
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotonic crystal protein hydrogel sensor materials enabled by conformationally induced volume phase transition†
Zhongyu
Cai
a,
Linda A.
Luck
b,
David
Punihaole
a,
Jeffry D.
Madura
c and
Sanford A.
Asher
*a
aDepartment of Chemistry, University of Pittsburgh, Pittsburgh, Pennsylvania 15260, USA. E-mail: asher@pitt.edu
bDepartment of Chemistry, State University of New York at Plattsburgh, Plattsburgh, NY 12901, USA
cDepartment of Chemistry and Biochemistry, Duquesne University, Pittsburgh, Pennsylvania 15282, USA
First published on 24th March 2016
Abstract
Hydrogels that change volume in response to specific molecular stimuli can serve as platforms for sensors, actuators and drug delivery devices. There is great interest in designing intelligent hydrogels for tissue engineering, drug delivery, and microfluidics that utilize protein binding specificities and conformational changes. Protein conformational change induced by ligand binding can cause volume phase transitions (VPTs). Here, we develop a highly selective glucose sensing protein photonic crystal (PC) hydrogel that is fabricated from genetically engineered E. coli glucose/galactose binding protein (GGBP). The resulting 2-D PC-GGBP hydrogel undergoes a VPT in response to glucose. The volume change causes the 2-D PC array particle spacing to decrease, leading to a blue-shifted diffraction which enables our sensors to report on glucose concentrations. This 2-D PC-GGBP responsive hydrogel functions as a selective and sensitive sensor that easily monitors glucose concentrations from ∼0.2 μM to ∼10 mM. This work demonstrates a proof-of-concept for developing responsive, “smart” protein hydrogel materials with VPTs that utilize ligand binding induced protein conformational changes. This innovation may enable the development of other novel chemical sensors and high-throughput screening devices that can monitor protein–drug binding interactions.
Introduction
Hydrogel volume phase transitions (VPTs) can be harnessed in a variety of applications, particularly in sensing and in drug delivery.1–3 Flory–Huggins theory proposes that hydrogel VPTs result from responses of hydrogels to osmotic pressures generated within the polymer–water network.4 The VPT of hydrogels can be employed for applications such as sensors, actuators, and drug delivery devices if the target analyte creates an osmotic pressure that forces hydrogel volume changes.5–7Recently, a number of studies utilized specific target recognition and protein conformational changes to design “smart” hydrogels for tissue engineering, drug delivery, and microfluidics.8–15 More interestingly, conformational changes induced by protein–ligand recognition were reported to trigger VPTs in polymer hydrogels containing calmodulin and glucose/galactose binding protein (GGBP).10,11,13,15 The use of these protein conformation-induced VPTs, however, have not been utilized for quantitative sensing applications.
Here, we report on a protein hydrogel VPT induced by GGBP's conformational change, which enables selective glucose sensing. GGBP is a member of the periplasmic binding superfamily of proteins; it selectively binds D-glucose and D-galactose.16–19Fig. 1a schematically illustrates the GGBP conformational changes that occur upon glucose binding. The glucose-binding site is located in the cleft between two domains. The crystal structures of E. coli GGBP in the D-glucose bound and unbound states16,17 show that upon glucose binding, the two domains rotate 31° toward each other and engulf the glucose inside the binding pocket (Fig. 1a).
