
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymyxins facilitate entry into mammalian cells†
Kristina M.
Hamill
a,
Lisa S.
McCoy
a,
Ezequiel
Wexselblatt
a,
Jeffrey D.
Esko
b and
Yitzhak
Tor
*a
aDepartment of Chemistry and Biochemistry, University of California, San Diego, La Jolla, CA 92093-0358, USA. E-mail: ytor@ucsd.edu
bCellular and Molecular Medicine, University of California, San Diego, La Jolla, CA 92093-0687, USA
First published on 25th April 2016
Abstract
Polymyxin B is an antibiotic used against multi-resistant Gram negative infections, despite observed nephrotoxicity. Here we report the synthesis of functionalized derivatives of polymyxin B and its per-guanidinylated derivative in order to further explore the structural requirements necessary to facilitate uptake of the antibiotic into mammalian cells. We also investigate the possibility of using these novel scaffolds as molecular transporters. At nanomolar concentrations, both are capable of delivering large cargo (>300 kDa) into living cells. Their uptake depends exclusively on cell surface heparan sulfate. Mechanistic studies indicate these novel transporters are internalized through caveolae-mediated pathways and confocal microscopy show colocalization with lysosomes. The polymyxin-based transporters demonstrate cytosolic delivery through the delivery of a ribosome-inactivating protein. Furthermore, the natural polymyxin scaffold can be incorporated into liposomes and enhance their intracellular uptake. In addition to demonstrating the ability of the polymyxin scaffold to facilitate internalization into mammalian cells, these observations suggest the potential use of polymyxin and guanidinopolymyxin for intracellular delivery.
Introduction
Several biological barriers stand between exogenous agents and their entry to cells and tissues. These barriers hamper the administration of therapeutic agents, limiting their delivery and therapeutic utility. For example, high molecular weight and highly charged biomolecules such as proteins and oligonucleotides display therapeutic potential but have limited cellular uptake.1 Thus, great interest exists in developing molecular transporters as tools for exploring cell entry pathways and for facilitating the delivery of impermeable agents.Certain natural macromolecules, for example the HIV-1 Tat protein, exhibit cellular uptake.2–4 When added exogenously, Tat efficiently crosses cell membranes and can facilitate the uptake of conjugated or fused cargo.4–7 The basic, arginine rich region of the protein is critical for uptake, mediated to a large extent through interaction with cell surface proteoglycans.8–10 Since these early observations, numerous guanidinium-rich molecular transporters of varying scaffolds have been described and exploited,11–13 and recent reports have shown that cyclic guanidinium-rich peptides have enhanced cellular uptake compared to linear peptides.14–18
Guanidinoglycosides constitute a distinct non-oligomeric, but multivalent, family of low MW cell penetrating scaffolds.19 Guanidinoglycosides are derived from the naturally occurring aminoglycosides by converting the ammonium to guanidinium groups. They display unique uptake features when compared to other guanidinium-rich transporters by their potency at nanomolar concentrations and dependence on cell surface heparan sulfate proteoglycans for cellular entry. A variety of analogues derived from different aminoglycosides enable uptake of large, bioactive cargo into cells.20–24
Recent observations suggesting similarities between aminoglycosides and polymyxins,25 in addition to their analogous self-promoted uptake mechanisms in bacterial cells,26–28 have provoked a hypothesis that guanidinylated derivatives of the latter may display useful cellular entry features in mammalian cells. Here we explore the cellular delivery and internalization mechanism of high molecular weight biomolecules and nano-assemblies using polymyxin (PMB) and guanidinopolymyxin (GPMB) derivatives as carriers. Unlike the guanidinoglycosides that show substantially enhanced cellular uptake, when compared to their parent aminoglycoside precursors, both PMB and GPMB display highly competent uptake in mammalian cells. Intriguingly, while the cellular uptake of PMB and GPMB remains highly dependent on cell surface