
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymeric Gd-DOTA amphiphiles form spherical and fibril-shaped nanoparticle MRI contrast agents†
Lyndsay M.
Randolph‡
a,
Clare L. M.
LeGuyader‡
a,
Michael E.
Hahn‡
*ab,
Christopher M.
Andolina
c,
Joseph P.
Patterson
a,
Robert F.
Mattrey
b,
Jill E.
Millstone
 c,
Mauro
Botta
d,
Miriam
Scadeng
b and
Nathan C.
Gianneschi
*a
c,
Mauro
Botta
d,
Miriam
Scadeng
b and
Nathan C.
Gianneschi
*a
aDepartment of Chemistry and Biochemistry, University of California, 9500 Gilman Dr., La Jolla, San Diego, CA 92093, USA. E-mail: ngianneschi@ucsd.edu; mehahn@ucsd.edu
bDepartment of Radiology, University of California, 9500 Gilman Dr., La Jolla, San Diego, CA 92093, USA
cDepartment of Chemistry, University of Pittsburgh, 4200 Fifth Ave, Pittsburgh, PA 15260, USA
dDipartimento di Scienze e Innovazione Tecnologica, Università del Piemonte Orientale “A. Avogadro”, Alessandria, Italy
First published on 22nd March 2016
Abstract
A Gd3+-coordinated polymerizable analogue of the MRI contrast agent Gd-DOTA was used to prepare amphiphilic block copolymers, with hydrophilic blocks composed entirely of the polymerized contrast agent. The resulting amphiphilic block copolymers assemble into nanoparticles (NPs) of spherical- or fibril-shape, each demonstrating enhanced relaxivity over Gd-DOTA. As an initial examination of their behavior in vivo, intraperitoneal (IP) injection of NPs into live mice was performed, showing long IP residence times, observed by MRI. Extended residence times for particles of well-defined morphology may represent a valuable design paradigm for treatment or diagnosis of peritoneal malignances.
Nanoparticle (NP) based drugs have reached the clinical mainstream, with established efficacy predominantly in oncology and infectious diseases.1–3 In addition, NPs are of interest as carriers of diagnostic agents capable of unique behavior and biodistribution patterns not observed for small molecule analogues.4 To this end, several strategies for the incorporation of imaging reporters within NPs have been introduced, allowing for NP visualization using, among other modalities, fluorescence,5 ultrasound,6 CT,7 MRI,8 and nuclear imaging.8,9 Of these modalities, MRI offers several advantages including exquisite anatomic co-registration with excellent tissue characterization, lack of ionizing radiation, and in general, provides high quality images at clinically relevant imaging depths. In turn, many NP-based MR imaging strategies have been explored using either inorganic6,7,10–14 or soft polymeric organic materials as contrast agents.15–18
Herein, we set out to prepare amphiphilic block copolymers for the formation of micellar NPs, in which the hydrophilic block would consist entirely of a Gd-based contrast agent for MRI. In developing this type of material, we sought a synthetic route through which the Gd3+ functionalized monomers are introduced directly into the polymer via a living polymerization approach, obviating the need for post-polymerization conjugation strategies, or ligand metallation processes.19–23 We then studied these materials as MRI contrast agents using in vitro techniques (nuclear magnetic resonance dispersion, NMRD) as well as in vivo, live animal imaging in mice.
Results and discussion
