
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and reactivity of a terminal uranium(IV) sulfide supported by siloxide ligands†
Julie
Andrez
a,
Jacques
Pécaut
bc,
Rosario
Scopelliti
a,
Christos E.
Kefalidis
d,
Laurent
Maron
d,
Michael W.
Rosenzweig
e,
Karsten
Meyer
e and
Marinella
Mazzanti
*a
aInstitut des Sciences et Ingénierie Chimiques Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland. E-mail: marinella.mazzanti@epfl.ch
bUniv. Grenoble Alpes, INAC-SyMMES, RICC, F-38000 Grenoble, France
cCEA, INAC-SyMMES, F-38000 Grenoble, France
dUniversité de Toulouse et CNRS INSA, UPS, CNRS, UMR 5215, LPCNO, 135 avenue de Rangueil, 31077 Toulouse, France
eDepartment of Chemistry and Pharmacy, Inorganic Chemistry, Friedrich-Alexander University Erlangen-Nürnberg, Egerlandstraße 1, 91058 Erlangen, Germany
First published on 10th May 2016
Abstract
The reactions of the tetrasiloxide U(III) complexes [U(OSi(OtBu)3)4K] and [U(OSi(OtBu)3)4][K18c6] with 0.5 equiv. of triphenylphosphine sulfide led to reductive S-transfer reactions, affording the U(IV) sulfide complexes [SU(OSi(OtBu)3)4K2]2, 1, and [{SU(OSi(OtBu)3)4K2}2(μ-18c6)], 2, with concomitant formation of the U(IV) complex [U(OSi(OtBu)3)4]. Addition of 1 equiv. of 2.2.2-cryptand to complex 1 resulted in the isolation of a terminal sulfide complex, [SU(OSi(OtBu)3)4K][Kcryptand], 3. The crucial role of the K+ Lewis acid in these reductive sulfur transfer reactions was confirmed, since the formation of complex 3 from the reaction of the U(III) complex [U(OSi(OtBu)3)4][Kcryptand] and 0.5 equiv. of PPh3S was not possible. Reactivity studies of the U(IV) sulfide complexes showed that the sulfide is easily transferred to CO2 and CS2 to afford S-functionalized products. Moreover, we have found that the sulfide provides a convenient precursor for the synthesis of the corresponding U(IV) hydrosulfide, {[(SH)U(OSi(OtBu)3)4][K18c6]}, 5, after protonation with PyHCl. Finally, DFT calculations were performed to investigate the nature of the U–S bond in complexes 1, 3 and 5. Based on various analyses, triple-bond character was suggested for the U–S bond in complexes 1 and 3, while double-bond character was determined for the U–SH bond in complex 5.
Introduction
Actinide complexes containing terminal oxide, sulfide or nitride ligands involved in multiple bonding with actinide ions are of high current interest, due to both their electronic structures and potential applications in atom transfer chemistry and catalysis.1–11 Though still considerably less numerous than complexes containing bridging oxo or uranyl (UO2n+) groups, the number of well-characterized terminal uranium oxo complexes has been rapidly increasing in recent years.3,9,12–16 In contrast, the majority of attempts to prepare terminal sulfides have resulted in the formation of di-uranium sulfide-bridged complexes17–22 or mononuclear disulfide complexes18,23 because of the nucleophilic character of the terminal sulfide. Such nucleophilic character has been demonstrated by reactivity studies of a bridging sulfide with heteroallenes, leading to thiocarbonates.24,25Chalcogenide complexes of actinides are also attracting increasing interest because of their importance in energy-related research. Additionally, the nature of the bonding between hard actinides and soft donor atoms and especially the involvement of f-orbitals in these bonds are of great relevance for academia and industry.26,27 Notably, the efficiency of sulfur-containing ligands in the selective extraction of actinides from spent nuclear fuel28 has been attributed to the presence of covalent An–S interactions, which remains a source of debate.29–34 However, to date, only one ligand system has been reported that enabled the isolation of a terminal uranium sulfide.16,35 Hayton and co-workers succeeded in synthesizing the terminal U(IV) sulfide complex [US(NR3)3] (R = SiMe3) by disfavouring the formation of bridging sulfide complexes with an ylide capping group during sulfur transfer from S8.16 The same complex was also prepared by cleavage of a trityl protecting group.35 A sodium-capped uranium mononuclear sulfide was also reported (with Cp* as a supporting ligand) that was isolated from the reduction of the thiolate [Cp*2U(StBu)2] with Na/Hg amalgam.36 While bulky ligands are usually used in transition metal chemistry to prevent the formation of bridging oxides and sulfides, this approach has not been successful so far in the preparation of a terminal uranium sulfide complex.
Recently, our group reported the synthesis of a terminal U(V) oxo complex, [UO(OSi(OtBu)3)4K], via cooperative two-electron reduction of carbon dioxide by the bulky heterobimetallic uranium(III) complex [U(OSi(OtBu)3)4K].14 Herein, we report the stabilization of a terminal uranium sulfide species by the bulky ligand environment created by four siloxide groups. Moreover, we found that sulfur transfer from the two-electron oxidizing agent Ph3PS to the highly hindered uranium centre is favoured by the presence of the Lewis acid K+. The reactivity of this U(IV) sulfide with different substrates was also investigated. Additionally, the bonding analysis of the terminal sulfide complex and of the potassium-bound sulfide complex revealed triple-bond character. Most importantly, calculations highlight that the participation of the f-orbitals in the bonding is indeed low.
Results and discussion
Syntheses and molecular structures of uranium(IV) sulfides
With the goal of preparing a terminal sulfide complex, the bulky U(III) complex [U(OSi(OtBu)3)4K] (Fig. 1, left) was reacted with elemental sulfur. This reaction led to multiple oxidation products, regardless of the applied stoichiometry (0.125 eq. or 0.25 eq. of S8). The 1H NMR spectra of the crude reaction mixtures show several resonances that are in agreement with the presence of multiple products. Among these products, we were able to crystallographically characterize a dimeric U(IV) persulfide complex, [(S2)U(OSi(OtBu)3)4K2]2, and a dimeric tris(siloxide) U(IV) complex, {[UK(OSi(OtBu)3)3]2(μ-S2)(μ-S3)}, containing both disulfide and trisulfide ligands (see ESI†). These results show that it is impossible to control the reaction stoichiometry by using elemental sulfur; thus, Ph3PS was used as the sulphur transfer agent.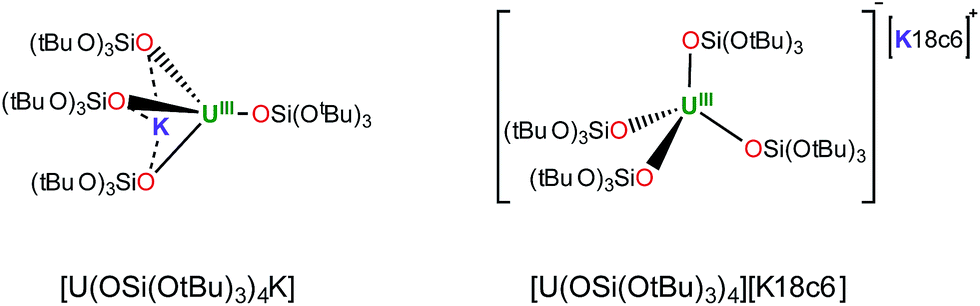 | ||
| Fig. 1 Drawing of complexes [U(OSi(OtBu)3)4K] (left) and [U(OSi(OtBu)3)4][K18c6] (right). | ||
The reaction of 0.5 equivalents of Ph3PS with a brown solution of [U(OSi(OtBu)3)4K] in toluene afforded a green solution from which the uranium(IV) sulfide complex [SU(OSi(OtBu)3)4K2]2, 1 (Scheme 1), was isolated analytically pure with yields up to 62%. Proton NMR studies reveal that complex 1 is formed with similar conversion rates when [U(OSi(OtBu)3)4K] is reacted with 1 or 0.5 equivalents of Ph3PS. The formation of 1 is accompanied by the formation of the uranium(IV) tetrasiloxide complex [U(OSi(OtBu)3)4],37 which was identified by NMR spectroscopy. The reaction of the U(III) complex [U(OSi(OtBu)3)4K] with the two-electron oxidizing agent Ph3PS led to two different U(IV) products rather than one U(V) species. This is due to the fact that U(III) complexes favour one-electron redox reactions to attain the thermodynamically more stable U(IV) ion. In this reaction, each Ph3PS oxidizes two U(III) complexes to U(IV) and transfers the sulfur atom to one of the two tetrasiloxide complexes, generating the U(IV) sulfide complex 1. Green single crystals of 1·toluene were obtained from the crude reaction mixture at room temperature.
