
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA supramolecularly tunable chiral diphosphine ligand: application to Rh and Ir-catalyzed enantioselective hydrogenation†
Xi-Chang
Zhang
ab,
Yi-Hu
Hu
a,
Chuan-Fu
Chen
a,
Qiang
Fang
a,
Li-Yao
Yang
a,
Ying-Bo
Lu
a,
Lin-Jie
Xie
a,
Jing
Wu
*a,
Shijun
Li
*a and
Wenjun
Fang
b
aCollege of Material, Chemistry and Chemical Engineering, Hangzhou Normal University, Hangzhou, China. E-mail: jingwubc@hznu.edu.cn; l_shijun@hznu.edu.cn
bDepartment of Chemistry, Zhejiang University, Hangzhou, China
First published on 31st March 2016
Abstract
A supramolecularly tunable chiral bisphosphine ligand bearing two pyridyl-containing crown ethers, (−) or (+)-Xyl-P16C6-Phos, was fabricated and utilized in the Rh-catalyzed asymmetric hydrogenation of α-dehydroamino acid esters and Ir-catalyzed asymmetric hydrogenation of quinolines in high yields with excellent enantioselectivities (90–99% ee). Up to a 22% enhancement in enantioselectivity was achieved by the addition of certain amounts of alkali ions (Li+, Na+ or K+), which could be selectively recognized and effectively complexed by the crown ethers on the chiral Xyl-P16C6-Phos.
Introduction
The design and synthesis of new chiral ligands play a pivotal role in the field of transition metal-catalyzed asymmetric reactions.1 It is well known that the performance of transition metal catalysts can be remarkably affected by subtle variations in either the geometric or electronic properties of chiral ligands. For instance, when chiral atropisomeric diphosphines2 are used as ligands, without altering the backbone structure, the catalytic properties can be tuned by attaching various P-substituents. Another strategy is the development of diphosphine ligands with adjustable dihedral angles,2d,e,3 such as o-Ph-hexaMeO-BIPHEP,4 Cn-TunaPhos5 and PQ-Phos,6 for different substrates or reactions. Although good-to-excellent results were obtained in some asymmetric catalytic reactions, these ligands often required individual and unique syntheses and thus are not really tunable by simply changing the catalytic reaction conditions.In the past decade, supramolecular chiral catalysis has attracted growing attention and fascination has been exhibited in both forming catalyst libraries and mimicking enzymes to achieve unexpected activities and stereoselectivities in relevant reactions.7 Some elegant supramolecular or combinatorial chiral catalyst systems have been designed, such as self-assembled bidentate ligands via intermolecular hydrogen bonding,8 ion pair catalysts,9 Co(II)–salen with a hydrogen-bonding network,10 bidentate ligands based on Zn(II) porphyrins,11 Zn(II) salphen12 or Zn(box)2 complexes13via metal–ligand interactions, supramolecular ligands based on a crown-ether appended phosphate or phosphoramidite skeleton,14,15 and bisphosphite ligands with oligo(ethylene glycol) as backbones,16 as well as self-supported chiral catalysts with coordination polymer frameworks.17
Employing supramolecular methods to impart tunable conformations, steric bulk and electronic properties to a given atropisomeric diphosphine is conceptually appealing since it could avoid the aforementioned individual tedious procedures associated with traditional syntheses of covalent bonds for acquiring enantiomerically pure ligands. To the best of our knowledge, examples of tunable ligands or catalysts illustrating this concept remained rare. Herein, we describe our research on introducing crown ethers, as host sites for recognition, to the scaffold of a well-established chiral dipyridylphosphine ligand Xyl-P-Phos2c,18,19 to form new ligands, (−) or (+)-Xyl-P16C6-Phos ((−) or (+)-7, Scheme 1). Utilizing the selective recognition and strong complexation between the crown ethers on 7 and different alkali cations,20 supramolecularly tunable chiral catalysts (Scheme 2) have been constructed and applied in both a Rh-catalyzed asymmetric hydrogenation of α-dehydroamino acid derivatives and an Ir-catalyzed asymmetric hydrogenation of quinolines.
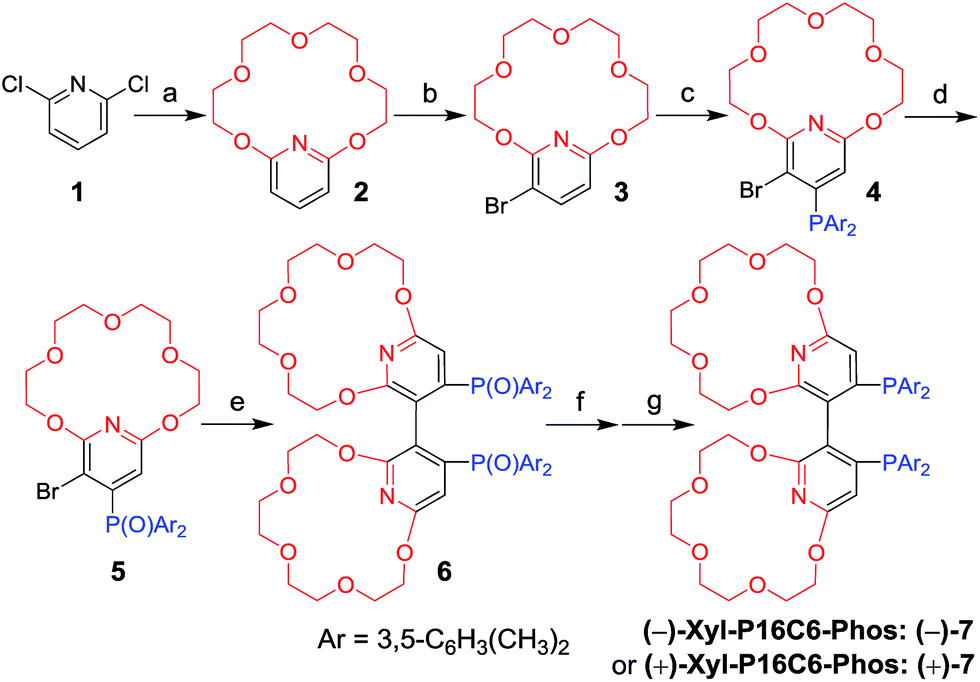 | ||
| Scheme 1 Synthesis of (−) or (+)-Xyl-P16C6-Phos (7). Reagents and conditions: (a) tetraethylene glycol, NaH, KPF6, THF, reflux, 65% yield; (b) NBS, CH2Cl2, −78 °C, 85% yield; (c) LDA, THF, then (3,5-Me2C6H3)2PCl, −78 °C, 73% yield; (d) H2O2, acetone, 0 °C, 85% yield; (e) Cu, DMF, 140 °C, 50% yield; (f) (i) (L or D)-DBTA, EtOAc/CHCl3, (ii) NaOH aq., 54–55% yield for two steps; (g) HSiCl3, Et3N, toluene, reflux, 87% yield for (−)-7, 91% yield for (+)-7. | ||
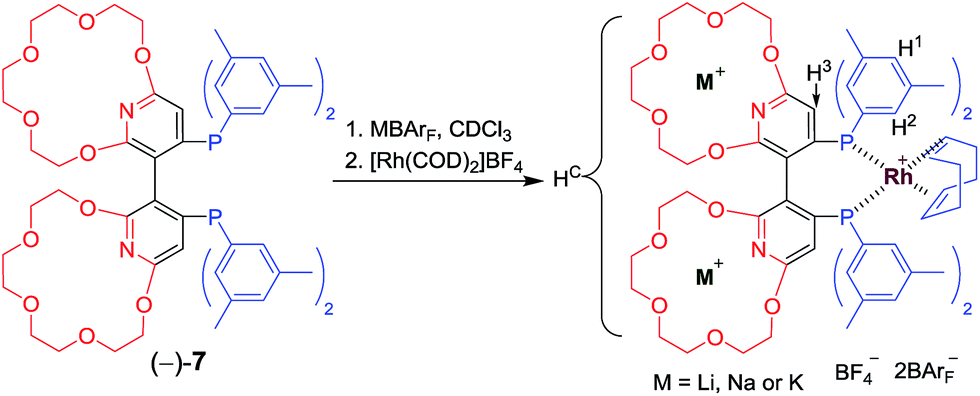 | ||
| Scheme 2 Preparation of Rh complexes with alkali ions. | ||
Results and discussion
The new ligand Xyl-P16C6-Phos (7) was synthesized as shown in Scheme 1.18 The cyclization of tetraethylene glycol and 2,6-dichloropyridine (1) afforded 2,6-pyrido-16-crown-6 (2), which was then brominated to yield 3. A regioselective lithiation of 3 followed by substitution with di(3,5-dimethylphenyl)phosphine chloride produced 4 in 73% yield. The oxidation of 4 furnished 5, which was further converted to the racemic diphosphine oxide 6via the copper-mediated Ullman coupling protocol. The resolution of racemate (±)-6 was realized by the use of (D) or (L)-O,O′-dibenzoyltartaric acid (DBTA). The enantiomerically pure (−) or (+)-6 was then reduced to the targeted atropisomeric ligand (−) or (+)-7, respectively.With the new, crown ether-attached chiral diphosphine ligand 7 in hand, the complexation of (−)-7 with alkali ions and coordination with [Rh(COD)2]BF4 (Scheme 2) were systematically investigated by 1H and 31P NMR spectroscopy. As compared with the 1H NMR spectrum of (−)-7 (Fig. 1a), the spectra of [(−)-7 + [Rh(COD)2]BF4] (Fig. 1b) and [(−)-7 + [Rh(COD)2]BF4 + MBArF (M = Li, Na or K, BArF− = (3,5-(CF3)2C6H3)4B−)] (Fig. 1c–e) displayed significant differences in either chemical shifts or peak shapes. After coordination with [Rh(COD)2]BF4, the signal of the H3 protons on the pyridyl group shifted downfield sharply (Fig. 1bvs.1a). Upon further binding alkali ions, the chemical shift of H3 changed slightly (Fig. 1c–evs.1b), while the signals of oxyethylene protons (Hc) changed as expected (Fig. 1c–evs.1a and 1b), owing to the complexation between crown ethers and alkali ions. The peaks of the oxyethylene protons exhibited marginal differences due to the effects of the different sizes and electronic properties of the alkali ions. The 31P NMR spectra also showed substantial shifts upon coordination of (−)-7 with [Rh(COD)2]BF4 (ESI, Fig. S1†). The lone singlet peak of the free ligand split into a doublet due to the coupling between Rh–P (Fig. S1b–e vs. S1a†). The obvious changes of both chemical shift and peak shapes in the 1H NMR spectra of [(−)-7 + NaBArF] also directly confirmed the complexation between (−)-7 and alkali ions (Fig. S2c vs. S2a†). Moreover, with the molar ratio of NaBArF to ligand increased from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 10:1, the 1H NMR peaks of the oxyethylene protons and 31P NMR spectra did not show distinct changes (Fig. S2e–g vs. S2d, S3e and S3f vs. S3d†). Additionally, the stoichiometries of the complexes of (±)-7 with LiBArF and (±)-6 with NaBArF or KBArF in solution were all determined to be 1:2 by Job plots using proton NMR data (Fig. S4–S6†). This binding ratio was further confirmed by low-resolution electrospray ionization mass spectroscopy (Fig. S7–S9†).
:1 to 10:1, the 1H NMR peaks of the oxyethylene protons and 31P NMR spectra did not show distinct changes (Fig. S2e–g vs. S2d, S3e and S3f vs. S3d†). Additionally, the stoichiometries of the complexes of (±)-7 with LiBArF and (±)-6 with NaBArF or KBArF in solution were all determined to be 1:2 by Job plots using proton NMR data (Fig. S4–S6†). This binding ratio was further confirmed by low-resolution electrospray ionization mass spectroscopy (Fig. S7–S9†).
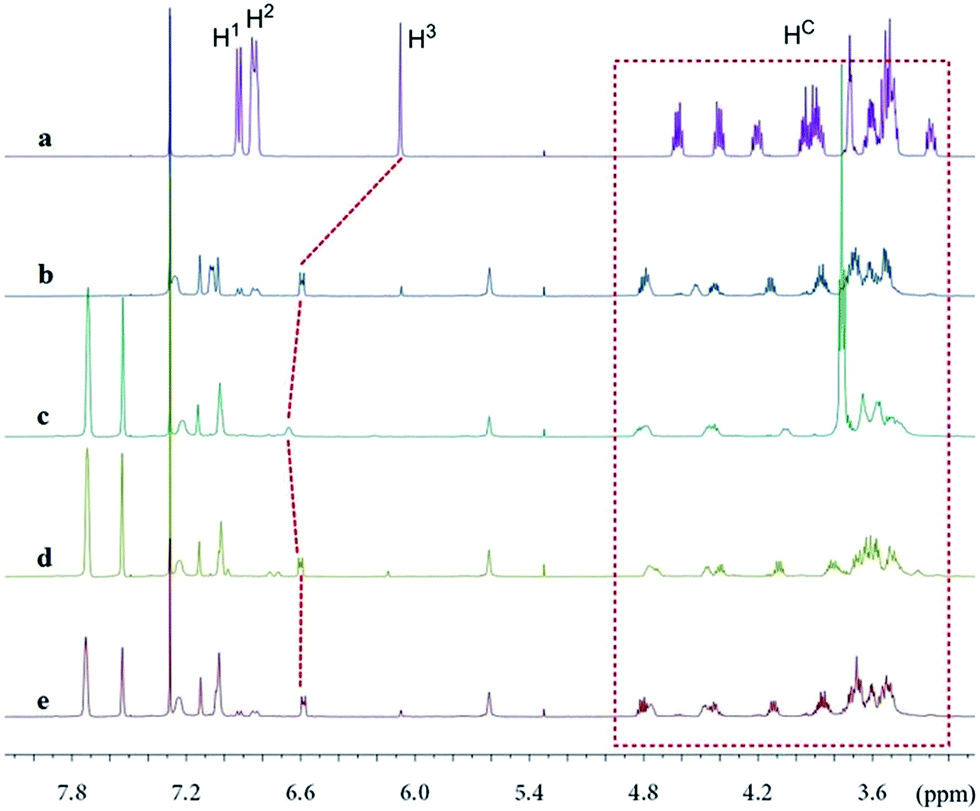 | ||
| Fig. 1 Partial 1H NMR (500 MHz, CDCl3) spectra of (a) (−)-7; (b) (−)-7 + [Rh(COD)2]BF4 (1:1 molar ratio); (c) (−)-7 + [Rh(COD)2]BF4 + LiBArF (1:1:2 molar ratio); (d) (−)-7 + [Rh(COD)2]BF4 + NaBArF (1:1:2 molar ratio); (e) (−)-7 + [Rh(COD)2]BF4 + KBArF (1:1:2 molar ratio). | ||
After the self-assembled chiral Rh complexes of 7 were prepared, these complexes were applied as catalysts for the asymmetric hydrogenation of α-dehydroamino acid derivatives to assess the host–guest effect between alkali ions and crown ethers on the catalytic reactions. The [Rh((−)-7)(COD)]BF4 complex was an effective catalyst for the enantioselective hydrogenation of the model substrate methyl-(Z)-2-acetamido-cinnamate (8a). The reaction proceeded smoothly in CH2Cl2 at an ambient temperature with 1 atm of initial H2 pressure for 6 h, resulting in full conversion to the chiral product (R)-9a with 82% ee (Table 1, entry 1).
| Entry | Additive [mol%] | Convb [%] | eec [%] |
|---|---|---|---|
| a Reaction conditions: 0.05–0.09 mol L−1 in CH2Cl2. b The conversions were determined by NMR and GC analysis. c The ee values were determined by chiral GC analysis. d (S)-Xyl-P-Phos was used as a ligand. e The reaction was carried out at 0 °C. | |||
| 1 | None | >99 | 82 |
| 2d | None | >99 | 88 |
| 3 | NaBArF (1) | >99 | 84 |
| 4 | NaBArF (2) | >99 | 88 |
| 5 | NaBArF (5) | >99 | 91 |
| 6 | LiBArF (2) | >99 | 84 |
| 7 | LiBArF (5) | >99 | 84 |
| 8 | KBArF (2) | >99 | 86 |
| 9 | KBArF (5) | >99 | 90 |
| 10 | NaBArF (10) | >99 | 93 |
| 11e | NaBArF (10) | >99 | 93 |
| 12 | NaBF4 (10) | 68 | 81 |
| 13 | NaPF6 (10) | 96 | 83 |
| 14 | (nBu)4NBArF (10) | 91 | 84 |
| 15d | NaBArF (10) | >99 | 88 |
Interestingly, the addition of certain amounts of alkali salts MBArF (M = Li, Na, K) as guests for the crown ethers of 7 had an obviously beneficial influence on the enantioselectivities of the catalysts while no decrease in activities was observed (Table 1, entries 3–10 vs. entry 1). Na+ or K+ appeared to be the preferential choice of cationic additives in terms of enantioselectivities (entries 4, 5, 8 and 9 vs. entries 6 and 7). Elevated ratios of alkali salts to (−)-7 facilitated an enhancement in the enantiomeric purity of the product (entries 4 and 5 vs. entry 3, entry 9 vs. entry 8). Finally, upon increasing the NaBArF loading to 10 mol%, 93% ee was achieved (entry 10). A lower reaction temperature (0 °C) did not render a higher ee (entry 11 vs. entry 10). Next, when using 10 mol% of Na+ as guest, various counter-ions were tested and the results indicated that the presence of a sterically bulkier and more weakly coordinating anion BArF− possessed superior levels of activity and asymmetric induction to those of BF4− and PF6− (entries 12 and 13 vs. entry 10), probably owing to the impact of anions on the complexation between crown ethers and Na+.
It is well-known that counter-ions may affect the outcomes of transition metal-mediated asymmetric reactions.19b,21 In order to investigate the contribution of anions on the present catalytic system, (nBu)4NBArF was used as a control additive (Table 1, entry 14), wherein the much more sterically demanding (nBu)4N+ cation could not complex with the pyridyl-containing crown ethers. When replacing Na+ with the (nBu)4N+ cation, only a slight increase in ee was observed (entry 14 vs. entry 1), which demonstrated that the host–guest interaction between (−)-7 and the alkali ions played a crucial role in the increase of the stereoselectivities. Additionally, a side by side comparison study showed that neither cationic or anionic additives had a pronounced effect on the reaction outcomes in the case of (S)-Xyl-P-Phos as the chiral ligand (entry 15 vs. entry 2).
Moreover, the present self-assembled catalyst system was strongly solvent-dependent (Table 2) and nonpolar c-hexane was much more effective for both the reactivity and enantioselectivity than other solvents such as CH2Cl2, MeOH and THF. Thus, 97% ee and full conversion were attained when the reaction was carried out in c-hexane (Table 2, entry 8), probably due to the stronger association between Na+ and (−)-7 in nonpolar solvents. In the absence of alkali additives, (R)-9a was furnished in 82% conversion with only 77% ee (Table 2, entry 9 vs. entry 8) in c-hexane under otherwise identical reaction conditions, which further confirmed the impact of the host–guest interaction between Na+ cations and crown ethers on the asymmetric induction.
|
|
|||
|---|---|---|---|
| Entry | Solvent | Convb [%] | eec [%] |
| a Reaction conditions: 0.05–0.09 mol L−1 in solvent. b The conversions were determined by NMR and GC analysis. c The ee values were determined by chiral GC analysis. The absolute configuration was determined by comparing the retention times with the known data. d n.d. = Not determined. e The isolated yield was 94%. f No NaBArF was added. | |||
| 1 | CH2Cl2 | >99 | 93 |
| 2 | Acetone | <2 | n.d.d |
| 3 | MeOH | <10 | n.d.d |
| 4 | Toluene | >99 | 96 |
| 5 | THF | >99 | 90 |
| 6 | Ethyl acetate | >99 | 90 |
| 7 | n-Hexane | 97 | 96 |
| 8e | c-Hexane | >99 | 97 |
| 9f | c-Hexane | 82 | 77 |
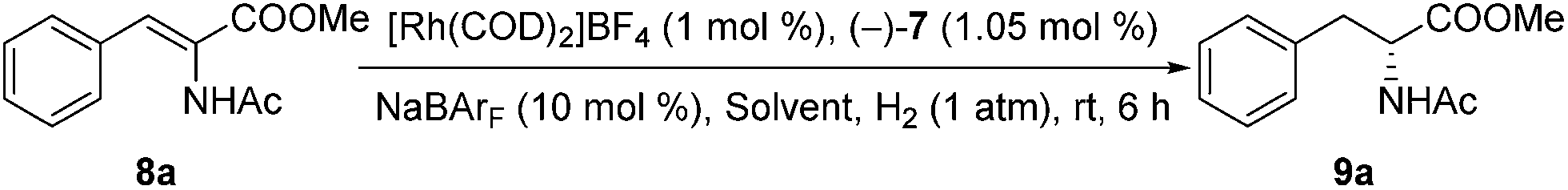
Having identified the optimized conditions, we set out to evaluate the general applicability of the self-assembled catalyst. As the findings in Scheme 3 depict, the asymmetric hydrogenation of a wide assortment of α-dehydroamino acid esters 8b–t proceeded to afford desired products 9b–t neatly in c-hexane under 1 atm of H2 at room temperature with excellent catalytic activities and enantioselectivities (95–99% ee). These results showed that the present supramolecular catalyst system was rather competitive in comparison with similar catalytic systems.16a,c,19a Further comparison between catalysts with or without the alkali salts for a few selected substrates indicated that only poor or moderate conversions and enantioselectivities were acquired when no NaBArF was added (Scheme 3, 9b, 9d, 9g, and 9m). For example, in the absence of Na+ as additives, 8d was hydrogenated to (R)-9d in only 33% conversion with 76% ee, whereas, full conversion and up to 22% enhancement in ee were achieved when 10 mol% NaBArF was added under otherwise identical conditions (Scheme 3).
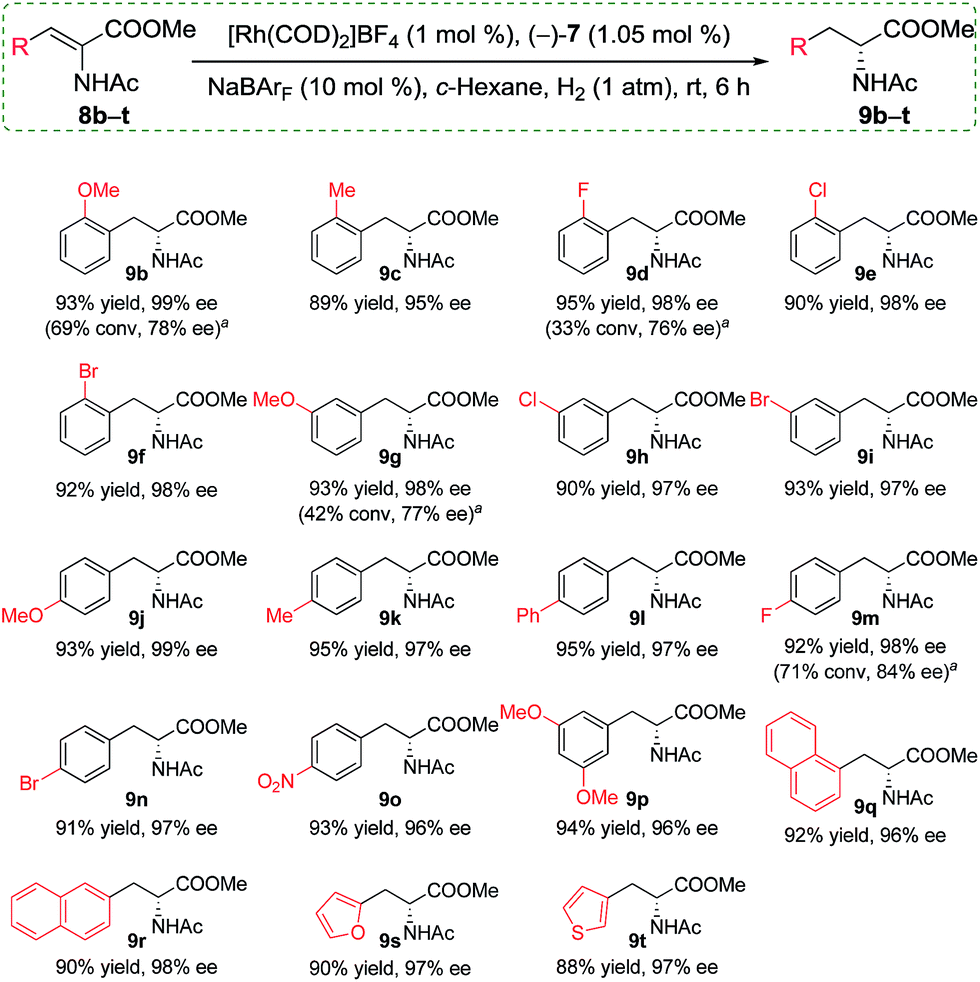 | ||
| Scheme 3 Asymmetric hydrogenation of α-dehydroamino acid esters 8b–t with the self-assembled chiral Rh catalyst. Reaction conditions: 0.05–0.09 mol L−1 in c-hexane; >99% conversions were observed in all cases. aNo NaBArF was added. | ||
Optically active tetrahydroquinoline derivatives are important synthetic intermediates and structural units for some natural products and biologically active compounds.22 The enantioselective hydrogenation of heteroaromatic compounds is a great challenge as harsh reaction conditions are usually necessary to overcome the aromaticity of the substrates. Although the asymmetric hydrogenation of aromatic compounds has been explored since 1987,23 there are but several elegant catalytic systems with high activity and enantioselectivity.24 As such, we were interested in the extension of our self-assembled catalyst design to the Ir-catalyzed asymmetric hydrogenation of substituted quinolines (Tables 3 and 4).
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Additive [mol%] | Convb [%] | eec [%] |
| a Reaction conditions: 0.2 mol L−1 in solvent. b The conversions were determined by NMR and GC analysis. c The ee values were determined by chiral HPLC analysis and the absolute configuration was determined by comparing the rotation sign with the reported data in the literature. d The isolated yield was 96%. | ||||
| 1 | c-Hexane | NaBArF (10) | 37 | 93 |
| 2 | CH2Cl2 | NaBArF (10) | 87 | 95 |
| 3 | Ethyl acetate | NaBArF (10) | 98d | 97 |
| 4 | Ethyl acetate | None | 97 | 87 |
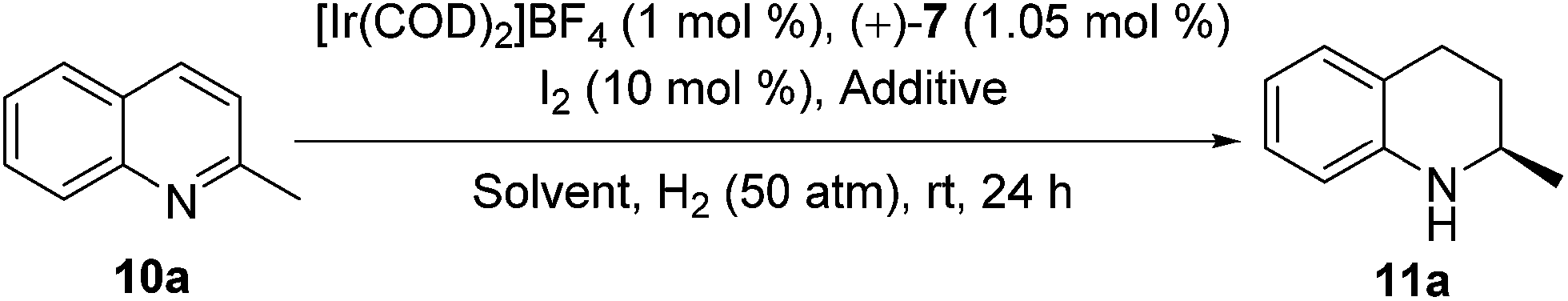
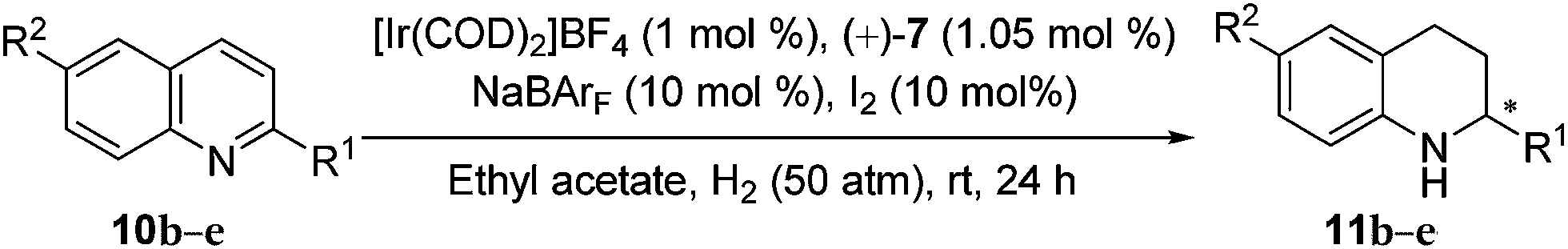
As the data in entry 3 of Table 3 indicate, when the model substrate 2-methylquinoline (10a) was treated with the self-assembled Ir catalyst generated from [Ir(COD)2]BF4, (+)-7 and NaBArF (1:1:10 molar ratio, Fig. S10 and S11†), the hydrogenation completed in ethyl acetate in the presence of 10 mol% of I2 under 50 atm of H2 at ambient temperature in 24 h furnished the chiral product in 96% yield and 97% ee, which were superior to those obtainable in the case of other solvents, such as c-hexane and CH2Cl2 (entry 3 vs. entries 1 and 2). In contrast, 87% ee was obtained if no NaBArF was added (entry 4 vs. entry 3). Further investigations (Table 4) showed that the present self-assembled Ir catalyst system gave rise to the formation of chiral tetrahydroquinolines 11b–e of high optical purities (90–97% ee) in quantitative yields (96–98%).
Conclusions
In conclusion, a supramolecularly tunable chiral bisphosphine ligand bearing a two pyridyl-containing crown ethers, (−) or (+)-Xyl-P16C6-Phos, was synthesized and successfully applied in the Rh-catalyzed asymmetric hydrogenation of α-dehydroamino acid esters and Ir-catalyzed asymmetric hydrogenation of quinolines, in the presence of certain amounts of alkali ions, in quantitative yields and with excellent enantioselectivities (90–99% ee). By finely regulating the host–guest interactions between the crown ethers of the chiral ligand and the alkali ion additives, up to 22% enhancement in enantioselectivities was achieved in comparison with those obtainable from the catalyst systems in the absence of the crown ether/alkali-metal ion recognition motifs. Studies aimed at elucidating the reaction mechanism and expanding these supramolecular catalysts to other asymmetric reactions are in progress in our laboratory.Acknowledgements
We thank the National Natural Science Foundation of China (21172049, 91127010, 21032003, and 21572042), the Program for Innovative Research Team in Chinese University (IRT 1231), the Zhejiang Provincial Natural Science Foundation of China (LZ13B030001 and LZ16B020002), the Public Welfare Technology and Application Program (2015C31141) and the Special Funds for Key Innovation Team of Zhejiang Province (2010R50017).Notes and references
- (a) Asymmetric Catalysis in Organic Synthesis, ed. R. Noyori, Wiley, New York, 1994 Search PubMed; (b) Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, Berlin, Germany, 1999 Search PubMed; (c) G. Q. Lin, Y. M. Li and A. S. C. Chan, Principles and Applications of Asymmetric Synthesis, Wiley & Sons, New York, 2001 CrossRef; (d) Catalytic Asymmetric Synthesis, ed. I. Ojima, John Wiley & Sons, New Jersey, 3rd edn, 2010 Search PubMed.
- For some reviews, see: (a) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029–3070 CrossRef CAS PubMed; (b) H. Shimizu, I. Nagasaki and T. Saito, Tetrahedron, 2005, 61, 5405–5432 CrossRef CAS; (c) J. Wu and A. S. C. Chan, Acc. Chem. Res., 2006, 39, 711–720 CrossRef CAS PubMed; (d) W. Zhang, Y. Chi and X. Zhang, Acc. Chem. Res., 2007, 40, 1278–1290 CrossRef CAS PubMed; (e) H. Shimizu, I. Nagasaki, K. Matsumura, N. Sayo and T. Saito, Acc. Chem. Res., 2007, 40, 1385–1393 CrossRef CAS PubMed.
- (a) T. Saito, T. Yokozawa, T. Ishizaki, T. Moroi, N. Sayo, T. Miura and H. Kumobayashi, Adv. Synth. Catal., 2001, 343, 264–267 CrossRef CAS; (b) P. Dierkes and P. W. N. M. Van Leeuwen, J. Chem. Soc., Dalton Trans., 1999, 1519–1530 RSC; (c) L. A. Van der Veen, M. D. K. Boele, F. R. Bregman, P. C. J. Kamer, P. W. N. M. Van Leeuwen, K. Goubitz, J. Fraanje, H. Schenk and C. Bo, J. Am. Chem. Soc., 1998, 120, 11616–11626 CrossRef CAS.
- W. Tang, Y. Chi and X. Zhang, Org. Lett., 2002, 4, 1695–1698 CrossRef CAS PubMed.
- (a) Z. Zhang, H. Qian, J. Longmire and X. Zhang, J. Org. Chem., 2000, 65, 6223–6226 CrossRef CAS PubMed; (b) S. Wu, W. Wang, W. Tang, M. Lin and X. Zhang, Org. Lett., 2002, 4, 4495–4497 CrossRef CAS PubMed.
- L. Q. Qiu, F. Y. Kwong, J. Wu, W. H. Lam, S. S. Chan, W.-Y. Yu, Y.-M. Li, R. W. Guo, Z. Y. Zhou and A. S. C. Chan, J. Am. Chem. Soc., 2006, 128, 5955–5965 CrossRef CAS PubMed.
- For some recent books, reviews and selected examples on supramolecular catalysis, see: (a) P. W. N. M. van Leeuwen, Supramolecular Catalysis, Wiley-VCH, 2008 Search PubMed; (b) B. Breit, Angew. Chem., Int. Ed., 2005, 44, 6816–6825 CrossRef CAS PubMed; (c) P. E. Goudriaan, P. W. N. M. van Leeuwen, M. N. Birkholz and J. N. H. Reek, Eur. J. Inorg. Chem., 2008, 19, 2939–2958 CrossRef; (d) J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615–621 CrossRef CAS PubMed; (e) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1660–1733 RSC; (f) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC; (g) D. A. Leigh, V. Marcos and M. R. Wilson, ACS Catal., 2014, 4, 4490–4497 CrossRef CAS; (h) K. Ohmatsu and T. Ooi, Tetrahedron Lett., 2015, 56, 2043–2048 CrossRef CAS; (i) V. Blanco, D. A. Leigh and V. Marcos, Chem. Soc. Rev., 2015, 44, 5341–5370 RSC; (j) J.-B. Wang and B. L. Feringa, Science, 2011, 331, 1429–1432 CrossRef CAS PubMed; (k) V. Blanco, A. Carlone, K. D. Hanni and D. A. Leigh, Angew. Chem., Int. Ed., 2012, 51, 5166–5169 CrossRef CAS PubMed; (l) M. Schmittel, S. De and S. Pramanik, Angew. Chem., Int. Ed., 2012, 51, 3832–3836 CrossRef CAS PubMed; (m) S. De, S. Pramanik and M. Schmittel, Angew. Chem., Int. Ed., 2014, 53, 14255–14259 CrossRef CAS PubMed; (n) V. Blanco and D. A. Leigh, J. Am. Chem. Soc., 2014, 136, 4905–4908 CrossRef CAS PubMed.
- (a) B. Breit and W. Seiche, J. Am. Chem. Soc., 2003, 125, 6608–6609 CrossRef CAS PubMed; (b) Y. Liu, C. A. Sandoval, Y. Yamaguchi, X. Zhang, Z. Wang, K. Kato and K. Ding, J. Am. Chem. Soc., 2006, 128, 14212–14213 CrossRef CAS PubMed; (c) J. Wieland and B. Breit, Nat. Chem., 2010, 2, 832–837 CrossRef CAS PubMed; (d) V. Agabekov, W. Seiche and B. Breit, Chem. Sci., 2013, 4, 2418–2422 RSC.
- (a) D. Uraguchi, Y. Ueki and T. Ooi, Science, 2009, 326, 120–123 CrossRef CAS PubMed; (b) F. W. Patureau, C. Worch, M. A. Siegler, A. L. Spek, C. Bolm and J. N. H. Reek, Adv. Synth. Catal., 2012, 354, 59–64 CrossRef CAS.
- J. Park, K. Lang, K. A. Abboud and S. Hong, J. Am. Chem. Soc., 2008, 130, 16484–16485 CrossRef CAS PubMed.
- (a) V. F. Slagt, M. Roder, P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, J. Am. Chem. Soc., 2004, 126, 4056–4057 CrossRef CAS PubMed; (b) T. Besset, D. W. Norman and J. N. H. Reek, Adv. Synth. Catal., 2013, 355, 348–352 CAS.
- (a) M. Kuil, P. E. Goudriaan, P. W. N. M. van Leeuwen and J. N. H. Reek, Chem. Commun., 2006, 4679–4681 RSC; (b) T. Gadzikwa, R. Bellini, H. L. Dekker and J. N. H. Reek, J. Am. Chem. Soc., 2012, 134, 2860–2863 CrossRef CAS PubMed.
- (a) J. M. Takacs, D. S. Reddy, S. A. Moteki, D. Wu and H. Palencia, J. Am. Chem. Soc., 2004, 126, 4494–4495 CrossRef CAS PubMed; (b) N. C. Thacker, S. A. Moteki and J. M. Takacs, ACS Catal., 2012, 2, 2743–2752 CrossRef CAS PubMed.
- G. Hattori, T. Hori, Y. Miyake and Y. Nishibayashi, J. Am. Chem. Soc., 2007, 129, 12930–12931 CrossRef CAS PubMed.
- Y. Li, Y. Feng, Y. M. He, F. Chen, J. Pan and Q. H. Fan, Tetrahedron Lett., 2008, 49, 2878–2881 CrossRef CAS.
- (a) Y. Li, B. Ma, Y. He, F. Zhang and Q.-H. Fan, Chem.–Asian J., 2010, 5, 2454–2458 CrossRef CAS PubMed; (b) G.-H. Ouyang, Y.-M. He and Q.-H. Fan, Chem.–Eur. J., 2014, 20, 16454–16457 CrossRef CAS PubMed; (c) G.-H. Ouyang, Y.-M. He, Y. Li, J.-F. Xiang and Q.-H. Fan, Angew. Chem., Int. Ed., 2015, 54, 4334–4337 CrossRef CAS PubMed; (d) A. Vidal-Ferran, I. Mon, A. Bauzá, A. Frontera and L. Rovira, Chem.–Eur. J., 2015, 21, 11417–11426 CrossRef CAS PubMed.
- (a) Z. Wang, G. Chen and K. Ding, Chem. Rev., 2009, 109, 322–359 CrossRef CAS PubMed; (b) L. Yu, Z. Wang, J. Wu, S. Tu and K. Ding, Angew. Chem., Int. Ed., 2010, 49, 3627–3630 CrossRef CAS PubMed.
- Xyl-P-Phos = 2,2′,6,6′-tetramethoxy-4,4′-bis[di(3,5-dimethyl-phenyl)phosphino]-3,3′-3,3-phosphino]-3,3-tetramethoxy-4,4′-bis-[di(3,5-dimethyl-phenyl)phosphino], J. Wu, H. Chen, W.-H. Kwok, C.-H. K.-H. Lam, Z.-Y. Zhou, C.-H. Yeung and A. S. C. Chan, Tetrahedron Lett., 2002, 43, 1539–1543 CrossRef CAS.
- For some representative applications of Xyl-P-Phos ligand in asymmetric catalytic reactions: (a) J. Wu, C. C. Pai, W. H. Kwok, R. W. Guo, T. T. L. Au-Yeung, C. H. Yeung and A. S. C. Chan, Tetrahedron: Asymmetry, 2003, 14, 987–992 CrossRef CAS; (b) J. Wu, J. X. Ji and A. S. C. Chan, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3570–3575 CrossRef CAS PubMed; (c) X.-C. Zhang, Y. Wu, F. Yu, F.-F. Wu, J. Wu and A. S. C. Chan, Chem.–Eur. J., 2009, 15, 5888–5891 CrossRef CAS PubMed; (d) F. Yu, X.-C. Zhang, F.-F. Wu, J.-N. Zhou, W. Fang, J. Wu and A. S. C. Chan, Org. Biomol. Chem., 2011, 9, 5652–5654 RSC; (e) F.-F. Wu, J.-N. Zhou, Q. Fang, Y.-H. Hu, S. Li, X.-C. Zhang, A. S. C. Chan and J. Wu, Chem.–Asian J., 2012, 7, 2527–2530 CrossRef CAS PubMed; (f) M. Li, B. Li, H.-F. Xia, D. Ye, J. Wu and Y. Shi, Green Chem., 2014, 16, 2680–2688 RSC.
- (a) M. Ewcomb, J. M. Timko, D. M. Walba and D. J. Cram, J. Am. Chem. Soc., 1977, 99, 6392–6398 CrossRef; (b) D. J. Cram and J. M. Cram, Acc. Chem. Res., 1978, 11, 8–14 CrossRef CAS; (c) B. M. Trost and R. Radinov, J. Am. Chem. Soc., 1997, 119, 5962–5963 CrossRef CAS.
- (a) I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef CAS PubMed; (b) K. Kallstrom, I. Munslow and P. G. Andersson, Chem.–Eur. J., 2006, 12, 3194–3200 CrossRef PubMed; (c) R. Schwenk and A. Togni, Dalton Trans., 2015, 44, 19566–19575 RSC.
- D. H. Barton, K. Nakanishi and O. Meth-Cohn, in Comprehensive Natural Products Chemistry, Elsevier, Oxford, 1999, vol. 1–9 Search PubMed.
- (a) C. W. Bird, Tetrahedron, 1992, 48, 335–340 CrossRef CAS; (b) S. Murata, T. Sugimoto and S. Matsuura, Heterocycles, 1987, 26, 763–766 CrossRef CAS.
- (a) W.-B. Wang, S.-M. Lu, P.-Y. Yang, X.-W. Han and Y.-G. Zhou, J. Am. Chem. Soc., 2003, 125, 10536–10537 CrossRef CAS PubMed; (b) Y.-G. Zhou, Acc. Chem. Res., 2007, 40, 1357–1366 CrossRef CAS PubMed; (c) R. Kuwano, Heterocycles, 2008, 76, 909–922 CrossRef CAS; (d) D.-S. Wang, Q.-A. Chen, S.-M. Lu and Y.-G. Zhou, Chem. Rev., 2012, 112, 2557–2590 CrossRef CAS PubMed; (e) C.-X. Zhuo, W. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 12662–12686 CrossRef PubMed; (f) L. Shi, Z.-S. Ye, L.-L. Cao, R.-N. Guo, Y. Hu and Y.-G. Zhou, Angew. Chem., Int. Ed., 2012, 51, 8286–8289 CrossRef CAS PubMed; (g) T. Nagano, A. Iimuro, R. Schwenk, T. Ohshima, Y. Kita, A. Togni and K. Mashima, Chem.–Eur. J., 2012, 18, 11578–11592 CrossRef CAS PubMed; (h) S. Urban, B. Beiring, N. Ortega, D. Paul and F. Glorius, J. Am. Chem. Soc., 2012, 134, 15241–15244 CrossRef CAS PubMed; (i) A. Iimuro, K. Yamaji, S. Kandula, T. Nagano, Y. Kita and K. Mashima, Angew. Chem., Int. Ed., 2013, 52, 2046–2050 CrossRef CAS PubMed; (j) Z.-S. Ye, R.-N. Guo, X.-F. Cai, M.-W. Chen, L. Shi and Y.-G. Zhou, Angew. Chem., Int. Ed., 2013, 52, 3685–3689 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data for the prepared compounds. See DOI: 10.1039/c6sc00589f |
| This journal is © The Royal Society of Chemistry 2016 |