We prepared amphiphilic block copolymers from hydrophobic monomer 1 and hydrophilic monomer 2 (Fig. 1 and S1†). Monomer 2 is a novel compound, synthesized specifically for direct incorporation of a modified Gd-DOTA moiety via living, ring-opening metathesis polymerization (ROMP).24,25 The norbornenyl-Gd-DOTA monoamide enables visualization with standard T1-weighted MRI. Importantly, monomer 2 is complexed to Gd3+ prior to its incorporation into synthetic polymers. To our knowledge, this represents the first demonstration of a Gd3+-labeled polymer prepared entirely by direct polymerization. Examples of Gd3+-labeled polymers and soft NPs to date have employed post-polymerization metal chelation, requiring subsequent purification steps and removal of excess Gd3+.26 Gratifyingly, we were able to avoid this, given that 2 was consumed during ROMP reactions (Fig. S2 and S3†).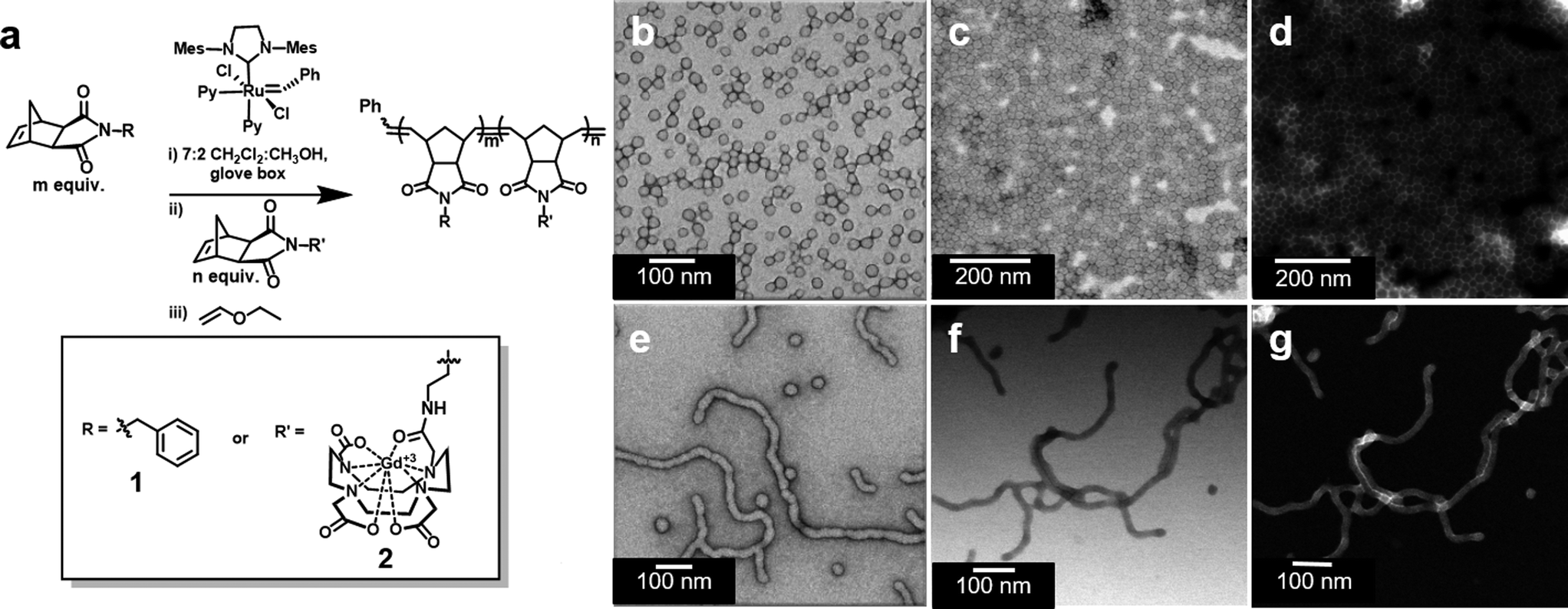 | ||
| Fig. 1 General synthetic scheme of Gd3+-containing block copolymer synthesis and particle characterization by electron microscopy of spherical micellar nanoparticles (SMNs) and fibril-shaped micellar nanoparticles (FMNs). (a) Polymer synthesis results in amphiphilic block copolymers. (b) Negative stained TEM of SMN. (c) Unstained BF-STEM of SMN. (d) Unstained HAADF-STEM of SMN. (e) Negative stained TEM of FMN. (f) Unstained BF-STEM of FMN. (g) Unstained HAADF-STEM of FMN. | ||
Following synthesis, solutions of polymers 1210-b-24 and 142-b-22 were subjected to slow transition from DMSO to water via dialysis, thus inducing the assembly of micellar NPs. NP structure was determined by transmission electron microscopy (TEM) and scanning TEM (STEM). For 1210-b-24, negative stain TEM revealed spherical micellar nanoparticles with low dispersity (with average diameter of 25 nm and standard deviation of 2.9 nm, see Fig. S4;† hereafter referred to as SMN, Fig. 1b). Unstained, bright field STEM (BF-STEM) demonstrated close packing of NPs with dark rings around the coronas, as would be expected for a NP bearing a heavy element (i.e. Gd3+) in the corona (Fig. 1c). This is supported by unstained high-angle annular dark field (HAADF) STEM coupled with energy-dispersive X-ray spectroscopy (EDS), which confirmed the presence of Gd3+ in the shell of the particle indicated by bright halos (Z-contrast) around the outer edges and characteristic X-ray edges of Gd3+ (Fig. 1d and S5†).
Similar microscopy characterization was performed on NPs derived from 142-b-22. Negative stain TEM, unstained BF-STEM, and unstained HAADF-STEM analysis revealed a predominantly fibrillar morphology, with Gd3+ present within the shell (Fig. 1e–g and S6†). We refer to these predominantly fibril-shaped NPs as fibrillar micellar nanoparticles (FMN), but note that the FMN formulation was not an entirely homogeneous phase, containing a minor component of spherical NPs. Indeed, mixed phases are common for cylindrical micelles.27 However, FMN represents approximately 95% of the sample, as determined by TEM image analysis (Fig. S7 and Table S1†). Importantly, the two NP formulations are stable as SMN or FMN for periods in excess of 14 months in aqueous media (Fig. S8 and S9†). Moreover, cryo-TEM revealed that NPs remain identifiable following incubation in blood serum at 37 °C for one week (Fig. S10 and S11†).
