(allylamine hydrochloride) encapsulated the photoNORM. NIR irradiation released NO, which unlike RBS can diffuse out of the nanocarrier. These proof-of-concept experiments showed that UCNPs offer the opportunity to deliver NO to specific targets using NIR light generated by a relatively inexpensive diode laser.
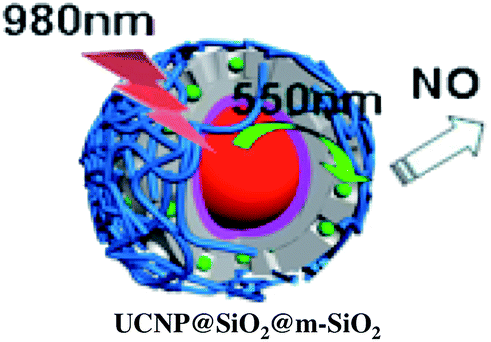 | ||
| Scheme 11 Nano-carrier with UCNP core and mesoporous silica shell impregnated with RBS (green dots) and coated with poly(allylamine). 980 nm irradiation leads to upconversion and NO release. | ||
The mechanism by which UCNPs sensitize NIR mediated NO release from photoNORMs has not yet been well defined. Although there have been reports of Förster resonance energy transfer involving UCNPs,135 the photosensitization described above likely occurs by the so-called “trivial” energy transfer mechanism,136 namely, reabsorption of upconverted emission by the precursor owing to overlapping emission and absorption spectra as in Fig. 16. Dong et al.137 have provided convincing arguments that heating may be responsible for certain reactions sensitized by UCNPs, and this issue needs to be more thoroughly explored using temperature sensors of the type described by those workers. Defining the sensitization mechanisms that successfully utilize NIR to trigger uncaging should provide guidelines for improving the efficiency of such systems.
Although the UCNP emission/photoNORM absorption may not be the most efficient, this pathway simply requires a precursor having strong absorption bands that overlap with the UCNP emissions. This concept led our laboratory to develop biocompatible polymer-based carriers in which the UCNP and the photoNORM are simply co-dissolved. The first examples are mm-sized polymer disks consisting of polydimethylsiloxane (PDMS) containing RBS and NaYF4:Yb/Er UCNPs.138 NO release was effected by 980 nm irradiation with a CW diode laser. Furthermore, analogous NIR excitation triggered photo-uncaging from these devices even after passing through a tissue filter. Subsequent experiments used the hydrophobic forms of Ru(salen-R)(NO)(X) and CrONO derivatives as photoNORMS in disks of various NuSIL silicone formulations.139 One can envision using such materials as drug delivering implants. It is also possible to miniaturize these polymer-based mini-carriers to micron and sub-micron sizes. Our on-going studies are addressing how to deliver the resulting micro- and nano-carriers with analogous formulations to physiological targets.
Another key issue in therapeutic NO delivery is detection and analysis. The Sievers NOA mentioned above measures NO that has been entrained into the gas phase from a solution or an organism. Although very accurate and sensitive, this method is transport-limited and not very useful for real time studies. There are also sensitive NO specific electrodes that are effective in solution media, although in our hands there were issues with reproducibility.98a Some years ago, derivatives of diaminofluorescein (DAF) were developed to provide sensitive detection of NO generation in cellular media,140 but it should be noted that this method actually senses the NO autoxidation product, NO2−. More direct NO detection is now available using copper(II) complexes with chelating ligands that have pendant luminophores.141 The Cu(II) largely quenches the emission from these species owing to its low energy LF transitions, but reaction with NO both modifies the ligand to enhance its PL and reduces the metal center to Cu(I), thereby deleting these low lying transitions.142
PhotoCORMS
Although our investment in developing photoCORMs has been less than with NO releasing analogs, the design principles carry over as do some problems, among them the need for reliable detection methods.103d,e The standard in vitro test for CO is the carboxymyoglobin (Mb-CO) assay, where CO is added to a solution of reduced deoxy-Mb and formation of Mb-CO is followed via changes in the absorption spectrum.143 However, this test is not effective in aerobic media. Several fluorescent sensors have recently been reported, and this is an area of growing development.144,145 In our laboratory, we have determined photoCORM CO release by headspace analysis using both gas-phase infra-red spectroscopy and gas chromatography techniques, but found the latter to be the more reliable quantitatively.108 Schiller and coworkers146 have described headspace analysis using a standard CO gas detector. These methods were reviewed recently103d,e,147 and will not be discussed further here.Our goals with photoCORMs are similar to those with the photoNORMs, namely to develop strategies to effect CO delivery to specific physiological targets. Initial studies at UCSB focused on water-soluble metal carbonyls and on improving methods for quantifying CO release.108,148 For selected group 6 carbonyls in aerated media, multiple CO's were released per complex, and the quantum yield for overall CO release (ΦCO) in some cases exceeded 1.0. The high yields of CO release can be attributed to subsequent reactions of initial photoproducts with O2 and/or the aqueous solvent (Scheme 12). In such instance, the primary photoproduct could be considered a “proCORM”, a molecular species that leads to slow CO release after the initial photoreaction.
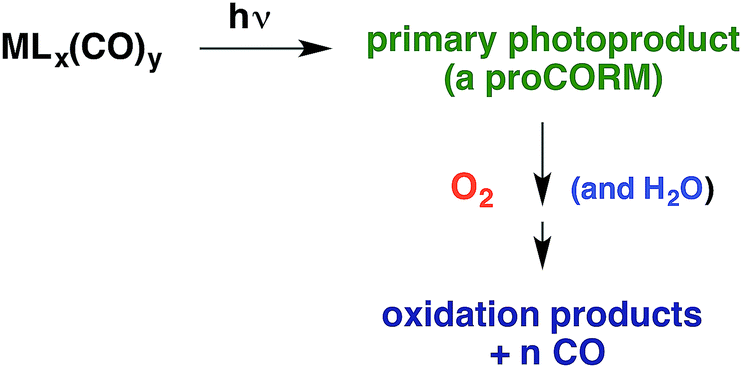 | ||
| Scheme 12 Generating a proCORM intermediate. | ||
A very interesting photoCORM developed at UCSB is the rhenium(I) complex Re(L)(CO)3(bpy)+ (L = P(CH2OH)3).149 This complex ion is strongly luminescent (τ = 400 ns) in solution and releases one CO upon photolysis (eqn (22), ΦCO = 0.11). The photoproduct Re(L)(CO)2(H2O)(bpy)+ is also luminescent. Neither Re(L)(CO)3(bpy)+ nor its photoproduct displayed any acute cellular toxicity up to 100 μM. Thus, we were able to use confocal microscopy to observe the PL of the rhenium species incorporated into cells (human prostatic carcinoma cell line PPC-1) before and after photolysis (Fig. 17). The ability to image the photoCORM in the cells and to determine whether it has undergone photoreaction is a highly desirable trait for uncaging.150 A major disadvantage, however, is the relatively short visible wavelengths needed to photolabilize the CO.