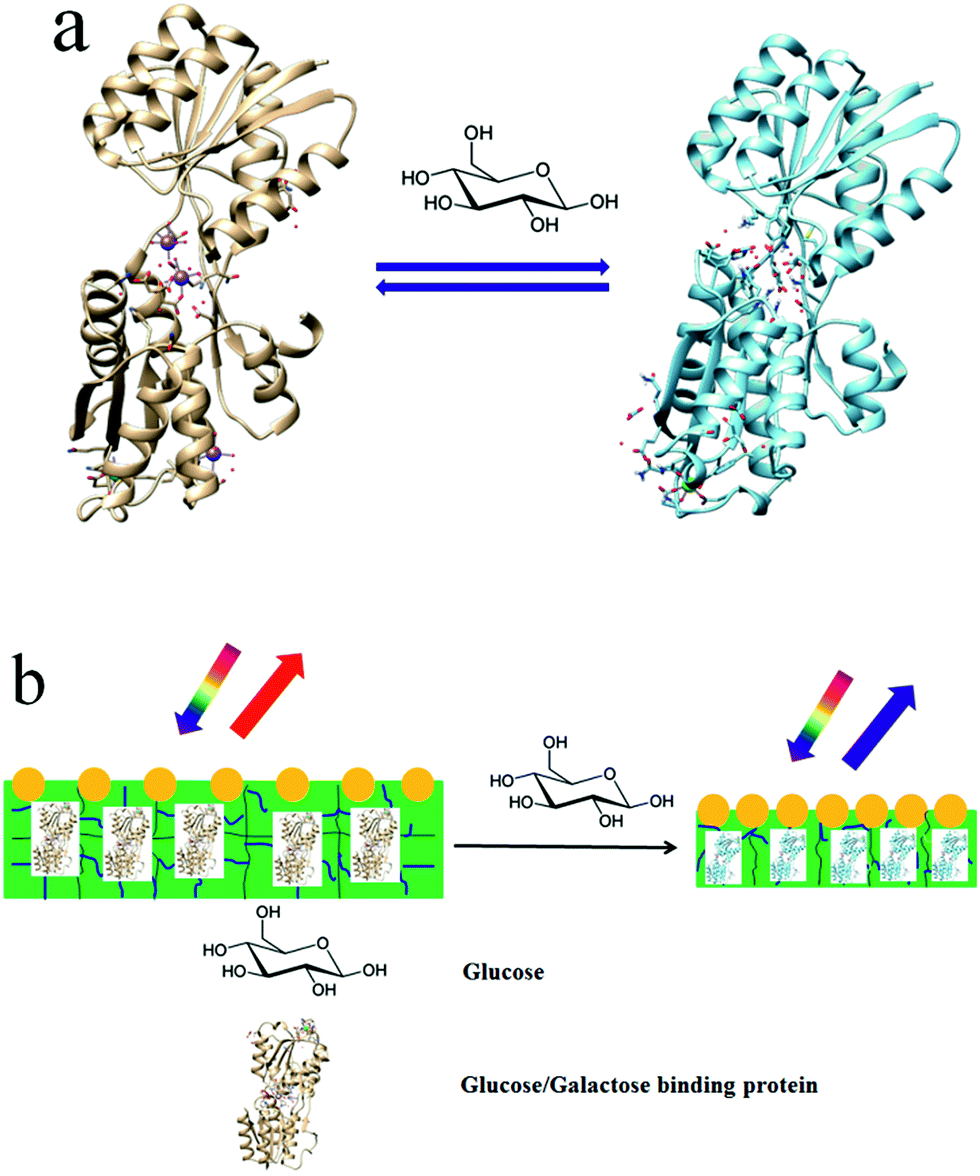 | ||
| Fig. 1 (a) Venus flytrap conformational change of GGBP induced by glucose. The figures were prepared by using the UCSF Chimera software package. (b) Schematic illustration showing that the glucose binding induced 2-D PC-GGBP hydrogel VPT causes diffraction blueshifts. | ||
We fabricated a GGBP hydrogel by cross-linking a GGBP solution with glutaraldehyde. This GGBP hydrogel contains a surface attached 2-D non-close packed photonic crystal (PC) whose diffraction serves as an optical readout of the hydrogel area. The glucose-induced conformational change induces a VPT that shrinks the protein hydrogel (Fig. 1b). This shrinkage decreases the attached 2-D PC particle spacing, which blue-shifts its diffraction. The glucose concentration can be quantitatively determined by measuring the diffraction shift.
Results and discussion
We fabricated the GGBP hydrogel by cross-linking a 50 mg mL−1 GGBP monomer solution (<5 vol%) with glutaraldehyde, similar to that previously demonstrated for serum albumin proteins and concanavalin A.20,21 The GGBP Lys groups react with glutaraldehyde to form Schiff base cross-links (the details of the reaction mechanism were shown in Fig. S1, S2, and ESI†). Fig. 2a shows the visually evident 2-D PC-GGBP hydrogel diffraction from the attached 2-D array. The Fig. 2a inset SEM image shows that the 2-D array is non-close packed and can respond to both hydrogel swelling and shrinking. | ||
Fig. 2 (a) Photograph of 2-D PC-GGBP sensing hydrogel diffracting white light; (b) UVRR spectra (excited at 204 nm) of GGBP monomer solution (shown in black) and hydrogel (shown in red). The UVRR monomer–hydrogel difference spectrum is shown in blue. (c) Photograph of Debye ring diffraction of 2-D PC-GGBP hydrogel sensor illuminated by a 532 nm green laser pointer along the normal; (d) Debye ring measurement. h is the distance between the 2-D array and the screen. D is the diameter of the Debye diffraction ring on the screen. The diffraction angle, α is calculated from tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) α = D/2h. α = D/2h. | ||
The GGBP hydrogels consist of essentially native conformation GGBP proteins, as indicated by the UV Resonance Raman (UVRR) difference spectrum in Fig. 2b, between the hydrogel and the native monomer solution. The Amide III3 region (∼1200–1300 cm−1) of the difference spectrum shows a small positive feature at ∼1225 cm−1 and a small negative feature at ∼1255 cm−1. The positive ∼1225 cm−1 feature indicates a slightly greater β-sheet content in the GGBP monomer than in the hydrogel. The negative ∼1255 cm−1 feature is likely indicative of somewhat more “disordered” PPII-like secondary structures in the hydrogel form. These two features suggest that β-strands in GGBP slightly disorder upon cross-linking into a hydrogel. The difference spectral features at ∼1180 cm−1, ∼1210 cm−1 and ∼1620 cm−1 from Tyr and Phe indicate that the Tyr and Phe residues experience a more hydrophilic environment in the GGBP hydrogels compared to the monomers in solution. We monitored the diffraction by irradiating the 2-D PC-GGBP hydrogel with a green laser pointer along the normal. The light is forward diffracted into a Debye ring (Fig. 2c) whose diameter reports on the 2-D array nearest neighbour spacing (Fig. 2d).22 The details on the Debye ring measurement are discussed in the Experimental section and are demonstrated well in refs. 20–22.
Fig. 3 compares the D-glucose, D-galactose and D-fructose concentration dependence of the particle spacing changes in a 2-D PC-GGBP hydrogel, and in a bovine serum albumin (BSA) protein hydrogel (2-D PC-BSA). The 2-D PC-GGBP sensor shows a 6 nm particle spacing decrease for a 0.2 μM free glucose concentration, whereas a ∼30 nm particle spacing decrease occurs upon exposure to 10 mM D-glucose. This particle spacing decrease is consistent with the diffraction wavelength maximum shift measured by using a fiber optic reflection probe in the Littrow configuration at an angle of 21.8° between the probe and the 2D array normal. We observe a blue shift from 662 to 642 nm as the glucose concentration increases from 0 to 10 mM (Fig. S4†).
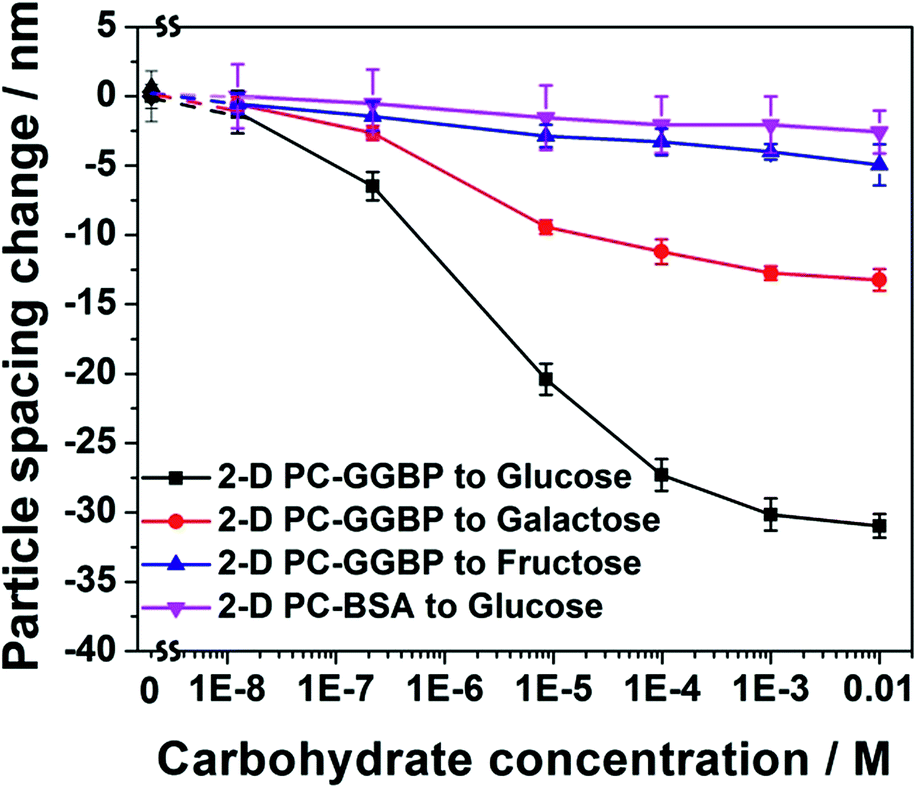 | ||
| Fig. 3 D-Glucose, D-galactose and D-fructose concentration dependencies of particle spacing changes for 2-D PC-GGBP and 2-D PC-BSA hydrogel PC. The original hydrogel PC has an initial particle spacing of 1028 nm. | ||
GGBP selectively binds D-glucose and D-galactose over D-fructose. As shown in Fig. 3, a 10 μM D-fructose solution only gives rise to a ∼3 nm particle spacing decease, while a 10 μM D-glucose concentration shows a ∼20 nm particle spacing decrease and a 10 μM D-galactose solution leads to a ∼9 nm particle spacing change at 4 °C. These results occur because GGBP does not bind D-fructose, and the binding affinity of GGBP to D-galactose is about half of that to D-glucose at 4 °C.23 Our negative control, a 2-D PC-BSA protein hydrogel that was fabricated in a manner similar to that of the 2-D PC-GGBP hydrogel, shows no significant response to D-glucose. This 2-D PC-GGBP hydrogel quantitatively and selectively detects D-glucose over a ∼0.2 μM to 10 mM concentration range. This is the concentration range that would enable the physiological monitoring of glucose in tear fluid.24 The glucose concentration of tear fluid is much lower than that of blood.25
The calculated glucose detection limit of our 2-D PC-GGBP hydrogel is 1.5 × 10−7 M. Half of the maximum particle spacing decrease occurs for a free glucose concentration of 2.0 × 10−6 M, which suggests an effective association constant (Ka) of 5.0 × 105 M−1 (see calculation details in ESI†). This Ka value is 10-fold smaller than the native monomer GGBP reported value of 5 × 106 M−1.23,26 This decrease is probably caused by the slight change in protein structure in the hydrogel, as indicated by the UVRR spectra.18
We investigated the reversibility of the sensing response to glucose by repeatedly exposing the 2-D PC-GGBP hydrogel to 100 μM D-glucose in 10 mM phosphate buffer at pH 8 followed by washing with a large amount of phosphate buffer (see Experimental section for details). As shown in Fig. 4a, the 2-D PC-GGBP sensor is highly reversible, even after 10 cycles of glucose exposure and washing.