SMN and FMN were further characterized by nuclear magnetic resonance dispersion (NMRD) analysis to investigate the basic magnetic properties as related to their utility as MRI contrast agents. At 37 °C and neutral pH, the NMRD profile of both NPs show the characteristic line-shape of macro-/supramolecular structures due to a reduction in rotational tumbling rate (Fig. 2);28,29 this is markedly distinct from the NMRD line-shape of Gd-DOTA. Notable features in the NMRD profile of the NPs include a region of constant relaxivity at low field strengths (∼0.01–0.5 MHz, 0.0002–0.01 T) for SMN and FMN, with FMN displaying an elevated per Gd3+ relaxivity over SMN at all field strengths. Both NPs have a maximum relaxivity at ∼20 MHz (0.5 T); SMN has a maximum per Gd3+r1p of 15.6 mM−1 s−1, while FMN demonstrates a favorably elevated r1p of 18.5 mM−1 s−1 (Table 1; for detailed description of NMRD fit parameters, see ESI and Fig. S12†). We note that the higher per Gd3+ relaxivities of SMN and FMN as compared to the mononuclear Gd-DOTA at 20 MHz (Table 1) are competitive (i.e. within the same order of magnitude) with other nanoparticle and polymer-based systems.30,31 Further, although all three of these systems converge to similar relaxivities at high frequencies, r1 enhancement for SMN and FMN over Gd-DOTA is still pronounced at 3 T (FMN: +210%; SMN: +169%). Such enhancement is largely due to the improved rotational dynamics for the two paramagnetic NPs combined with an exchange rate of coordinated water sufficiently fast as not to strongly limit the relaxivity.
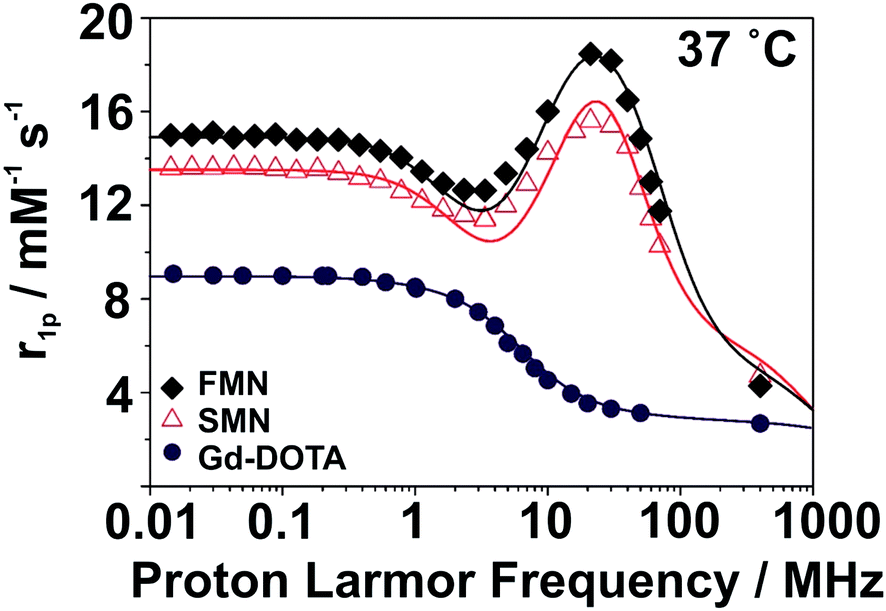 | ||
| Fig. 2 1H NMRD profiles for Gd-DOTA, SMN, and FMN. | ||
| 20 r 1p (mM−1 s−1) | Δ 2 (1019 s−2) | τ V (ps) | τ RL (ns) | τ RG (ns) | S 2 | τ M (ns) | |
|---|---|---|---|---|---|---|---|
| a The fitting parameters a, 310D, q and r were fixed to the values of 4.0 Å, 3.10 × 10−5 cm2 s−1, 1 and 3.0 Å, respectively. b Let to vary between 100 and 900 ns. | |||||||
| SMN | 15.6 | 0.9 | 52 | 0.19 | 5.9 | 0.20 | 560 |
| FMN | 18.5 | 0.7 | 53 | 0.15 | 2.8 | 0.25 | 350 |
| Gd-DOTA | 3.5 | 1.0 | 7 | — | 0.04 | — | 122 |
Next, we performed a pilot study to determine if the Gd3+-labeled NPs would serve as MRI contrast agents in vivo following IP injection in live mice (Fig. 3). For these studies, mice were first anesthetized and imaged by MRI prior to injection. For all formulations of contrast agent studied (Gd-DOTA, SMN, and FMN, n = 3 for each material), T1-weighted images obtained immediately after injection demonstrate hyperintense IP signal due to the Gd3+ reporter displayed on all injected samples (Fig. S13†). Each animal was then imaged up to several hours post-injection to track retention in the peritoneal cavity. At two hours following IP injection, no enhanced signal was observed in the IP space of animals post injection of Gd-DOTA (Fig. 3a and S14a†). However, enhanced signal was clearly observed in the IP spaces of animals injected with either SMN or FMN (Fig. 3b, c, S14b and c†). This enhancement demonstrates that the NP formulations are retained in the IP space longer than Gd-DOTA. In addition, MRI of the urinary bladder of animals following injection of Gd-DOTA demonstrated rapid accumulation of Gd-DOTA, indicating clearance through renal excretion, as expected (Fig. S15†). This was markedly delayed and less intense in the case of SMN or FMN (Fig. S15†).