 | (22) |
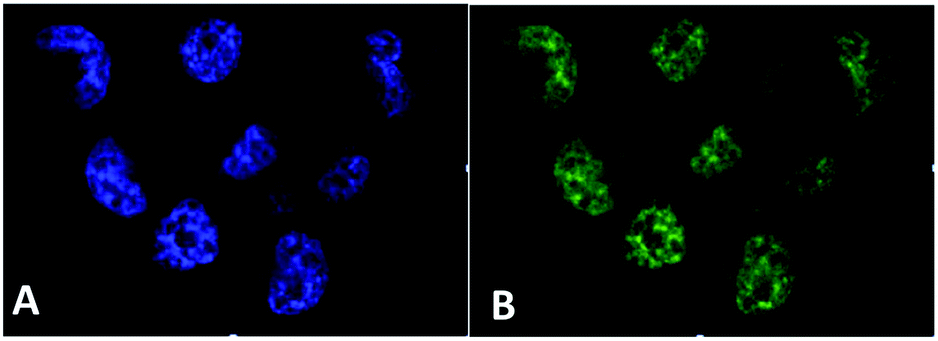 | ||
| Fig. 17 Confocal fluorescence microscopy images of PPC-1 cells incubated with Re(P(CH2OH)3)(CO)3(bpy)+ (50 μM) (A) and after photolysis at 405 nm (B) (figure adapted from ref. 149). | ||
The task remains therefore to develop a mechanism to release CO with NIR light. In this context, the photophysics of metal carbonyls offers a challenge. The stronger lower energy absorptions and the PL of diimine complexes such as Re(L)(CO)3(bpy)+ can be attributed to metal-to-bipyridine charge transfer states. However, CO photolability occurs primarily from metal-centered ligand field states.151 Substituents can be used to tune the MLCT absorptions of the Re(I) complexes to the red, but excitation at those longer wavelength bands does not lead to significant CO release, since this does not populate the higher-energy MC states necessary for CO labilization. In fact, the story is quite analogous to the photoreactivity of the Ru(NH3)5(L)2+ complexes discussed above.10
We have addressed these issues by designing a nanocarrier that incorporates both a photoCORM and a UCNP antenna to mediate multi-photon NIR labilization of CO. The photoCORM is a Mn(I) complex similar to the Re(I) complex just described. Manganese, however, has smaller d-orbital splitting than the heavier transition element analogs. Thus, it has inherently lower energy LF states, thereby making CO photodissociation energetically more accessible.
For example, the Mn(I) photoCORM trans-Mn(CO)2(PPh3)2(bpy)+ displays strong visible range absorptions, photolysis of which leads to facile CO release. This spectrum is well suited for matching the upconverted emissions from a NaYF4(Yb20Tm0.2) UCNP. In collaboration with Nanfeng Zheng of Xiamen U (China), we assembled nano-carriers consisting of a UCNP core coated with a phospholipid-functionalized with poly-(ethylene glycol) (DSPE-PEG 2000) (Scheme 13).152 The amphiphilic polymer confers water solubility to the nano-carrier while providing a lipid-like interior into which hydrophobic compound trans-[Mn(CO)2(PPh3)2(bpy)](CF3SO3) is readily infused. NIR excitation (980 nm) of an aq. solution containing the loaded nano-carrier leads to CO release. With these hydrophobic components, leaching is minimized, while encapsulation isolates them from the medium, thereby reducing potential toxicity and promoting higher loading. This remarkable nano-carrier brings together the UCNP NIR antenna and a hydrophobic photoCORM in a water-soluble ensemble.
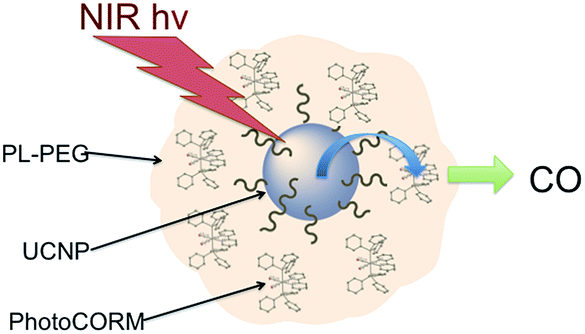 | ||
| Scheme 13 Water-soluble photoCORM nano-carriers with diameters 16–30 nm according to dynamic light scattering. | ||
We are extending these studies to examine the efficacies of such nano-carriers with other photoCORMS and photoNORMs and with UCNPs responsive to 800 nm excitation. In addition, surface modifications should be well suited for the control of targeting and other properties of these units.
Better targeting
While photo-excitation allows one to define location and timing of SMB uncaging in tissue, a key remaining task is to improve transport of the precursor–antenna conjugates to the desired targets. Although one can envision implanting a device such as the mm-sized PDMS mini-disks described above, a more elegant strategy would be to employ biological mechanisms for this purpose. We are embarking on this quest. One approach is to use targeting peptides that, depending on the amino acid sequence, can direct water-soluble nano-carriers to specific cell types both in vitro and in vivo.153 For example, Tirrell et al., used micelles constructed of peptide terminated polymers for the targeted delivery of chemotherapeutic agents to malignant gliomas, thereby minimizing other side effects.154In collaboration with UCSB colleague Norbert Reich, we have constructed nano-carriers with hollow gold nanoparticle (HGN) cores (∼40 nm diameters).155 The surfaces of these HGN cores were decorated with thiocupferron (TCF), a molecule that when heated releases one equivalent of NO. Also decorating the surface was a 5 kDa thiolated polyethylene glycol (TPEGRP), which was terminated by a C-end Rule peptide, RPARPAR that targets cells with the neuropilin-1 receptor common to certain cancers. The TCF-HGN-TPEGRP conjugates prepared in this manner undergo neuropilin-1 receptor mediated endocytosis156 by PPC-1 and 22RV1 prostate cancer cells but not with HeLa cells, which lack that receptor. NIR irradiation with a pulsed laser at a wavelength (800 nm) resonant with the surface plasmon of the gold nanoparticles rapidly heats the nanoparticle.157 In solution the resulting NO release (Scheme 14) was quantified using the NOA, while intracellular NO release in 22RV1 cells was demonstrated by using the DAF-2 assay.140
 | ||
| Scheme 14 HGN surfaces are coated by co-absorption of TCF and a thiolated PEG, which enhances aqueous solubility and in the case of TPEGRP provides the cell-targeting peptide. 800 nm laser excitation of the conjugates heat the HGN surfaces and NO is released. | ||
Based on this precedent, our premise is that decorating photoCORM and photoNORM nano-carriers with a targeting polypeptide will greatly enhance delivery specificity. The novelty here lies not in using these targeting polypeptides, but in combining this methodology with the uncaging of SMBs from NIR responsive conjugates with UCNP cores. The residual PL from UCNPs offers imaging opportunities as well. Such devices will provide imageable and unprecedented spatio-temporal control of SMB delivery to specific cell types.