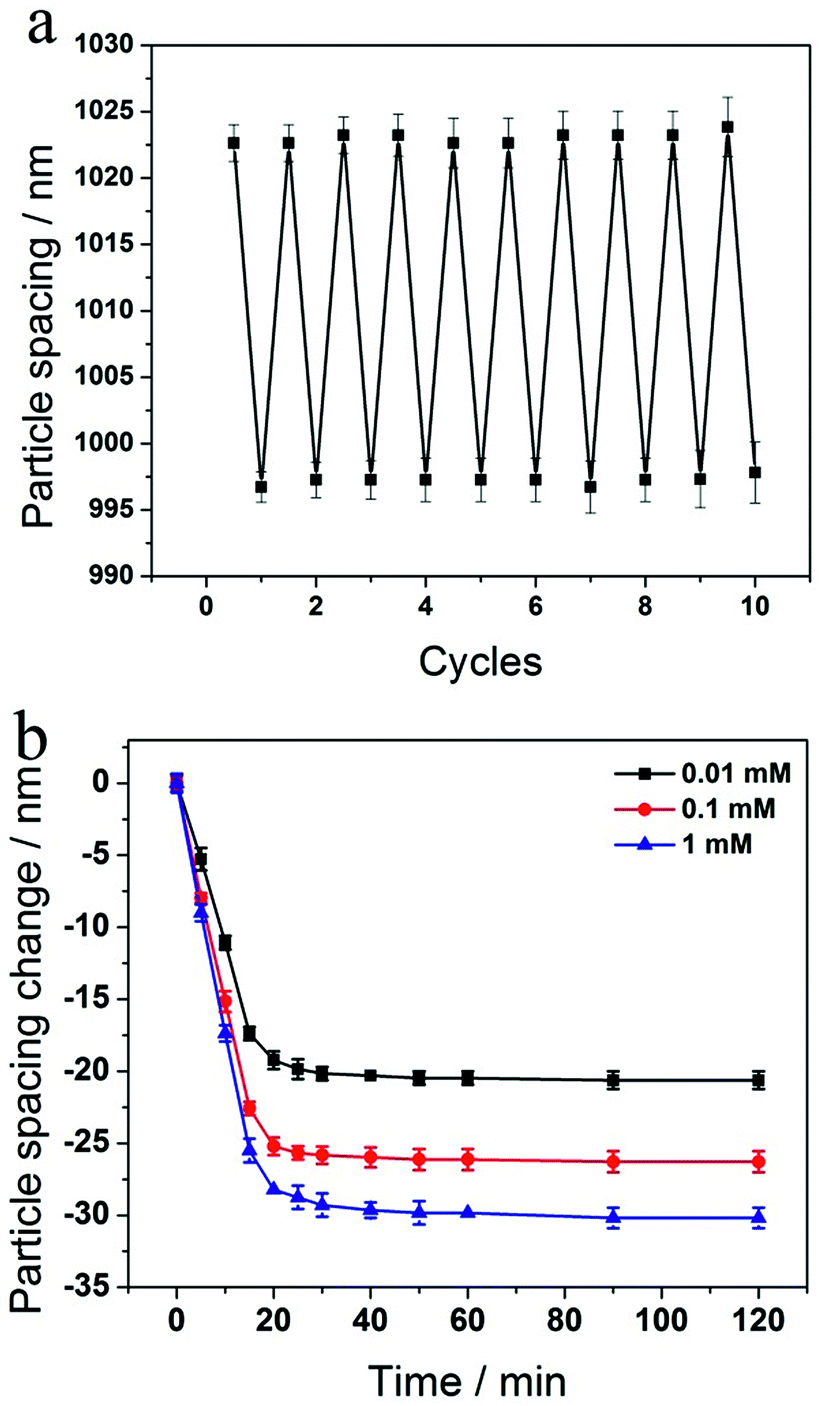 | ||
| Fig. 4 (a) Reversibility of 2-D PC-GGBP hydrogel response to 100 μM glucose solutions that contain 10 mM phosphate buffer at pH 8, followed by washing. (b) Kinetics of 2-D PC-GGBP hydrogel sensor responses to 0.01, 0.1 and 1.0 mM glucose solutions, respectively. | ||
The kinetics of the 2-D PC-GGBP hydrogel responses to 0.01, 0.1 and 1.0 mM glucose concentrations are shown in Fig. 4b. The 2-D PC-GGBP sensor saturates within ∼20 min after immersion into 20 mL of these glucose solutions. The 2-D PC-GGBP sensor shows a response time constant of k ∼ (9 min)−1 for 1.0 mM glucose solution. These kinetics are ∼100-fold slower than would be expected from the glucose diffusion time through the ∼60 μm thick sensing hydrogel, given the expected glucose in water diffusion constant of 6.8 × 10−6 cm2 s−1.27 The slow response probably results from the slower limiting collective response of the GGBP hydrogel protein.
GGBP-glucose binding induces a conformation change, which causes a VPT that decreases the hydrogel volume. We calculate from the X-ray structures that the GGBP undergoes a 0.07% volume decrease upon glucose binding by using the MSP program (http://www.biohedron.com, ESI†).17,28 In contrast, the GGBP hydrogel volume shrinkage calculated from the 2-D array particle spacing change is much larger, at ∼8.7%.
The small volume change of GGBP is consistent with our UVRR measurements (see ESI, Fig. S3†), which show no significant secondary structural changes in GGBP upon binding glucose. The X-ray crystal structures indicate that the only significant structural changes in GGBP upon glucose binding are rigid body movements of the two domains relative to each other (Fig. S5†).
We conclude that the larger hydrogel volume decrease of the 2-D PC-GGBP hydrogel upon glucose binding does not derive from structural changes within the individual cross-linked proteins. Instead, the hydrogel volume decrease results from a macroscopic hydrogel VPT which decreases the GGBP protein hydrogel volume by decreasing the hydrogel water content. This could be caused, for example, by a decreased exposure of hydrophilic groups and/or an increased exposure of hydrophobic groups upon glucose binding.
We calculate from the X-ray structures that upon glucose binding the GGBP protein shows a 4.7% decreased water accessible surface area (calculated from the MSP program, see ESI†). The protein hydrogel volume is dominated by the water content that is stabilized in the hydrogel by interactions with the protein surface. We roughly expect that a decreased water accessible surface area would cause a proportional water content decrease.
In contrast, we find that glucose binding causes a ∼2-fold greater water content decrease. This larger decrease may result from additional alterations in the protein surface hydrophilicity. We looked for evidence for a less hydrophilic protein surface but were unable to clearly document this surface alteration. The other possibility is that the hydrogel GGBP, which is less constrained than that in the crystal, shows larger conformational changes. These changes could be more similar to that of the native protein monomer in solution.
Thus, the magnitude of the protein hydrogel VPT volume shrinkage is more than that expected from the D-glucose bound and unbound GGBP X-ray structures.17 The amplified volume change may enable us to examine subtle aspects of protein structural changes that result from protein–ligand binding. Protein–ligand binding hydrogel VPT studies similar to those carried out for GGBP here, may enable physiologically relevant investigations of subtle drug-induced protein binding conformational transitions, which may be helpful for guiding drug discovery.29
Conclusions
In summary, we fabricated a glucose-responsive protein hydrogel by gently cross-linking a GGBP monomer solution with glutaraldehyde. Glucose binding to the GGBP initiates a VPT that decreases the hydrogel volume. We sensitively monitored the hydrogel volume by monitoring the diffraction of a surface attached 2-D colloidal crystalline array. This 2-D PC-GGBP hydrogel glucose sensing response demonstrates a proof-of-concept for fabricating protein hydrogel sensors that utilize a protein conformational induced hydrogel VPT to specifically detect analytes. The protein–ligand binding hydrogel studies described here may also enable physiologically relevant investigations of drug-induced protein binding conformational transitions.Experimental
Materials
Styrene, D-glucose, D-galactose, D-fructose, D-(+)-fucose, phenylmethylsulfonyl fluoride (PMSF), sodium dodecyl sulfate (SDS), Tris, KCl, ethylenediaminetetraacetic acid disodium salt dehydrate (EDTA) and bovine serum albumin (BSA, essentially fatty acid free) were purchased from Sigma-Aldrich. 1-Propanol, sodium phosphate monobasic dihydrate and sodium dihydrogen phosphate monohydrate were purchased from J. T. Baker Inc. These chemicals were used as received. Glutaraldehyde (50 wt% in water) was purchased from Sigma-Aldrich and diluted to a 2.5 wt% aqueous stock solution prior to use. Ultrapure water with a resistivity > 18.2 MΩ cm−1 was obtained from an ultrapure water system (Barnstead). Glass slides (25 mm × 75 mm × 1 mm) were purchased from Fisher Scientific.Expression and purification of GGBP