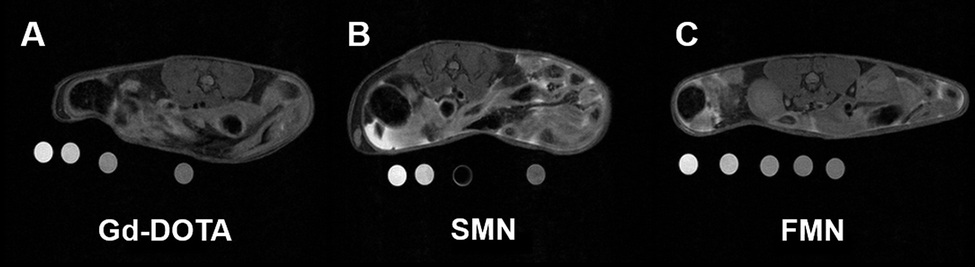 | ||
| Fig. 3 Axial T1-weighted images of the abdomen approximately 2 hours following IP injection of (a) Gd-DOTA, (b) SMN, and (c) FMN. See S14† for additional images of IP space at 2 hours. Phantoms, containing dilutions of the same material, were placed underneath the animal during scanning and used to correct for inter-scan variability. See ESI† for further description of analysis. | ||
After the pilot study, we performed a preliminary ex vivo biodistribution analysis to gain insight as to where NPs localized in vivo following IP injection. As whole body MRI demonstrated signal enhancement in the liver following injection of NPs (Fig. S16†), we designed a quantitative analysis of Gd3+ in the liver, spleen, and bowel to delineate the accumulation of NPs following IP injection. Notably, this observation of hepatic uptake is consistent with that of others who have observed NP accumulation in the liver following IP injection.32,33
Animals were sacrificed at 2 or 24 hours following IP-injection. The liver, spleen and bowel were harvested and analyzed via ICP-MS or ICP-OES for Gd3+ content (Fig. 4; see Fig. S17 and S18† for individual animal Gd accumulation profiles). In these initial in vivo analyses of the materials, SMN appeared to accumulate in the spleen and liver (n = 3) at higher concentrations than the bowel at 2 and 24 hours (Fig. 4). FMN showed less accumulation in the spleen or liver as compared to SMN at 2 hours post-injection (n = 3). These data suggest that both NPs are cleared relatively slowly from the IP space.
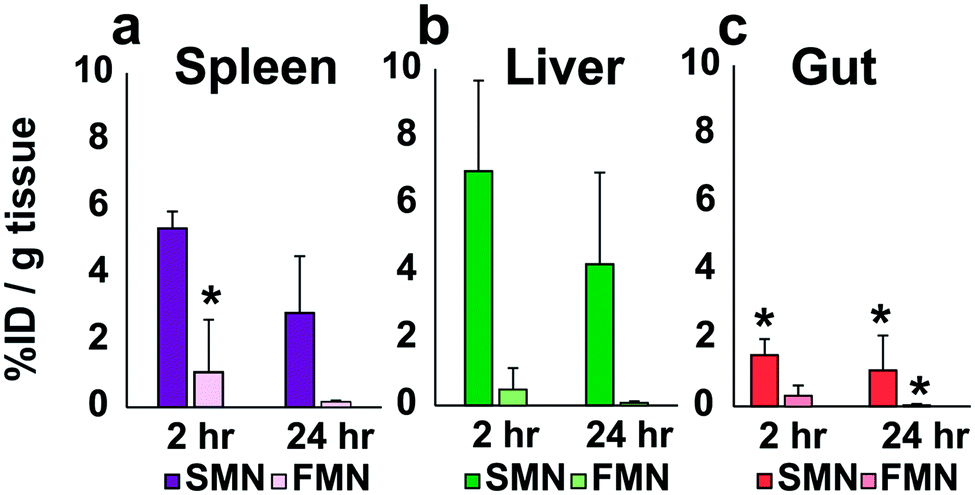 | ||
| Fig. 4 Accumulation of SMN and FMN in the spleen (a), liver (b), and bowel (c), at 2 h or 24 h following IP injections as detected by the presence of Gd by ICP-MS and ICP-OES. For each time point n = 3, except where indicated with *, where n = 2. Error bars represent standard deviations. | ||
Conclusions
We report a new synthetic method for labeling polymers and NPs with a Gd3+-based contrast agent through direct polymerization. This procedure ensures that the entire shell of the NPs consists of Gd3+-labeled monomers. This route led to the preparation of two different phases of NP, one entirely spherical (SMN), the other predominantly fibrillar (FMN). Preliminary in vivo MRI and ex vivo elemental analyses suggest that these differently shaped NPs derived from chemically identical starting materials show promise for imaging following IP injection. We note that the concept of shape dependent in vivo behavior of NPs has been observed in the context of intravenous (IV) injection by Discher, Caruso, Mitragotri, and others,34–41 but to the best of our knowledge, this report represents the first attempt to begin to address this issue following the clinically important IP injection route. Indeed, direct delivery of small molecule chemotherapeutic agents via intraperitoneal injection is a well-established therapeutic paradigm in the treatment of human metastatic ovarian, pancreatic and gastric malignancies42–46 and with this method in hand, future studies will build on these initial in vivo observations to further explore and exploit this concept in the intraperitoneal space.Experimental methods
Monomer and polymer synthesis and characterization
Monomer 1, (N-benzyl)-5-norbornene-exo-2,3-dicarboximide was prepared as previously described.47Polymerizations were performed under a dry dinitrogen atmosphere with anhydrous, degassed solvents in a glove box. Initiator (IMesH2)(C5H5N)2(Cl)2Ru![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh was prepared as described by Sanford et al.48