Concluding remarks
This article illustrates the evolution of research into the photochemistry of transition metal chemistry by tracing the author's interest and involvement in this area over four decades. Initial studies focused on correlating reaction product characterizations and quantum yields with spectroscopic properties to identify the excited states responsible for specific photochemical processes. This information was then used to tune excited state energies in order to correspondingly direct the resulting photoreactivities. Within the same time frame, related studies were directed toward elucidating specific mechanisms for excited state reactions and deactivation pathways. The availability of flash photolysis methods provided access to the dynamics and trajectories of excited state pathways as well as of subsequent reactions of transient intermediates formed. Notably, such species are often analogous to intermediates proposed for certain thermo-chemical processes, so pulsed photolysis offers an important mechanistic tool in catalysis and bioinorganic chemistry. Our personal journey in this area has turned to using photochemical methodologies for the uncaging of bioactive small molecules, with the eventual goal of establishing guidelines for therapeutic application.Another researcher would undoubtedly provide a different perspective, given that individual's choice of other research problems and tools employed. Regardless, it is clear that the successful use of inorganic photochemistry, whether in energy science, biomedicine, or other foreseen or unforeseen application, needs to be based on a sound understanding of the fundamental principles that define the mechanisms of these systems. I am convinced that, as new tools and ideas are introduced, fascinating and unexpected developments in the photochemistry and photophysics of transition metal compounds will continue to be forthcoming.
Acknowledgements
The research described in this manuscript was largely funded by a series of grants from the Chemistry Division of the US National Science Foundation. The students and postdoctoral fellows involved in this research over the lengthy period surveyed deserve the principal credit for seeing it come to fruition. The cage image in the table of contents illustration is by Luis Martin (123RF Stock Photo).Notes and references
- P. C. Ford, D. F. P. Rudd, R. Gaunder and H. Taube, J. Am. Chem. Soc., 1968, 90, 1187–1194 CrossRef CAS.
- C. K. Jørgensen, Absorption Spectra and Chemical Bonding in Complexes, Pergamon Press, London, 1962 Search PubMed.
- V. Balzani and V. Carassiti, Photochemistry of Coordination Compounds, Academic Press, New York, 1970 Search PubMed.
- A. W. Adamson and P. D. Fleischauer, Concepts of Inorganic Photochemistry, Wiley-Interscience, New York, 1975 Search PubMed.
- C. G. Hatchard and C. A. Parker, Proc. R. Soc. London, Ser. A, 1956, 235, 518–536 CrossRef CAS.
- P. S. Wagenknecht and P. C. Ford, Coord. Chem. Rev., 2011, 255, 591–616 CrossRef CAS.
- T. Matsubara and P. C. Ford, Inorg. Chem., 1978, 17, 1747 CrossRef CAS.
- D. A. Chaisson, R. E. Hintze, D. H. Stuermer, J. D. Petersen, D. P. McDonald and P. C. Ford, J. Am. Chem. Soc., 1972, 94, 6665 CrossRef CAS.
- (a) C. N. Tsai, Y.-H. Tian, X. Shi, R. L. Lord, H. B. Schlegel, Y. J. Chen and J. F. Endicott, Inorg. Chem., 2013, 52, 9774–9790 CrossRef CAS PubMed; (b) C. N. Tsai, S. Mazumder, X. Z. Zhang, H. B. Schlegel, Y. J. Chen and J. F. Endicott, Inorg. Chem., 2015, 54, 8495–8508 CrossRef CAS PubMed.
- G. Malouf and P. C. Ford, J. Am. Chem. Soc., 1974, 96, 601–604 CrossRef CAS.
- G. Malouf and P. C. Ford, J. Am. Chem. Soc., 1977, 99(22), 7213 CrossRef CAS.
- (a) S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, Phototchemistry and Photophysics of Coordination Compounds: Ruthenium, Top. Curr. Chem., 2007, 280, 117–214 CrossRef CAS; (b) D. L. Ashford, C. R. K. Glasson, M. R. Norris, J. J. Concepcion, S. Keinan, M. K. Brennaman, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2014, 53, 5637–5646 CrossRef CAS PubMed.
- (a) T. Sajoto, P. I. Djurovich, A. B. Tamayo, J. Oxgaard, W. A. Goddard and M. E. Thompson, J. Am. Chem. Soc., 2009, 131, 9813–9822 CrossRef CAS PubMed; (b) A. F. Rausch, M. E. Thompson and H. Yersin, J. Phys. Chem. A, 2009, 113, 5927–5932 CrossRef CAS PubMed.
- (a) J. N. Schrauben, K. L. Dillman, W. F. Beck and J. K. McCusker, Chem. Sci., 2010, 1, 405–410 RSC; (b) E. A. Juban, A. L. Smeigh, J. E. Monat and J. K. McCusker, Coord. Chem. Rev., 2006, 250, 1783–1791 CrossRef CAS.
- G. B. Porter , “Kinetics of Photophysical Processes” cpt 2 in ref. 4.
- J. R. Winkler, T. L. Netzel, C. Creutz and N. Sutin, J. Am. Chem. Soc., 1987, 109, 2381–2392 CrossRef CAS.
- R. Santana da Silva, M. S. P. Marchesi, A. C. Tedesco, A. Mikhailovsky and P. C. Ford, Photochem. Photobiol. Sci., 2007, 6, 515–518 CAS.
- J. D. Petersen and P. C. Ford, J. Phys. Chem., 1974, 78, 1144 CrossRef CAS.
- J. D. Petersen, R. J. Watts and P. C. Ford, J. Am. Chem. Soc., 1976, 98, 3188 CrossRef CAS.
- M. A. Bergkamp, R. J. Watts, P. C. Ford, J. Brannon and D. Magde, Chem. Phys. Lett., 1978, 59, 125 CrossRef CAS.
- M. A. Bergkamp, J. Brannon, D. Magde, R. J. Watts and P. C. Ford, J. Am. Chem. Soc., 1979, 101, 4549 CrossRef CAS.
- M. E. Frink, D. Magde, D. Sexton and P. C. Ford, Inorg. Chem., 1984, 23, 1238 CrossRef CAS.
- W. Weber, R. van Eldik, H. Kelm, J. DiBenedetto, Y. Ducommun, H. Offen and P. C. Ford, Inorg. Chem., 1983, 22, 623 CrossRef CAS.
- T. L. Kelly and J. F. Endicott, J. Phys. Chem., 1974, 78, 1144 CrossRef.
- D. A. Sexton, P. C. Ford and D. Magde, J. Phys. Chem., 1983, 87, 197 CrossRef CAS.
- T. R. Thomas, R. J. Watts and G. A. Crosby, J. Chem. Phys., 1973, 59, 2123 CrossRef CAS.
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145 CrossRef CAS.
- M. E. Frink and P. C. Ford, Inorg. Chem., 1985, 24, 1033–1035 CrossRef CAS.
- L. H. Skibsted, Inorg. Chem., 1985, 24, 3791 CrossRef CAS.
