
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembled, nanostructured coatings for water oxidation by alternating deposition of Cu-branched peptide electrocatalysts and polyelectrolytes†
Enikő
Farkas
ab,
Dávid
Srankó
c,
Zsolt
Kerner
c,
Bartosz
Setner
d,
Zbigniew
Szewczuk
d,
Wiesław
Malinka
e,
Robert
Horvath
 *a,
Łukasz
Szyrwiel
*ef and
József S.
Pap
*c
*a,
Łukasz
Szyrwiel
*ef and
József S.
Pap
*c
aNanobiosensorics Group, MTA Centre for Energy Research – MFA, H-1121 Budapest, Hungary. E-mail: horvathr@mfa.kfki.hu
bDoctoral School of Molecular- and Nanotechnologies, Faculty of Information Technology, University of Pannonia, Egyetem u. 10, H-8200 Veszprém, Hungary
cSurface Chemistry and Catalysis Department, MTA Centre for Energy Research, Konkoly Thege str. 29-33, H-1121 Budapest, Hungary. E-mail: pap.jozsef@energia.mta.hu
dFaculty of Chemistry, Univ. of Wrocław, ul. F. Joliot-Curie 14, 50-383 Wrocław, Poland
eDept of Chemistry of Drugs, Wrocław Medical Univ., ul. Borowska 211, 50-552 Wrocław, Poland. E-mail: lukszyr@wp.pl
fCNRS/UPPA, LCABIE, UMR5254, Helioparc, 2, av. Pr. Angot, Pau, F-64053, France
First published on 21st April 2016
Abstract
This work demonstrates the heterogenization of homogeneous water oxidation electrocatalysts in surface coatings produced by combining the substances with a suitable polyelectrolyte. The electrocatalysts i.e. Cu(II)-branched peptide complexes involving a 2,3-L-diaminopropionic acid junction unit are heterogenized by building composite layers on indium-tin-oxide (ITO) electrode surface. Alternating deposition of the peptide complexes and poly(L-lysine) or poly(allylamine hydrochloride) were carried out in the presence of phosphate in a pH range of 7.5–10.5. Discussion of the results is divided to (1) characteristics of composite layer buildup and (2) electrocatalytic water oxidation and accompanying changes of these layers. For (1), optical waveguide lightmode spectroscopy (OWLS) has been applied to reveal the layer-by-layer formation of a Cu-ligand/polyelectrolyte/phosphate coating. The fabricated structures had a nanoporous topography (atomic force microscopy). As for (2), electrochemistry employing coated ITO substrates indicated improved water oxidation electrocatalysis vs. neat ITO and dependence of this improvement on the presence or absence of a histidine ligand in the deposited Cu(II)-complexes equally, as observed in homogeneous systems. Electrochemical OWLS revealed changes in the coatings in operando, upon alternating positive–zero–positive etc. polarization: after some initial loss of the coating mass steady-state electrolysis was sustained by a compact and stable layer. According to X-ray photoelectron spectroscopy Cu remains in an N-donor ligand environment after electrolysis.
Introduction
Water oxidation catalysts (WOCs, also referred to as catalysts for the oxygen-evolving reaction i.e. OER) have received considerable attention as a key subunit of prospective systems that can generate renewable chemical energy carriers by the conversion of solar energy.1 According to the general view, in systems where a light-harvesting unit is coupled to water electrolysis, WOCs facilitate the anodic half-reaction as electrocatalyst and supply the cathodic half-reaction with reducing equivalents to produce the energy carrier H2.2 Heterogeneous WOC materials built on inorganic precursors of first-row transition metals stand out with their robustness and cost-effective fabrication that can be tuned to reduce the overpotential (η) of the OER.3–6 On the other hand, the OER can be boosted by molecular electrocatalysts, too, where the turnover frequency (TOF, s−1) or η can be correlated with ligand effects, thus allowing mechanistic conclusions. Since high TOF values were reported for molecular catalysts, their heterogenization on an electrode surface and, ultimately, their combination with light-harvesting units to directly support the system with energy are attractive possibilities.7 Several complexes with first-row transition metals can act as homogeneous catalysts of the OER including Cu as the most recent addition to this group.8–22 Electrodeposited thin films of nanostructured copper oxide with enhanced activity were prepared, too, by applying Cu complex precursors,13 similarly to cobalt.22,23 For all these reasons Cu complexes represent a valuable contribution to artificial photosynthesis research24,25 and has become highly pursued.A novel family of branched peptides has recently been reported to form neutral 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes with Cu(II) in the basic pH region, which could be thoroughly characterized in solution.26 The complexes undergo Cu(II) → Cu(III) oxidation and proved to be effective water oxidation electrocatalysts in phosphate electrolyte.27 The applied branched ligands are shown in Scheme 1 as bound in the catalytically competent complex forms. As far as light harvesting or heterogenization is concerned, the branching of peptides (although synthetically challenging) offers unique options, for example, to combine within the same molecule one or more peptide fragments designed for specific metal binding with free arms reserved for another purpose e.g. to support self-assembling. This requires the appropriate extension of each arm.
:1 complexes with Cu(II) in the basic pH region, which could be thoroughly characterized in solution.26 The complexes undergo Cu(II) → Cu(III) oxidation and proved to be effective water oxidation electrocatalysts in phosphate electrolyte.27 The applied branched ligands are shown in Scheme 1 as bound in the catalytically competent complex forms. As far as light harvesting or heterogenization is concerned, the branching of peptides (although synthetically challenging) offers unique options, for example, to combine within the same molecule one or more peptide fragments designed for specific metal binding with free arms reserved for another purpose e.g. to support self-assembling. This requires the appropriate extension of each arm.
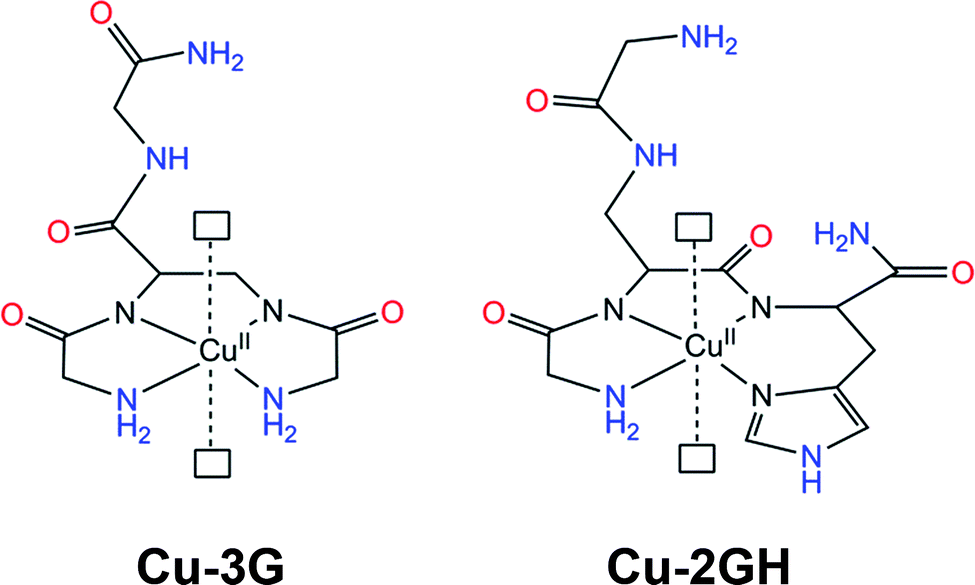 | ||
| Scheme 1 Structural representation27 of the Cu(II) complexes Cu-3G and Cu-2GH of the present study. Open rectangles represent sites of water coordination. Ligands are H-Gly-Dap(H-Gly)-Gly-NH2 (3G) and H-Gly-Dap(H-Gly)-His-NH2 (2GH). Potential H-bonding sites are marked with red and blue. | ||
The schematic structures of the copper branched peptide complexes studied in water oxidation are presented in Scheme 1 to exemplify the complexity of the effects occurring upon the exchange of only one amino acid. The histidine at the C-terminus modifies the coordination sphere from {NH2, N−, N−, NH2} to {NH2, N−, N−, Nim} leaving a different arm (C-terminus instead of N-terminus) to supply second-sphere functions. The localization of His at the N-terminus favours Cu binding at lower pH ((ligand H-His-Dap(H-His)-Gly-NH2) = 2HG), at the C-terminus at higher pH (ligand 2GH), while use of both the N- and the C-termini allows Cu binding over the entire pH range (ligand H-His-Dap(H-His)-His-NH2) = 3H)28). The presence of a Dap junction unit suppresses dimeric or oligomeric complex forms allowing the stabilization of metal binding exclusively near the branching group. Competition studies between fragment-by-fragment modified branched ligands and their linear components for metal ion binding demonstrated that branching can increase stability.26 Above all, the C-terminal substitution of Gly with His affects homogeneous water oxidation catalysis positively through the equatorial ligand set, but H-bonding interactions can also contribute to the overall performance. Peptides are exceptionally rich in such groups, which has a profound effect in enzyme catalysis either by means of contributing to the active site structure or to protein dynamics.29 With respect to the complexes Cu-3G and Cu-2GH, the high number of potential donor/acceptor sites for H-bonding (Scheme 1) could be exploited to anchor these molecules as catalytic centres in a composite system on a working electrode surface. This kind of attachment could preserve the coordination sphere along with the associated catalytic activity, yet entrap catalyst molecules at the surface. Note that biological studies have shown that this unique group of peptides can carry out targeted transport of selected metal ions possibly involving specific interactions between the surface of biomolecules and the freely available arms of the peptide ligands.30
These considerations led to the idea that positively charged polymers such as poly(L-lysine) (PLL) could be candidates as a support medium for catalyst heterogenization. PLL itself has been used for functionalization of solid substrates, for example that of indium-tin-oxide (ITO) with biomolecules for a number of purposes.31 The layer-by-layer (LbL) deposition of composite systems consisting of polyelectrolytes of alternating charges is a well-established cost-effective methodology.32 The notion that certain complexes with pincer ligands can be attached to polypeptides by coordinative bonds33 and metal receptors can be imprinted into multilayer polyelectrolytes34 made this strategy even more attractive, since the accessible axial sites in complexes Cu-3G and Cu-2GH and the amine or amide groups of the polyelectrolytes should allow this kind of attachment (Scheme 2b). However, catalytic applications of such systems would require analytical methods that can detect rather small changes in mass on the surface.