CHPh was prepared as described by Sanford et al.48
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 CH2Cl2:CH3OH (0.129 mL) was added to a stirred solution of 1 (68 mg, 0.268 mmol) in a mixture of anhydrous 7:2 CH2Cl2:CH3OH (2.97 mL) in a glove box. The reaction was left to stir in a glove box for 20 min, after which an analytical aliquot (approximately 32% by volume) was removed and mixed with an excess of ethyl vinyl ether for 30 min, then dried under high vacuum to give a homopolymer of 1 as a solid. To the remaining reaction mixture, a solution of 2 (25 mg, 0.0335 mmol) in anhydrous 7:2 CH2Cl2:CH3OH (0.08 mL) was added immediately following analytical aliquot removal. The reaction was monitored by analytical RP-HPLC (using the same conditions described above for compound 2, see Fig. S2†) to ensure complete consumption of 2. The mixture was left to stir in a glove box for 90 min and then quenched with excess ethyl vinyl ether (0.0251 mL) for 20 minutes, which was concentrated to dryness to give a greenish solid. This was used without further purification until particle formation. The homopolymer and block-copolymer were analyzed by SEC-MALS as shown in Fig. S2.†
:2 CH2Cl2:CH3OH (0.129 mL) was added to a stirred solution of 1 (68 mg, 0.268 mmol) in a mixture of anhydrous 7:2 CH2Cl2:CH3OH (2.97 mL) in a glove box. The reaction was left to stir in a glove box for 20 min, after which an analytical aliquot (approximately 32% by volume) was removed and mixed with an excess of ethyl vinyl ether for 30 min, then dried under high vacuum to give a homopolymer of 1 as a solid. To the remaining reaction mixture, a solution of 2 (25 mg, 0.0335 mmol) in anhydrous 7:2 CH2Cl2:CH3OH (0.08 mL) was added immediately following analytical aliquot removal. The reaction was monitored by analytical RP-HPLC (using the same conditions described above for compound 2, see Fig. S2†) to ensure complete consumption of 2. The mixture was left to stir in a glove box for 90 min and then quenched with excess ethyl vinyl ether (0.0251 mL) for 20 minutes, which was concentrated to dryness to give a greenish solid. This was used without further purification until particle formation. The homopolymer and block-copolymer were analyzed by SEC-MALS as shown in Fig. S2.†
Nanomaterial formulation and characterization
:1 DMSO:H2O (2.5 mL) was added drop wise. The cloudy polymer mixture was sonicated for 15 minutes, then transferred to a 3500 MWCO snakeskin dialysis tubing (Pierce) and dialyzed against a 1:1 DMSO:H2O solution (2 L). After 24 hours, the milky solution was removed from dialysis and H2O was added drop wise (2.5 mL) to the polymer mixture. This mixture was then transferred to a 10000 MWCO snakeskin dialysis tubing and dialyzed against 2 L of H2O. After 24 hours, H2O (2.5 mL) was added to the dialysis bag and the dialysate was refreshed with 2 L of H2O. Dialysis was allowed to continue for 24 hours. The sample was removed from dialysis and the tubing was rinsed with H2O (3 × 1 mL) into the polymer sample. The sample was further diluted with 25 mL of H2O, which was then sonicated for 9 hours. The sample was placed in a centrifuge at 4000 rcf for 6 min. The supernatant was used for all further analysis.
:2 CH2Cl2:CH3OH. This solution was placed into a 3500 MWCO snakeskin dialysis tubing and dialyzed against DMSO for 4 hours. The sample was then dialyzed against 4:1 DMSO:H2O for 4 hours, followed by 3:2 DMSO:H2O for 4 hours, followed by 2:3 DMSO:H2O for 4 hours, followed by 1:4 DMSO:H2O for 4 hours, then 2 × H2O for 4 hours each to yield an opalescent sample, which was used as is for all further analysis.