- L. H. Skibsted and P. C. Ford, Inorg. Chem., 1983, 22, 2749 CrossRef CAS.
- J. S. Svendsen and L. H. Skibsted, Acta Chem. Scand., 1983, 37a, 443–445 CrossRef.
- F. Basolo and R. G. Pearson, Mechanisms of Inorganic Reactions, A study of metal complexes in solution, John Wiley and Sons, Inc., New York, 2nd edn, 1967 Search PubMed.
- M. A. Bergkamp, R. J. Watts and P. C. Ford, J. Am. Chem. Soc., 1980, 102, 2627 CrossRef CAS.
- M. Hoshino, Y. Nagashima, H. Seki, M. DeLeo and P. C. Ford, Inorg. Chem., 1998, 37, 2464–2469 CrossRef CAS.
- H. Ishida, S. Tobita, Y. Hasegawa, R. Katoh and K. Nozaki, Coord. Chem. Rev., 2010, 254, 2449–2458 CrossRef CAS.
- (a) N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–1573536 CrossRef CAS PubMed; (b) M. Graetzel, Acc. Chem. Res., 2009, 42, 1788–1798 CrossRef PubMed; (c) J. J. Concepcion, J. W. Jurss, M. K. Brennaman, P. G. Hoertz, A. O. T. Patrocinio, N. Y. M. Iha, J. L. Templeton and T. J. Meyer, Acc. Chem. Res., 2009, 42, 1954–1965 CrossRef CAS PubMed.
- (a) Y. Yamazaki, H. Takeda and O. Ishitani, J. Photochem. Photobiol., C, 2015, 25, 106–137 CrossRef CAS; (b) K. Sakai and H. Ozawa, Coord. Chem. Rev., 2007, 251, 2753–2766 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- (a) J. D. Knoll and C. Turro, Coord. Chem. Rev., 2015, 282, 110–126 CrossRef PubMed; (b) C. Mari, V. Pierroz, S. Ferrari and G. Gasser, Chem. Sci., 2015, 6, 2660–2686 RSC; (c) J. S. Butler and P. J. Sadler, Curr. Opin. Chem. Biol., 2013, 17, 175–188 CrossRef CAS PubMed; (d) K. K.-W. Lo, Acc. Chem. Res., 2015, 48, 2985–2995 CrossRef CAS PubMed.
- (a) M. W. Blaskie and D. R. McMillin, Inorg. Chem., 1980, 19, 3519–3522 CrossRef CAS; (b) D. G. Cuttell, S. M. Kuang, P. E. Fanwick, D. R. McMillin and R. A. Walton, J. Am. Chem. Soc., 2002, 124, 6–7 CrossRef CAS PubMed; (c) W. H. Lam, E. C. C. Cheng and V. W.-W. Yam, Inorg. Chem., 2006, 45, 9434–9441 CrossRef CAS PubMed; (d) K. Tsuge, Y. Chishina, H. Hashiguchi, Y. Sasaki, M. Kato, S. Ishizaka and N. Kitamura, Coord. Chem. Rev., 2016, 206, 636–651 CrossRef.
- (a) Z. Liu, M. F. Qayyum, C. Wu, M. T. Whited, P. I. Djurovich, K. O. Hodgson, B. Hedman, E. I. Solomon and M. E. Thompson, J. Am. Chem. Soc., 2011, 133, 3700–3703 CrossRef CAS PubMed; (b) D. Volz, M. Wallesch, C. Flechon, M. Danz, A. Verma, J. M. Navarro, D. M. Zink, S. Braese and T. Baumann, Green Chem., 2015, 17, 1988–2011 RSC; (c) E. Cariati, E. Lucenti, C. Botta, U. Giovanella, D. Marinotto and S. Righetto, Coord. Chem. Rev., 2016, 206, 566–614 CrossRef.
- (a) K. R. Kyle, C. K. Ryu, J. A. DiBenedetto and P. C. Ford, J. Am. Chem. Soc., 1991, 113, 2954 CrossRef CAS; (b) P. C. Ford and A. Vogler, Acc. Chem. Res., 1993, 26, 220 CrossRef CAS; (c) P. C. Ford, E. Cariati and J. Bourassa, Chem. Rev., 1999, 99, 3625–3647 CrossRef CAS PubMed.
- A. Vogler and H. Kunkely, J. Am. Chem. Soc., 1986, 108, 7211 CrossRef CAS.
- (a) M. Vitale, C. K. Ryu, W. E. Palke and P. C. Ford, Inorg. Chem., 1994, 33, 561 CrossRef CAS; (b) F. De Angelis, S. Fantacci, A. Sgamellotti, E. Cariati, R. Ugo and P. C. Ford, Inorg. Chem., 2006, 45, 10576–10584 CrossRef CAS PubMed.
- (a) A. Dossing, C. K. Ryu, S. Kudo and P. C. Ford, J. Am. Chem. Soc., 1993, 115, 5132–5137 CrossRef CAS; (b) D. Tran, C. K. Ryu and P. C. Ford, Inorg. Chem., 1994, 33, 5957–5958 CrossRef CAS.
- R. E. Gamache, R. A. Rader and D. R. McMillin, J. Am. Chem. Soc., 1985, 107, 1141–1146 CrossRef CAS.
- N. Sutin, Prog. Inorg. Chem., 1983, 30, 441–497 CrossRef CAS.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1986, 811, 265 CrossRef.
- V. A. Durante and P. C. Ford, Inorg. Chem., 1979, 18, 588 CrossRef CAS.
- (a) J. A. Osborn, F. H. Jardine, J. F. Young and G. Wilkinson, J. Chem. Soc. A, 1966, 1711 RSC; (b) J. P. Collman, L. S. Hegedus, J. R. Norton and R. G. Finke, Principles and Applications of Organo-transition Metal Chemistry, University Science Books, Mill Valley, CA, 1987, ch. 10 Search PubMed.
- (a) J. Halpern, Acc. Chem. Res., 1970, 3, 386–392 CrossRef CAS; (b) J. Halpern and C. S. Wong, J. Chem. Soc., Chem. Commun., 1973, 629–630 RSC.
- D. A. Wink and P. C. Ford, J. Am. Chem. Soc., 1987, 109, 436–442 CrossRef CAS.
- J. S. Bridgewater, T. L. Netzel, J. R. Schoonover, S. M. Massick and P. C. Ford, Inorg. Chem., 2001, 40, 1466–1476 CrossRef CAS PubMed.
- P. C. Ford, J. S. Bridgewater and B. Lee, Photochem. Photobiol., 1997, 65, 57–64 CrossRef CAS.
- M. W. George and J. J. Turner, Coord. Chem. Rev., 1998, 177, 201–217 CrossRef CAS.
- J. P. Lomont, S. C. Nguyen and C. B. Harris, Acc. Chem. Res., 2014, 47, 1634–1642 CrossRef CAS PubMed.
- J. A. Calladine, R. Horvath, A. J. Davies, A. Wriglesworth, X. Z. Sun and M. W. George, Appl. Spectroscopy, 2015, 69, 519–524 CrossRef CAS PubMed.
- A. Vlcek Jr, H. Kvapilova, M. Towrie and S. Zalis, Acc. Chem. Res., 2014, 47, 1634–1642 CrossRef PubMed.
- J. DiBenedetto, D. W. Ryba and P. C. Ford, Inorg. Chem., 1989, 28, 3503–3507 CrossRef CAS.
- W. T. Boese and P. C. Ford, Organometallics, 1994, 13, 3525–3531 CrossRef CAS.
- J. M. Kelly, J. M. Long and R. Bonneau, J. Phys. Chem., 1983, 87, 3344 CrossRef CAS.
- C. Hall and R. N. Perutz, Chem. Rev., 1996, 96, 3125–3146 CrossRef CAS PubMed.
- B. Cornils, W. A. Herrman, C.-H. Wong and H.-W. Zanthoff, Catalysis from A to Z: A Concise Encyclopedia, 2408 Seiten, Verlag Wiley-VCH Verlag GmbH & Co., 2013 Search PubMed.
- (a) R. J. Mawby, F. Basolo and R. G. Pearson, J. Am. Chem. Soc., 1964, 86, 3994–3999 CrossRef CAS; (b) F. Calderezzo, Angew. Chem., Int. Ed. Engl., 1977, 16, 299 CrossRef; (c) T. C. Flood, Top. Stereochem., 1981, 12, 37–118 CAS; (d) S. Webb, C. Giandomenico and J. Halpern, J. Am. Chem. Soc., 1986, 108, 345 CrossRef CAS.
- W. T. Boese and P. C. Ford, J. Am. Chem. Soc., 1995, 117, 8381–8391 CrossRef CAS.
- S. M. Massick, V. Mertens, J. Marhenke and P. C. Ford, Inorg. Chem., 2002, 41, 3553–3559 CrossRef CAS PubMed.
- K. L. McFarlane, B. Lee, W. Fu, R. van Eldik and P. C. Ford, Organometallics, 1998, 17, 1826–1834 CrossRef CAS.
- S. M. Massick, J. G. Rabor, S. Elbers, J. Marhenke, S. Bernhard, J. R. Schoonover and P. C. Ford, Inorg. Chem., 2000, 39, 3098–3106 CrossRef CAS PubMed.
- S. M. Massick, T. Büttner and P. C. Ford, Inorg. Chem., 2003, 42, 575–580 CrossRef CAS PubMed.
- P. C. Ford and S. M. Massick, Coord. Chem. Rev., 2002, 226, 39–49 CrossRef CAS.
- M. Volpe, G. Wu, A. Iretskii and P. C. Ford, Inorg. Chem., 2006, 45, 1861–1870 CrossRef CAS PubMed.
- J. H. Jones, The Cativa Process for the Manufacture of Acetic Acid, Platinum Met. Rev., 2000, 44, 94–105 CAS.
- R. F. Heck and D. S. Breslow, J. Am. Chem. Soc., 1961, 83, 4023 CrossRef.
- D. C. Roe, Organometallics, 1987, 6, 942–946 CrossRef CAS.
- M. Sola and T. Zeigler, Organometallics, 1996, 15, 2611–2618 CrossRef CAS.
- L. J. Ignarro, Nitric Oxide: Biology and Pathobiology, Elsevier Inc., Burlington, MA, 2nd edn, 2010 Search PubMed.
- M. Hoshino, K. Ozawa, H. Seki and P. C. Ford, J. Am. Chem. Soc., 1993, 115, 9568 CrossRef CAS.
- P. C. Ford, Inorg. Chem., 2010, 49, 6226–6239 CrossRef CAS PubMed.
- (a) B. M. Hoffman and Q. H. Gibson, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 21–25 CrossRef CAS PubMed; (b) E. J. Rose and B. M. Hoffman, J. Am. Chem. Soc., 1983, 105, 2866–2873 CrossRef CAS.
- (a) K. N. Walda, X. Y. Liu, V. S. Sharma and D. Magde, Biochemistry, 1994, 33, 2198–2209 CrossRef CAS PubMed; (b) K. A. Jongeward, D. Magde, D. J. Taube, J. C. Marsters, T. G. Traylor and V. S. Sharma, J. Am. Chem. Soc., 1988, 110, 380–387 CrossRef CAS; (c) V. G. Kharitonov, V. S. Sharma, D. Magde and D. Koesling, Biochemistry, 1997, 36, 6814–6818 CrossRef CAS PubMed.
- (a) J. S. Olson and G. N. Phillips, J. Biol. Chem., 1996, 271, 17593 CrossRef CAS PubMed; (b) M. Ikeda-Saito, Y. Dou, T. Yonetani, J. S. Olson, T. Li, R. Regan and Q. H. Gibson, J. Biol. Chem., 1993, 268, 6855 CAS; (c) D. Ionascu, F. Gruia, X. Ye, A. Yu, F. Rosca, C. Beck, A. Demidov, J. S. Olson and P. M. Champion, J. Am. Chem. Soc., 2005, 127, 16921–16934 CrossRef CAS PubMed.
- (a) E. A. Morlino and M. A. J. Rodgers, J. Am. Chem. Soc., 1996, 118, 11798–11804 CrossRef CAS; (b) N. Purwar, J. M. McGarry, J. Kostera, A. A. Pacheco and M. Schmidt, Biochemistry, 2011, 50, 4491–4503 CrossRef CAS PubMed; (c) H. R. Lucas, G. Meyer and K. D. Karlin, J. Am. Chem. Soc., 2009, 131, 13924–13925 CrossRef CAS PubMed.
- L. E. Laverman and P. C. Ford, J. Am. Chem. Soc., 2001, 123, 11614 CrossRef CAS PubMed.
- L. E. Laverman, A. Wanat, J. Oszajca, G. Stochel, P. C. Ford and R. van Eldik, J. Am. Chem. Soc., 2001, 123, 285–293 CrossRef CAS PubMed.
- J. N. Armor, H. A. Scheidegger and H. Taube, J. Am. Chem. Soc., 1968, 90, 5928 CrossRef CAS.
- J. N. Armor and S. D. Pell, J. Am. Chem. Soc., 1973, 95, 7625 CrossRef.
- A. Czap and R. van Eldik, Dalton Trans., 2003, 665–671 RSC.
- I. J. Ostrich, L. Gordon, H. W. Dodgen and J. P. Hunt, Inorg. Chem., 1980, 19, 61 CrossRef.
- T. Schneppensieper, A. Zahl and R. van Eldik, Angew. Chem., Int. Ed., 2001, 40, 1678 CrossRef CAS.
- (a) V. S. Sharma and D. Magde, Methods-A Companion to Methods in Enzymology, 1999, vol. 19, p. 494 Search PubMed; (b) V. G. Kharitonov, M. Russwurm, D. Madge, V. S. Sharma and D. Koesling, Biochem. Biophys. Res. Commun., 1997, 239, 284 CrossRef CAS PubMed.
- (a) T. C. Bellamy, C. Griffiths and J. Garthwaite, J. Biol. Chem., 2002, 277, 31801 CrossRef CAS PubMed; (b) J. Garthwaite, Mol. Cell. Biochem., 2010, 334, 221 CrossRef CAS PubMed.
- (a) B. G. Hill, B. P. Dranka, S. M. Bailey, J. R. Lancaster Jr and V. M. Darley-UsmarJ, Biol. Chem., 2010, 285, 19699–19704 CrossRef CAS PubMed; (b) T. A. Heinrich, R. S. da Silva, K. M. Miranda, C. H. Switzer, D. A. Wink and J. M. Fukuto, Br. J. Pharmacol., 2013, 169, 1417–1429 CrossRef CAS PubMed; (c) D. Basudhar, L. A. Ridnour, R. Cheng, A. H. Kesarwala, J. Heinecke and D. A. Wink, Coord. Chem. Rev., 2016, 306, 708–723 CrossRef CAS PubMed.
- (a) S. Mocellin, V. Bronte and D. Nitti, Med. Res. Rev., 2007, 27, 317–352 CrossRef CAS PubMed; (b) D. D. Thomas, L. A. Ridnour, J. S. Isenburg, W. Flores-Santana, C. Switzer, S. Donzelli, P. Hussain, C. Vecoli, N. Paolocci, S. Ambs, C. A. Colton, C. C. Harris, D. D. Roberts and D. A. Wink, Free Radical Biol. Med., 2008, 45, 18–31 CrossRef CAS PubMed.
- J. Bourassa, W. DeGraff, S. Kudo, D. A. Wink, J. B. Mitchell and P. C. Ford, J. Am. Chem. Soc., 1997, 119, 2853–2862 CrossRef CAS.
- P. Wardman, Clin. Oncol., 2007, 19, 397–417 CrossRef CAS PubMed.
- Examples are: (a) J. B. Mitchell, D. A. Wink, W. DeGraff, J. Gamson, L. Keefer and M. C. Krishna, Cancer Res., 1993, 53, 5845–5848 CAS; (b) B. F. Jordan, P. Sonveaux, O. Feron, V. Gregoire, N. Beghein, C. Dessy and B. Gallez, Int. J. Cancer, 2004, 109, 768–773 CrossRef CAS PubMed; (c) M. Nagane, H. Yasui, T. Yamamori, S. Zhao, Y. Kuge, N. Tamaki, H. Kameya, H. Fujii and O. Inanami, Biochem. Biophys. Res. Commun., 2013, 437, 420–425 CrossRef CAS PubMed; (d) H. Jiang, V. N. Verovski, W. Leonard, K. Law, M. Vermeersch, G. Storme, D. Van den Berge, T. Gevaert, A. Sermeus and M. De Ridder, Int. J. Radiat. Oncol., Biol., Phys., 2013, 85, 820–827 CrossRef CAS PubMed; (e) S. Ning, M. Bednarski, B. Oronsky, J. Scicinski and S. J. Knox, Biochem. Biophys. Res. Commun., 2014, 447, 537–542 CrossRef CAS PubMed; (f) Y. Samuni, D. A. Wink, M. Krishna, J. B. Mitchell and S. Goldstein, Free Radical Biol. Med., 2014, 73, 291–298 CrossRef CAS PubMed.
- M. L. Roussin, Ann. Chim. Phys., 1858, 52, 285 Search PubMed.
- (a) S. Kudo, J. L. Bourassa, S. E. Boggs, Y. Sato and P. C. Ford, Anal. Biochem., 1997, 247, 193–202 CrossRef CAS PubMed; (b) J. Bourassa, B. Lee, S. Bernhard, J. R. Schoonover and P. C. Ford, Inorg. Chem., 1999, 38, 2947–2952 CrossRef CAS PubMed.
- (a) I. M. Lorkovic, K. M. Miranda, B. Lee, S. Bernhard, J. R. Schoonover and P. C. Ford, J. Am. Chem. Soc., 1998, 120, 11674 CrossRef CAS; (b) C. F. Works and P. C. Ford, J. Am. Chem. Soc., 2000, 122, 7592 CrossRef CAS.
- M. De Leo and P. C. Ford, J. Am. Chem. Soc., 1999, 121, 1980–1981 CrossRef CAS.
- F. W. Flitney, I. L. Megson, J. L. M. Thomson, G. D. Kennovin and A. R. Butler, Br. J. Pharmacol., 1996, 117, 1549–1557 CrossRef CAS PubMed.
- T. Sjostrand, Scand. J. Clin. Lab. Invest., 1949, 1, 201 CrossRef CAS , also Nature 1949, 164, 580–581.
- Examples are: (a) L. E. Otterbein, B. S. Zuckerbraun, M. Haga, F. Liu, R. Song, A. Usheva, C. Stachulak, N. Bodyak, R. N. Smith, E. Csizmadia, S. Tyagi, Y. Akamatsu, R. J. Flavell, T. R. Billiar, E. Tzeng, F. H. Bach, A. M. Choi and M. P. Soares, Nat. Med., 2003, 9, 183 CrossRef CAS PubMed; (b) D. J. Kaczorowski and B. S. Zuckerbraun, Curr. Med. Chem., 2007, 14, 2720 CrossRef CAS PubMed; (c) R. Motterlini and L. E. Otterbein, Nat. Rev. Drug Discovery, 2010, 9, 728–743 CrossRef CAS PubMed; (d) C. C. Romão, W. A. Blaettler, J. D. Seixas and G. J. L. Bernardes, Chem. Soc. Rev., 2012, 41, 3571 RSC; (e) S. H. Heinemann, T. Hoshi, M. Westerhausen and A. Schiller, Chem. Commun., 2014, 50, 3644 RSC; (f) S. García-Gallego and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2014, 53, 9712 CrossRef PubMed; (g) U. Schatzschneider, Br. J. Pharmacol., 2015, 172, 1638 CrossRef CAS PubMed.
- (a) T. S. Murray, C. Okegbe, Y. Gao, B. I. Kazmierczak, R. Motterlini, L. E. P. Dietrich and E. M. Bruscia, PLoS One, 2012, 7, e35499S Search PubMed; (b) S. J. Lee, A. A. Coronata, L. E. Fredenburgh, S. W. Chung, M. A. Perrella, K. Nakahira, S. W. Ryter and A. M. K. Choi, Antioxid. Redox Signaling, 2014, 20, 432–442 CrossRef CAS PubMed; (c) D. Nguyen, T.-K. Nguyen, S. A. Rice and C. Boyer, Biomacromolecules, 2015, 16, 2776–2786 CrossRef CAS PubMed.