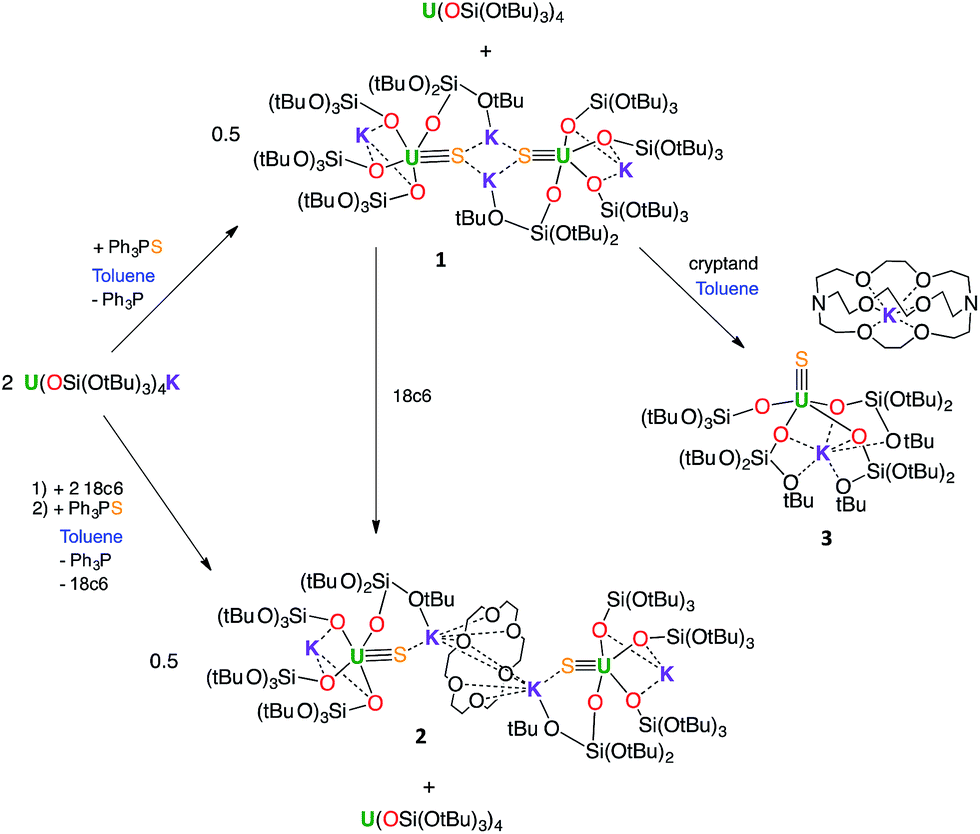 | ||
| Scheme 1 Synthesis of U(IV) terminal and potassium-capped sulfide complexes. | ||
Complex 1·toluene crystallized as a dimer in the centrosymmetric, triclinic space group, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The solid-state structure of complex 1 (Fig. 2) shows that two [SU(OSi(OtBu)3)4K2] moieties are bridged by two potassium cations, capping the sulfides to yield a dimer. Two potassium cations (each one also bound to a OtBu group) and the two sulfides form an SKSK diamond core around the inversion centre. The second potassium ion of the asymmetric unit is located in an O6 coordination pocket formed by three siloxide ligands. The five-coordinate uranium centre is ligated by four oxygen atoms of the siloxide ligands and one sulfide. The coordination geometry can best be described as distorted trigonal bipyramidal. The U–S bond length (2.5440(8) Å) is significantly longer than those found in the previously reported sodium- or potassium-capped U(IV) sulfide complexes (2.4805(5)–2.4463(6) Å),35,36 probably due to steric hindrance. The S1–K2 bond length of 3.0455(12) Å is comparable to those found in the U(IV) complex [K(18c6)][U(S)(NR2)3] (R = SiMe3) (3.0684(8) Å and 3.1551(8) Å),35 in which the sulfide is capped by the K(18c6)+ cation. The average U–Osiloxide bond length (2.22(1) Å) falls in the range of U–O bond lengths reported for uranium(IV) siloxide complexes.14,37–40
. The solid-state structure of complex 1 (Fig. 2) shows that two [SU(OSi(OtBu)3)4K2] moieties are bridged by two potassium cations, capping the sulfides to yield a dimer. Two potassium cations (each one also bound to a OtBu group) and the two sulfides form an SKSK diamond core around the inversion centre. The second potassium ion of the asymmetric unit is located in an O6 coordination pocket formed by three siloxide ligands. The five-coordinate uranium centre is ligated by four oxygen atoms of the siloxide ligands and one sulfide. The coordination geometry can best be described as distorted trigonal bipyramidal. The U–S bond length (2.5440(8) Å) is significantly longer than those found in the previously reported sodium- or potassium-capped U(IV) sulfide complexes (2.4805(5)–2.4463(6) Å),35,36 probably due to steric hindrance. The S1–K2 bond length of 3.0455(12) Å is comparable to those found in the U(IV) complex [K(18c6)][U(S)(NR2)3] (R = SiMe3) (3.0684(8) Å and 3.1551(8) Å),35 in which the sulfide is capped by the K(18c6)+ cation. The average U–Osiloxide bond length (2.22(1) Å) falls in the range of U–O bond lengths reported for uranium(IV) siloxide complexes.14,37–40
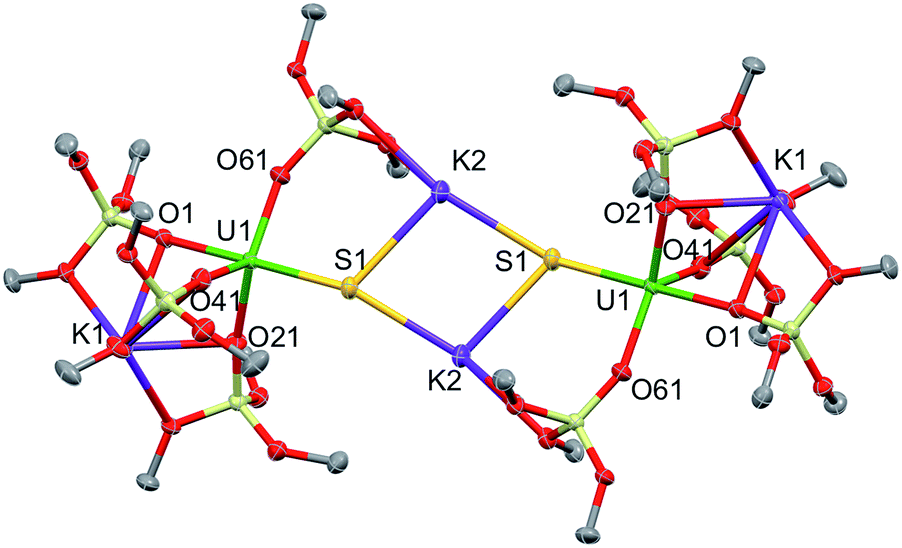 | ||
| Fig. 2 Thermal ellipsoid diagram of [SU(OSi(OtBu)3)4K2]2 (1) (50% probability ellipsoids). The methyl groups and lattice solvent molecules are omitted for clarity. | ||
The 1H NMR spectrum of crystals of 1 in deuterated toluene shows a very broad signal between 3 and 1 ppm, in agreement with the presence of a fluxional behaviour of the siloxide ligands in solution. Decreasing the temperature down to 233 K did not lead to a fully resolved spectrum. Complex 1 is stable in the solid state at room temperature but slowly decomposes in toluene at room temperature, leading to the formation of [U(OSi(OtBu)3)4], free ligand and unidentified species (decomposition products are observed after 24 hours). Complex 1 can be isolated analytically pure from toluene due to its lower solubility compared to [U(OSi(OtBu)3)4].
In order to identify the role of the potassium cation in the stabilization of the U(IV)–S species, and to prepare a terminal sulfide, complex 1 was reacted with 18c6 and 2.2.2-cryptand. The addition of 1 equivalent of 18c6 to 1 in toluene led to the formation of [{SU(OSi(OtBu)3)4K2}2(μ-18c6)], 2, containing a U(IV)–S group capped by a K(18c6)+ ion.
Blue-green single crystals of 2 were obtained from a toluene solution of the reaction mixture at 233 K. 2·tol crystallized in the centrosymmetric, monoclinic space group, P21/n. In the structure of 2, a 18c6 bridges two [SU(OSi(OtBu)3)4K2] units to yield a dimer with the inversion centre located in the middle of the crown ether (Fig. 3). The coordination environment of the U(IV) ion in 2 is very similar to that found in 1. In contrast, the US2K2 core present in the structure of 1 is disrupted by the presence of the bridging 18c6. Each potassium cation capping the sulfides in 2 is also bound to two OtBu groups from a siloxide ligand and to four oxygen atoms of the bridging crown ether. Thus, the crown ether is coordinated to two different potassium cations, and adopts a non-planar conformation.
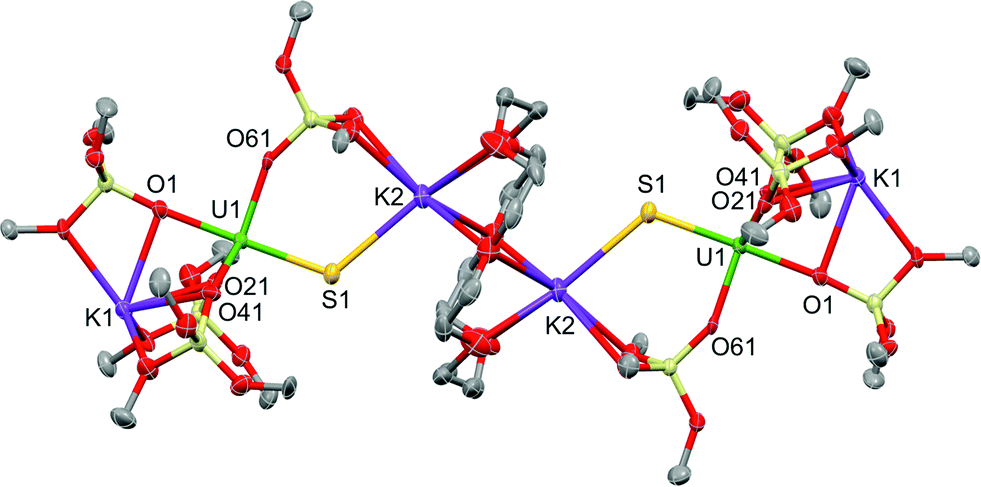 | ||
| Fig. 3 Thermal ellipsoid diagram of [{SU(OSi(OtBu)3)4K2}2(μ-18c6)] (2·tol) (50% probability ellipsoids). Methyl groups and lattice solvent molecule are omitted for clarity. | ||
The U–S distance (2.534(2) Å) in 2 is similar to that found in complex 1, indicating that the presence of the crown ether coordinated to the potassium cation does not significantly affect the U–S bonding interaction. The S1–K2 bond length (3.128(3) Å) in 2 is slightly elongated compared to 1 because of the presence of the crown ether (Table 1).
| Structural parameters | 1·tol | 2·tol | 3·hex | 5·tol |
|---|---|---|---|---|
| U–S | 2.5440(8) | 2.534(2) | 2.5220(14) | 2.834(3) |
| S–K | 3.0455(12) | 3.128(3) | — | 3.229(5) |
| Av. U–Obridging | 2.22(1) | 2.22(2) | 2.26(2) | — |
| Av. U–Oterminal | — | — | 2.197(4) | 2.15(3) |
Complex 2 can also be prepared by the reaction of 0.5 or 1 equivalent of Ph3PS with the U(III) complex [U(OSi(OtBu)3)4][K18c6] (Fig. 1, right)37 in toluene for 12 hours.