Transmission electron microscopy
TEM and cryo-TEM (see Fig. 1b, e, and Fig. S8–S11) was performed on a FEI Sphera microscope operating at 200 keV. TEM grids were prepared by depositing small (3.5 μL) aliquots of sample onto grids (formvar stabilized with carbon (5–10 nm) on 400 copper mesh, Ted Pella Inc.) that had previously been glow discharged using an Emitech K350 glow discharge unit and plasma-cleaned for 90 s in an E.A. Fischione 1020 unit. The sample grid was then stained with 1% uranyl acetate solution, rinsed with water (∼5 μL), and excess liquid was removed. Micrographs were recorded on a 2k × 2k Gatan CCD camera. Samples for cryo-TEM were prepared by depositing 3.5 μL of sample to a freshly glow discharged Quantifoil R2/2 TEM grid. The grids were blotted with filter paper under high humidity to create thin films and rapidly plunged into liquid ethane. The grids were transferred to the microscope under liquid nitrogen and kept at <−175 °C while imaging.STEM and STEM-EDS
Images were acquired on a JEOL JEM 2100F TEM equipped with an INCA (Oxford) EDS detector at the University of Pittsburgh, PA. Samples were prepared by drop-casting 5 μL of sample onto TEM grids (ultrathin 5 nm A-type carbon with 400 mesh Copper) followed by slow drying covered on the bench top for at least 3 hours. Samples were then dried under vacuum for 24–48 hours to remove contamination that would interfere with STEM-EDS. STEM-EDS data was collected for 180–600 s at specific points, using the largest probe size (1.5 nm electron beam diameter) with a 200 kV accelerating voltage. Images were collected in bright field (BF) and high-angle annular dark field (HAADF) modes, and grid background subtraction taken.1H NMRD profiles, fitting parameters and description
See ESI† for instrumental set-up details. The nuclear magnetic relaxation dispersion (NMRD) profiles allow a detailed characterization of the paramagnetic solutes in terms of a large set of structural and dynamic parameters. The experimental profiles were measured in aqueous solution at 310 K and neutral pH. The best fit parameters, listed in Table 1, were obtained using the standard equations for the inner sphere (IS) and outer sphere (OS) relaxation contributions and by fixing the values of the following parameters: the hydration number (q = 1), the Gd-Hw distance (r = 3.0 Å), the distance of closest approach of the bulk water molecules (a = 4.0 Å) to the metal ion, and the relative diffusion coefficient (D = 3.1 × 10−5 cm2 s−1).28,29,49In the case of paramagnetic nanoparticles, analysis of NMRD profiles takes into account the occurrence of a relatively fast local rotation of the metal complex superimposed to the global motion of the nanoparticle (Lipari–Szabo approach).50,51 This model allows separating the contribution of the overall global rotation of the paramagnetic nanoparticle (τRG = 5.9 and 2.8 ns for SMN and FMN, respectively) from the contribution of a faster local motion (τRL = 0.19 and 0.15 ns for SMN and FMN, respectively) associated with the rotation of the coordination polyhedron about the linker connecting the complex to the nanoparticle. The large difference between the two correlation times indicates that the system is rather flexible and its relaxivity limited by the relatively short value of τRL. The correlation of the two types of motions is described by the parameter S2 whose value is comprised between zero (completely independent motions) and one (totally correlated motions). The parameters for electronic relaxation (Δ2, τV) were used as empirical fitting parameters and do not have a precise physical meaning for these macromolecular systems.52,53 The temperature dependence of proton relaxivity was also measured at 0.5 T in order to obtain an independent evaluation of the residence lifetime τM of the coordinated water molecule (Fig. S12†).
In vitro and in vivo MRI and analysis of T1 data
See ESI† for MRI instrumental set-up details and parameters. MR images were acquired on a Bruker 7.0 T magnet, and all animal procedures were approved by University of California, San Diego's institutional animal care and use committee, protocol S10145.Female mice (C57Bl/6) weighing 18 grams were purchased from Harlan Sprague Dawley were anesthetized with 3% isoflurane in O2 and subjected to baseline MRI imaging before injection. A total of nine mice (three sets of three) were injected with 550 μL of an aqueous 0.4 mM Gd-DOTA, SMN, or FMN intraperitoneally and imaged continuously under anesthesia for two hours and then again at selected time points of approximately 3 h, 4 h, 5 h, 6 h, 7 h, 8 h, 24 h, 48 h, and 1 week (each measurement made under anesthesia). Images of successful IP injections can be found in Fig. S13.† To correct for minor scan-to-scan variations due to noise, T1 was normalized to pre-injection phantom relaxivities (see ESI† for detailed description of T1 data analysis).
Ex vivo ICP-MS analysis of organs
Mice were sacrificed using a lethal overdose of >5% isofluorane and selected organs harvested (see Fig. S16 and S17†). The liver, bowel and spleen were dissected, placed in separate tubes and their masses were recorded individually. Mass of the entire liver, bowel and spleen was recorded, added to concentrated nitric acid (900 μL), and placed on a shaker overnight, vented. The following morning, concentrated H2O2 (50 μL) was added to each of the organ solutions and placed back on the shaker, vented, for approximately 30 min. An aliquot (200 μL) of the digested organs was added to distilled DI water (800 μL) and submitted to Exova for ICP-MS analysis to determine Gd3+ concentration. Final concentration of Gd3+ in each organ was normalized to the organ mass.Acknowledgements
We acknowledge support from the NIH through a Director's New Innovator Award (1DP2OD008724), through the NIBIB (1R01EB011633) and for a Transformative Award (NHLBI R01HL117326). C. L. M. L. is supported by a training grant from the Department of Education (P200A120223–14). M. E. H. is supported by a training grant from the NIH (5T32EB005970). N. C. G. acknowledges the Alfred P. Sloan Foundation for a fellowship. We acknowledge the use of the UCSD Cryo-Electron Microscopy Facility which is supported by NIH grants to Dr Timothy S. Baker and a gift from the Agouron Institute to UCSD. We acknowledge the UCSD Chemistry and Biochemistry Molecular MS Facility for mass analysis of compounds.Notes and references
- B. Y. Kim, J. T. Rutka and W. C. Chan, N. Engl. J. Med., 2010, 363, 2434–2443 CrossRef CAS PubMed.