- (a) I. Nassour, B. Kautza, M. Rubin, D. Escobar, J. Luciano, P. Loughran, H. Gomez, J. Scott, D. Gallo, J. Brumfield, L. E. Otterbein and B. S. Zuckerbraun, Shock, 2015, 43, 166–171 CrossRef CAS PubMed; (b) D. Babu, R. Motterlini and R. A. Lefebvre, Br. J. Pharmacol., 2015, 172, 1557–1573 CrossRef CAS PubMed; (c) I. Andreadou, E. K. Iliodromitis, T. Rassaf, R. Schulz, A. Papapetropoulos and P. Ferdinandy, Br. J. Pharmacol., 2015, 172, 1587–1606 CrossRef CAS PubMed.
- (a) K. Konig, J. Microsc., 2000, 200, 83–104 CrossRef CAS PubMed; (b) A. M. Smith, M. C. Mancini and S. Nie, Nat. Nanotechnol., 2009, 4, 710–711 CrossRef CAS PubMed.
- B. P. Timko, M. Arruebo, S. A. Shankarappa, J. B. McAlvin, O. S. Okonkwo, B. Mizrahi, C. F. Stefanescu, L. Gomez, J. Zhu, A. Zhu, J. Santamaria, R. Langer and D. S. Kohane, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1349–1354 CrossRef CAS PubMed.
- R. Dale Rimmer, H. Richter and P. C. Ford, Inorg. Chem., 2010, 49, 1180–1185 CrossRef PubMed.
- Examples are: (a) P. C. Ford, J. Bourassa, K. M. Miranda, B. Lee, I. Lorkovic, S. Boggs, S. Kudo and L. Laverman, Coord. Chem. Rev., 1998, 171, 185–202 CrossRef CAS; (b) E. Tfouni, M. Krieger, B. R. McGarvey and D. W. Franco, Coord. Chem. Rev., 2003, 236, 57–69 CrossRef CAS; (c) C. M. Pavlos, H. Xu and J. P. Toscano, Curr. Top. Med. Chem., 2005, 5, 635–647 CrossRef; (d) M. J. Rose and P. K. Mascharak, Coord. Chem. Rev., 2008, 252, 2093–2114 CrossRef CAS PubMed; (e) P. C. Ford, Acc. Chem. Res., 2008, 41, 190–200 CrossRef CAS PubMed; (f) S. Sortino, Chem. Soc. Rev., 2010, 39, 2903–2913 RSC; (g) P. C. Ford, Nitric Oxide, 2013, 34, 56–64 CrossRef CAS PubMed; (h) A. W. Carpenter and M. H Schoenfisch, Chem. Soc. Rev., 2012, 41, 3742–3752 RSC; (i) B. Heilman and P. K. Mascharak, Philos. Trans. R. Soc., A, 2013, 371(1995), 20120368 CrossRef PubMed; (j) B. J. Heilman, M. A. Gonzalez and P. K. Mascharak, Prog. Inorg. Chem., 2014, 58, 185–224 CrossRef CAS; (k) A. E. Pierri, D. A. Muizzi, A. D. Ostrowski and P. C. Ford, Struct. Bonding, 2015, 165, 1–45 CrossRef.
- (a) A. D. Ostrowski, S. J. Deakin, B. Azhar, T. W. Miller, N. Franco, M. M. Cherney, A. J. Lee, J. N. Burstyn, J. M. Fukuto, I. L. Megson and P. C. Ford, J. Med. Chem., 2010, 53, 715–722 CrossRef CAS PubMed; (b) A. D. Ostrowski, R. O. Absalonson, M. A. De Leo, G. Wu, J. G. Pavlovich, J. Adamson, B. Azhar, A. V. Iretskii, I. L. Megson and P. C. Ford, Inorg. Chem., 2011, 50, 4453–4462 CrossRef CAS PubMed.
- M. Yamaji, Y. Hama, Y. Miyazaki and M. Hoshino, Inorg. Chem., 1992, 31, 932–934 CrossRef CAS.
- M. A. De Leo and P. C. Ford, Coord. Chem. Rev., 2000, 208, 47–59 CrossRef CAS.
- C. Kutal and A. W. Adamson, Inorg. Chem., 1973, 12, 1990 CrossRef CAS.
- F. DeRosa, X. Bu and P. C. Ford, Inorg. Chem., 2005, 44, 4157–4165 CrossRef CAS PubMed.
- (a) I. L. Medintz and H. Mattoussi, Phys. Chem. Chem. Phys., 2009, 11, 17 RSC; (b) T. L. Doane and C. Burda, Chem. Soc. Rev., 2012, 41, 2885 RSC.
- D. Neuman, A. D. Ostrowski, A. A. Mikhailovsky, R. O. Absalonson, G. F. Strouse and P. C. Ford, J. Am. Chem. Soc., 2008, 130, 168–175 CrossRef CAS PubMed.
- P. T. Burks, A. D. Ostrowski, A. A. Mikhailovsky, E. M. Chan, P. S. Wagenknecht and P. C. Ford, J. Am. Chem. Soc., 2012, 134, 13266 CrossRef CAS PubMed.
- C. L. Conrado, S. Wecksler, C. Egler, D. Magde and P. C. Ford, Inorg. Chem., 2004, 43, 5543–5549 CrossRef CAS PubMed.
- S. R. Wecksler, J. Hutchinson and P. C. Ford, Inorg. Chem., 2006, 45, 1192–1200 CrossRef CAS PubMed.
- R. J. Gui, A. Wan, Y. L. Zhang, H. L. Li and T. T. Zhao, RSC Adv., 2014, 4, 30129–30136 RSC.
- F. N. Castellano, H. Malak, I. Gryczynski and J. R. Lakowicz, Inorg. Chem., 1997, 36, 5548–5551 CrossRef CAS.
- (a) G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245–1330 CrossRef CAS PubMed; (b) A. Gollmer, F. Besostri, T. Breitenbach and P. R. Ogilby, Free Radical Res., 2013, 47, 718–730 CrossRef CAS PubMed.
- J.-H. Olivier, Y. S. Bai, H. Uh, H. Yoo, M. J. Therien and F. N. Castellano, J. Phys. Chem. A, 2015, 119, 5642–5649 CrossRef CAS PubMed.
- M. Rumi, S. Barlow, J. Wang, J. W. Perry and S. R. Marder, Adv. Polym. Sci., 2008, 213, 1–95 CAS.
- S. Dayal and C. Burda, J. Am. Chem. Soc., 2008, 130, 2890–2891 CrossRef CAS PubMed.
- S. R. Wecksler, A. Mikhailovsky and P. C. Ford, J. Am. Chem. Soc., 2004, 126, 13566–13567 CrossRef CAS PubMed.
- S. R. Wecksler, A. Mikhailovsky, D. Korystov and P. C. Ford, J. Am. Chem. Soc., 2006, 128, 3831–3837 CrossRef CAS PubMed.
- (a) S. R. Wecksler, A. Mikhailovsky, D. Korystov, F. Buller, R. Kannan, L.-S. Tan and P. C. Ford, Inorg. Chem., 2007, 46, 395–402 CrossRef CAS PubMed; (b) Q. Zheng, A. Bonoiu, T. Y. Ohulchanskyy, G. S. He and P. N. Prasad, Mol. Pharm., 2008, 5, 389–398 CrossRef CAS PubMed.