heparan sulfate, a fine mechanistic analysis suggests unique internalization pathways and more efficient cytosolic delivery than other well-studied molecular transporters. Our major conclusions include: (1) polymyxin and its guanidinylated form effectively enter mammalian cells, (2) the cyclic peptide and not the hydrophobic tail is the entry-facilitating module, and (3) these molecules can facilitate the cellular delivery of large biomolecules and liposomal assemblies. We further speculate that the effective entry of parent natural antibiotics into mammalian cells, coupled to earlier observations indicating their ability to interfere with eukaryotic translation,25 may contribute to their adverse cytotoxic effects in mammals. These observations, in addition to enriching the repertoire of cellular delivery vehicles, illustrate the wide landscape and potential utility of new ammonium and guanidinium rich transporters.
Results
Synthetic strategy
We envisioned a convergent synthesis, wherein the PMB molecular transporters were made via amide formation from two fragments, a biotinylated linker and the PMB core (Scheme 1). Relying on steric hindrance of the amine of Thr2, the primary amines on Dab residues of the polymyxin core could be tetra-Boc protected29 or guanidinylated. Instead of synthesizing these PMB derivatives de novo, enzymatic hydrolysis of the fatty acyl chain-Dab1 portion of the commercially available PMB can generate the polymyxin B nonapeptide (PMBN).30–32 The linker can be formed by a 1,3-dipolar cycloaddition of an alkyne portion and an azide-containing biotinylated linker.21 The enzymatically cleaved Dab residue can be reintroduced as part of the linker by amide bond formation to hexynoic acid so the final product will be similar to the natural PMB scaffold containing five Dab moieties (and the same number of primary amines).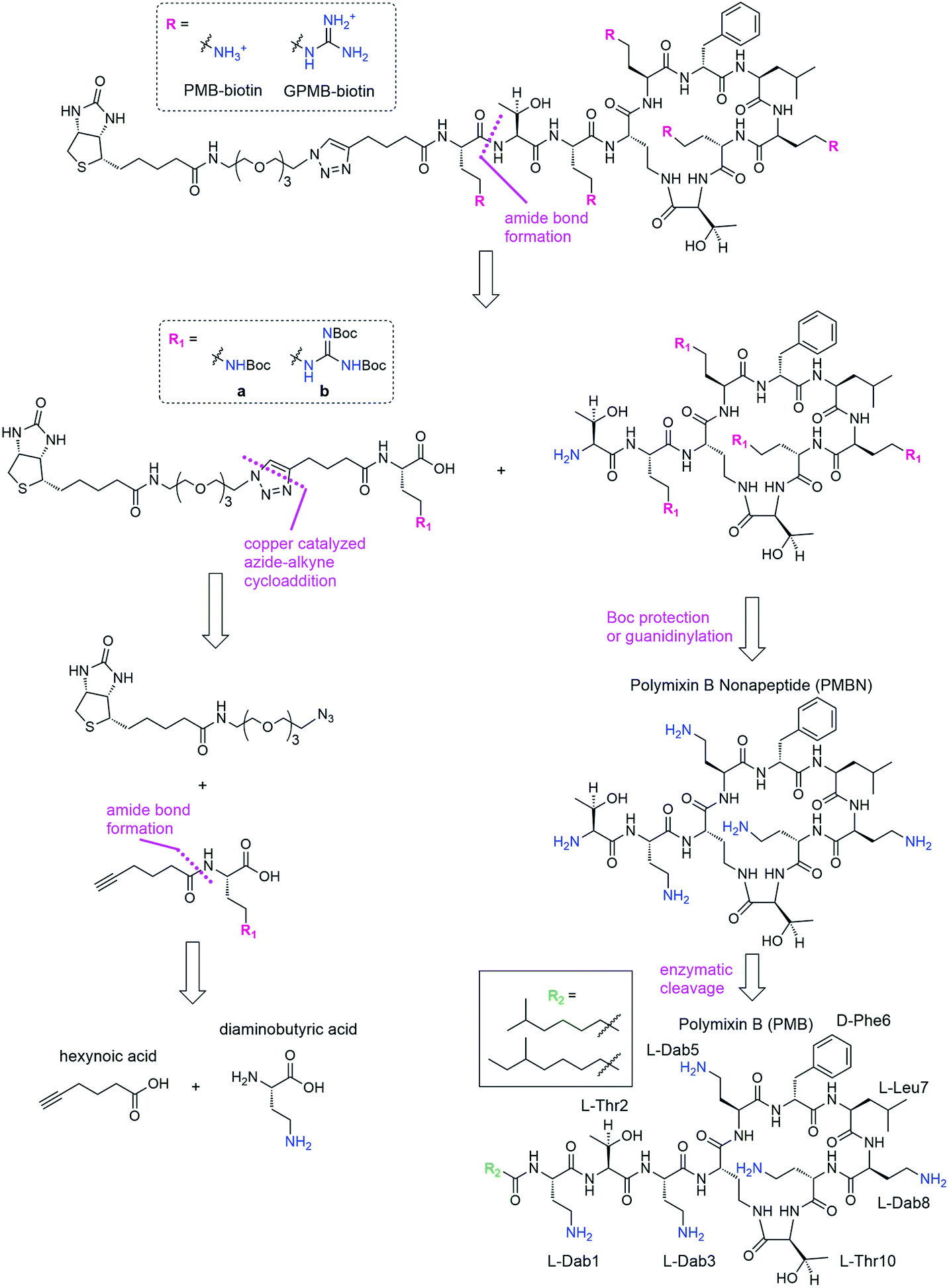 | ||
| Scheme 1 Retrosynthesis of biotinylated PMB and GPMB. | ||
Synthesis of transporters
The fatty acid chain and Dab1 residue on PMB were cleaved with ficin to generate PMBN (1, Scheme S1†).30–32 PMBN (1) was subsequently tetra-Boc-protected29 using Boc-ON or Boc-guanidinylated using N,N′-di-Boc-1H-pyrazole-1-carboxamidine to afford 2a and 2b, respectively (Scheme 2). | ||
| Scheme 2 Synthesis of Boc protected PMBN and Boc guanidinylated PMBN. | ||
The biotinylated Dab linker (3a or 3b, Scheme S2†) was then coupled to Boc-protected PMBN (2a) or Boc-protected guanidinylated PMBN (2b), and subsequently deprotected using trifluoroacetic acid (TFA). HPLC purification afforded the analytically pure bPMB (4a) and bGPMB (4b) (Scheme 3).
 | ||
| Scheme 3 Synthesis of biotinylated polymyxin and guanidinopolymyxin transporters. | ||
The same biotin linker was also clicked to the previously reported alkyne-tobramycin or alkyne-guanidinotobramycin derivatives21 to yield biotinylated tobramycin (bTob) and guanidinotobramycin (bGTob) (Fig. 1a and Scheme S4†). For comparison, biotinylated octaarginine (bArg8, Fig. 1b) was synthesized using standard solid phase peptide synthesis protocols. An aminohexanoic acid spacer was introduced at the N-terminus and coupled to biotin-NHS.
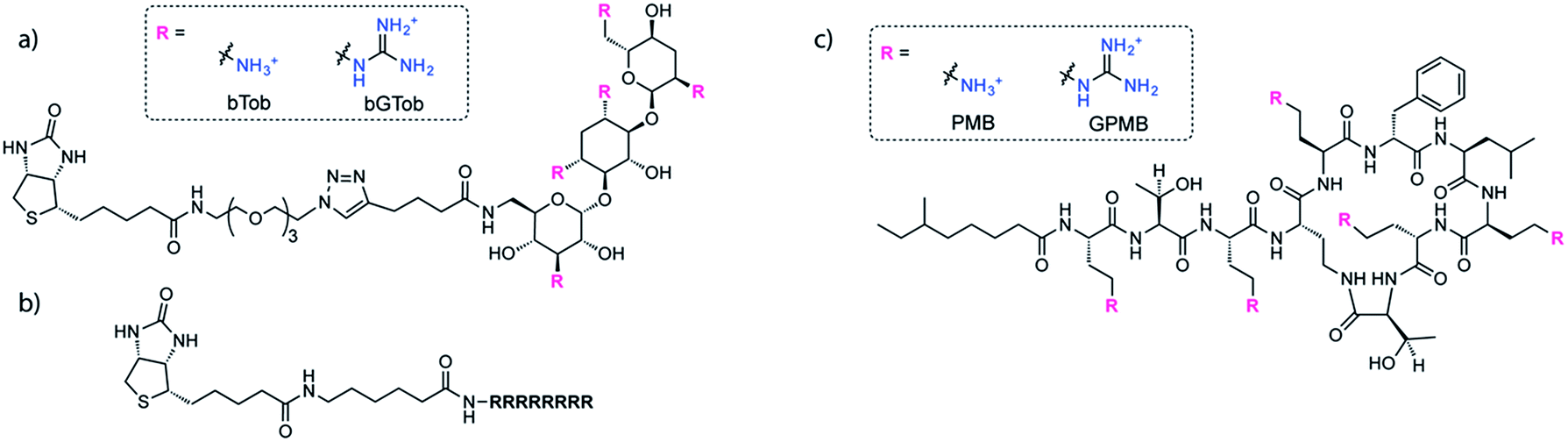 | ||
| Fig. 1 Structures of (a) bTob and bGTob, (b) bArg8, and (c) PMB and GPMB. | ||
To obtain the PMB and GPMB transporters (Fig. 1c), commercially available PMB was HPLC purified to isolate PMB terminated with 6-methyloctanoic acid. Purified PMB was then guanidinylated using N,N′-di-Boc-1H-pyrazole-1-carboxamidine followed by TFA deprotection to yield GPMB.
Formation of biotin–streptavidin complexes
Biotinylated transporters allow conjugation of the carriers to fluorescent proteins and the ability to test a variety of analogs in different assays.33–36 Biotinylated carriers were incubated in a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio with streptavidin derivatives for 20 min at room temperature. Conjugates were then diluted in culture medium to the final streptavidin concentration. Streptavidin conjugated to R-phycoerythrin (PE) coupled to the cyanine dye Cy5 (ST–PE–Cy5) was used as a high molecular weight model protein (MW = 300 kDa), streptavidin–Cy5 (ST–Cy5) was used for microscopy experiments, and streptavidin–saporin was used to analyze cytosolic delivery.
:1 molar ratio with streptavidin derivatives for 20 min at room temperature. Conjugates were then diluted in culture medium to the final streptavidin concentration. Streptavidin conjugated to R-phycoerythrin (PE) coupled to the cyanine dye Cy5 (ST–PE–Cy5) was used as a high molecular weight model protein (MW = 300 kDa), streptavidin–Cy5 (ST–Cy5) was used for microscopy experiments, and streptavidin–saporin was used to analyze cytosolic delivery.
Cellular uptake and cell surface binding in wild type and heparan sulfate-deficient CHO cells
Wild type CHO-K1 cells were incubated with the carrier–ST–PE–Cy5 conjugate for 1 h at 37 °C, detached with trypsin/EDTA, and analyzed by flow cytometry. Uptake of the fluorescent conjugate occurred at concentrations as low as 2 nM and increased in a dose dependent manner (Fig. 2a). bGTob showed uptake behavior similar to bPMB and bGPMB whereas bTob exhibited a ten-fold reduction in uptake. bArg8 showed approximately three-fold higher uptake than bPMB and bGPMB. Similar uptake patterns for bPMB and bGPMB were also observed in the human embryonic kidney cell line HEK-293 and the human hepatocyte cell line HEP-3B (Fig. S1†).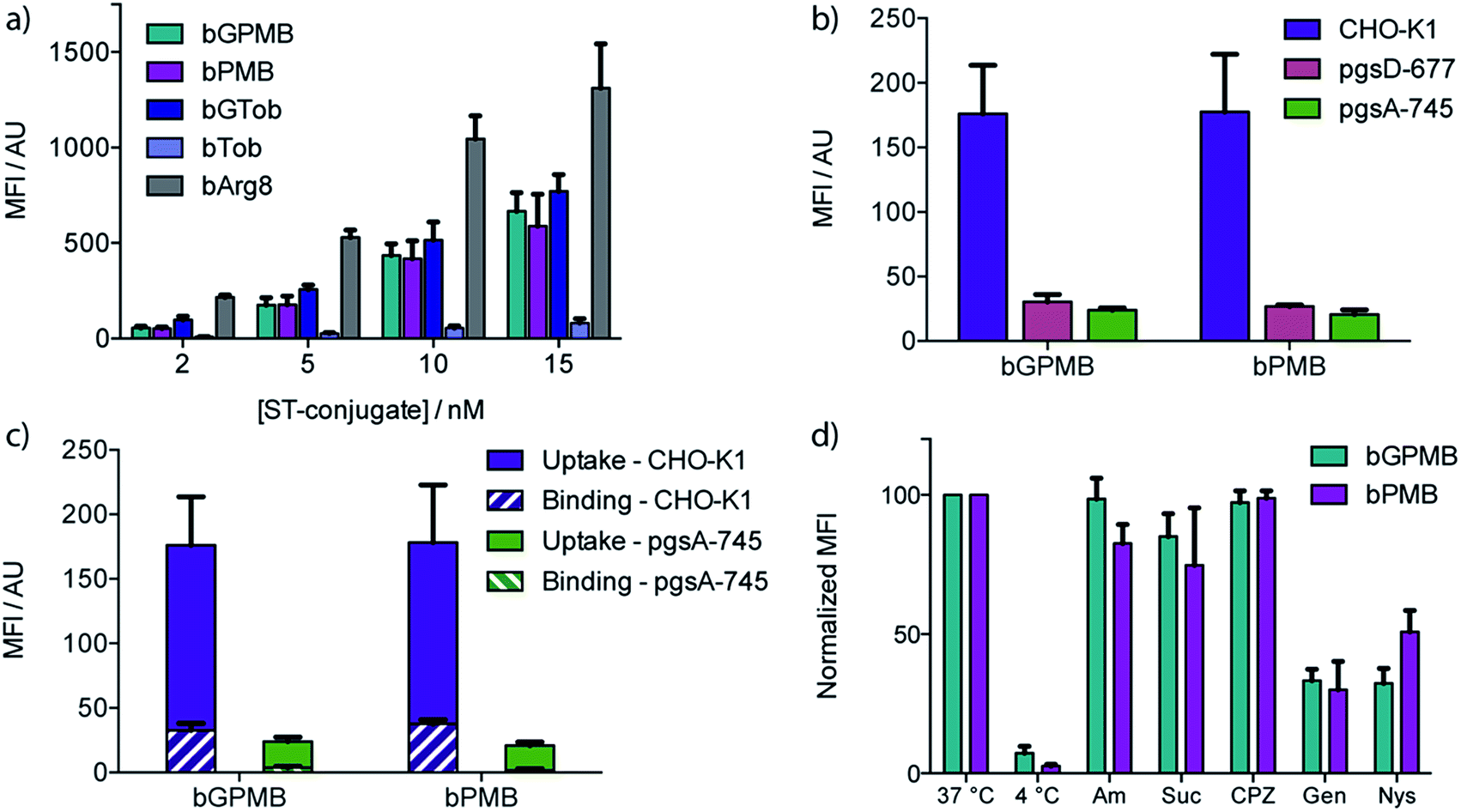 | ||
| Fig. 2 Cellular uptake, binding, and effects of inhibitors. Mean fluorescence intensity (MFI) was measured by flow cytometry. The background signal from untreated cells was subtracted. (a) Cellular uptake of biotinylated guanidinopolymyxin (bGPMB), polymyxin (bPMB), guanidinotobramycin (bGTob), tobramycin (bTob), and octarginine (bArg8) conjugated to ST–PE–Cy5. CHO-K1 cells were incubated with the conjugates at the indicated concentrations at 37 °C for 1 h and lifted with EDTA/trypsin. (b) bGPMB and bPMB streptavidin–PE–Cy5 conjugates (5 nM) were incubated with WT and mutant CHO cell lines for 1 h at 37 °C then lifted with EDTA/trypsin. (c) CHO-K1 and pgsA-745 cells were incubated with bGPMB and bPMB streptavidin–PE–Cy5 conjugates (5 nM) for 1 h at 37 °C. Cells were lifted with EDTA only (binding + uptake) or EDTA/trypsin (uptake). Binding values were determined by subtracting the MFI values obtained for cells lifted with EDTA/trypsin from those obtained for cells lifted with EDTA only. (d) CHO-K1 cells were incubated with bGPMB and bPMB conjugated to streptavidin–PE–Cy5 (5 nM) for 1 h at 37 °C or 4 °C. For inhibition experiments, cells were pretreated with amiloride (Am, 10 min, 5 mM), sucrose (Suc, 30 min, 400 mM), chlorpromazine (CPZ, 30 min, 20 μM), genistein (Gen, 30 min, 200 μM), or nystatin (Nys, 30 min, 5 μM) at 37 °C prior to incubation with the conjugates (5 nM) for 1 h at 37 °C in the presence of inhibitor (except Am). The MFI was normalized. | ||
CellTiter Blue assays indicate that bPMB and bGPMB showed no cytotoxicity when incubated with either CHO-K1 or HEK-293 cell lines for 72 h at concentrations as high as 0.5 μM (Fig. S2†). To demonstrate that the formation of a stable biotin–streptavidin complex is necessary for cellular delivery, ST–PE–Cy5 was incubated with PMB and GPMB, which do not contain the biotin moiety. Fig. S3† shows no enhanced uptake of ST–PE–Cy5 in the absence of the biotin-linked transporter.
The dependence of bPMB and bGPMB on cell surface heparan sulfate (HS) proteoglycans for cellular entry was evaluated using two mutant CHO cell lines, pgsA-745 which does not express HS or chondroitin sulfate/dermatan sulfate (CS/DS), and pgsD-677 which does not express HS but expresses 2 to 3-fold higher levels of CS/DS. Fig. 2b shows uptake in HS-deficient cell lines was reduced to <20% of that observed in wild-type cells.
To investigate cell-surface binding, CHO-K1 cells were incubated with the fluorescent conjugates at 37 °C and harvested using EDTA to prevent cleavage of cell-surface bound compounds. The fluorescence signal was compared to that observed when cells were lifted with EDTA/trypsin, which removes cell surface proteoglycans. Fig. 2c shows that for both bPMB and bGPMB, binding accounts for about 15% of the signal. Greater binding was observed in CHO-K1 cells compared to mutant pgsA-745 cells for both transporters (Fig. 2c).
Mechanism(s) of uptake
To shed light on the internalization mechanism(s), the contribution of various endocytotic pathways was evaluated pharmacologically. CHO-K1 cells were incubated with fluorescent bPMB or bGPMB conjugates at 4 °C to assess the contribution of energy-dependent processes. Further, uptake was evaluated in cells pretreated with inhibitors of macropinocytosis (amiloride), clathrin-mediated endocytosis (sucrose and chlorpromazine) or caveolae-mediated endocytosis (genistein and nystatin) (Fig. 2d). Their inhibitory effect on the uptake of bGTob was also evaluated for comparison. While low temperature practically abolished internalization, treatment with amiloride, sucrose or chlorpromazine did not affect the cellular uptake of bPMB or bGPMB high MW conjugates. However, in cells treated with either genistein or nystatin, cellular uptake was reduced by ∼50–60%, indicating that bPMB and bGPMB conjugates internalize through energy-dependent processes, presumably through caveolae-mediated pathways. bGTob, on the other hand, showed reduced uptake in the presence of sucrose, chlorpromazine and genistein and no change in uptake when cells were treated with amiloride or nystatin (Fig. S4†), indicating a different mechanism of uptake.Intracellular localization
To learn about the intracellular localization of these transporters, wild type CHO-K1 cells were incubated with the carrier–ST–Cy5 conjugates (20 nM) for 1 h at 37 °C and imaged using confocal laser scanning microscopy (CLSM). Cells were further treated with the nuclear stain Hoechst 33342 and the lysosomal marker LysoTracker Green DND-26 (Fig. 3). The images show transporter accumulation in punctate vesicles. Overlaying the images from the green and far red (pseudo-colored in red) channels reveals a moderate degree of co-localization for the conjugates and LysoTracker-stained compartments, resulting in a Pearson's correlation of 0.62 for both bPMB and bGPMB (Fig. 3). FACS analyses of carrier–ST–Cy5 conjugates up to 20 nM (Fig. S5†) were consistent with ST–PE–Cy5 delivery. | ||
| Fig. 3 Intracellular localization of bGPMB and bPMB. Confocal microscopy images of CHO K1 cells incubated for 1 h at 37 °C with bGPMB (upper panels) or bPMB (lower panels) conjugated to streptavidin–Cy5 (20 nM) (a) LysoTracker Green (b) ST–Cy5 conjugate (c) merged images with nuclear Hoechst dye. | ||
Cytoplasmic delivery
The ability of bPMB and bGPMB to deliver cargo to the cytosol was evaluated by conjugating the transporter to streptavidin–saporin (ST–SAP) and incubating the conjugate with CHO-K1 or pgsA cells at various concentrations. After four days, the CellTiter-Blue assay was used to asses the number of viable cells. Fig. 4 shows bPMB and bGPMB have LD50 values of 3.1 nM and 3.3 nM respectively. bGTob has an LD50 value of 13.2 nM and bArg8 of 3.1 nM. Additionally, ST–SAP without transporter or saporin without streptavidin incubated with bPMB and bGPMB were unable to induce cell death (Fig. S6†) indicating the need to form the biotin–ST complex. ST–SAP–transporter conjugates showed no toxicity in pgsA cells (Fig. S6†). | ||
| Fig. 4 Delivery of saporin. CHO-K1 cells were incubated with transporter–streptavidin–saporin conjugates at 37 °C. After four days, the number of viable cells was determined using CellTiter-Blue and measuring fluorescence intensity at 560/590. | ||
Cellular uptake of PMB and GPMB modified liposomes
To examine the significance of the cyclic peptide as a delivery module, rather than the lipophilic tail, and the versatility of PMB and GPMB as transporters, liposomes containing the fluorescent Cy5 dye were mixed with 10 mol% PMB or GPMB. Uptake was evaluated using flow cytometry and the size and zeta potential of the liposomes were measured using dynamic light scattering (DLS) and are reported in Table S1.† Both carriers showed the ability to enhance delivery of cargo containing liposomes into wild-type CHO cells with similar efficacy (Fig. 5). | ||
| Fig. 5 Cellular uptake of liposomes. CHO-K1 cells were incubated with plain and PMB or GPMB decorated liposomes at the indicated concentration for 1 h at 37 °C. The background signal from untreated cells was subtracted and the MFI was normalized. | ||
Discussion
Polymyxin B is a cyclic polypeptide antibiotic containing five primary amines and an eight carbon fatty acid chain, used against Gram-negative bacteria.37 The proposed antibacterial mechanism of action first involves electrostatic interactions between the positively charged 2,4-diaminobutyric acid (Dab) residues with the negatively charged bacterial lipopolysaccharide phosphate groups.38–41 This interaction facilitates insertion of the lipophilic moieties and disruption of the outer membrane.42 Internalized polymyxin, through self-promoted uptake, can then interact with the cytoplasmic membrane, a pathway that remains poorly characterized.38,39 Clinical use of polymyxin has diminished due to observed neurotoxicity and nephrotoxicity. Recently, however, with the increase in resistant Gram-negative infections, the clinical use of polymyxins has been resurrected.43–46Interestingly, the cellular uptake of polymyxins in eukaryotic cells has predominantly been studied in renal tubular cells in order to better understand their observed nephrotoxicity.47–52 While the entry pathway in renal cells is still not completely known, it has been suggested that at low micromolar concentrations it is carrier mediated, possibly by the multiligand receptor megalin, and is similar to the uptake mechanism of aminoglycoside antibiotics.47,49 As mentioned previously, guanidinylation of aminoglycosides transforms these potent antibiotics into a family of highly efficient eukaryotic cell-transporters.19,20 Their unique 3D display of guanidinium groups has proven to be key to their cellular uptake,21 and their multivalency has been linked to cell surface aggregation of heparan sulfate proteoglycans leading to endocytosis.23 Cargo–carrier complexes accumulate in the lysosomes through this endocytotic heparan sulfate-dependent uptake pathway, providing useful transporters for lysosomal delivery.22,24 Inspired by the observed similarities between aminoglycosides and polymyxins,25–28 as well as the enhanced uptake observed for other guanidinium-rich cyclic peptides,14–18 we synthesized both polymyxin and guanidinylated polymyxin derived transporters and investigated their uptake in CHO cells.
To evaluate the ability of PMB and GPMB to deliver large, bioactive cargo into mammalian cells, biotinylated derivatives were synthesized and conjugated to streptavidin–PE–Cy5, a fluorescently tagged, high molecular weight protein (MW 300 kDa). Uptake of bPMB and bGPMB in wild-type CHO cells was observed at low nanomolar transporter concentrations. While conversion of ammonium groups to guanidinium groups generally enhances cellular uptake rather dramatically,10,53 as seen with the conversion of bTob to bGTob (Fig. 2a) and after guanidinylation of other aminoglycosides,19,20 bPMB and bGPMB exhibit comparable cellular uptake (Fig. 2a). This surprising observation suggests that both bPMB and bGPMB are able to form related interactions with the extracellular components that facilitate their internalization. When compared to bGTob, which has five guanidinium groups, these novel polymyxin-based carriers show the same cellular uptake efficiencies. Furthermore, similar uptake patterns were observed in two human cell lines, HEK-293 and HEP-3B, suggesting that this phenomenon is cell-type independent. Of significance for potential applications as a cellular delivery tool, we showed that bPMB and bGPMB are nontoxic to mammalian cells at sub-micromolar concentrations using the CellTiter-Blue assay (Fig. S2†). Treated cells were >90% viable compared to untreated control cells after incubation for 72 h at concentrations as high as 0.5 μM, six-fold higher than the highest concentrations used in uptake assays. This is not necessarily unexpected, as previous studies have shown that polymyxin analogs lacking the fatty acid tail, for example PMBN, have less acute toxicity and reduced nephrotoxicity.42,54–56 This suggests that the cyclic peptide core can potentially serve as a non-toxic transporting module.
To investigate the involvement of cell surface glycosaminoglycans in the cellular uptake of these transporters, two mutant CHO cell lines were used, pgsA-745 and pgsD-677.57,58 The cellular uptake of bPMB and bGPMB is significantly diminished in the absence of HS by about seven-fold (Fig. 2b). These experiments imply that the internalization of both bPMB and bGPMB rely on the presence of HS on the cell surface. The polycationic scaffold must therefore preferentially bind HS over other negatively charged cell surface glycans.
To examine cell-surface binding, cells were incubated with the fluorescent conjugates and harvested using only EDTA to prevent cleavage of cell-surface bound compounds. The fluorescence signal was compared to that arising from cells lifted with EDTA/trypsin, which effectively cleaves the cell surface bound components and represents only internalized conjugate. Fig. 2c shows that although surface binding is observed (ca. 15% of the signal), these conjugates are efficiently internalized into wild type cells. bPMB and bGPMB derivatives display a comparable ratio between cellular uptake and surface binding. Additionally, greater binding was observed in CHO-K1 cells compared to mutant pgsA-745 cells for both transporters (Fig. 2c). This is consistent with the observation that bPMB and bGPMB rely on interactions with cell surface heparan sulfate proteoglycans for internalization.
To gain insight into the potential internalization pathway(s) of these transporters, various endocytosis mechanisms were probed. Cellular uptake at low temperatures was initially evaluated to determine the contribution of energy-dependent processes. Minimal uptake of bPMB and bGPMB at low temperatures suggests endocytosis as the predominant mode of uptake. Known inhibitors of various endocytotic pathways were then used to analyze the uptake mechanisms of the high MW bPMB or bGPMB conjugates, including amiloride, sucrose, chlorpromazine, genistein and nystatin. Amiloride inhibits macropinocytosis by impairing Na+/H+ exchange.59,60 Hypertonic sucrose and chlorpromazine are known inhibitors of clathrin-mediated endocytosis through dissociation of clathrin lattices on the plasma membrane and inhibition of clathrin-coated pit formation, respectively.61,62 Genistein and nystatin were used to investigate caveolin-dependent endocytosis. Genistein is a tyrosine kinase inhibitor that inhibits caveolar endocytosis by preventing vesicle fusion.63–65 Nystatin sequesters membrane cholesterol and inhibits lipid raft/caveolae mediated uptake.66,67 Treatment of cells with amiloride, sucrose, or chlorpromazine resulted in little to no effect on uptake, whereas genistein and nystatin reduced cellular uptake of bPMB and bGPMB conjugates by over 50% compared to untreated cells (Fig. 2d). Taken together, these results suggest that bPMB and bGPMB conjugates internalize predominantly through caveolae-mediated pathways. Although bGTob has five guanidinium groups and shows similar uptake to bPMB and bGPMB, the mechanisms of its cellular uptake suggest internalization through both clathrin-dependent and -independent endocytosis. Imaging studies (Fig. 3) further support that bPMB and bGPMB are internalized through endocytosis.
To establish whether bPMB and bGPMB can deliver cargo to the cytosol, ST–SAP conjugated bPMB and bGPMB were added to CHO K1 or pgsA cells and cell death was monitored using the CellTiter-Blue assay. Saporin, a type I ribosome-inactivating protein from Saponaria officinalis seeds, is an ideal candidate for evaluating cytosol delivery because it exhibits minimal cellular uptake on its own and needs to reach its cytosolic targets (ribosomal RNA) to exert its cytotoxic activity.68 As shown in Fig. 4, both bPMB- and bGPMB–saporin complexes killed CHO-K1 cells with an LD50 of ∼3 nM. Although bGTob shows similar uptake efficiency as bPMB and bGPMB, its LD50 is almost seven-fold higher. Likewise, bArg8 showed three-fold higher uptake but a similar LD50 as bPMB and bGPMB suggesting a higher ability of bPMB and bGPMB to escape the endosomal uptake pathway and deliver a bioactive protein to the cytosol. While a majority of the cargo resides in lysosomes (Fig. 3), there are differences in endosomal escape between the different transporters and only a low concentration of the enzyme is likely necessary to inhibit ribosomal activity. The resistance of pgsA cells to transporter–toxin conjugates (Fig. S6†) once again confirms the role of heparan sulfate in the uptake of bPMB and bGPMB.