The transport and binding of adsorbing molecules in the close vicinity of the coated surface can be investigated by means of optical waveguide lightmode spectroscopy (OWLS)35,36 and its combination with electrochemistry (EC-OWLS).
OWLS and EC-OWLS have excellent sensitivities to monitor molecular adsorptions in real-time, and have already evolved into reliable analytical methods (Scheme 2a). OWLS itself is ranked as a high performance surface-sensitive technique allowing real-time monitoring of processes accompanied by refractive index changes in the close proximity of the surface of a waveguide sensor chip (typically <200 nm depth into the solution above the sensor surface). The waveguide sensor consists of a high refractive index waveguide layer with a shallow diffraction grating embossed into its structure supported by a thicker optical glass slide. Incoupling of a linearly polarized monochromatic He–Ne laser beam via the grating results in sharp resonance peaks when plotting the intensity of the incoupled light against the incident angle of the illuminating beam. The sensing principle relies on the perturbation of the evanescent optical waves of the guided lightmodes. Refractive index variations in the close vicinity of the sensing surface alter the discrete incoupling angles, thus shift the positions of the resonance peaks.37,38 The waveguide film can be coated by a thin layer of any transparent material required by the application. For example, when a thin coating of the n-type semiconductor ITO (a good electric conductor) is employed the method can be extended to EC-OWLS. Electrochemical methods, such as controlled potential electrolysis (CPE), can be performed in parallel with the OWLS measurements.
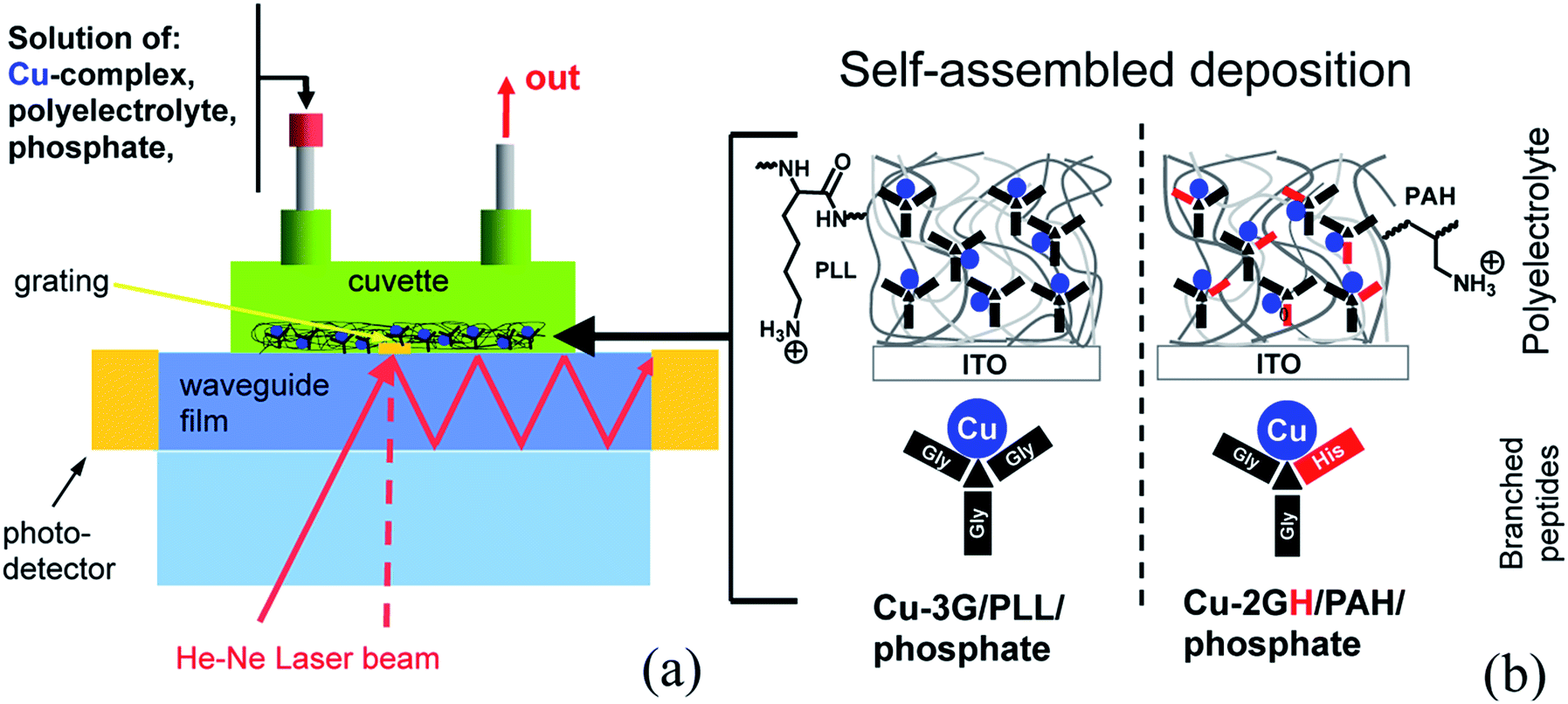 | ||
| Scheme 2 (a) Parts and operation principles of the OWLS flow-through cell, (b) components for the self-assembled catalyst deposition on ITO surface. | ||
The aim to anchor Cu complexes and this way activate ITO electrodes toward the OER has urged us to explore the LbL build-up method as an option for the heterogenization with suitable polyelectrolytes and follow the process by means of (EC-)OWLS. Note that in principle this means only one extra component to the Cu complex–electrolyte–ITO triformity of the homogeneous system (Scheme 2b). To our knowledge the application of branched peptide complexes in self-assembled electrode materials is unprecedented. Reported below are the results of the alternating depositions of Cu-3G or Cu-2GH and PLL or poly(allylamine hydrochloride) (PAH), resulting in LbL-ITO electrodes sensitized toward electrocatalytic water oxidation. The fabricated composite layers were investigated by detailed electrochemical experiments.