The 1H NMR spectrum of 2 in deuterated toluene features two signals for the siloxide ligands at −0.9 ppm and −10.3 ppm, respectively, with an integration ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. This is in agreement with the presence of a C3-symmetric species and a fluxional binding of the potassium cation in solution.
:3. This is in agreement with the presence of a C3-symmetric species and a fluxional binding of the potassium cation in solution.
The solid-state structure of 2 shows that the addition of crown ether to complex 1 does not prevent the binding of potassium to the U(IV)–S. The addition of excess crown ether does not afford a more symmetric solution species, indicating that the binding of the potassium cannot be prevented by crown ether in solution.
In order to inhibit the coordination of potassium to the sulfide, we resorted to the use of 2.2.2-cryptand.
The addition of 1 equivalent of cryptand to a solution of 1 in toluene (Scheme 1) afforded the U(IV) terminal sulfide complex [SU(OSi(OtBu)3)4K][Kcryptand], 3.
Green single crystals of 3 were obtained from hexane at room temperature. Complex 3·hex crystallized in the centrosymmetric, triclinic space group, P, as a separated ion pair, consisting of the [Kcryptand]+ cation and the [SU(OSi(OtBu)3)4K]− anion (Fig. 4). In 3, the five-coordinate uranium ion is ligated by four siloxide ligands and a terminal sulfide, giving a distorted trigonal bipyramidal coordination geometry. One potassium atom remains encapsulated in the O6 pocket made by three bridging siloxide ligands. The U–S bond length in 3 (2.5220(14) Å) is comparable to the U–S bond distance in complex 1 (2.5440(8) Å) (Table 1). This indicates that potassium binding and dimer formation only lead to a slight lengthening of the U–S bond. The U–O bond length is 2.197(4) Å for the terminal siloxide oxygen, while the average for the three UK-bridging siloxide ligands is 2.26(2) Å, which is in the range of previously reported U–Osiloxide bond lengths.14,37–40
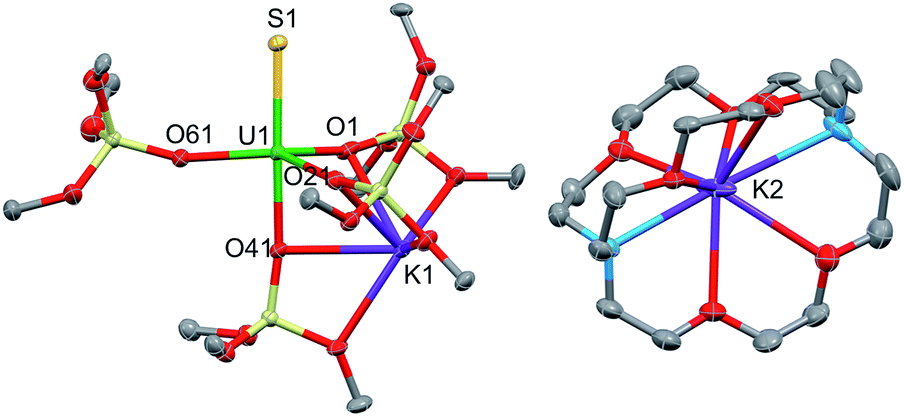 | ||
| Fig. 4 Thermal ellipsoid diagram of [SU(OSi(OtBu)3)4K][Kcryptand] (3·hex) (50% probability ellipsoids). Methyl groups and lattice solvent molecule are omitted for clarity. | ||
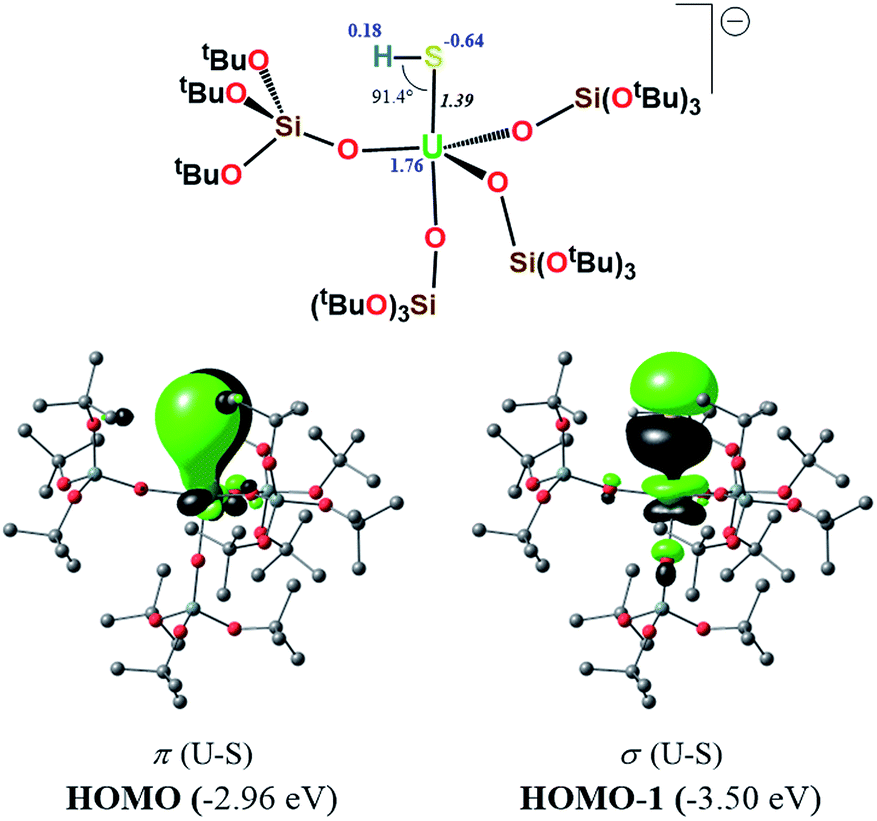
The 1H NMR spectrum of 3 in deuterated toluene shows two signals (ratio 1:3) for the siloxide ligands, in agreement with a C3-symmetric species in solution within the NMR timescale. This suggests the presence of a unique chemical environment for the three siloxide ligands that compose the equatorial plane giving rise to one signal and one environment for the siloxide trans to the U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S giving rise to the second signal. This can be interpreted in terms of a fluxional binding of the potassium cation. Three additional paramagnetically shifted signals assigned to the cryptand protons are also observed. The paramagnetic shift of the cryptand signals strongly suggests that the terminal sulfide could be in fast exchange in solution with a potassium-capped sulfide species.41
S giving rise to the second signal. This can be interpreted in terms of a fluxional binding of the potassium cation. Three additional paramagnetically shifted signals assigned to the cryptand protons are also observed. The paramagnetic shift of the cryptand signals strongly suggests that the terminal sulfide could be in fast exchange in solution with a potassium-capped sulfide species.41
The terminal sulfide complex 3 is stable in the solid state at room temperature, but slowly decomposes in toluene (50% decomposition after 1 month), as well as in THF solution (decomposition products already visible in the NMR spectrum after 24 hours), affording the U(IV) complex [U(OSi(OtBu)3)4] and a mixture of other decomposition products. One of the decomposition products of 3 in THF has been identified as the U(IV)–U(IV) complex [S3U2(OSi(OtBu)3)6K3][Kcryptand], 4 (20% conversion in 4 after 7 months determined by NMR spectroscopy). Complex 4 crystallizes as an ion pair consisting of the [U2(μ-S)3(OSi(OtBu)3)6K3]− anion and the [Kcryptand]+ cation. In the [U2(μ-S)3(OSi(OtBu)3)6K3]− anion, three S2− anions bridge the two uranium atoms (see ESI†). Thus, the decomposition of complex 3 leads to the loss of one siloxide ligand from each uranium complex, as well as sulfide redistribution to afford a sulfide-bridged diuranium(IV) complex. Although a terminal U(IV) sulfide is stabilized by the presence of the sterically hindered environment provided by the four siloxide ligands, this complex can slowly eliminate one siloxide ligand and further react to afford a sulfide-bridged diuranium(IV) complex.
Influence of cation binding on the S-transfer reaction
Interestingly, no reaction is observed when Ph3PS is added to a solution of the U(III) complex [U(OSi(OtBu)3)4K] that has been pre-treated with cryptand (Scheme 2). This unambiguously shows that the presence of a bound potassium cation is crucial in the S-transfer reaction between [U(OSi(OtBu)3)4K] and Ph3PS.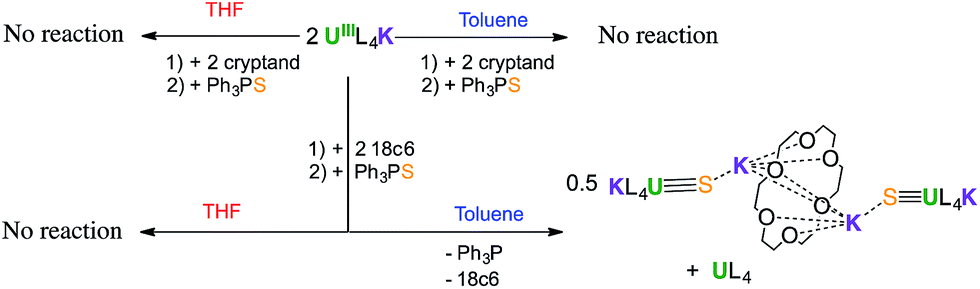 | ||
| Scheme 2 Reactivity of [U(OSi(OtBu)3)4K] with Ph3PS in the presence of cryptand and 18C6. | ||
Notably, in toluene solution, the potassium cation remains bound to the siloxide ligands in complex [U(OSi(OtBu)3)4K], while [U(OSi(OtBu)3)4][Kcryptand] exists as separated ion pair. The solid-state structure of [U(OSi(OtBu)3)4][K18c6]37 reveals an ion pair with (K18c6)+ located in the outer coordination sphere of the complex, but the coordination of potassium is probably still possible in solution.
To confirm the important role of potassium for the reactivity of the complexes, the reactions were studied in the more polar solvent THF. In THF, both [U(OSi(OtBu)3)4][K18c6] and [U(OSi(OtBu)3)4][Kcryptand] most likely exist as ion pairs and therefore do not react with Ph3PS. Reactivity is only observed in THF for the [U(OSi(OtBu)3)4K] complex.
To some extent, such pronounced differences in reactivity arise from steric differences that result in reduced access of the substrate to the metal centre in [U(OSi(OtBu)3)4][Kcryptand]. However, the possible role of cooperative binding of the potassium to the sulfur might also be important. Comparing the O–U–O angles in the reported X-ray structures of the heterobimetallic UK complex [U(OSi(OtBu)3)4K] and the ion pair [U(OSi(OtBu)3)4][K18c6],37 a significant difference is observed. In the ion pair [U(OSi(OtBu)3)4][K18c6] the four ligands form a weakly distorted tetrahedron with three angles having a mean value of 110.6(6)° and three angles having a mean value of 108.3(3)°. In the UK complex, the coordination tetrahedron is highly distorted. The potassium cation coordinates three of the four ligands and brings them closer together, resulting in an average value of the three O–U–O angles of 94.91(7)°. In contrast, the O–U–O angles between the bridging siloxides and the terminal one are significantly larger (127.2(3)°, 122.5(3)° and 115.0(3)°) than those found in [U(OSi(OtBu)3)4][K18c6], rendering the metal centre more accessible to the substrate.
In both complexes, the steric hindrance provided by the four siloxide ligands prevents the rapid formation of sulfide-bridged complexes previously observed when reacting neutral U(III) silylamide- or tacn-based amido or aminophenolato complexes with sulfur transfer agents (S8 or Ph3PS).17–19,21,42
However, the presence of the potassium cation in the oxygen pocket of three siloxide ligands in the heterobimetallic UK complex results in easier access to the uranium centre than in the ion pair complex, in which the potassium is encapsulated in the crown ether (Fig. 1 and 5).
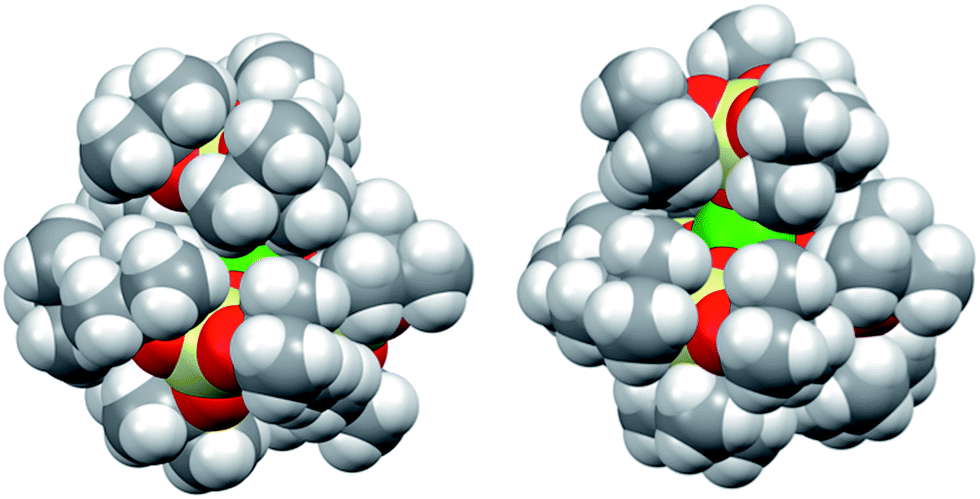 | ||
| Fig. 5 Space-filling representation of [U(OSi(OtBu)3)4][K18c6] (left) and [U(OSi(OtBu)3)4K] (right). The lattice solvent molecule and the [K18c6]+ cation in [U(OSi(OtBu)3)4][K18c6] are omitted for clarity. | ||
On the other hand, due to the low accessibility of the metal centre in these tetrasiloxide complexes, cooperative binding of potassium might be also important for the S-transfer process to occur. Binding of the sulfur atom to the potassium cation during the sulfur transfer reaction may also reduce the nucleophilic character of the sulfur, rendering reaction pathways leading to bridging sulfides less favourable. The important role of cooperative UK binding in the reduction of CO2 and CS2 by [U(OSi(OtBu)3)4K] has been reported previously.14,39
Reactivity of the U(IV) sulfide
Previous reactivity studies have shown that sulfide-bridged diuranium(IV) complexes can undergo nucleophilic addition of chalcogens, CS2 and CO2, to afford stable disulfide-,43 trithiocarbonate-25 and monothiocarbonate-bridged (CO2S2−)24 diuranium(IV) complexes, respectively. In order to probe the possibility of sulfur transfer in complexes containing a U–S multiple bond, we investigated the reactivity of the sulfide complex 1 with various substrates (Scheme 3). To date, only chalcogen atom transfer reactivity has been investigated for terminal U(IV) sulfides, affording di-chalcogenide and tri-chalcogenide complexes depending on the stoichiometry.44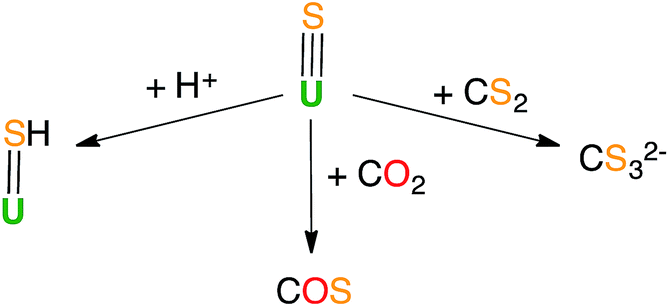 | ||
| Scheme 3 Reactivity of U(IV) sulfide species. | ||
Complex 1 reacts rapidly with 13CS2 (1 equivalent per U atom) in toluene. The 1H NMR spectrum of the reaction mixture shows the presence of signals assigned to exchanging species that we were unable to identify due to the low stability of the corresponding compound.
After several hours, the presence of [U(OSi(OtBu)3)4] was observed by 1H NMR spectroscopy. Dissolution of the evaporated reaction mixture in DMSO-d6 led to complete dissociation of the reaction products. The 1H NMR spectrum in DMSO-d6 only contains one signal, which was assigned to [U(OSi(OtBu)3)4].
The 13C NMR spectrum of the reaction products in DMSO-d6 shows the presence of only one signal at 267 ppm, which was assigned to free thiocarbonate.45 This indicates that, in toluene, the 13CS2 molecule inserts into the U–S bond to afford a U(IV) thiocarbonate complex (Scheme 4), which dissociates into [U(OSi(OtBu)3)4] and K2CS3 in DMSO. The labile trithiocarbonate complex [U(OSi(OtBu)3)4(μ3κ2:κ2:κ2CS3)K2(18c6)2]39 was isolated previously from the reaction of [U(OSi(OtBu)3)4][K18c6] with CS2. This complex quickly dissociates in solution, affording [U(OSi(OtBu)3)4] and K2CS3.
 | ||
| Scheme 4 CS2 insertion into complex 1. | ||
Complex 3 displays the same reactivity towards 13CO2 and 13CS2 as 1. The 13C NMR spectrum in DMSO-d6 of the reaction of 3 with 13CS2 only shows the signals of thiocarbonate and 2.2.2-cryptand. The presence of the cryptand in the reaction mixture does not affect the insertion of CS2 into the U–S bond and a terminal thiocarbonate U(IV) complex is also likely to be formed in this case. Formation of a stable trithiocarbonate-bridged di-uranium(IV) complex from the nucleophilic addition of a sulfide-bridged diuranium(IV) complex to CS2 has been previously reported by Meyer et al.,25 but such reactivity has never been reported for terminal sulfides.
In light of the observed fast addition of CS2 to 1 and 3, we also decided to explore the reaction of 1 with CO2. Complex 1 reacted immediately with an excess of 13CO2 in toluene, affording a new labile U(IV) species that decomposes rapidly at room temperature. Attempts to crystallize the reaction products were not successful. The 13C NMR spectrum of the reaction mixture in deuterated toluene shows the presence of a peak at 153.5 ppm that increases over time. This chemical shift corresponds to free COS.46 The formation of COS can be interpreted as arising from the decomposition of a hypothetical U–CO2S intermediate.
A sulfide complex seemed the ideal precursor for the straightforward synthesis of a U(IV) hydrosulfide complex and therefore we investigated the reactivity of complex 2 with pyHCl. This is a known strategy in transition metal chemistry for the synthesis of hydrosulfide complexes.47,48 Hydrosulfide complexes of transition metals have attracted considerable attention because of their relevance to metalloenzymes and metal sulfide catalysts for industrial hydrodesulfurization.48–51 The only crystallographically characterized uranium hydrosulfido complexes to date have been prepared through reduction of H2S by tacn- and N-anchored tris(aryl oxide) U(III) complexes.52
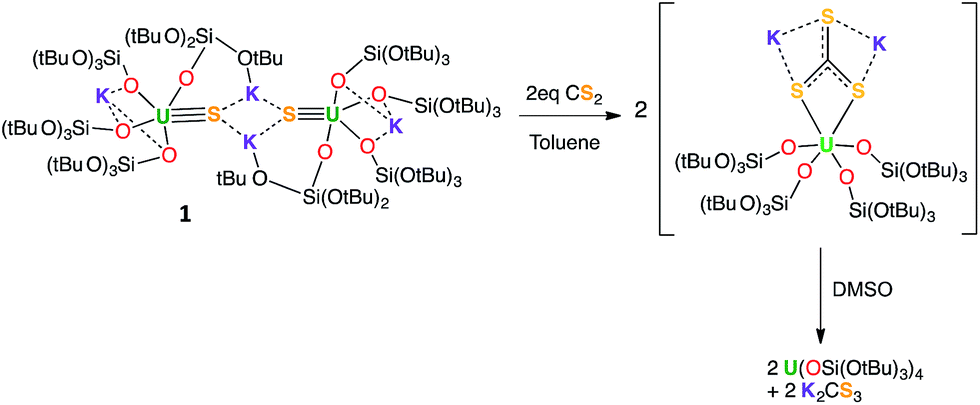
The addition of one equivalent (per U atom) of pyHCl to the [{SU(OSi(OtBu)3)4K2}2(μ-18c6)] complex 2 in THF led to the formation of {[(SH)U(OSi(OtBu)3)4][K18c6]}, 5, with 53% conversion determined by 1H NMR spectroscopy. Light blue-green single crystals were obtained from the crude toluene mixture at 233 K (Fig. 6). Complex 5·tol crystallized in the non-centrosymmetric, monoclinic space group, Cc. The five-coordinate uranium atom is ligated by four terminal siloxide ligands and one SH− moiety in a distorted trigonal bipyramidal geometry. The 18c6-encapsulated potassium counter-ion is bound to the sulfur atom with a S1–K1 bond length of 3.229(5) Å. The U1–S1 distance was measured to be 2.834(3) Å, which is much longer than the U–S bond distance in 3 (2.5220(14) Å), but is very similar to that found in the only other example of a mononuclear U(IV)–SH complex that was reported by Meyer et al. (2.797(1) Å).52 The average U–Osiloxide bond length (2.15(3) Å) is similar in length to that found for the terminal siloxides in the terminal sulfide complex 3 (2.197(4) Å).
 | ||
| Fig. 6 Thermal ellipsoid diagram of {[(SH)U(OSi(OtBu)3)4][K18c6]} (5·tol) (50% probability ellipsoids). Methyl groups and lattice solvent molecule are omitted for clarity. | ||
Complex 5 can also be prepared from the reaction of the U(III) complex [U(OSi(OtBu)3)4][K18c6] with H2S, but in much lower yield, independent of the U:H2S ratio (17% yield and 22% overall conversion was determined by quantitative NMR spectroscopy). Moreover, this alternative synthetic route requires the handling of a solution of toxic H2S. X-ray quality crystals of a dimeric by-product were also isolated from this reaction. This structure clearly shows the presence of two U(IV) and [U(OSi(OtBu)3)3K] moieties, bridged by three sulfur atoms (complex 6 in ESI†). The bond valence sum and the intermediate average value of the U–S distances (2.83(2) Å) fall in between the reported values for bridging S2− (2.59(1)–2.736(2) Å)17,21 and bridging SH− (2.877(1)–2.964(1) Å)52 moieties, suggesting that 2 SH− and 1 S22− groups are bridging the uranium centres. The hydrogens of the SH− moieties are probably fluxional, and therefore very difficult to locate in the crystal structure. The isolation of this by-product suggests that the low yield of the reaction with H2S is probably associated with the fast reaction of the hydrosulfide with more than one equivalent of H2S (even using stoichiometric conditions). The further aggregation of the initially formed mononuclear hydrosulfide complexes leads to polynuclear sulfide complexes. This represents a well-recognized problem in the preparation of mononuclear hydrosulfide complexes.48 Therefore, the protonation of the sulfide 2 provides a more convenient route to the preparation of the hydrosulfide complex 5.
Computational bonding analysis
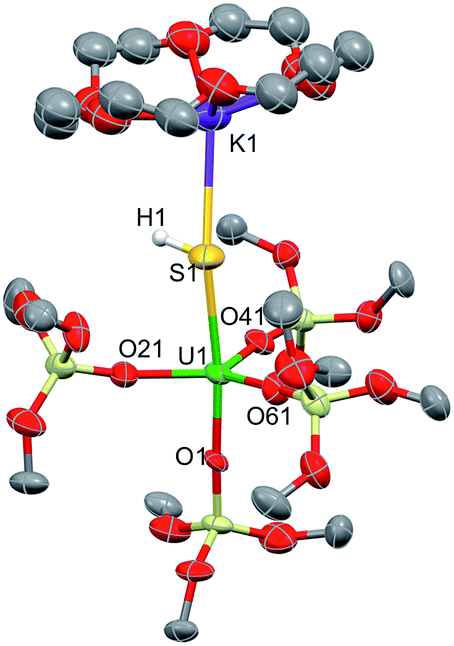
In order to investigate the nature of the U–S bond in complexes 1, 3 and 6, we performed calculations at the B3PW91 level. In particular for complex 1, a small core pseudopotential basis set was chosen for the uranium atom, in which the f-electrons are included in the valence shell. Moreover, the monomeric form of complex 1 was considered for the sake of computational time. To verify the validity of such a theoretical protocol, we compared some important geometrical features to the available X-ray data and found that they are in close agreement (see ESI†). In particular, we found that the calculated values of the U–S bond distances were in agreement with the experimental ones (Fig. S.D.1†). Henceforth, we proceed into the analysis by firstly inspecting the related molecular orbitals (MOs).As expected, for the U(IV) electron configuration (triplet multiplicity), the (SOMO)α and (SOMO−1)α orbitals correspond to pure non-bonding f-orbitals. Interestingly, the subsequent three MOs correspond to two π- (HOMO, HOMO−1) and one σ-type (HOMO−2) singly-occupied MOs of the U–S bond, as depicted in Fig. 9. It is worth noting that in (HOMO−2) the orbital is polarized towards the potassium atom, indicating small, but not negligible, overlap between the K and the S atom. This molecular orbital picture between the dianionic sulfide and the U(IV) centre, which is unconventional for the transition metals, is also found in H2US gas phase compounds.33 Similarly, they closely resemble the shape of the MOs responsible for the U![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Nterminal triple bond in TrenTIPS-based complexes,53,54 as well as of the recently reported Th(IV) chalcogenide tris(amide) system.11 The peculiar triple character bonding situation is also evident based on the natural bond orbital (NBO) analysis. In particular, the contribution of the uranium atom to the σ-bond is 18%, whereas the contribution of the sulfur is 77%. For the two π combinations, the contributions are 23% and 15% for the uranium atom, and 72% and 78% for the sulfur atom, respectively. Additionally, the two highest singly-occupied orbitals based on NBO analysis are almost pure 5f orbitals, with the composition of (SOMO−1)α being 92% of U, and of (SOMO)α being 84% of U and 8% of S. In the same way, the Wiberg bond order analysis in a Löwdin orthogonalized basis gave a bond order of 2.25 for the U–S bond, indicating partial triple bond character. In addition, the natural charges of the U and S are 1.39 and −0.86 |e|, respectively. The natural electron configuration (NEC) of the uranium (see Fig. 7) corresponds to what is expected for a formal f2 configuration. At first sight, this seems to indicate a crucial role of the f-orbitals in the bonding.
Nterminal triple bond in TrenTIPS-based complexes,53,54 as well as of the recently reported Th(IV) chalcogenide tris(amide) system.11 The peculiar triple character bonding situation is also evident based on the natural bond orbital (NBO) analysis. In particular, the contribution of the uranium atom to the σ-bond is 18%, whereas the contribution of the sulfur is 77%. For the two π combinations, the contributions are 23% and 15% for the uranium atom, and 72% and 78% for the sulfur atom, respectively. Additionally, the two highest singly-occupied orbitals based on NBO analysis are almost pure 5f orbitals, with the composition of (SOMO−1)α being 92% of U, and of (SOMO)α being 84% of U and 8% of S. In the same way, the Wiberg bond order analysis in a Löwdin orthogonalized basis gave a bond order of 2.25 for the U–S bond, indicating partial triple bond character. In addition, the natural charges of the U and S are 1.39 and −0.86 |e|, respectively. The natural electron configuration (NEC) of the uranium (see Fig. 7) corresponds to what is expected for a formal f2 configuration. At first sight, this seems to indicate a crucial role of the f-orbitals in the bonding.
 | ||
| Fig. 7 σ and π molecular orbitals (α-spin) calculated for the monomer-1 using the small-core pseudopotential. The numbers in blue correspond to the natural charges and those in italics to the Wiberg bond orders. Hydrogen atoms are omitted for clarity. | ||
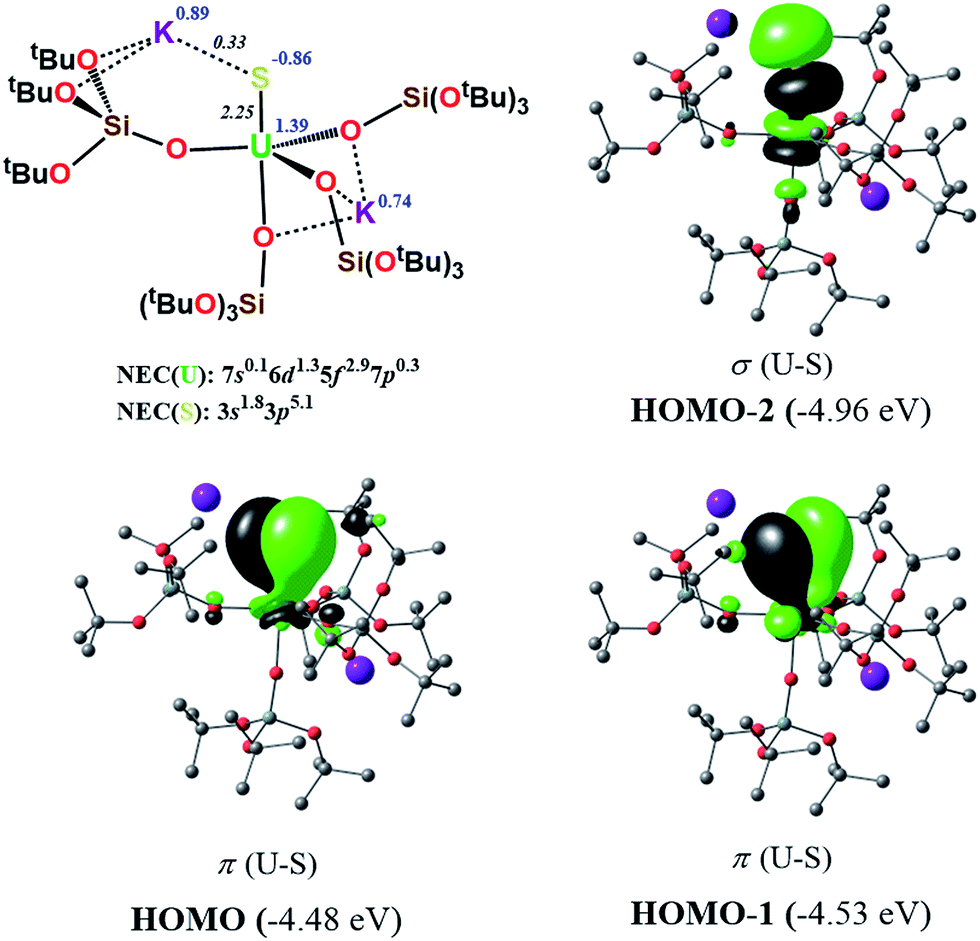
In order to further investigate the role of the f-orbitals in the bonding, we considered a different pseudopotential for the uranium(IV) atom, the so-called “large-core”. By using this f-in-core pseudopotential, the f-electrons are now explicitly included in the core shell configuration, and hence not available for any mixture with other orbitals, with the d-orbitals being the only ones available for bonding. However, even with such a computational strategy, the bonding picture remains essentially the same, as illustrated in Fig. 8. It should be noted that the NBO analysis also predicts that the U–S bond features triple-bond character as well, in line with the corresponding small-core calculations.
 | ||
| Fig. 8 σ and π molecular orbitals (α-spin) calculated for the monomer-1 using the “large-core” pseudopotential. Hydrogen atoms are omitted for clarity. | ||
Therefore, the triple-bond character is not due to 5f involvement in the bonding. Finally, the spin density (SD) of uranium was found to be 2.19 (being depopulated by 0.19), with most of this residual being donated to the sulfide atom (SDsulfur = −0.12).
In order to investigate the effect of the presence of S-bound potassium on the U–S bond, we also calculated the electronic structure of complex 3. The cryptand moiety was excluded from the calculations, and consequently a negative charge was placed on the overall complex. The DFT-predicted structure of the triplet state, which is imposed by the uranium +IV oxidation state, is in close agreement with that found in the solid-state structure (see ESI†). Molecular orbital analysis gave the same picture for the bonding situation between the U and S atoms as for the monomeric structure 1, as clearly depicted in Fig. 9.
 | ||
| Fig. 9 σ and π molecular orbitals (α-spin) calculated for 3, using the small-core pseudopotential. The numbers in blue correspond to the natural charges and those in italics to the Wiberg bond orders. Hydrogen atoms are omitted for clarity. | ||
Interestingly, despite the absence of a second potassium atom in the vicinity of the sulfide, the total picture of the bonding remains unchanged with respect to monomer-1. Minor differences are apparent, which are mostly linked to the absence of the polarization induced by the potassium atom on the σ-orbital (HOMO−2), and to the slightly smaller natural charge located on the sulfide group. Moreover, there are virtually no changes to the charges of the uranium and potassium atom in 3 with respect to the corresponding ones in monomer-1. NBO analysis also predicted a σ2π4 U–S configuration, in line with a triple bond. Specifically, the σ-bond has 20% uranium and 76% sulfur character, whereas the contributions of each atom to the two π bonds are 29/18% for U and 67/75% for S, respectively. Again, as found for monomer-1, the exclusion of the f-electrons from the valence shell does not change the bonding picture (see ESI†). The bond order of the U–S bond was found to be 2.55, slightly higher than that calculated for monomer-1. In addition, the spin density (SD) of uranium was found to be 2.21 and that of the sulfur was found to be −0.14.
In order to gain insights into the bonding situation in complex 5, we proceeded to the optimization of the X-ray structure at the B3PW91 level, once again using the “small-core” basis set for the uranium atom. The potassium crown ether (K18C6) cation was excluded from the calculations, and consequently a negative charge was placed on the overall complex. Molecular orbital analysis is consistent with the presence of a double bond between the U and S atoms, as depicted in Fig. 10. This is in line with the previous MO picture found in the monomer-1 and 3 models, since here the protonation of the strongly nucleophilic sulfide results in the breaking of one of the two π bonds. In particular, the HOMO−1 and HOMO orbitals possess 14% and 12% uranium character, respectively, and 82% sulfur character in both cases. The two SOMOs are mainly composed of pure f-orbitals. This further highlights the strongly polarized nature of this bond. Interestingly, the Wiberg bond orders in a Löwdin orthogonalized basis gave a bond order of 1.39 for the U–S bond, a value that is significantly smaller than in the other complexes, and is fully consistent with the partial double character of such an interaction.
 | ||
| Fig. 10 σ and π molecular orbitals (α-spin) calculated for complex 5 using the small-core pseudopotential. The numbers in blue correspond to the natural charges and those in italics to the Wiberg bond orders. Hydrogen atoms are omitted for clarity. | ||
Conclusions
In summary, we have isolated a new series of complexes containing a U–S bond from the reaction of the bulky U(III) tetrasiloxide complex [U(OSi(OtBu)3)4K] with Ph3PS.This is the first example of a terminal uranium(IV) sulfide complex, which was directly obtained from the reaction of U(III) and a sulfur-transfer agent without the addition of protecting groups. The bulk provided by the four siloxide ligands prevents the formation of sulfide-bridged complexes. Moreover, the presence of bound potassium is essential for the reaction to occur, probably due to both steric effects and cooperative binding of the sulfur-transfer reagent to the K and U centres.
The terminal sulfide can easily be transferred to CO2 and CS2 to afford new thiocarbonates. Moreover, the terminal sulfide provides a convenient precursor for the synthesis of the corresponding hydrosulfide complex upon protonation with PyHCl.
DFT calculations carried out for the potassium-bound sulfide complex 1, and for the terminal sulfide complex [SU(OSi(OtBu)3)4K][Kcryptand], 3, showed that the U–S interactions in both complexes consist of three bonding pairs (σ + 2π bonds) with a Wiberg bond order of 2.25 for 1 and a bond order of 2.55 for 3. However, the use of the “large-core” pseudopotential indicates that the triple-bond character is not due to 5f involvement in the bonding. For the hydrosulfide complex 5, the molecular orbital analysis is consistent with the presence of a double bond between the U and S atoms with a Wiberg bond order of 1.39, a value significantly smaller than those found in complexes 1 and 3.
Future studies will be directed to explore the reactivity of the terminal sulfide with other organic molecules and to isolate terminal sulfide complexes containing uranium in oxidation states higher than +IV and to investigate the nature of the U–S bond in these systems.
Experimental
General procedures
Unless otherwise noted, all experiments were carried out at ambient temperature under an inert atmosphere using Schlenk techniques and an MBraun glovebox equipped with a purifier unit. Water and oxygen levels were always kept at less than 1 ppm. Glassware was dried overnight at 150 °C prior to use. Syntheses were performed using glass-covered stirring bars.Starting materials
Solvents were purchased in their anhydrous form from Aldrich or Cortecnec (deuterated solvents), conditioned under argon and vacuum distilled from K/benzophenone (toluene, THF) or sodium dispersion (hexane) or dried over molecular sieves for one week (DMSO). All reagents were dried under high-vacuum for 5 days prior to use. Dry 13CO2 was prepared by storing over molecular sieves for one week prior to use. HOSi(OtBu)3 ligand was purchased from Aldrich and purified by sublimation prior to use. Depleted uranium turnings were purchased from the “Société Industrielle du Combustible Nucléaire” of Annecy (France). [U(OSi(OtBu)3)4K]14 and [U(OSi(OtBu)3)4][18c6]37 were prepared according to the published procedures.1H NMR experiments
NMR spectra were performed in J. Young NMR tubes. 1H and 13C NMR spectra were recorded on a Bruker 400 MHz spectrometer. NMR chemical shifts are reported in ppm and are referenced to the residual 1H and 13C signals of the deuterated solvents.Elemental analyses
They were performed under argon by Analytische Laboratorien GMBH at Lindlar (Germany) or by the elemental analyses department of the EPFL using a Thermo Scientific Flash 2000 Organic Elemental Analyzer.X-ray analyses
Experimental details for X-ray data collections of all complexes are given in Table S1.† Figure graphics were generated using MERCURY 3.6 supplied by the Cambridge Structural Database; CCDC: Cambridge, U.K., 2001–2015. Diffraction data were taken using Oxford-Diffraction Xcalibur S or Bruker APEX II CCD kappa geometry diffractometers (Mo-Kα radiation, graphite monochromator, λ = 0.71073 Å). To prevent evaporation of co-crystallized solvent molecules the crystals were coated with light hydrocarbon oil and the data were collected at 150 K or 100 K. The datasets were reduced by CrysAlis55 or EvalCCD56 and then corrected for absorption.57The structure resolutions were performed with SHELXS or Superflip and the structure refinement was performed with SHELXL.58,59 The crystal structures were refined using full-matrix least-squares based on F2 with all non-hydrogen atoms anisotropically defined. Hydrogen atoms were placed in calculated positions by means of the “riding” model. Additional electron density found in the difference Fourier map (due to highly disordered solvent) was eventually treated by the SQUEEZE algorithm of PLATON.60
Synthesis of complex 1
A colourless solution of PPh3S (26.3 mg, 0.089 mmol, 0.5 eq.) in toluene (2 mL) was added to a stirred brown solution of [U(OSi(OtBu)3)4K] (238.1 mg, 0.179 mmol, 1 eq.) in toluene (4 mL). The mixture was stirred at room temperature for 18 h. The resulting green solution was concentrated to approximately 3 mL and big green crystals formed overnight from toluene at room temperature. The crystals were filtered and dried for 2 hours (79.3 mg, 62% yield in 2 crops). The yield can be increased by recovering additional crops but it leads to co-crystallization of small amounts of the byproduct [U(OSi(OtBu)3)4]. 1H NMR (400 MHz, Tol-d8, 298 K): δ [ppm] ≈ 1 (very broad, 216H). Anal. calcd for 1·(tol)0.6 C100.2H220.8O32Si8S2K4U2: C, 42.09; H, 7.78; S, 2.24. Found C, 42.17; H, 7.68; S, 2.10. Green single crystals of 1·tol suitable for X-ray diffraction were obtained from a concentrated toluene solution of the complex at room temperature.Reaction of [U(OSi(OtBu)3)4][K18c6] with 0.5 eq. of Ph3PS: isolation of 2
A colourless solution of PPh3S (2.1 mg, 0.007 mmol, 0.5 eq.) in toluene (1 mL) was added to a stirred brown suspension of [U(OSi(OtBu)3)4][K18c6] (22.3 mg, 0.014 mmol, 1 eq.) in toluene (0.5 mL). The mixture was stirred at room temperature for 18 h to yield a green solution. The 1H NMR spectrum shows the presence of PPh3, [U(OSi(OtBu)3)4] and complex 2 (90% conversion determined by 1H NMR spectroscopy using naphthalene as an internal standard) in the reaction mixture. 1H NMR (400 MHz, Tol-d8, 298 K): δ [ppm] = −0.9 (s, 162H), −10.3 (s, 54H). Blue-green single crystals of 2·tol were obtained from the toluene reaction mixture at 233 K. Due to the similar solubility of [U(OSi(OtBu)3)4] and complex 2, 2 can only be isolated in low yield from this reaction (25%).Complex 2 can also be obtained with similar conversion rates by addition of 1 eq. of 18c6 per U atom to complex 1 in toluene.
Anal. Calcd for 2 C108H240O38Si8S2K4U2: C, 42.28; H, 7.88; found C, 42.33; H, 8.07.
Synthesis of complex 3
A colourless solution of 2.2.2-cryptand (7.8 mg, 0.021 mmol, 2 eq., 1 eq. per U) in toluene (1 mL) was added to a stirred green solution of complex 1 (29.5 mg, 0.010 mmol, 1 eq.) in toluene (1 mL). After 20 min. of stirring, toluene was removed and hexane (1 mL) was added. Green single crystals of complex 3·hex formed from hexane at room temperature. The crystals were filtered, washed with hexane and dried under vacuum for 1 h (27 mg, 74%). 1H NMR (400 MHz, Tol-d8, 298 K): δ [ppm] = 13.1 (bs, 12H), 12.9 (bs, 12H), 11.8 (bs, 12H), −0.5 (s, 81H), −10.6 (s, 27H). Anal. calcd for 3 C66H144N2O22Si4SK2U: C, 44.57; H, 8.16; N, 1.58. Found C, 44.16; H, 8.15; N, 1.65.Reaction of complex 1 with 2 eq. of 13CS2
To a green solution of complex 1 (8.5 mg, 0.003 mmol, 1 eq.) in deuterated toluene (0.5 mL), 9.3 μL of a 636.5 mM solution of 13CS2 (0.006 mmol, 2 eq.) in deuterated toluene was added. The reaction mixture immediately turned light yellow, affording a labile species. 1H NMR (400 MHz, Tol-d8, 298 K): δ [ppm] = 11.1 (bs, 27H), −2.8 (bs, 81H). The solution was periodically monitored by 1H NMR spectroscopy over the course of a week, and it showed a decrease in the intensity of the two broad peaks and an increase in the intensity of the signal corresponding to the U(IV) complex [U(OSi(OtBu)3)4]. This evolution shows the lability of the likely formed thiocarbonate complex, resulting in the release of the thiocarbonate anion. When toluene was removed and DMSO-d6 added, the 13C NMR spectrum only showed the signal assigned to thiocarbonate. 13C{1H} NMR (100 MHz, DMSO-d6, 298 K): δ [ppm] = 267.4 (s, CS32−).Reaction of complex 1 with an excess of 13CO2
An excess (1 atm) of 13CO2 was added to a frozen green solution of complex 1 (11.7 mg, 0.004 mmol, 1 eq.) in deuterated toluene (0.5 mL). The solution was allowed to warm up to room temperature to yield a light pink solution. 1H NMR (400 MHz, Tol-d8, 298 K): δ [ppm] = 8.7 (bs, 27H), −2.9 (bs, 81H). 13C{1H} NMR (100 MHz, Tol-d8, 298 K): δ [ppm] = 153.5 (s, CSO).Synthesis of complex 5
A colourless solution of 18c6 (5.2 mg, 0.020 mmol, 2 eq.) in THF (1 mL) was added to a stirred green solution of complex 1 (28.2 mg, 0.010 mmol, 1 eq.) in THF (1 mL). The resulting green solution of complex 2 was stirred for ten minutes and was added to a stirred white suspension of PyHCl (2.3 mg, 0.020 mmol, 2 eq.) in THF (1 mL). The resulting yellow suspension was stirred for 2 h to yield a light green suspension. The 1H NMR spectrum of the crude reaction mixture in THF-d8 at 298 K showed the formation of complex 5 with 53% conversion (determined by NMR spectroscopy using naphthalene as an internal standard) as the main reaction product. Blue-green single crystals of complex 5·tol were obtained by storing the toluene reaction mixture at 233 K. The crystals were collected and dried under vacuum for 2 h (11.1 mg, 34% yield). 1H NMR (400 MHz, THF-d8, 298 K): δ [ppm] = 3.07 (s, 24H, 18c6), 0.74 (s, 108H). Anal. calcd for 5·(tol)0.5 C63.5H137O22Si4SKU: C, 45.55; H, 8.25. Found C, 45.66; H, 8.61.Complex 5 was also obtained in lower yield (17%) (22% overall conversion determined by NMR spectroscopy using naphthalene as an internal standard) by addition of 1 eq. of H2S to the U(III) complex [U(OSi(OtBu)3)4][K18c6] in THF.
The reaction of complex 1 with PyHCl was carried out under analogous conditions. The 1H NMR spectrum of the crude reaction mixture performed in THF-d8 at 298 K after addition of 18c6 showed the formation of 5 with 48% conversion.
The reaction of 3 with PyHCl affords {[(SH)U(OSi(OtBu)3)4][Kcrypt]} with 52% conversion as determined by 1H NMR. 1H NMR (400 MHz, THF-d8, 298 K): δ [ppm] = 3.36–3.33 (m, crypt), 2.35 (s, crypt), 0.70 (s, 108H) (the identity of this complex was confirmed by comparing this 1H NMR spectrum to that obtained after adding cryptand to a solution of complex 5).
Computational details
All the structures reported in this study were fully optimized with the Becke's 3-parameter hybrid functional combined with the non-local correlation functional provided by Perdew/Wang (denoted as B3PW91).61,62 The Stuttgart-Dresden RECP (relativistic effective core potential) 5f-in-valence was used for the uranium atom, in combination with its adapted basis set.63–65 However, in some cases, the 5f-in-core ECP augmented by a f polarization function (α = 1.0) was used for the fixed oxidation state IV of the uranium atom.66 In addition, silicon atoms were treated with the corresponding Stuttgart-Dresden RECP in combination with its adapted basis sets,67 each one augmented by an extra set of polarization functions.68 For the rest of the atoms, the 6-31G(d,p) basis set was used.69–71 For analysing the bonding situation in the complexes of interest, we mainly used natural bond orbital analysis (NBO) using Weinhold's methodology.72,73 Also, the Multiwfn program74 was used for obtaining the composition of the molecular orbitals, based on the natural atomic orbital method,75 as well as the Wiberg bond order analysis in a Löwdin orthogonalized basis. The Chemcraft program was used for the visualization of the molecular orbitals.76 Finally, the GAUSSIAN09 program suite was used in all calculations.77Acknowledgements
We would like to thank Dr Oliver Cooper for preliminary experiments and Dr Euro Solari for elemental analysis. LM is a member of the Institut Universitaire de France. CAlmip is acknowledged for the generous grant of computing time. This work was supported by EPFL, by the Swiss National Science Foundation and by funds of the Joint DFG-ANR projects (ANR-14-CE35-0004-01 and ME1754/7-1).Notes and references
- D. S. J. Arney and C. J. Burns, J. Am. Chem. Soc., 1995, 117, 9448–9460 CrossRef CAS.
- D. S. J. Arney and C. J. Burns, J. Am. Chem. Soc., 1993, 115, 9840–9841 CrossRef CAS.
- G. F. Zi, L. Jia, E. L. Werkema, M. D. Walter, J. P. Gottfriedsen and R. A. Andersen, Organometallics, 2005, 24, 4251–4264 CrossRef CAS.
- T. W. Hayton, Dalton Trans., 2010, 39, 1145–1158 RSC.
- S. T. Liddle, Angew. Chem., Int. Ed. Engl., 2015, 54, 8604–8641 CrossRef CAS PubMed.
- M. B. Jones and A. J. Gaunt, Chem. Rev., 2013, 113, 1137–1198 CrossRef CAS PubMed.
- T. Andrea and M. S. Eisen, Chem. Soc. Rev., 2008, 37, 550–567 RSC.
- M. S. Eisen, Top. Organomet. Chem., 2010, 31, 157–184 CrossRef CAS.
- T. W. Hayton, Chem. Commun., 2013, 49, 2956–2973 RSC.
- W. S. Ren, G. F. Zi, D. C. Fang and M. D. Walter, J. Am. Chem. Soc., 2011, 133, 13183–13196 CrossRef CAS PubMed.
- D. E. Smiles, G. Wu, P. Hrobarik and T. W. Hayton, J. Am. Chem. Soc., 2016, 138, 814–825 CrossRef CAS PubMed.
- S. J. Kraft, J. Walensky, P. E. Fanwick, M. B. Hall and S. C. Bart, Inorg. Chem., 2010, 49, 7620–7622 CrossRef CAS PubMed.
- B. Kosog, H. S. La Pierre, F. W. Heinemann, S. T. Liddle and K. Meyer, J. Am. Chem. Soc., 2012, 134, 5284–5289 CrossRef CAS PubMed.
- O. Cooper, C. Camp, J. Pecaut, C. E. Kefalidis, L. Maron, S. Gambarelli and M. Mazzanti, J. Am. Chem. Soc., 2014, 136, 6716–6723 CrossRef CAS PubMed.
- S. Fortier, N. Kaltsoyannis, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2011, 133, 14224–14227 CrossRef CAS PubMed.
- J. L. Brown, S. Fortier, R. A. Lewis, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2012, 134, 15468–15475 CrossRef CAS PubMed.
- O. P. Lam, F. W. Heinemann and K. Meyer, Chem. Sci., 2011, 2, 1538–1547 RSC.
- C. Camp, M. A. Antunes, G. Garcia, I. Ciofini, I. C. Santos, J. Pecaut, M. Almeida, J. Marcalo and M. Mazzanti, Chem. Sci., 2014, 5, 841–846 RSC.
- J. L. Brown, G. Wu and T. W. Hayton, Organometallics, 2013, 32, 1193–1198 CrossRef CAS.
- D. E. Smiles, G. Wu and T. W. Hayton, New J. Chem., 2015, 39, 7563–7566 RSC.
- J. G. Brennan, R. A. Andersen and A. Zalkin, Inorg. Chem., 1986, 25, 1761–1765 CrossRef CAS.
- L. P. Spencer, P. Yang, B. L. Scott, E. R. Batista and J. M. Boncella, Inorg. Chem., 2009, 48, 11615–11623 CrossRef CAS PubMed.
- E. M. Matson, M. D. Goshert, J. J. Kiernicki, B. S. Newell, P. E. Fanwick, M. P. Shores, J. R. Walensky and S. C. Bart, Chem.–Eur. J., 2013, 19, 16176–16180 CrossRef CAS PubMed.
- O. P. Lam, S. M. Franke, F. W. Heinemann and K. Meyer, J. Am. Chem. Soc., 2012, 134, 16877–16881 CrossRef CAS PubMed.
- O. P. Lam, L. Castro, B. Kosog, F. W. Heinemann, L. Maron and K. Meyer, Inorg. Chem., 2012, 51, 781–783 CrossRef CAS PubMed.
- M. L. Neidig, D. L. Clark and R. L. Martin, Coord. Chem. Rev., 2013, 257, 394–406 CrossRef CAS.
- M. Mazzanti, R. Wietzke, J. Pécaut, J.-M. Latour, P. Maldivi and M. Remy, Inorg. Chem., 2002, 41, 2389–2399 CrossRef CAS PubMed.
- H. H. Dam, D. N. Reinhoudt and W. Verboom, Chem. Soc. Rev., 2007, 36, 367–377 RSC.
- A. J. Gaunt, B. L. Scott and M. P. Neu, Chem. Commun., 2005, 3215–3217 RSC.
- K. I. M. Ingram, N. Kaltsoyannis, A. J. Gaunt and M. P. Neu, J. Alloys Compd., 2007, 444, 369–375 CrossRef.
- C. C. L. Pereira, C. J. Marsden, J. Marcalo and J. K. Gibson, Phys. Chem. Chem. Phys., 2011, 13, 12940–12958 RSC.
- M. B. Jones, A. J. Gaunt, J. C. Gordon, N. Kaltsoyannis, M. P. Neu and B. L. Scott, Chem. Sci., 2013, 4, 1189–1203 RSC.
- X. F. Wang, L. Andrews, K. S. Thanthiriwatte and D. A. Dixon, Inorg. Chem., 2013, 52, 10275–10285 CrossRef CAS PubMed.
- L. Karmazin, M. Mazzanti and J. Pécaut, Chem. Commun., 2002, 654–655 RSC.
- D. E. Smiles, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2014, 136, 96–99 CrossRef CAS PubMed.
- L. Ventelon, C. Lescop, T. Arliguie, P. C. Leverd, M. Lance, M. Nierlich and M. Ephritikhine, Chem. Commun., 1999, 659–660 RSC.
- C. Camp, J. Pecaut and M. Mazzanti, J. Am. Chem. Soc., 2013, 135, 12101–12111 CrossRef CAS PubMed.
- V. Mougel, C. Camp, J. Pecaut, C. Coperet, L. Maron, C. E. Kefalidis and M. Mazzanti, Angew. Chem., Int. Ed. Engl., 2012, 51, 12280–12284 CrossRef CAS PubMed.
- C. Camp, O. Cooper, J. Andrez, J. Pecaut and M. Mazzanti, J. Chem. Soc., Dalton Trans., 2015, 44, 2650–2656 RSC.
- C. Camp, C. E. Kefalidis, J. Pecaut, L. Maron and M. Mazzanti, Angew. Chem., Int. Ed. Engl., 2013, 52, 12646–12650 CrossRef CAS PubMed.
- B. M. Gardner, G. Balazs, M. Scheer, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed. Engl., 2014, 53, 4484–4488 CrossRef CAS PubMed.
- L. R. Avens, D. M. Barnhart, C. J. Burns, S. D. McKee and W. H. Smith, Inorg. Chem., 1994, 33, 4245–4254 CrossRef CAS.
- S. M. Franke, F. W. Heinemann and K. Meyer, Chem. Sci., 2014, 5, 942–950 RSC.
- D. E. Smiles, G. Wu and T. W. Hayton, Inorg. Chem., 2014, 53, 12683–12685 CrossRef CAS PubMed.
- D. Stueber, D. Patterson, C. L. Mayne, A. M. Orendt, D. M. Grant and R. W. Parry, Inorg. Chem., 2001, 40, 1902–1911 CrossRef CAS PubMed.
- W. Gombler, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1981, 36, 1561–1565 Search PubMed.
- S. J. Smith, C. M. Whaley, T. B. Rauchfuss and S. R. Wilson, Inorg. Chem., 2006, 45, 679–687 CrossRef CAS PubMed.
- S. Kuwata and M. Hidai, Coord. Chem. Rev., 2001, 213, 211–305 CrossRef CAS.
- M. Peruzzini, I. De los Rios and A. Romerosa, Prog. Inorg. Chem., 2001, 49, 169–453 CrossRef CAS.
- C. Bianchini and A. Meli, Acc. Chem. Res., 1998, 31, 109–116 CrossRef CAS.
- M. R. Dubois, Chem. Rev., 1989, 89, 1–9 CrossRef CAS.
- (a) S. M. Franke, M. W. Rosenzweig, F. W. Heinemann and K. Meyer, Chem. Sci., 2015, 6, 275–282 RSC; (b) M. W. Rosenzweig, A. Scheurer, F. W. Heinemann, C. A. Lamsfus, L. Maron, J. Andrez, M. Mazzanti and K. Meyer, Chem. Sci., 2016 10.1039/C6SC00677A.
- Q.-Y. Wu, J.-H. Lan, C.-Z. Wang, Y.-L. Zhao, Z.-F. Chai and W.-Q. Shi, J. Phys. Chem. A, 2015, 119, 922–930 CrossRef CAS PubMed.
- D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Science, 2012, 337, 717–720 CrossRef CAS PubMed.
- CrysAlisPro CCD, CrysAlisPro RED and ABSPACK, CrysAlis PRO. Agilent Technologies, Yarnton, England., Agilent, 2010.
- A. J. M. Duisenberg, L. M. J. Kroon-Batenburg and A. M. M. Schreurs, J. Appl. Crystallogr., 2003, 36, 220–229 CrossRef CAS.
- R. H. Blessing, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 33–38 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef.
- W. Kuchle, M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1994, 100, 7535–7542 CrossRef.
- X. Y. Cao, M. Dolg and H. Stoll, J. Chem. Phys., 2003, 118, 487–496 CrossRef CAS.
- X. Y. Cao and M. Dolg, J. Mol. Struct.: THEOCHEM, 2004, 673, 203–209 CrossRef CAS.
- A. Moritz, X. Y. Cao and M. Dolg, Theor. Chem. Acc., 2007, 118, 845–854 CrossRef CAS.
- A. Bergner, M. Dolg, W. Kuechle, H. Stoll and H. Preuss, Mol. Phys., 1993, 80, 1431–1441 CrossRef CAS.
- A. W. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Höllwarth, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 111–114 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- F. Weinhold, in The Encyclopedia of Computational Chemistry, ed. P. v. R. Schleyer, John Wiley & Sons, Chichester, 1998, p. 1792 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- T. Lu and F. Chen, Acta Chim. Sin., 2011, 69, 2393–2406 CAS.
- http://www.chemcraftprog.com .
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full computational details, 1H NMR spectra, and detailed X-ray crystallographic data in CIF format. CCDC 1447982–1447990. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc00675b |
| This journal is © The Royal Society of Chemistry 2016 |