The versatility of PMB and GPMB as transporters was further illustrated by incorporating these amphiphiles into preformed, cargo-containing liposomes. The hydrophobic tail of PMB is known to interact with phospholipids;69,70 therefore, we used a post-insertion method to incorporate PMB or GPMB into premade and preloaded liposomes. Fig. 5 shows that compared to unmodified liposomes, PMB and GPMB-containing liposomes exhibit enhanced cellular uptake. As with the chemically conjugated cargo, no difference between PMB and GPMB was observed.
Lipid vesicles have previously been modified with cell-penetrating peptides and used to improve delivery of various drugs and cargo while avoiding covalent modification of the cargo.71–77 The unique angle presented here is that we exploit the innate amphiphilic feature of PMB and its ability to act as a molecular transporter to assemble a novel delivery system utilizing a commercially available transporter. Unlike other transporter-modified liposomes, synthesis of a carrier–lipid or covalent modification of the liposome surface is not required. Furthermore, our observations with the decorated lipososmes further corroborate that the cyclic, polycationic portion of PMB is able to facilitate its entry into mammalian cells despite the hydrophobic tail. These observations may prove useful in advancing the understanding of the nephrotoxic effects impart by PMB and perhaps lead to novel safer polymyxins analogs with potent antibacterial features and minimal accumulation in human cells.
Conclusions
In summary, we have synthesized two new transporter modules derived from polymyxin B that can internalize and deliver large biomolecules, such as proteins, into mammalian cells via interactions with cell surface heparan sulfate. Inhibition studies indicate that these transporters are internalized through endocytosis, primarily caveolae-mediated mechanisms, and predominantly localize in the lysosomes. Delivery of a ribosome-inactivating protein demonstrates higher endosomal escape of these transporters when compared to bGTob or bArg8, which exhibit equal or greater overall uptake respectively. Furthermore, the natural polymyxin scaffold can spontaneously be incorporated into liposomes and substantially enhance their intracellular uptake. This suggests the potential for PMB and GPMB to expand our ability to selectively deliver bioactive cargo to intracellular targets and organelles. Additionally, the effective cellular uptake of the polymyxin-based transporter in eukaryotic cells presented here could help explain the observed human toxicity of the parent antibiotic.Experimental section
Synthesis
Detailed synthetic procedures and characterization of biotin–polymyxin, biotin–guanidinopolymyxin, and biotin–tobramycin are presented in the ESI.†Cell culture
Wild-type Chinese hamster ovary cells (CHO-K1) were obtained from the American Type Culture Collection (CCL-61). Mutant pgsA745 and pgsD677 were described previously.41,42 All cells were grown under an atmosphere of 5% CO2 in air and 100% relative humidity. CHO-K1, pgsA, and pgsD cells were grown in F-12 medium (Life Technologies) supplemented with fetal bovine serum (10% v/v, Gemini Bio-Products) and penicillin/streptomycin solution (1% v/v). The Hep3B cell line was obtained from ATCC (HB-8064) and cultured in MEM (Invitrogen) supplemented with 10% fetal bovine serum, nonessential amino acids, and 1% penicillin/streptomycin. HEK293T cells were obtained from ATCC and maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin.Quantifying cellular uptake/binding
The polymyxin derivatives (2.5 μM in PBS) were incubated with ST–PE–Cy5 (0.5 μM in PBS) for 20 min at ambient temperature then diluted with F-12 cell culture medium to give final conjugate solutions.Cells were plated onto 24-well plates (100000 cells per well) and grown for 24 h to about 80% confluence. Cells were washed with PBS and incubated with 300 μL of the corresponding conjugate for 1 h at 37 °C under an atmosphere of 5% CO2. Cells were washed twice with PBS, detached with 50 μL of trypsin–EDTA for uptake studies or 100 μL Versene (EDTA) for binding studies, diluted with PBS containing 0.1% BSA, and analyzed by FACS. Cellular uptake was quantified by the mean fluorescence intensity; raw data was interpreted by using FlowJo v8.8.6.
Evaluation of uptake dependency on temperature
Cells were grown for 24 h in 24-well plates as described above. Cells were incubated at 4 °C for 15 min in F-12, washed with cold PBS, then incubated with the precooled conjugate solution for 30 min at 4 °C. Cells were washed, detached with trypsin–EDTA, and analyzed as described above.Evaluation of endocytosis mechanisms
Cells were grown for 24 h in 24-well plates as described above, washed with PBS, and incubated with 5 mM amiloride for 10 min or 400 mM sucrose, 20 μM chlorpromazine, 200 μM genistein, or 5 μM nystatin for 30 min at 37 °C. Cells were then washed with PBS and treated with the conjugate solution in F-12 for the cells pretreated with amiloride, or the conjugate solution in the presence of the inhibitor, using the same concentration used for pretreatment, for 1 h at 37 °C under an atmosphere of 5% CO2. Cells were washed, detached with trypsin–EDTA, and analyzed as described above.Fluorescence microscopy
CHO-K1 cells were grown for 24 h in 35 mm dishes equipped with a glass bottom coverslip coated with poly-D-lysine. Cells were washed with PBS, treated with 1.5 mL of transporter conjugated to ST–Cy5 (20 nM) and incubated at 37 °C for 1 h under an atmosphere of 5% CO2. Cells were washed with PBS and stained with Hoescht stain and LysoTracker. Images were processed and analyzed using Nikon Imaging Software Elements and ImageJ. Pearson's correlations were calculated for individual cells in three separate images from two different experiments and then averaged (n = 30).Saporin delivery
The biotinylated transporters were incubated with ST–SAP in a 5:1 molar ratio for 20 min at ambient temperature then diluted with F-12 cell culture medium to give final conjugate solutions. CHO-K1 and pgsA cells were incubated with 100 μL of the corresponding conjugate for 4 days at 37 °C under an atmosphere of 5% CO2. CellTiter-Blue (20 μL) was added to the medium and incubated for an additional 4 h to measure viability.
Preparation of liposomes
A mixture (30 mg total) of DOPC, DOPE, and cholesterol (73:11:16) was dissolved in chloroform to a final volume of 1 mL and evaporated in a round flask and further dried under high vacuum overnight to form a thin lipid layer. The resulting film was hydrated for 15 min at 37 °C with 1 mL of PBS containing 100 μM Cy5. The mixture was sonicated for 30 s, subjected to six freeze/thaw cycles using a dry ice/acetone bath and a water bath at 37 °C. Lastly, the suspension was extruded 17 times through a polycarbonate membrane (pore size 100 nm) at room temperature. Non-encapsulated dye was removed by gravitational gel filtration (Sephadex G-50). Lipid concentration was determined adapting the Stewart method.78 Plain liposomes were diluted to 3 mg mL−1 and mixed with 10 mol% PMB or GPMB for 1 h at room temperature to obtain PMB- and GPMB-decorated liposomes, respectively. Unincorporated PMB or GPMB was removed via centrifuge gel filtration (Sephadex G-50).79
Acknowledgements
We are grateful to the W. M. Keck Foundation, the National Science Foundation (Grant No. CHE-1303554) and the National Institutes of Health (Grant No. GM077471) for supporting research in our laboratories. We thank Y. Su (Mass Spectrometry Facility, Chemistry and Biochemistry, UCSD) for help with sample analysis and K. Pestonjamasp and the UCSD Specialized Cancer Center Support Grant P30 2P30CA023100-28 for confocal microscopy.References
- R. J. Y. Ho and J. Y. Chien, J. Pharm. Sci., 2012, 101, 2668–2674 CrossRef CAS PubMed.