Experimental
Materials
All salts were purchased from commercial sources and were of puriss p.a. grade. All Fmoc amino acids, including Fmoc-L-Dap(Fmoc)-OH used as a branching amino acid, solvents and reagents were purchased from Iris Biotech GmbH (Marktredwitz, Germany) and used as received. The synthetic processes of the ligands 3G and 2GH, their complexation properties with Cu(II) are discussed elsewhere, as well as the detailed spectroscopic characterization (UV/VIS, CD, EPR and ESI-MS) of the different pH-dependent species.27,28 PAH was purchased from Alfa Aesar (Johnson Matthey Company) (MW = 120000–200000 g mol−1), while PLL from Sigma Aldrich (MW = 150000–300000 g mol−1).
Optical wavegiude lightmode spectroscopy (OWLS) experiments
OWLS210 from MicroVacuum Ltd, Budapest, is an optical (bio)sensor instrument, which is based on integrated optics that can be used to investigate the adsorption of various substances from their solution at the solid (waveguide)–liquid interface in real-time. During the deposition experiments a glass supported TiO2–SiO2 waveguide with ITO coating was applied (e.g. OW2400c, MicroVacuum Ltd, Budapest) as the bottom of a liquid flow cell. The employed waveguides support two modes with orthogonal polarizations (TE0 and TM0) and enable to detect the adsorbed material up to a depth of 100–150 nm inside the aqueous solutions flowing over the waveguide. From the measured incoupling angles the effective refractive indices of the excited waveguide modes (NTE and NTM) can be calculated. These values were further used to determine the optogeometrical parameters (thickness and refractive index) of the adsorbed layers. The adsorbed mass was then calculated by using Feijter's formula.39 Prior to every experiment the waveguide sensor chips underwent a standard cleaning routine e.g. immersed into chromic acid (Merck) for 3 min, followed by rinsing with Milli-Q water, KOH and Milli-Q again. The chips were then placed into an ultrasonic bath for at least 30 min and the bathing Milli-Q water was changed in every 5 min. The cleaned waveguides were incubated in phosphate electrolyte at the pH of the subsequent experiment overnight.Deposition experiments
The general principles of the LbL technique can be found in the literature.32,40 The experiments were carried out under various conditions by employing one of the following two methods:(A) 14 × 14 mm pieces of ITO electrodes (∼20 ohm sq−1, Präzisions Glas & Optic) were first immersed into the polyelectrolyte solution (PLL, 0.01 mg mL−1 or PAH, 0.5 mg mL−1) then rinsed in phosphate buffer. The ITO was then dipped into the solution of the Cu complex (Cu-3G or Cu-2GH, 0.05–1 mM) and washed with buffer again. These steps were repeated 0–20 times. Each step took 5 min and this period was set to mimic the OWLS experiments. The pH of the concomitant solutions was identical, set by titration of 0.1 M phosphate electrolyte by NaOH. The solutions of the components were distributed in a multiwell sterile plate, the volumes were adjusted to cover half of the ITO, this way 0.98 (±10%) cm2 of the electrode surface could be exposed to LbL deposition. The ITO pieces modified by method (A) were then used as working electrodes in a standard electrochemistry cell.
(B) LbL was performed in the flow cell of the OWLS instrument to follow the build-up process in real-time. An ITO coated optical chip (MicroVacuum Ltd, Budapest) was used for either electrochemical i.e. EC-OWLS or OWLS experiments. The first polyelectrolyte solution (PLL or PAH) was injected into the cell. After no further changes were detected by OWLS, buffer was let through the cell by using a peristaltic pump with 1 μL s−1 flow rate to eliminate the remaining polyelectrolytes until, again, no further change was observed in the OWLS signal. Next, buffered Cu complex solution (Cu-3G or Cu-2GH) was injected trough a septum injection port, that was followed by buffer rinsing again. These steps were repeated in the case of multiple deposition cycles and each step took ∼10 min. The concentration and pH of the applied solutions were identical to those used in method (A). The complex solutions were made before the experiments by using slight excess of ligands (0.9:1.0 Cu:ligand ratio); these solutions were then titrated with 1 M NaOH to the desired pH and mixed with appropriate amounts of phosphate solution. Lower concentrations were set by dilution. All experiments were performed at 25 °C and the formation of the complexes was confirmed by measuring the electronic absorption spectra26 with an Agilent Cary 60 spectrophotometer coupled to the immersion probe.
EC-OWLS experiments
The cell was filled with electrolyte after LbL treatment of the ITO coated optical chip that was set as the working electrode in a three-electrode (Pt aux. and Ag/AgCl ref.) system. The ITO was then set under +1.1 V potential for 10 min and after this the potential was removed for 10 min. This was repeated three times and the Cu solution was supplied with injection again. This step and the concomitant rinse lasted 10 min. After the Cu re-supplement step a potential of +1.1 V was set to the ITO chip for 10 min and the potential was removed. The current was measured in parallel with the optical signal.Electrochemistry
Cyclic voltammetry (CV) and controlled potential electrolysis (CPE) measurements were performed under air on a general purpose potentiostat (BioLogic SP-150). A standard three-electrode setup was used including an ITO working electrode layered by method (A), Pt spring auxiliary electrode (∼7 cm2) and Ag/AgCl (3 M KCl, 0.2 V vs. SHE) reference electrode. The cell was equipped with a pH microelectrode (Mettler-Toledo) or a fluorescent O2 sensor (Ocean Optics NeoFox) through slot with o-ring.X-Ray photoelectron spectroscopy (XPS)
Surface compositions of the film deposited on the ITO electrode were determined by a KRATOS XSAM 800 XPS machine equipped with an atmospheric reaction chamber. Al-Kα characteristic X-ray line, 40 eV pass energy (energy steps 0.1 eV) and FAT mode were applied for recording the XPS lines of Cu2p, CuLMM, O1s, Sn3d, In3d, N1s, C1s and P2p. The C1s binding energy at 284.8 eV was used as reference for charge compensation. The surface concentrations of the elements were calculated from the integral intensities of the XPS lines using sensitivity factors given by the manufacturer.
Atomic force microscopy (AFM) experiments
AFM was used to obtain morphological and homogeneity data of the polyelectrolyte composite layers with Cu content. All images were captured by an AIST-NT Digiscope 1000 AFM instrument using tapping mode. AFM samples were prepared analogously to method (A) at pH 10.Results and discussion
Deposition monitored by OWLS
Initially the effect of the polyelectrolyte on the adsorption of the Cu complexes on an ITO-coated OWLS sensor chip was investigated at pH 7.5 in phosphate buffer (PB) (Fig. 1A). In the basic pH region the neutral form of the two Cu-peptide complexes is present predominantly, as confirmed by means of potentiometry earlier.26 | ||
| Fig. 1 (A) Variation in surface density (PB, 0.1 M, pH = 7.5) with [Cu-3G] (for concentrations see legend) on ITO coated chip followed by OWLS (vertical grid lines stand for approx. time of injection of the different components as indicated); (B) mass development of Cu-3G (mCu-3G) with the number of LbL cycles (same as in A), inset: mCu-3G normalized with mPLL and plotted against the number of LbL cycles (used for calculating Γ0cat in Table S2†); (C) variation in surface density growth ([Cu-3G] = 0.5 mM) with pH (see legend); (D) variation in LbL development of Cu-2GH/PAH with [Cu-2GH] (PB, 0.1 M, pH = 7.5); (E) mass development of Cu-2GH (mCu-2GH) on ITO with the number of LbL cycles (same as in D), inset: mCu-2GH normalized with mPAH and plotted against the number of LbL cycles (used for calculating Γ0cat in Table S2†); (F) variation in surface density growth ([Cu-2GH] = 0.5 mM) with pH (as indicated by the legend). | ||
The pH optimum for LbL deposition was found at ∼9–10 (Fig. 1C), overlapped with the pH domain of the [CuH−23G] species.26 Note that pH strongly affects adhesion of PLL reaching a maximum at pH 10 (see the first 20 min in Fig. 1C, where the surface density of the 1st PLL layer was normalized to ∼300 ng cm−2 uniformly). For this reason pH 10 and [Cu-3G] = 0.5 mM represent optimum conditions for the LbL deposition of Cu-3G/PLL in PB. XPS analysis on bare ITO and after LbL deposition of Cu-3G and PLL by method (A) confirmed that the surface is composed of organic material with peptide bonds, contains Cu(II) in N donor environment and phosphate (for a detailed discussion see ESI, Fig. S1–S3†). The detected surface composition of the LbL-ITO is included in Table S1.†
Obviously, the number, nature and accessibility of the potential hosting sites can be expected to fundamentally determine the LbL build-up, but the sharp difference between Cu-3G and Cu-2GH is still surprising, since these complexes have very similar properties. However, this highlights the significance of the branched peptide structure that can be related to their interactions with biomolecules.30 Therefore the preference of Cu-3G to PLL is tentatively assigned to the available C-terminal glycyl amide unit that could form strong H-bonds with the peptide functions on PLL. Cu-2GH, on the other hand, has a C-terminal histidine coordinated to Cu(II). The non-coordinated arm here is the N-terminal glycine and this ligand arrangement is apparently more selective to allylamine functions with respect to H-bond formation. Electrostatic interactions are thought to have much less role (if any) in the adhesion of the Cu complexes, since PDDA or PSS did not support deposition. The role of electrostatic interactions involving phosphate anions is rather crucial in holding together the supporting polyelectrolytes, with ITO thus building the host surface arrangement for the catalytically active complexes. Note that in biological systems the affinity of the cationic side chain of lysine for the negatively charged phosphate backbone of DNA is well known42 and serves as a model for the interaction between PLL or PAH and phosphate.
The pH optimum for layering is ∼10 (Fig. 1F) resulting from parallel growing speciation% of the [CuH−22GH] species and the adsorption preference of PAH (i.e. although Cu-2GH is still adsorbed at pH 11, multiple deposition cycles with PAH is not possible). The pH also strongly affects adhesion of PAH to ITO (see the first 20 min section of Fig. 1F, the surface density of 1st PAH layer is normalized to ∼200 ng cm−2 for comparison). For the above experimental findings and for the sake of comparable conditions, during electrochemistry with LbL-ITO [Cu-3G] = [Cu-2GH] = 0.5 mM was set.
The above discussed OWLS experiments showed that the Cu complexes can be layered with polyelectrolytes in a highly selective manner, governed by the ligand structure. Optimum layering pH approximates that of catalysis in homogeneous solution, while multiple deposition cycles help attach increasing amounts of Cu complexes. These are important features from the viewpoint of electrocatalytic applications.
Surface topography by atomic force microscopy (AFM)
It has been demonstrated that multiple deposition cycles lead to layers that are enriched in catalyst. However, since OWLS averages over 1 mm2 area, it is less informative about incomplete or patchy coatings. Therefore films of Cu-3G/PLL and Cu-2GH/PAH formed in 0.1 M PB at pH 10 on an ITO surface were morphologically described by 3D-AFM (Fig. 2, for comparison neat ITO is shown in Fig. S15†). Typical AFM topographic images of Cu-3G/PLL and Cu-2GH/PAH (Fig. 2A, B and D, E, respectively) show that the composite layers are inhomogeneous and interspersed by pores. Fig. 2C and F present the roughness of a typical profilometric section of the surface (indicated with white lines across the 2D views). Pores are typically made by the evaporating water content of the film.43 This feature may imply retrospectively the importance of hydration in the LbL formations along with the sparsely observed salient formations that are probably phosphate salt residues, or vertical threads of complex–polyelectrolyte assemblies. The Cu-3G/PLL coating has a rougher surface than that of Cu-2GH/PAH. While the typical perpendicular expansion of the formations is <5 nm for Cu-2GH/PAH it can approximate 30 nm in the case of Cu-3G/PLL, indicating a more compact arrangement for the former. This can originate from the structural differences between PLL and PAH. According to AFM, formations created by the LbL method can be depicted as fully dispersed on the surface and arranged in interpenetrating layers for Cu-3G/PLL44 and smoother, horizontal deposits for Cu-2GH/PAH.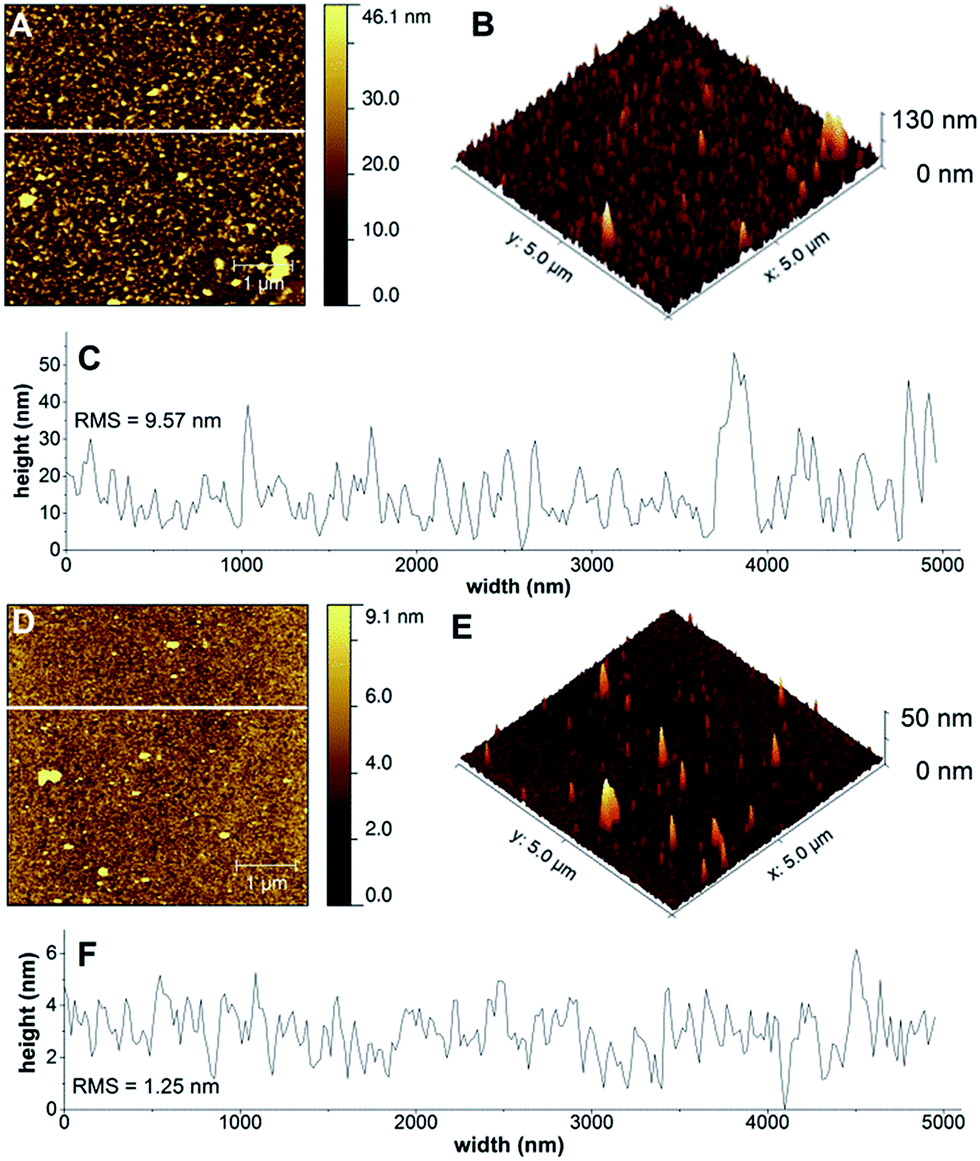 | ||
| Fig. 2 (A) Representative AFM image (5 × 5 μm) of the Cu-3G/PLL/phosphate composite layers; (B) 3D view of the same area; (C) profilometric section of the AFM image; (D)–(F) the same images for the Cu-2GH/PAH/phosphate LbL formations. Eight deposition cycles were conducted in both cases according to method (A) at pH = 10. The places of profilometric cross-sections are indicated by white lines in (A) and (D). | ||
The combination of OWLS, XPS and AFM allowed gathering information about the surface density and chemical environment of the heterogenized Cu complexes and surface topography of the composite layers that was considered in the course of electrochemistry. This information served also as a starting point for the preparation of the LbL-ITO electrodes for water oxidation electrocatalysis.