- Examples are (a) K. Hishikawa, H. Nakagawa, T. Furuta, K. Fukuhara, H. Tsumoto, T. Suzuki and N. Miyata, J. Am. Chem. Soc., 2009, 131, 7488 CrossRef CAS PubMed; (b) C. Fowley, A. P. McHale, B. McCaughan, A. Fraix, S. Sortino and J. F. Callan, Chem. Commun., 2015, 51, 81 RSC; (c) Z. Xu, Z. Y. Wu, J. Sun and R. J. Gui, Mater. Chem. Phys., 2015, 162, 286 CrossRef CAS.
- (a) F. Wang and X. Liu, Chem. Soc. Rev., 2009, 38, 976–989 RSC; (b) J. V. Garcia, F. Zhang and P. C. Ford, Philos. Trans. R. Soc., A, 2013, 371(1995), 20120129/1–20120129/25 CrossRef CAS PubMed.
- F. Zhang, Y. Wan, T. Yu, F. Q. Zhang, Y. Shi, S. Xie, Y. Li, L. Xu, B. Tu and D. Y. Zhao, Angew. Chem., Int. Ed., 2007, 46, 7976–7979 CrossRef CAS PubMed.
- C. J. Carling, F. Nourmohammadian, J. C. Boyer and N. R. Branda, Angew. Chem., Int. Ed., 2010, 49, 3782–3785 CrossRef CAS PubMed.
- Y. Yang, Q. Shao, R. Deng, C. Wang, X. Teng, K. Cheng, Z. Cheng, L. Huang, Z. Liu, X. Liu and B. Xing, Angew. Chem., Int. Ed., 2012, 51, 3125–3129 CrossRef CAS PubMed.
- J. V. Garcia, J. Yang, D. Shen, C. Yao, X. Li, G. D. Stucky, D. Y. Zhao, P. C. Ford and F. Zhang, Small, 2012, 8, 3800–3805 CrossRef CAS PubMed.
- S. Zhang, J. Wang, W. Xu, B. Chen, W. Yu, L. Xu and H. Song, J. Lumin., 2014, 147, 278–283 CrossRef CAS.
- Principles of Molecular Photochemistry, An Introduction, ed. N. J. Turro, V. Ramamurthy and J. C. Scaiano, University Science Books, Sausalito, CA, 2009, ch. 7 Search PubMed.
- J. Dong and J. I. Zink, Small, 2015, 11, 4165–4172 CrossRef CAS PubMed.
- P. T. Burks, J. V. Garcia, R. GonzalezIrias, J. T. Tillman, M. Niu, J. Zhang, F. Zhang and P. C. Ford, J. Am. Chem. Soc., 2013, 135, 18145–18152 CrossRef CAS PubMed.
- J. V. Garcia, M. A. Crisalli, A. Fenwick, P. C. Ford, manuscript in preparation.
- (a) H. Kojima, N. Nakatsubo, K. Kicuchi, S. Kawahara, Y. Kirino, H. Nagaoshi, Y. Hirata and T. Nagano, Anal. Chem., 1998, 70, 2446–2453 CrossRef CAS PubMed; (b) L. A. Ridnour, J. E. Sim, M. A. Hayward, D. A. Wink, S. M. Martin, G. R. Buettner and D. R. Spitz, Anal. Biochem., 2000, 281, 223–229 CrossRef CAS PubMed.
- (a) L. D. Xu and S. J. Lippard, Nat. Chem. Biol., 2006, 2, 375–380 CrossRef PubMed; (b) Z. J. Tonzetich, L. E. McQuade and S. J. Lippard, Inorg. Chem., 2010, 49, 6338–6348 CrossRef CAS PubMed.
- (a) K. Tsuge, F. DeRosa, M. D. Lim and P. C. Ford, J. Am. Chem. Soc., 2004, 126, 6564–6565 CrossRef CAS PubMed; (b) C. Khin, M. Lim, K. Tsuge, A. Iretskii, G. Wu and P. C. Ford, Inorg. Chem., 2007, 46, 9323–9331 CrossRef CAS PubMed.
- A. J. Atkin, J. M. Lynam, B. E. Moulton, P. Sawle, R. Motterlini, N. M. Boyle, M. T. Pryce and I. J. S. Fairlamb, Dalton Trans., 2011, 40, 5755–5761 RSC.
- B. W. Michel, A. R. Lippert and C. J. Chang, J. Am. Chem. Soc., 2012, 134, 15668–15671 CrossRef CAS PubMed.
- Y. Cao, D.-W. Li, L.-J. Zhao, X.-Y. Liu, X.-M. Cao and Y.-T. Long, Anal. Chem., 2015, 87, 9696–9701 CrossRef CAS PubMed.
- C. Bohlender, S. Glaeser, M. Klein, J. Weisser, S. Thein, U. Neugebauer, J. Popp, R. Wyrwa and A. Schiller, J. Mater. Chem. B, 2014, 2, 1454–1463 RSC.
- X. Zhou, S. Lee, Z. C. Xu and J. Yoon, Chem. Rev., 2015, 115, 7944–8000 CrossRef CAS PubMed.
- R. D. Rimmer, A. E. Pierri and P. C. Ford, Coord. Chem. Rev., 2012, 256, 1509–1519 CrossRef CAS.
- A. E. Pierri, A. Pallaoro, G. Wu and P. C. Ford, J. Am. Chem. Soc., 2012, 134, 18197–18200 CrossRef CAS PubMed.
- A. Fraix and S. Sortino, Chem.–Asian J., 2015, 10, 1116–1125 CrossRef CAS PubMed.
- P. C. Ford, D. Wink and J. DiBenedetto, Prog. Inorg. Chem., 1983, 30, 213–271 CrossRef CAS.
- A. Pierri, P.-J. Huang, J. V. Garcia, J. Stanfill, M. Chui, N. Zheng and P. C. Ford, Chem. Commun., 2015, 51, 2072–2075 RSC.
- E. Ruoslahti, Adv. Mater., 2012, 24, 3747–3756 CrossRef CAS PubMed.
- E. J. Chung, Y. Cheng, R. Morshed, K. Nord, Y. Han, M. L. Wegscheid, B. Auffinger, D. A. Wainwright, M. S. Lesniak and M. V. Tirrell, Biomaterials, 2014, 35, 1249–1256 CrossRef CAS PubMed.
- E. S. Levy, D. P. Morales, J. V. Garcia, N. O. Reich and P. C. Ford, Chem. Commun., 2015, 51, 17692–17695 RSC.
- T. Sugahara, K. N. Kotamraju and E. Ruoslahti, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16157 CrossRef PubMed.
- (a) P. K. Jain, W. Qian and M. A. El-Sayed, J. Am. Chem. Soc., 2006, 128, 2426–2433 CrossRef CAS PubMed; (b) Z. Qin and J. C. Bischof, Chem. Soc. Rev., 2012, 41, 1191–1217 RSC.
| This journal is © The Royal Society of Chemistry 2016 |