The GGBP was obtained by expression of plasmid pSF5 in the strain E. coli NM303 cells as described previously.30 In this study, the Luria–Bertani (LB) media for the growth of the protein contained 0.5 mM D-(+)-fucose, an inducer of the protein promoter and 100 μg mL−1 ampicillin. Cultures were grown with vigorous aeration at 37 °C for 24 h. After harvesting the cells, standard osmotic shock procedures were used to lyse the E. coli outer membrane to release the contents of the periplasm, including the proteins. PMSF was added to the osmotic shock fluid to reach a concentration of 0.5 mM and the resulting supernatant was concentrated to 15 mL by ultrafiltration (Amicon apparatus and YM10 membranes). The protein was then dialyzed against two types of buffers, each for 6–12 h at 4 °C in a volume at least 50-fold larger than that of the sample. The unfolding buffer was first used to denature the protein and release bound substrate. This unfolding buffer contained 3.0 M guanidine hydrochloride, 100 mM KCl, 20 mM EDTA, 10 mM Tris, pH 7.1, 0.5 mM PMSF. This was performed twice. The second buffer was the refolding buffer which contained 10 mM phosphate pH 7.0. The protein was dialyzed against this four times. Dialyzed samples were further concentrated to yield protein at the specific concentrations required. Protein purity was 99% according to SDS page electrophoresis. The GGBP has been well characterized and one may refer to the references for more information.16–18 The information on GGBP molecular weight and protein sequence can be seen in the ESI.†Preparation of 2-D PC-GGBP hydrogels
The 2-D PC-GGBP hydrogels were prepared as follows. We synthesized monodisperse 650 nm polystyrene (PS) colloidal spheres via an emulsifier free emulsion polymerization method.31 The PS colloidal spheres self-assembled on a water surface via a tip spreading technique to form 2-D array PCs.32 The 2-D PCs were transferred onto a glass slide and dried in air. Then a solution of GGBP (50 mg mL−1) in 0.1 M phosphate buffer (PB) mixed with a aqueous glutaraldehyde solution (2.5 wt%) was layered on the surface of the 2-D PCs.20 The cross-linking reaction was conducted at 4 °C in a refrigerator. After 24 h, the 2-D PC-GGBP hydrogel was carefully peeled off the glass slide. Samples were equilibrated in 0.01 M PB at pH 8 for 24 h prior to use, during which the PB was frequently changed. The hydrogel sensors should be stored in a refrigerator at 4 °C. The 2-D PC-BSA hydrogel was fabricated by using a similar method as that for 2-D PC-GGBP hydrogel. The hydrogel sensors were cut into 6 mm × 6 mm squares for analyte detection.UV resonance Raman (UVRR) spectroscopy
Fresh GGBP monomer solutions were dissolved in 10 mM PB at pH 8. GGBP hydrogels without 2-D arrays were fabricated using an identical method to that for 2-D PC-GGBP hydrogels. The UVRR measurements were performed using ∼204 nm excitation using a tunable Ti:sapphire laser (Photonics Industries) that operated at a 1 kHz repetition rate. The ∼204 nm light was generated by mixing the third harmonic with the ∼816 nm fundamental. The laser light was focused onto a spinning Suprasil quartz NMR tube containing the sample. An ∼165° backscattering geometry was used, and the scattered light was imaged using a double monochromator, modified for use in the UV region,33 and detected with a liquid N2 cooled back-thinned CCD camera (Spec-10:400B, Princeton Instruments) with a Lumogen E coating. After the UVRR measurements, the monomer and hydrogel samples were incubated in glucose solutions for 2 h. We then collected UVRR spectra of the glucose bound GGBP monomer solutions and hydrogels under the same conditions as for the unbound samples.The UVRR spectra were processed using the GRAMS/AI (ver. 8.0) software suite (Thermo Fisher Scientific) and home-written MATLAB scripts. The contributions of Suprasil quartz, phosphate buffer, and glucose were subtracted from the UVRR spectra. We compared the UVRR spectra of the GGBP hydrogels with and without glucose (Fig. S3a†) and the monomer solution-state spectra (with and without glucose, Fig. S3b†). To compare these spectra, we normalized the glucose bound monomer (hydrogel) spectrum to the unbound monomer (hydrogel) spectrum. The spectra were normalized by finding a factor, κ, which scales the glucose-bound spectra to the unbound spectra. i.e.,
| Sunbound(ν) = κSbound(ν) |
Debye ring measurement
Debye diffraction ring measurements were used to directly monitor the 2-D PC array particle spacing changes.20,22,34 The 2-D PC-GGBP hydrogel was excited with a 532 nm laser pointer along its normal. The photograph and scheme for the measurement of the Debye diffraction ring diameter are shown in Fig. 2c and d, respectively. The 2-D PC array diffracts light at an angle that depends on both the particle spacing and the laser wavelength. The rotationally disordered small 2-D array domains (20 μm × 20 μm) diffract a Debye ring pattern. The first-order diffraction angle, α, depends upon the particle spacing: sinα = 2λlaser/(31/2d), where α is the interior angle of the Debye diffraction ring. λlaser is the laser wavelength, and d is the particle spacing. The diffraction angle α, is determined from the Debye ring diameter: α = tan−1(D/2h), where D is the Debye ring diameter and h is the distance between the 2-D array and screen. We monitor the 2-D array particle spacing by measuring D and h:  .20 In this study, h value was set as 82 mm. For each concentration, 3 identical samples were used and each sample was measured at 3 different positions. The average and standard deviation of d were obtained from the 9 values. The standard deviation of the particle spacing is calculated using the following equation:
.20 In this study, h value was set as 82 mm. For each concentration, 3 identical samples were used and each sample was measured at 3 different positions. The average and standard deviation of d were obtained from the 9 values. The standard deviation of the particle spacing is calculated using the following equation: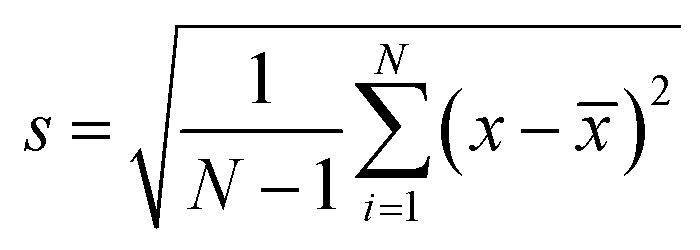
We also measured the diffraction of our 2-D PC-GGBP hydrogel sensors by using an Ocean Optics USB 2000-UV-Vis spectrometer, a LS-1 tungsten halogen light source, and an R-series fiber optic reflection probe. The diffraction measurements were carried out in a Littrow configuration with the fiber at a ∼21.8° angle from the array normal. The Debye diffraction ring diameter measurement involves the same diffraction as that measured by the UV-Vis reflection probe fiber optic spectrometer in a Littrow configuration. In a Littrow configuration, the 2-D Bragg diffraction relationship is mλ = 31/2dsinθ, where m is the diffraction order, λ is the diffracted wavelength (in vacuum), d is the 2-D particle spacing, and θ is the angle of the light relative to the normal to the 2-D PC arrays.35,36 The Debye diffraction ring diameter measurement is more convenient.
For the reversibility study, the 2-D PC-GGBP samples were immersed into a 20 mL 100 μM D-glucose solution containing 10 mM phosphate buffer at pH 8 for 4 h before measurement. After Debye diffraction ring diameter measurement, the samples were washed with a large amount of 10 mM phosphate buffer at pH 8 for the next round of measurement.
Microscopy characterization
The PS 2-D PCs and the 2-D PC-GGBP hydrogel sensors were sputter coated with a thin layer of Au and then studied by using a scanning electron microscope (SEM, Joel JSM6390LV).Acknowledgements
We are thankful for helpful discussions with Professor Milan Mrksich, Professor William Murphy and Mr Andrew Eagle Cookouma. The authors gratefully acknowledge HDTRA for financial support (Grant No. 1-10-1-0044 and 1-15-1-0038 to SAA). This work has been supported by NSF funding for the LC-MS instrumentation in the Chemistry Department at the University of Vermont (CHE MRI-0821501, LAL Co-PI), SUNY at Plattsburgh President's Award and Mini Grant (LAL).Notes and references
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS.
- L. Ionov, Mater. Today, 2014, 17, 494–503 CrossRef.
- D. Buenger, F. Topuz and J. Groll, Prog. Polym. Sci., 2012, 37, 1678–1719 CrossRef CAS.
- P. J. Flory, Principles of Polymer Chemistry, 1953 Search PubMed.
- J. H. Holtz and S. A. Asher, Nature, 1997, 389, 829–832 CrossRef CAS PubMed.
- Z. Cai, N. L. Smith, J.-T. Zhang and S. A. Asher, Anal. Chem., 2015, 87, 5013–5025 CrossRef CAS PubMed.
- Y. Gao, X. Li and M. J. Serpe, RSC Adv., 2015, 5, 44074–44087 RSC.
- W. L. Murphy, Soft Matter, 2011, 7, 3679–3688 RSC.
- J. D. Ehrick, S. K. Deo, T. W. Browning, L. G. Bachas, M. J. Madou and S. Daunert, Nat. Mater., 2005, 4, 298–302 CrossRef CAS PubMed.
- W. L. Murphy, W. S. Dillmore, J. Modica and M. Mrksich, Angew. Chem., Int. Ed., 2007, 46, 3066–3069 CrossRef CAS PubMed.
- Z. Sui, W. J. King and W. L. Murphy, Adv. Funct. Mater., 2008, 18, 1824–1831 CrossRef CAS.
- J. D. Ehrick, S. Stokes, S. Bachas-Daunert, E. A. Moschou, S. K. Deo, L. G. Bachas and S. Daunert, Adv. Mater., 2007, 19, 4024–4027 CrossRef CAS.
- Z. Sui, W. J. King and W. L. Murphy, Adv. Mater., 2007, 19, 3377–3380 CrossRef CAS.
- W. Yuan, J. Yang, P. Kopečková and J. i. Kopeček, J. Am. Chem. Soc., 2008, 130, 15760–15761 CrossRef CAS PubMed.
- J. D. Ehrick, M. R. Luckett, S. Khatwani, Y. Wei, S. K. Deo, L. G. Bachas and S. Daunert, Macromol. Biosci., 2009, 9, 864–868 CrossRef CAS PubMed.
- N. Vyas, M. Vyas and F. Quiocho, Science, 1988, 242, 1290–1295 CAS.
- M. J. Borrok, L. L. Kiessling and K. T. Forest, Protein Sci., 2007, 16, 1032–1041 CrossRef CAS PubMed.
- G. Ortega, D. Castaño, T. Diercks and O. Millet, J. Am. Chem. Soc., 2012, 134, 19869–19876 CrossRef CAS PubMed.
- S. Andreescu and L. A. Luck, Anal. Biochem., 2008, 375, 282–290 CrossRef CAS PubMed.
- Z. Cai, J.-T. Zhang, F. Xue, Z. Hong, D. Punihaole and S. A. Asher, Anal. Chem., 2014, 86, 4840–4847 CrossRef CAS PubMed.
- Z. Cai, D. H. Kwak, D. Punihaole, Z. Hong, S. S. Velankar, X. Liu and S. A. Asher, Angew. Chem., Int. Ed., 2015, 54, 13036–13040 CrossRef CAS PubMed.
- J.-T. Zhang, X. Chao, X. Liu and S. A. Asher, Chem. Commun., 2013, 49, 6337–6339 RSC.
- R. S. Zukin, P. G. Strange, L. R. Heavey and D. E. Koshland, Biochemistry, 1977, 16, 381–386 CrossRef CAS PubMed.
- V. L. Alexeev, S. Das, D. N. Finegold and S. A. Asher, Clin. Chem., 2004, 50, 2353–2360 CAS.
- K. M. Daum and R. M. Hill, Invest. Ophthalmol. Visual Sci., 1982, 22, 509–514 CAS.
- D. M. Miller, J. S. Olson and F. A. Quiocho, J. Biol. Chem., 1980, 255, 2465–2471 CAS.
- H. Tanaka, M. Matsumura and I. A. Veliky, Biotechnol. Bioeng., 1984, 26, 53–58 CrossRef CAS PubMed.
- http://biohedron.drupalgardens.com/ .
- S. J. Teague, Nat. Rev. Drug Discovery, 2003, 2, 527–541 CrossRef CAS PubMed.
- E. E. Snyder, B. W. Buoscio and J. J. Falke, Biochemistry, 1990, 29, 3937–3943 CrossRef CAS PubMed.
- C. E. Reese and S. A. Asher, J. Colloid Interface Sci., 2002, 248, 41–46 CrossRef CAS PubMed.
- J.-T. Zhang, L. Wang, D. N. Lamont, S. S. Velankar and S. A. Asher, Angew. Chem., Int. Ed., 2012, 51, 6117–6120 CrossRef CAS PubMed.
- S. Bykov, I. Lednev, A. Ianoul, A. Mikhonin, C. Munro and S. A. Asher, Appl. Spectrosc., 2005, 59, 1541–1552 CrossRef CAS PubMed.
- J.-T. Zhang, Z. Cai, D. H. Kwak, X. Liu and S. A. Asher, Anal. Chem., 2014, 86, 9036–9041 CrossRef CAS PubMed.
- I. M. Krieger and F. M. O'Neill, J. Am. Chem. Soc., 1968, 90, 3114–3120 CrossRef CAS.
- A. Tikhonov, N. Kornienko, J.-T. Zhang, L. Wang and S. A. Asher, J. Nanophotonics, 2012, 6, 063509 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Mechanism of the crosslinking reaction, protein sequence of GGBP, UVRR spectra, UV-Vis reflection spectra of 2-D PC-GGBP sensor at different glucose concentrations and additional information of crystal structures of GGBP. See DOI: 10.1039/c6sc00682e |
| This journal is © The Royal Society of Chemistry 2016 |