- I. F. Uchegbu and A. Siew, J. Pharm. Sci., 2013, 102, 305–310 CrossRef CAS PubMed.
- A. Wicki, D. Witzigmann, V. Balasubramanian and J. Huwyler, J. Controlled Release, 2015, 200, 138–157 CrossRef CAS PubMed.
- H. S. Choi and J. V. Frangioni, Mol. Imaging, 2010, 9, 291–310 CAS.
- M. Srinivas, L. J. Cruz, F. Bonetto, A. Heerschap, C. G. Figdor and I. J. M. de Vries, Biomaterials, 2010, 31, 7070–7077 CrossRef CAS PubMed.
- C.-H. Wang, S.-T. Kang, Y.-H. Lee, Y.-L. Luo, Y.-F. Huang and C.-K. Yeh, Biomaterials, 2012, 33, 1939–1947 CrossRef CAS PubMed.
- J. M. Kinsella, R. E. Jimenez, P. P. Karmali, A. M. Rush, V. R. Kotamraju, N. C. Gianneschi, E. Ruoslahti, D. Stupack and M. J. Sailor, Angew. Chem., Int. Ed., 2011, 50, 12308–12311 CrossRef CAS PubMed.
- Q. Fan, K. Cheng, X. Hu, X. Ma, R. Zhang, M. Yang, X. Lu, L. Xing, W. Huang, S. S. Gambhir and Z. Cheng, J. Am. Chem. Soc., 2014, 136, 15185–15194 CrossRef CAS PubMed.
- C. Kaittanis, T. M. Shaffer, D. L. Thorek and J. Grimm, Crit. Rev. Oncog., 2014, 19, 143–176 CrossRef PubMed.
- D. P. Cormode, B. L. Sanchez-Gaytan, A. J. Mieszawska, Z. A. Fayad and W. J. Mulder, NMR Biomed., 2013, 26, 766–780 CrossRef CAS PubMed.
- B. H. Kim, N. Lee, H. Kim, K. An, Y. I. Park, Y. Choi, K. Shin, Y. Lee, S. G. Kwon, H. B. Na, J. G. Park, T. Y. Ahn, Y. W. Kim, W. K. Moon, S. H. Choi and T. Hyeon, J. Am. Chem. Soc., 2011, 133, 12624–12631 CrossRef CAS PubMed.
- M. H. Mendonca Dias and P. C. Lauterbur, Magn. Reson. Med., 1986, 3, 328–330 CrossRef CAS PubMed.
- H. B. Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133–2148 CrossRef CAS.
- J. Xie, G. Liu, H. S. Eden, H. Ai and X. Chen, Acc. Chem. Res., 2011, 44, 883–892 CrossRef CAS PubMed.
- P. Besenius, J. L. Heynens, R. Straathof, M. M. Nieuwenhuizen, P. H. Bomans, E. Terreno, S. Aime, G. J. Strijkers, K. Nicolay and E. W. Meijer, Contrast Media Mol. Imaging, 2012, 7, 356–361 CrossRef CAS PubMed.
- M. A. Bruckman, X. Yu and N. F. Steinmetz, Nanotechnology, 2013, 24, 462001 CrossRef PubMed.
- G. L. Davies, I. Kramberger and J. J. Davis, Chem. Commun., 2013, 49, 9704–9721 RSC.
- P. Mi, D. Kokuryo, H. Cabral, M. Kumagai, T. Nomoto, I. Aoki, Y. Terada, A. Kishimura, N. Nishiyama and K. Kataoka, J. Controlled Release, 2014, 174, 63–71 CrossRef CAS PubMed.
- M. P. Chien, A. S. Carlini, D. Hu, C. V. Barback, A. M. Rush, D. J. Hall, G. Orr and N. C. Gianneschi, J. Am. Chem. Soc., 2013, 135, 18710–18713 CrossRef CAS PubMed.
- C. R. James, A. M. Rush, T. Insley, L. Vukovic, L. Adamiak, P. Kral and N. C. Gianneschi, J. Am. Chem. Soc., 2014, 136, 11216–11219 CrossRef CAS PubMed.
- A. M. Rush, D. A. Nelles, A. P. Blum, S. A. Barnhill, E. T. Tatro, G. W. Yeo and N. C. Gianneschi, J. Am. Chem. Soc., 2014, 136, 7615–7618 CrossRef CAS PubMed.
- M. P. Thompson, L. M. Randolph, C. R. James, A. N. Davalos, M. E. Hahn and N. C. Gianneschi, Polym. Chem., 2014, 5, 1954–1964 RSC.
- M. E. Hahn, L. M. Randolph, L. Adamiak, M. P. Thompson and N. C. Gianneschi, Chem. Commun., 2013, 49, 2873–2875 RSC.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- A. Leitgeb, J. Wappel and C. Slugovc, Polymer, 2010, 51, 2927–2946 CrossRef CAS.