Electrochemistry with LbL-ITOs
First cyclic voltammetry (CV) has been performed on LbL-ITOs in order to detect redox processes associated with the Cu content and record behaviour upon repeated polarization cycles. ITO is a popular choice for a working electrode in water oxidation research, since OER starts at relatively high η in the absence of any catalyst. Therefore the contribution to the overall performance is usually very low. Evaluation of CVs sped up finding sustainable conditions for controlled potential electrolysis (CPE). LbL-ITO electrodes were layered according to method (A) and set as the working electrode in a standard three-electrode setup, in phosphate electrolyte, similarly to the setup for the earlier homogeneous experiments.27Fig. 3A shows the effect of electrolyte pH on the cyclic voltammetry (CV) responses. A symmetric oxidation peak occurs and shifts from ∼0.71 to ∼0.59 V vs. Ag/AgCl upon changing the pH from 9.1 to 11.0. This behaviour is analogous to that of Cu-3G in solution and therefore the current peak can be assigned as the Cu(III/II) transition of adsorbed complex. Further polarization yields a current peak that is pH-sensitive and evolves into a catalytic current, again, very similarly to the homogeneous system with the complex in solution. Upon reverse polarization no reduction can be detected, which indicates that the oxidation peaks attributed to the [CuII–OH2] → [CuIII–OH] → [CuIII–O˙] PCET transitions (numbers in red circles in Fig. 3A), respectively, may originate from a portion of Cu-3G that is non-reversibly transformed upon oxidation. This portion could theoretically dissociate to the bulk or yield another complex form (or both processes could take place). After the 3rd cycle the CV current response becomes steady and corresponds at 1.1 V vs. Ag/AgCl to the CPE current value after 5 min (Fig. 3B, for description of CPE experiments at different pH values and the effect of the number of deposition cycles see Fig. S5 and S6,† respectively) e.g. these CV conditions represent diffusion controlled rates (see also Fig. S7† to compare two different scan rates on LbL-ITO previously used for CPE for 10 min). Apparently, the portion of Cu-3G that is responsible for the current peaks in the first cycle has no further role in sustained electrolysis (EC-OWLS and XPS experiments are supportive of this presumption).
 | ||
| Fig. 3 (A) First cycle of CVs of LbL-ITO electrodes treated with eight deposition cycles of Cu-3G/PLL (pH = 10) in 0.1 M phosphate solution at different pH values as indicated, inset: blown up picture of the current peak attributed to the Cu(II)/Cu(III) transition of the surface adsorbed Cu-3G; (B) third cycle of CVs of ITO electrodes with or without Cu-3G/PLL (deposited at pH = 10) in 0.1 M phosphate solution at pH = 10.64 (see legend and text for details); (C) third cycle of CVs of LbL-ITO treated with eight deposition cycles of Cu-2GH/PAH (pH = 10) in 0.1 M phosphate solution at pH = 10.55 (for colour codes see the legend, open circles stand for CPE current after 5 min). Scan rates were 25 mV s−1 uniformly. | ||
In Fig. 3B the 3rd cycles of CVs taken with differently fabricated LbL-ITOs are compared. The neat (orange curve) or PLL coated ITO (green curve) cannot compete with the current growth on the electrode coated by Cu-3G/PLL (purple curve). When the final deposition cycle is that of PLL the current response is lower at the potential of the Cu(III/II) transition of Cu-3G in the first cycle (Fig. S8†), while the catalytic current corresponds to that of CPE at 1.1 V from the 3rd cycle (Fig. 3B, dark blue curve, PLL–Cu-3G/PLL). This can be explained by the existence of a complex fraction that behaves like Cu-3G in solution and corresponds by amount to roughly 38% of the total deposited amounts at most. This is supported by the quantitative analysis of the Cu(III/II) current peaks (see Fig. S8,† caption) and calculated from OWLS data on the Cu-3G/deposition cycle quantity (values are listed in Table S2†).
The optimum pH of the deposition from the electrochemistry perspective was investigated by comparison of the performance of LbL-ITO electrodes layered at different pH values and immersed into a phosphate electrolyte at pH = 10.6 (Fig. S9†). It can be concluded that LbL-ITOs layered at pH 9–10 show the highest catalytic currents by good reproducibility of CPE currents (Fig. S10†). However, further anodic polarization of the electrodes to above 1.2 V vs. Ag/AgCl results in gradual loss of catalytic capacity (Fig. S11†) therefore EC-OWLS experiments and FOWA (vide infra) were done on LbL-electrodes polarized to a maximum of 1.2 V.
ITO pieces modified with PAH and Cu-2GH and used as working electrodes in CV experiments behave similarly to the Cu-3G/PLL system. The results are in favour of pH ∼ 10 as the optimum for LbL build-up (Fig. S12† shows the effect of deposition pH). The peak associated with the Cu(III/II) transition in the first cycles shifts from ∼0.77 to ∼0.61 V vs. Ag/AgCl as the pH is set from 9 to 11 (Fig. S13a and b†). The peak cannot be observed upon consecutive cycles and the reduction peak is also missing from the reverse polarization curve (Fig. S13c†), which points to the same conclusions as for the Cu-3G/PLL system. Anodic polarization to 1.2 V yields a catalytic current response, which, after the 3rd cycle is similar to CPE current after 5 min at the corresponding potential (Fig. 3C, for CPE experiments see Fig. S14†). The exception is CPE at above pH ∼10.6, where the development in current with time is associated with unexplored side events.
The first cycle of CV experiments revealed two consecutive oxidation steps upon anodic polarization of LbL-ITOs preceding electrocatalysis at above ∼1 V that were assigned to a fraction of Cu-3G and Cu-2GH that dissociates from the surface. The catalytic current became steady upon repeated cycles between 0 and 1.2 V and the oxidation step triggering catalysis shifted to the anodic direction. This and the absence of the initial oxidation steps indicated a substantial change in the mechanism of catalysis compared to the homogeneous systems.
In operando electrochemical OWLS (EC-OWLS) experiments
In order to get insight into surface density changes on a functioning LbL-ITO surface under CPE conditions, EC-OWLS experiments were performed in a cell where the ITO-coated chip could be set as the working electrode (the setup is analogous to that in Scheme 2). First, the optical chip was treated according to method (B) with three deposition cycles of Cu-3G/PLL (Fig. 4A) or Cu-2GH/PAH (Fig. 4B) at pH 10.12 (note that OWLS is very sensitive to changes in pH, so a compromise value was set, at which layering is reasonable and catalysis is still detectable), and covered either with the corresponding polyelectrolyte, or with the complex as indicated in the figures. In the case of Cu-3G, 0.9 × 10−10 mol cm−2 of complex could be deposited and concomitantly exposed to CPE at 1.1 V vs. Ag/AgCl for periods indexed with red asterisks (Fig. 4A), followed by periods of zero polarization. During the three turns of CPE ∼2.7 × 10−8 moles of electrons were counted, while the mass change after the second and third round was only 8 and 4 ng cm−2 and the surface density was apparently stabilized.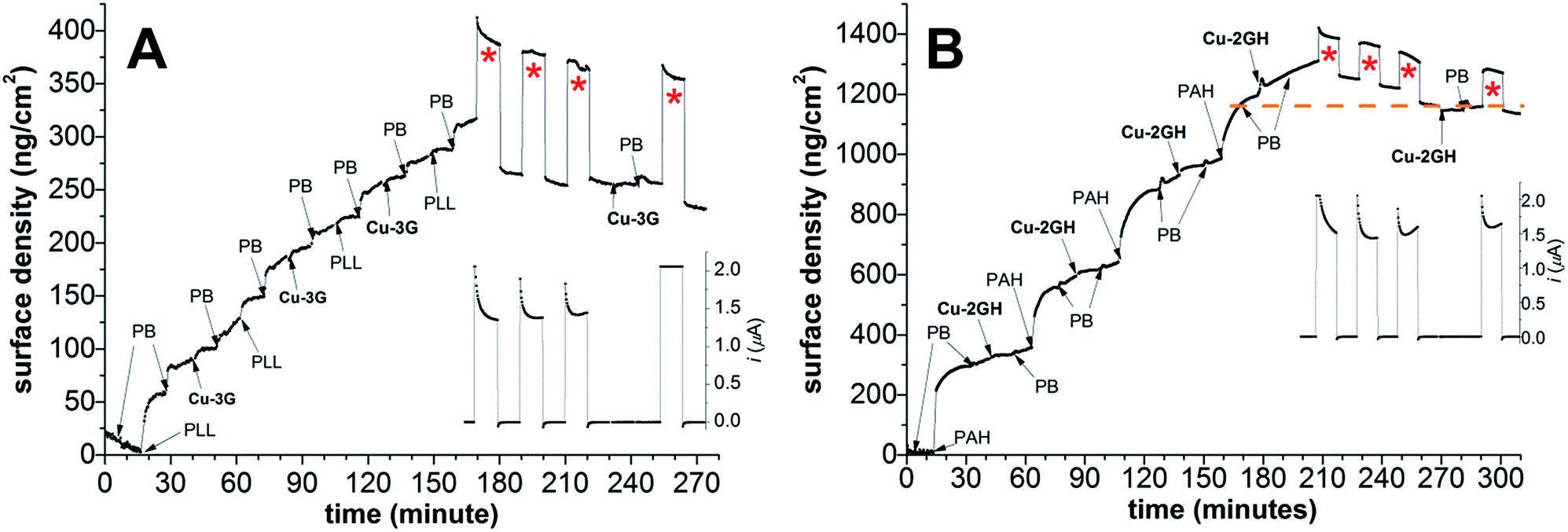 | ||
| Fig. 4 (A) Three deposition cycles of PLL and Cu-3G (0.5 mM) in PB (0.1 M) at pH = 10.12 and concomitant 3 × 10 min runs of CPE at 1.1 V vs. Ag/AgCl with intermittent breaks of 10 min and concomitant attempt to supply the electrolyzed surface with Cu-3G at an ITO-coated chip placed in an EC-OWLS cell; (B) the same sequence applied to PAH and Cu-2GH layers. | ||
Most importantly, after the initial change in mass the current during CPE became steady and caused no further loss of surface density. The question emerged whether the LbL-ITO exposed to CPE will accommodate additional Cu-3G. In an attempt to re-supply the LbL film with Cu-3G only minimal (<0.1 × 10−10 nmol cm−2) complex was adsorbed indicating the formation of a very compact LbL-ITO surface upon electrolysis. The follow-up CPE shows higher current, however, this may be caused by remaining Cu-3G in the bulk dead-space of the cell (in the case of Cu-2GH no change in the follow-up CPE current was experienced).
According to XPS analysis of Cu and N content on Cu-3G/PLL LbL-ITO pieces as prepared and after exposure to CPE at pH 10.64 (Fig. 5A) the Cu remains in the Cu(II) state in a peptide environment that is rich in N (Fig. 5B).34 This is evident from the Cu2p3/2 peaks that are present at nearly the same energy (with no shake-up peaks typical for Cu-oxide/hydroxide formations27). Modelling the N1s peak with amine (NH), quaternary ammonium (NH+) and amide (OCNH) component peaks yields a somewhat changed ratio and enrichment in NH+. In parallel, the N/Cu ratio is higher in the sample exposed to CPE, suggesting that the surface structure of the Cu-3G/PLL layers changes upon electrolysis and 31% less Cu remains in the deposited film. This is in accordance with CV results (absence of the [CuII–OH2] → [CuIII–OH] current peaks typical for the original complex Cu-3G after repeated cycles, indicating ∼38% initial loss of Cu-3G, Fig. S8†) and with the EC-OWLS findings (some change in surface density and no increase in mass with follow-up addition of Cu-3G).
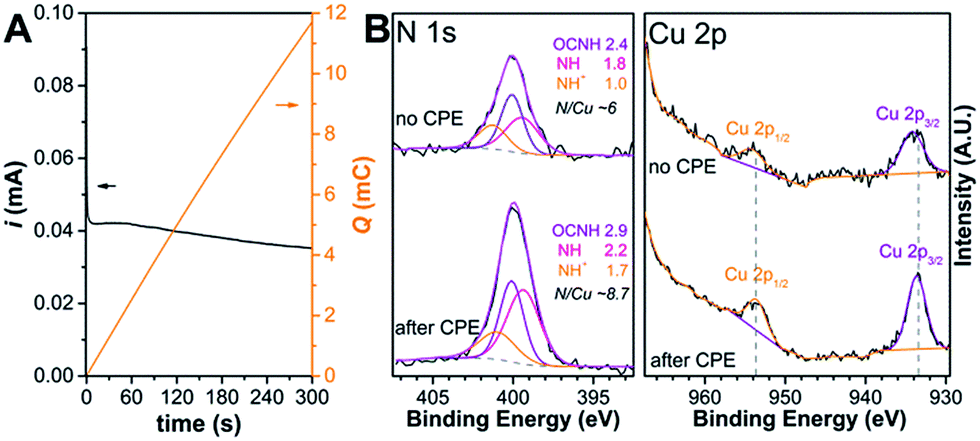 | ||
| Fig. 5 (A) CPE current (i at 1.1 V) and charge on an LbL-ITO treated with 8 Cu-3G/PLL/phosphate deposition cycles (surface area ∼ 1.4 cm2); (B) XPS analysis of N and Cu on the electrode after CPE and the same analysis on another LbL-ITO prepared analogously, but unused (numbers indicate relative contributions). | ||
The same general observations could be highlighted in the case of Cu-2GH/PAH (Fig. 4B) judged from a similar set of experiments. After three deposition cycles finished with PAH, 1.6 × 10−10 mol cm−2 complex could be anchored in total and the electrode could traffic 3 × 10−8 moles of electrons in the course of 3 × 10 min of CPE at 1.1 V (periods indexed with red asterisks). Remarkably, the loss in surface density ceased at ∼1160 ng cm−2, where the last PAH deposition after 160 min was finished (dashed orange line). No further changes upon addition of Cu-2GH and follow-up CPE could be detected, in support of a compact electrocatalyst film on ITO.
Taken together, these findings indicate that the fraction of the Cu-3G and Cu-2GH complexes responsible for catalysis changes coordination mode upon deposition and oxidative polarization of the LbL-ITO. The resulting Cu is still bound in N donor set according to XPS however. Leaching of the complexes from LbL-ITO can be estimated based on CV and XPS results, and supported by EC-OWLS. This means that an altered complex form of Cu should be responsible for the sustained CPE.
Catalytic rates from foot-of-the-wave analysis (FOWA) of CV data and long-term electrolysis
In homogeneous solution the TOF values determined for Cu-3G and Cu-2GH showed strong dependence on the presence of His ligand. The question emerged whether this effect was still traceable on LbL-ITO, among heterogeneous conditions, after several cycles of CV. Therefore apparent rate constants (kcat, s−1 analogue to turnover frequency, TOF) were obtained by FOWA of catalytic currents45 at pH = 10.55 for the Cu-3G/PLL and Cu-2GH/PAH systems. The methodology described for heterogeneous reactions was followed i.e. for the case of film deposited catalytic layers on an electrode support45,46 (for further details see ESI†).According to the FOWA results (Fig. S17 and S19†), the catalysis is less disturbed by side phenomena below the formal potential of the redox transition initiating catalysis for Cu-3G/PLL and Cu-2GH/PAH in good agreement with the CPE experiments that show steady currents on LbL-ITO electrodes at pH ≤ 10.6. The potential of the catalysis-initiating oxidation step was detected by square wave voltammetry (SWV) for both systems as illustrated in Fig. S16.†
Long-term CPE experiments for Cu-3G/PLL and Cu-2GH/PAH at pH = 10.6 (Fig. S18†) were performed to decide whether the catalytic current was associated with O2 production. The data suggest that O2 is produced and the stability of the Cu-3G/PLL systems lags behind that for Cu-2GH/PAH even at lower potential. This is associated with the strongly overlapping onset potential for PLL with that of catalysis (Fig. S18 inset†). The steady current (i.e. the linear increase of charge, Fig. S18†) of the long-term electrolysis experiments indicate no change in the catalyst for a ∼20 min (Cu-3G/PLL/phosphate) and ∼40 min (Cu-2GH/PAH/phosphate) period during which several turnovers of O2 production takes place with acceptably good faradaic efficiency. CV performed after CPE experiments confirmed that the LbL-ITO still exhibited catalytic current over several cycles and current peaks associated with the Cu content were present (Fig. S18A inset and B†). On the other hand, when pH or the potential (see Fig. S11†) are further increased the catalysis is affected by side events, which is traced in the shifting CPE currents with time (Fig. S5 and S14†).
The comparison of kcat values from FOWA should reflect dominantly the inner-sphere structural differences in the two different catalytic centres that are anchored at the LbL-ITO. The TOF values from the homogeneous catalysis study between Cu-3G (TOF = 24 s−1, pH = 11) and Cu-2GH (TOF = 53 s−1, pH = 11)27 and especially their ratio of 2.2 therefore represent a good reference for comparison (note that direct numerical comparison is not viable since TOF values were determined by a different method at a glassy carbon electrode e.g. GCE in the earlier study). Remarkably, the kcat(Cu-2GH)/kcat(Cu-3G) ratios in Table S2† (2.32 in average) are in good agreement with the value for the molecular catalysts, where the better catalytic performance of Cu-2GH was attributed to the coordinated C-terminal His. This (and the high preference of His over amines toward Cu) supports the hypothesis that His should be found within the first coordination sphere of Cu in the LbL built system. Finally, if one compares TOF values obtained by different numbers of deposition cycles (n, Table S2†) it becomes apparent that the TOF values drop when n > 8 in spite of increasing Γ0cat. In accordance, saturation in the steady-state current can be observed during CPE with increasing n (Fig. S6†). This suggests that only a certain fraction of the anchored complex molecules is activated toward catalysis that is limited by the LbL film thickness.
Proposed mechanism
In accordance with the experimental observations a general mechanism can be proposed for catalysis (Scheme 3). Freshly prepared LbL-ITO electrodes show current peaks (circled red numbers 1 and 2 in Fig. 3A) that correspond to the [CuII–OH2] → [CuIII–OH] → [CuIII–O˙] transitions, respectively, of the original complexes (ligands are omitted for clarity). These concomitant oxidation steps lead to a catalytically active intermediate and in the homogeneous system that initiates water oxidation by closing cycle A as it has been proposed by others and in our earlier communication.9,10,27 However, when the complexes are deposited, the current peaks corresponding to cycle A are absent from the 2nd CV cycle indicating the change of the original catalytic centres. In Fig. S16† we have shown that an oxidation peak can be detected by SWV (circled red number 3, this peak is hidden under the catalytic response in CVs) that we tentatively associate with step 3 of cycle B in Scheme 3. The difference between cycle A and cycle B is presumably the coordination of L′ (a supporting ligand) to the metal. This may be favoured in the polyelectrolyte/phosphate environment, where a large excess of potential donor groups is present. Upon repeated catalytic cycles depletion of water molecules from the vicinity of the catalytic centres can further drive the system towards cycle B. Thus [CuII–L′], a supposedly five-coordinate Cu(II) site, is formed within the layers that is less apt to oxidation and water ligation. As a consequence, step 3, oxidation of the Cu(II) assigned as the rate limiting step of cycle B, in accordance (a) with the high potential of the catalysis initiating oxidation step (>1.1 V, Fig. S16†), and (b) with the steady-state CPE currents that correlate with [OH−] (Fig. S20†). The overall suggested mechanism is a type of chemical inactivation–redox reactivation mechanism.46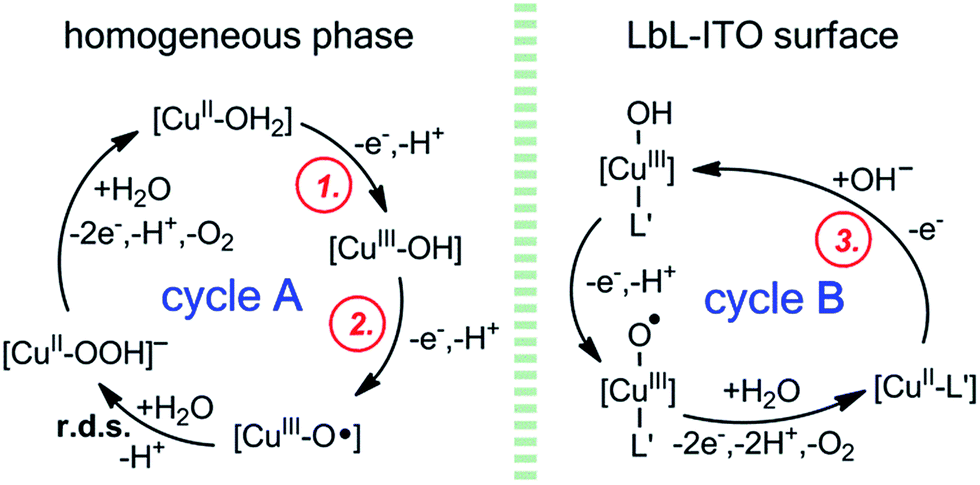 | ||
| Scheme 3 Proposed mechanism for the water oxidation catalysis at LbL-ITO in accordance with experimental findings and in comparison with the homogeneous systems (3G and 2GH ligands are omitted). | ||
Conclusions
The presented work combines synthetic and spectroscopic methods in a novel way for the functionalization and characterization of LbL-ITO activated toward water oxidation electrocatalysis. To functionalize the surface, three main components have been combined including: (1) small molecule electrocatalysts (Cu-branched peptides), (2) large molecule polyelectrolytes and (3) supporting electrolyte (phosphate). The optimization of some easy-to-set parameters (pH, concentrations) to promote bottom-up self-assembly was possible by means of OWLS that could monitor the changes in surface density in real-time. Note, since OWLS measures refractive index differences, it does not measure the bound water molecules, and therefore supplies the surface adhered “dry mass” only, with a ng cm−2 resolution.40 OWLS is therefore capable of the detection of processes involving rather low amounts of the complexes. While the kinetics could be followed through OWLS, layering among the same conditions can be easily performed by the sequential immersion of ITO electrode pieces into solutions of the components. Simplicity and low cost make this method very attractive for scanning a wide variety of component combinations. Although LbL self-assembly of the Cu-3G/PLL/phosphate or Cu-2GH/PAH/phosphate combinations on ITO largely relies on electrostatic interactions between the oxide surface, the cationic polymers and the phosphate anions, these alone would be insufficient for the incorporation of the complexes as a third component as it is implied by experiments with different polyelectrolytes. Anchoring the complex molecules at the macromolecules apparently requires specific H-bonding or coordinating N donor ligand options, which makes the self-assembly very selective with respect to complex–polyelectrolyte combinations.It has been shown by means of CV and CPE experiments that water oxidation can be performed and sustained for longer periods on LbL-ITO, if pH and potential are kept at values below those resulting in high initial rates, but conflicting with layer stability. In operando EC-OWLS analysis revealed mass transport events that ceased after initial loss in surface density. Failed efforts to supplement the LbL-ITO used in EC-OWLS experiments with Cu-peptide and the observed transformation of CV curves altogether imply that structural changes take place upon polarization of the electrode. Most likely the Cu complexes act as nodules, to form (in part) catalytic centres and get wrapped into microcapsules of the polymeric chains through coordinative bonds, similarly to literature examples with pincer ligand complexes.47 However, in the present case these events are completed by electrochemistry. The nanostructured surface assemblies can accommodate no additional catalyst and follow a different operating mechanism in comparison with the homogeneous system. The method represents a viable strategy to incorporate molecular catalysts by self-assembly into functional layers. Based on these initial results a PLL-type polylysine extension of the branched metal binding peptide site (head–tail structure) is currently being investigated in order to advance layering properties. Although it was neither exploited nor emphasized, PLL and both catalysts were enantiomerically pure isomers that could be utilized in future enantioselective electrocatalytic applications. It was also out of the scope of this study to pursue electrolytic transformations at high pH and E (with simple precursors, of course) that could serve as templates of nanostructured patterns for metal-oxide–hydroxide WOCs with enhanced catalytic activity.19,48
Acknowledgements
This work was supported by the Lendület (Momentum) Program of the Hungarian Academy of Sciences, the ERC_HU Program of NKFIH and, by the Polish Foundation of Science within the POMOST program co-financed by the European Union within European Regional Development Fund (POMOST/2012-5/9). Ł. Szyrwiel acknowledges the support of the European Commission under the Marie Curie Intra-European Fellowship (PIEF-GA-2012-329969). J. S. Pap is grateful for the useful remarks from Prof. Róbert Schiller.References
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729 CrossRef CAS PubMed.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767 CrossRef CAS PubMed.
- F. Jiao and H. Frei, Energy Environ. Sci., 2010, 3, 1018 CAS.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072 CrossRef CAS PubMed.
- D. K. Bediako, B. Lassalle-Kaiser, Y. Surendranath, J. Yano, V. K. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2012, 134, 6801 CrossRef CAS PubMed.
- X. Liu, S. Cui, Z. Sun, Y. Ren, X. Zhang and P. Du, J. Phys. Chem. C, 2016, 120, 831 CAS.
- D. L. Ashford, M. K. Gish, A. K. Vannucci, M. K. Brenneman, J. L. Templeton, J. M. Papanikolas and T. J. Meyer, Chem. Rev., 2015, 115, 13006 CrossRef CAS PubMed.
- S. M. Barnett, K. I. Goldberg and J. M. Mayer, Nat. Chem., 2012, 4, 498 CrossRef CAS PubMed.
- M. T. Zhang, Z. Chen, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 2048 CrossRef CAS PubMed.
- M. K. Coggins, M. T. Zhang, Z. Chen, N. Song and T. J. Meyer, Angew. Chem., Int. Ed., 2014, 53, 12226 CrossRef CAS PubMed.
- X. J. Su, M. Gao, L. Jiao, R. Z. Liao, P. E. Siegbahn, J. P. Cheng and M. T. Zhang, Angew. Chem., Int. Ed., 2015, 54, 4909 CrossRef CAS PubMed.
- T. Zhang, C. Wang, S. Liu, J.-L. Wang and W. Lin, J. Am. Chem. Soc., 2014, 136, 273 CrossRef CAS PubMed.
- X. Liu, H. Jia, Z. Sun, H. Chen, P. Xu and P. Du, Electrochem. Commun., 2014, 46, 1 CrossRef CAS.
- L.-Z. Fu, T. Fang, L.-L. Zhou and S.-Z. Zhan, RSC Adv., 2014, 4, 53674 RSC.
- D. L. Gerlach, S. Bhagan, A. A. Cruce, D. B. Burks, I. Nieto, H. T. Truong, S. P. Kelley, C. J. Herbst-Gervasoni, K. L. Jernigan, M. K. Bowman, S. Pan, M. Zeller and E. T. Papish, Inorg. Chem., 2014, 53, 12689 CrossRef CAS PubMed.
- W.-B. Yu, Q.-Y. He, X.-F. Ma, H.-T. Shi and X. Wei, Dalton Trans., 2015, 44, 351 RSC.
- T.-T. Li, S. Cao, C. Yang, Y. Chen, X.-J. Lu and W.-F. Fu, Inorg. Chem., 2015, 54, 3061 CrossRef CAS PubMed.
- P. Garrido-Barros, I. Funes-Ardois, S. Drouet, J. Benet-Buchholz, F. Maseras and A. Llobet, J. Am. Chem. Soc., 2015, 137, 6758 CrossRef CAS PubMed.
- C. Lu, J. Du, X.-J. Su, M.-T. Zhang, X. Xu, T. J. Meyer and Z. Chen, ACS Catal., 2016, 6, 77 CrossRef CAS.
- Z. Chen and T. J. Meyer, Angew. Chem., Int. Ed., 2013, 52, 700 CrossRef CAS PubMed.
- F. Yu, F. Li, B. Zhang, H. Li and L. Sun, ACS Catal., 2015, 5, 627 CrossRef CAS.
- A. Han, H. Wu, Z. Sun, H. Jia, Z. Yan, H. Ma, X. Liu and P. Du, ACS Appl. Mater. Interfaces, 2014, 6, 10929 CAS.
- D. Hong, J. Jung, J. Park, Y. Yamada, T. Suenobu, Y. M. Lee, W. Nam and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 7606 CAS.
- T. R. Crook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474 CrossRef PubMed.
- M. D. Kärkäs, O. Verho, E. V. Johnston and B. Akermark, Chem. Rev., 2014, 114, 11863 CrossRef PubMed.
- Ł. Szyrwiel, Ł. Szczukowski, J. S. Pap, B. Setner, Z. Szewczuk and Z. Malinka, Inorg. Chem., 2014, 53, 7951 CrossRef PubMed.
- J. S. Pap, Ł. Szyrwiel, D. Srankó, Z. Kerner, B. Setner, Z. Szewczuk and Z. Malinka, Chem. Commun., 2015, 51, 6322 RSC.
- Ł. Szyrwiel, J. S. Pap, Ł. Szczukowski, Z. Kerner, J. Brasun, B. Setner, Z. Szewczuk and Z. Malinka, RSC Adv., 2015, 5, 56922 RSC.
- Dynamics in Enzyme Catalysis, Top. Curr. Chem., ed. J. Klinman and S. Hammes-Schiffer, Springer-Verlag, Berlin, Heidelberg, 2013, vol. 337 Search PubMed.
- Ł. Szyrwiel, M. Shimura, J. Shirataki, S. Matsuyama, A. Matsunaga, B. Setner, Ł. Szczukowski, Z. Szewczuk, K. Yamauchi, W. Malinka, L. Chavatte and R. Łobinski, Metallomics, 2015, 7, 1155 RSC.
- C. Shan, H. Yang, D. Han, Q. Zhang, A. Ivaska and I. Niu, Langmuir, 2009, 25, 12030 CrossRef CAS PubMed.
- G. Decher, in Comprehensive Supramolecular Chemistry, ed. J. P. Sauvage and M. W. Hosseini, Pergamon Press, Oxford, 1996, vol. 9, ch. 14 Search PubMed.
- R. Zou, Q. Wang, J. Wu, J. Wu, C. Schmuck and H. Tian, Chem. Soc. Rev., 2015, 44, 5200 RSC.
- S. R. Puniredd, D. Janczewski, D. P. Go, X. Zhu, S. Guo, S. L. M. Teo, S. S. C. Lee and G. J. Vancso, Chem. Sci., 2015, 6, 372 RSC.
- N. Orgovan, D. Patko, C. Hos, S. Kurunczi, B. Szabó, J. J. Ramsden and R. Horvath, Adv. Colloid Interface Sci., 2014, 211, 1 CrossRef CAS PubMed.
- N. Kovacs, D. Patko, N. Orgovan, S. Kurunczi, J. J. Ramsden, F. Vonderviszt and R. Horvath, Anal. Chem., 2013, 85, 5382 CrossRef CAS PubMed.
- K. Tiefenthaler and W. Lukosz, J. Opt. Soc. Am. B, 1989, 6, 209 CrossRef CAS.
- J. Vörös, J. J. Ramsden, G. Csúcs, I. Szendrő, S. M. De Paul, M. Textor and N. D. Spencer, Biomaterials, 2002, 23, 3699 CrossRef.
- J. Vörös, Biophys. J., 2004, 87, 553 CrossRef PubMed.
- G. Decher, Science, 1997, 277, 1232 CrossRef CAS.
- M. Morga, Z. Adamczyk, S. Godrich, M. Oćwieja and G. Papastavrou, J. Colloid Interface Sci., 2015, 456, 116 CrossRef CAS PubMed.
- C. Harford and B. Sarkar, Acc. Chem. Res., 1997, 30, 123 CrossRef CAS.
- R. A. McAloney, M. Sinyor, V. Dudnik and M. C. Goh, Langmuir, 2001, 17, 6655 CrossRef CAS.
- S. T. Dubas and J. B. Schlenoff, Macromolecules, 1999, 32, 8153 CrossRef CAS.
- C. Costentin, S. Douet, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2012, 134, 11235 CrossRef CAS PubMed.
- B. Limoges and J.-M. Savéant, J. Electroanal. Chem., 2004, 562, 43 CrossRef CAS.
- R. Zou, Q. Wang, J. Wu, J. Wu, C. Schmuck and H. Tian, Chem. Soc. Rev., 2015, 44, 5200 RSC.
- V. Artero and M. Fontecave, Chem. Soc. Rev., 2013, 42, 2338 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1 and S2, X-ray photoelectron spectroscopy (XPS) on LbL-ITO with Cu-3G and PLL, Fig. S1–S20. See DOI: 10.1039/c6sc00595k |
| This journal is © The Royal Society of Chemistry 2016 |