- H. J. Ryser, Science, 1968, 159, 390–396 CAS.
- M. Green and P. M. Loewenstein, Cell, 1988, 55, 1179–1188 CrossRef CAS PubMed.
- A. D. Frankel and C. O. Pabo, Cell, 1988, 55, 1189–1193 CrossRef CAS PubMed.
- D. A. Mann and A. D. Frankel, EMBO J., 1991, 10, 1733–1739 CAS.
- S. Fawell, J. Seery, Y. Daikh, C. Moore, L. L. Chen, B. Pepinsky and J. Barsoum, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 664–668 CrossRef CAS.
- R. B. Pepinsky, E. J. Androphy, K. Corina, R. Brown and J. Barsoum, DNA Cell Biol., 1994, 13, 1011–1019 CrossRef CAS PubMed.
- E. Vivès, P. Brodin and B. Lebleu, J. Biol. Chem., 1997, 272, 16010–16017 CrossRef.
- E. Vivès, C. Granier, P. Prevot and B. Lebleu, Letters in Peptide Science, 1997, 4, 429–436 CrossRef.
- P. A. Wender, D. J. Mitchell, K. Pattabiraman, E. T. Pelkey, L. Steinman and J. B. Rothbard, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13003–13008 CrossRef CAS PubMed.
- E. G. Stanzl, B. M. Trantow, J. R. Vargas and P. A. Wender, Acc. Chem. Res., 2013, 46, 2944–2954 CrossRef CAS PubMed.
- A. Chugh, F. Eudes and Y.-S. Shim, IUBMB Life, 2010, 62, 183–193 CrossRef CAS PubMed.
- E. Wexselblatt, J. D. Esko and Y. Tor, J. Org. Chem., 2014, 79, 6766–6774 CrossRef CAS PubMed.
- D. Mandal, A. Nasrolahi Shirazi and K. Parang, Angew. Chem., Int. Ed., 2011, 50, 9633–9637 CrossRef CAS PubMed.
- Z. Qian, T. Liu, Y.-Y. Liu, R. Briesewitz, A. M. Barrios, S. M. Jhiang and D. Pei, ACS Chem. Biol., 2013, 8, 423–431 CrossRef CAS PubMed.
- G. Lättig-Tünnemann, M. Prinz, D. Hoffmann, J. Behlke, C. Palm-Apergi, I. Morano, H. D. Herce and M. C. Cardoso, Nat. Commun., 2011, 2, 453 CrossRef PubMed.
- N. Nischan, H. D. Herce, F. Natale, N. Bohlke, N. Budisa, M. C. Cardoso and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2015, 54, 1950–1953 CrossRef CAS PubMed.
- M. Li, M. Ehlers, S. Schlesiger, E. Zellermann, S. K. Knauer and C. Schmuck, Angew. Chem., Int. Ed., 2016, 55, 598–601 CrossRef CAS PubMed.
- N. W. Luedtke, P. Carmichael and Y. Tor, J. Am. Chem. Soc., 2003, 125, 12374–12375 CrossRef CAS PubMed.
- L. Elson-Schwab, O. B. Garner, M. Schuksz, B. E. Crawford, J. D. Esko and Y. Tor, J. Biol. Chem., 2007, 282, 13585–13591 CrossRef CAS PubMed.
- A. V. Dix, L. Fischer, S. Sarrazin, C. P. H. Redgate, J. D. Esko and Y. Tor, ChemBioChem, 2010, 11, 2302–2310 CrossRef CAS PubMed.
- S. Sarrazin, B. Wilson, W. S. Sly, Y. Tor and J. D. Esko, Mol. Ther., 2010, 18, 1268–1274 CrossRef CAS PubMed.
- M. Inoue, W. Y. Tong, J. D. Esko and Y. Tor, ACS Chem. Biol., 2013, 8, 1383–1388 CrossRef CAS PubMed.
- E. Wexselblatt, J. D. Esko and Y. Tor, ACS Nano, 2015, 9, 3961–3968 CrossRef CAS PubMed.
- L. S. McCoy, K. D. Roberts, R. L. Nation, P. E. Thompson, T. Velkov, J. Li and Y. Tor, ChemBioChem, 2013, 14, 2083–2086 CrossRef CAS PubMed.
- R. E. W. Hancock, J. Antimicrob. Chemother., 1981, 8, 429–445 CrossRef CAS PubMed.
- R. E. Hancock, V. J. Raffle and T. I. Nicas, Antimicrob. Agents Chemother., 1981, 19, 777–785 CrossRef CAS PubMed.
- H. Nikaido, Microbiol. Mol. Biol. Rev., 2003, 67, 593–656 CrossRef CAS PubMed.
- H. O'Dowd, B. Kim, P. Margolis, W. Wang, C. Wu, S. L. Lopez and J. Blais, Tetrahedron Lett., 2007, 48, 2003–2005 CrossRef.
- R. L. Danner, K. A. Joiner, M. Rubin, W. H. Patterson, N. Johnson, K. M. Ayers and J. E. Parrillo, Antimicrob. Agents Chemother., 1989, 33, 1428–1434 CrossRef CAS PubMed.
- S. Chihara, T. Tobita, M. Yahata, A. Ito and Y. Koyama, Agric. Biol. Chem., 1973, 37, 2455–2463 CAS.
- S. Chihara, A. Ito, M. Yahata, T. Tobita and Y. Koyama, Agric. Biol. Chem., 1974, 38, 521–529 CrossRef CAS.
- J. M. Hyman, E. I. Geihe, B. M. Trantow, B. Parvin and P. A. Wender, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13225–13230 CrossRef CAS PubMed.
- S. Console, C. Marty, C. Garcia-Echeverria, R. Schwendener and K. Ballmer-Hofer, J. Biol. Chem., 2003, 278, 35109–35114 CrossRef CAS PubMed.
- G. Gasparini and S. Matile, Chem. Commun., 2015, 51, 17160–17162 RSC.
- J. Fu, C. Yu, L. Li and S. Q. Yao, J. Am. Chem. Soc., 2015, 137, 12153–12160 CrossRef CAS PubMed.
- D. R. Storm, K. S. Rosenthal and P. E. Swanson, Annu. Rev. Biochem., 1977, 46, 723–763 CrossRef CAS PubMed.
- T. Velkov, P. E. Thompson, R. L. Nation and J. Li, J. Med. Chem., 2010, 53, 1898–1916 CrossRef CAS PubMed.
- A. Clausell, M. Garcia-Subirats, M. Pujol, M. A. Busquets, F. Rabanal and Y. Cajal, J. Phys. Chem. B, 2007, 111, 551–563 CrossRef CAS PubMed.
- M. Wu, E. Maier, R. Benz and R. E. Hancock, Biochemistry, 1999, 38, 7235–7242 CrossRef CAS PubMed.
- P. Pristovsek and J. Kidric, J. Med. Chem., 1999, 42, 4604–4613 CrossRef CAS PubMed.
- M. Vaara, Microbiol. Rev., 1992, 56, 395–411 CAS.
- M. E. Falagas and S. K. Kasiakou, Clin. Infect. Dis., 2005, 40, 1333–1341 CrossRef CAS PubMed.
- B. Kadar, B. Kocsis, K. Nagy and D. Szabo, Curr. Med. Chem., 2013, 20, 3759–3773 CrossRef CAS PubMed.
- D. Yahav, L. Farbman, L. Leibovici and M. Paul, Clin. Microbiol. Infect., 2012, 18, 18–29 CrossRef CAS PubMed.
- R. L. Nation and J. Li, Curr. Opin. Infect. Dis., 2009, 22, 535–543 CrossRef CAS PubMed.
- K. Abdelraouf, K. T. Chang, T. J. Yin, M. Hu and V. H. Tam, Antimicrob. Agents Chemother., 2014, 58, 4200–4202 CrossRef PubMed.
- K. Abdelraouf and V. H. Tam, Antimicrob. Agents Chemother., 2014, 58, 6339 CrossRef PubMed.
- S. K. Moestrup, S. Cui, H. Vorum, S. E. Rn, K. Norris, J. Gliemann and E. I. Christensen, J. Clin. Invest., 1995, 96, 1404–1413 CrossRef CAS PubMed.
- M. A. K. Azad, K. D. Roberts, H. H. Yu, B. Liu, A. V. Schofield, S. A. James, D. L. Howard, R. L. Nation, K. Rogers, M. D. de Jonge, P. E. Thompson, J. Fu, T. Velkov and J. Li, Anal. Chem., 2015, 87, 1590–1595 CrossRef CAS PubMed.
- M. A. K. Azad, B. Yun, K. D. Roberts, R. L. Nation, P. E. Thompson, T. Velkov and J. Li, Antimicrob. Agents Chemother., 2014, 58, 6337–6338 CrossRef PubMed.
- M. P. Mingeot-Leclercq, P. M. Tulkens, S. Denamur, T. Vaara and M. Vaara, Peptides, 2012, 35, 248–252 CrossRef CAS PubMed.
- D. J. Mitchell, D. T. Kim, L. Steinman, C. G. Fathman and J. B. Rothbard, J. Pept. Res., 2000, 56, 318–325 CrossRef CAS PubMed.
- P. Viljanen and M. Vaara, Antimicrob. Agents Chemother., 1984, 25, 701–705 CrossRef CAS PubMed.
- C. Vingsbo Lundberg, T. Vaara, N. Frimodt-Moller and M. Vaara, J. Antimicrob. Chemother., 2010, 65, 981–985 CrossRef CAS PubMed.
- M. Vaara and T. Vaara, Nature, 1983, 303, 526–528 CrossRef CAS PubMed.
- J. D. Esko, T. E. Stewart and W. H. Taylor, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 3197–3201 CrossRef CAS.
- K. Lidholt, J. L. Weinke, C. S. Kiser, F. N. Lugemwa, K. J. Bame, S. Cheifetz, J. Massague, U. Lindahl and J. D. Esko, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 2267–2271 CrossRef CAS.
- M. A. West, M. S. Bretscher and C. Watts, J. Cell Biol., 1989, 109, 2731–2739 CrossRef CAS PubMed.
- M. Koivusalo, C. Welch, H. Hayashi, C. C. Scott, M. Kim, T. Alexander, N. Touret, K. M. Hahn and S. Grinstein, J. Cell Biol., 2010, 188, 547–563 CrossRef CAS PubMed.
- G. Daukas and S. H. Zigmond, J. Cell Biol., 1985, 101, 1673–1679 CrossRef CAS PubMed.
- L. H. Wang, K. G. Rothberg and R. G. Anderson, J. Cell Biol., 1993, 123, 1107–1117 CrossRef CAS PubMed.
- N. S. Dangoria, W. C. Breau, H. A. Anderson, D. M. Cishek and L. C. Norkin, J. Gen. Virol., 1996, 77(9), 2173–2182 CrossRef CAS PubMed.
- Y. Chen and L. C. Norkin, Exp. Cell Res., 1999, 246, 83–90 CrossRef CAS PubMed.
- L. Pelkmans, D. Puntener and A. Helenius, Science, 2002, 296, 535–539 CrossRef CAS PubMed.
- K. G. Rothberg, Y. S. Ying, B. A. Kamen and R. G. Anderson, J. Cell Biol., 1990, 111, 2931–2938 CrossRef CAS PubMed.
- A. Ros-Baro, C. Lopez-Iglesias, S. Peiro, D. Bellido, M. Palacin, A. Zorzano and M. Camps, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12050–12055 CrossRef CAS PubMed.
- D. J. Flavell, Curr. Top. Microbiol. Immunol., 1998, 234, 57–61 CAS.
- M. Imai, K. Inoue and S. Nojima, Biochim. Biophys. Acta, Biomembr., 1975, 375, 130–137 CrossRef CAS.
- C. C. HsuChen and D. S. Feingold, Biochemistry, 1973, 12, 2105–2111 CrossRef CAS PubMed.
- V. P. Torchilin, Biopolymers, 2008, 90, 604–610 CrossRef CAS PubMed.
- C. Marty, C. Meylan, H. Schott, K. Ballmer-Hofer and R. A. Schwendener, Cell. Mol. Life Sci., 2004, 61, 1785–1794 CAS.
- M. Furuhata, H. Kawakami, K. Toma, Y. Hattori and Y. Maitani, Bioconjugate Chem., 2006, 17, 935–942 CrossRef CAS PubMed.
- Y. L. Tseng, J. J. Liu and R. L. Hong, Mol. Pharmacol., 2002, 62, 864–872 CrossRef CAS PubMed.
- V. P. Torchilin, R. Rammohan, V. Weissig and T. S. Levchenko, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8786–8791 CrossRef CAS PubMed.
- M. M. Fretz, G. A. Koning, E. Mastrobattista, W. Jiskoot and G. Storm, Biochim. Biophys. Acta, Biomembr., 2004, 1665, 48–56 CrossRef CAS PubMed.
- S.-A. Cryan, M. Devocelle, P. J. Moran, A. J. Hickey and J. G. Kelly, Mol. Pharmaceutics, 2006, 3, 104–112 CrossRef CAS PubMed.
- J. C. Stewart, Anal. Biochem., 1980, 104, 10–14 CrossRef CAS PubMed.
- D. W. Fry, J. C. White and I. D. Goldman, Anal. Biochem., 1978, 90, 809–815 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of new compounds, uptake in other cell lines, cell viability, control experiments, and characterization of liposomes. See DOI: 10.1039/c6sc00488a |
| This journal is © The Royal Society of Chemistry 2016 |