- M. J. Allen, R. T. Raines and L. L. Kiessling, J. Am. Chem. Soc., 2006, 128, 6534–6535 CrossRef CAS PubMed.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- S. Aime, M. Botta and E. Terreno, in Advances in Inorganic Chemistry, Academic Press, 2005, vol. 57, pp. 173–237 Search PubMed.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed.
- A. J. Villaraza, A. Bumb and M. W. Brechbiel, Chem. Rev., 2010, 110, 2921–2959 CrossRef CAS PubMed.
- Y. Liu and N. Zhang, Biomaterials, 2012, 33, 5363–5375 CrossRef CAS PubMed.
- C. Jung, M. G. Kaul, O. T. Bruns, T. Dučić, B. Freund, M. Heine, R. Reimer, A. Meents, S. C. Salmen, H. Weller, P. Nielsen, G. Adam, J. Heeren and H. Ittrich, Circ. Cardiovasc. Imaging, 2014, 7, 303–311 CrossRef PubMed.
- M. Abdelhalim and M. Mady, Lipids Health Dis., 2011, 10, 1–9 CrossRef PubMed.
- D. A. Christian, S. Cai, O. B. Garbuzenko, T. Harada, A. L. Zajac, T. Minko and D. E. Discher, Mol. Pharmaceutics, 2009, 6, 1343–1352 CrossRef CAS PubMed.
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS PubMed.
- M. Mullner, S. J. Dodds, T. H. Nguyen, D. Senyschyn, C. J. Porter, B. J. Boyd and F. Caruso, ACS Nano, 2015, 9, 1294–1304 CrossRef CAS PubMed.
- J. A. Champion and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4930–4934 CrossRef CAS PubMed.
- X. Hu, J. Hu, J. Tian, Z. Ge, G. Zhang, K. Luo and S. Liu, J. Am. Chem. Soc., 2013, 135, 17617–17629 CrossRef CAS PubMed.
- X. Hu, G. Liu, Y. Li, X. Wang and S. Liu, J. Am. Chem. Soc., 2015, 137, 362–368 CrossRef CAS PubMed.
- Z. Liu, W. Cai, L. He, N. Nakayama, K. Chen, X. Sun, X. Chen and H. Dai, Nat. Nanotechnol., 2007, 2, 47–52 CrossRef CAS PubMed.
- L. Tao, W. Hu, Y. Liu, G. Huang, B. D. Sumer and J. Gao, Exp. Biol. Med., 2011, 236, 20–29 CrossRef CAS PubMed.
- M. T. Basel, S. Balivada, H. Wang, T. B. Shrestha, G. M. Seo, M. Pyle, G. Abayaweera, R. Dani, O. B. Koper, M. Tamura, V. Chikan, S. H. Bossmann and D. L. Troyer, Int. J. Nanomed., 2012, 297–306 CrossRef CAS PubMed.
- A. J. Di Pasqua, J. E. Huckle, J. K. Kim, Y. Chung, A. Z. Wang, M. Jay and X. Lu, Small, 2012, 8, 997–1000 CrossRef CAS PubMed.
- H. Ishigami, J. Kitayama, S. Kaisaki, A. Hidemura, M. Kato, K. Otani, T. Kamei, D. Soma, H. Miyato, H. Yamashita and H. Nagawa, Ann. Oncol., 2010, 21, 67–70 CrossRef CAS PubMed.
- M. Markman, Crit. Rev. Oncol. Hematol., 1999, 31, 239–246 CrossRef CAS PubMed.
- Y. Xie, Q. Long, Q. Wu, S. Shi, M. Dai, Y. Liu, L. Liu, C. Gong, Z. Qian, Y. Wei and X. Zhao, RSC Adv., 2012, 2, 7759–7771 RSC.
- M. P. Chien, A. M. Rush, M. P. Thompson and N. C. Gianneschi, Angew. Chem., Int. Ed., 2010, 49, 5076–5080 CrossRef CAS PubMed.
- M. S. Sanford, J. A. Love and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 6543–6554 CrossRef CAS PubMed.
- É. Tóth, L. Helm and A. Merbach, in The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, ed. A. Merbach, L. Helm and É. Tóth, John Wiley & Sons, Ltd, Chichester, UK, 2013, ch. 2, pp. 25–81 Search PubMed.
- G. Lipari and A. Szabo, J. Am. Chem. Soc., 1982, 104, 4546–4559 CrossRef CAS.
- G. Lipari and A. Szabo, J. Am. Chem. Soc., 1982, 104, 4559–4570 CrossRef CAS.
- M. F. Ferreira, B. Mousavi, P. M. Ferreira, C. I. O. Martins, L. Helm, J. A. Martins and C. F. G. C. Geraldes, Dalton Trans., 2012, 41, 5472–5475 RSC.
- Z. Zhang, M. T. Greenfield, M. Spiller, T. J. McMurry, R. B. Lauffer and P. Caravan, Angew. Chem., Int. Ed., 2005, 44, 6766–6769 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc00342g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |