
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDivergent enantioselective synthesis of hapalindole-type alkaloids using catalytic asymmetric hydrogenation of a ketone to construct the chiral core structure†
Yang
Liu‡
a,
Li-Jie
Cheng‡
a,
Hai-Tao
Yue
a,
Wen
Che
a,
Jian-Hua
Xie
*a and
Qi-Lin
Zhou
ab
aState Key Laboratory and Institute of Elemento-organic Chemistry, Nankai University, Tianjin 300071, China. E-mail: jhxie@nankai.edu.cn
bCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Nankai University, Tianjin 300071, China
First published on 12th April 2016
Abstract
A divergent enantioselective approach to hapalindole-type alkaloids is described. The route features a ruthenium-catalyzed asymmetric hydrogenation of a ketone via DKR to construct the chiral trans-1-indolyl-2-isopropenylcyclohexane skeleton and a switchable sequence of methylation and acetylation/aldol reaction to access a chiral quaternary stereocenter. (+)-Hapalindole Q (1, 13 steps, 5.9% overall yield), (−)-12-epi-hapalindole Q isonitrile (2, 15 steps, 5.5% overall yield), (−)-hapalindole D (3, 14 steps, 2.3% overall yield), and (+)-12-epi-fischerindole U isothiocyanate (4, 14 steps, 3.0% overall yield) were synthesized in 13–15 steps from a commercially available material to demonstrate the application of this approach.
Introduction
Owing to the unique and diverse molecular architectures of hapalindole-type alkaloids and their broad range of biological activities, they have recently attracted great interest as synthetic targets.1 Since these hybrid isoprenoid–indole alkaloids derived from tryptophan and geraniol pyrophosphate were first isolated by Moore et al. in 1984 from the Stigonemataceae family of cyanobacteria,2 more than 70 have been identified.3 Because they exhibit diverse chirality and have a quaternary stereocenter, they are challenging targets for enantioselective synthesis.4 Several hapalindole-type alkaloids have been synthesized in enantiomerically pure form by means of chiral pool5 and chiral resolution approaches.6 For example, Vaillancourt and Albizati synthesized (+)-hapalindole Q (1, Scheme 1) from (+)-camphor.5a Fukuyama and Chen5b and Natsume et al.5c independently synthesized (−)-hapalindoles G and O from (−)-carvone. Baran et al. synthesized (+)-hapalindole Q (1), (−)-12-epi-fischerindole U isothiocyanate, (−)-fischerindole G, (+)-fischerindole I, and (+)-welwitindolinone A from (−)-carvone,5d–gent-12-epi-fischerindole G and 12-epi-fischerindole I from (+)-carvone,5g and (−)-hapalindole U and (+)-ambiguine H from (+)-p-menth-1-en-9-ol.3,5f However, only one catalytic enantioselective synthesis of a hapalindole-type alkaloid has been reported: Kinsman and Kerr7 used an organocatalyzed asymmetric Diels–Alder reaction as a key step in the synthesis of (+)-hapalindole Q (1).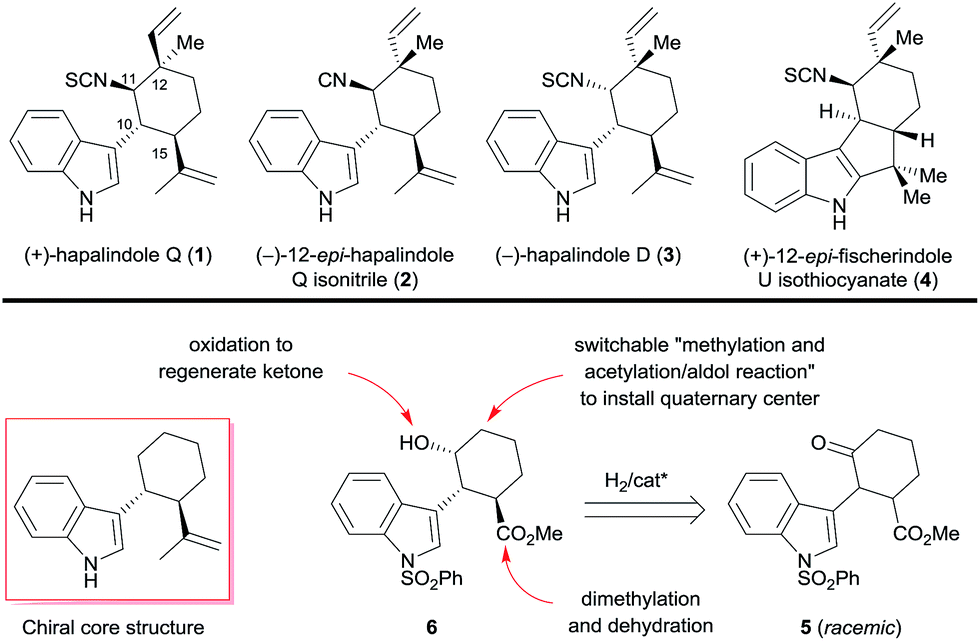 | ||
| Scheme 1 Selected hapalindole-type alkaloids and our divergent enantioselective synthetic strategy. | ||
Recently, we developed a protocol for the catalytic asymmetric hydrogenation of racemic α-substituted ketones via dynamic kinetic resolution (DKR), allowing for the highly efficient enantioselective synthesis of natural products such as galantamine, lycorane, and Δ9-THC.8 During our ongoing work on the enantioselective construction of quaternary and/or contiguous stereocenters, we noted that a chiral trans-1-indolyl-2-isopropenylcyclohexane skeleton is a core structure present in various hapalindole-type alkaloids (Scheme 1). Interestingly, both enantiomers of the skeleton are found among the naturally occurring hapalindole-type alkaloids, and alkaloids with inverted stereochemistry at the chiral quaternary center at C12 of the cyclohexane ring are also common in nature. These unique stereochemical characteristics present great challenges for the divergent enantioselective synthesis of hapalindole-type alkaloids. We envisioned that the asymmetric hydrogenation of 2-(1H-indol-3-yl)-3-(methoxycarbonyl)cyclohexanone (5) could be used to access both enantiomers of the core skeleton by changing the configuration of the chiral catalyst; switching the order of the two carbon–carbon bond-forming reactions at C12 would install the chiral quaternary stereocenter (Scheme 1). Dimethylation of the ester group of the hydrogenation product 6 followed by a dehydration would be a practical way to generate the isopropenyl group. Construction of the quaternary stereocenter and subsequent introduction of an isonitrile or isothiocyanate group could be accomplished via a carbonyl group, which could be generated by oxidation of the hydroxyl group of 6.
We herein report that we successfully employed the above-described strategy for the divergent enantioselective syntheses of (+)-hapalindole Q (1), (−)-12-epi-hapalindole Q isonitrile (2), (−)-hapalindole D (3), and (+)-12-epi-fischerindole U isothiocyanate (4) by using catalytic asymmetric hydrogenation of a ketone via DKR to construct the chiral core skeleton.
Results and discussion
We began by investigating the enantioselective synthesis of chiral alcohol 6, which has three contiguous stereocenters, via catalytic asymmetric hydrogenation of ketone 5 (Scheme 2). Commercially available methyl 3-oxocyclohex-1-enecarboxylate (7) was treated with iodine in the presence of trimethylsilyl azide (TMSN3) and pyridine using Sha and Huang's procedure9 to yield iodide 8 in 83% yield. Coupling of 8 with indolylboronic acid 910 and subsequent hydrogenation, both steps with Pd/C as the catalyst, afforded racemic ketone 5 in 74% yield over two steps. We investigated the asymmetric hydrogenation of 5 by using our chiral spiro ruthenium catalysts 11.11 Careful optimization of the reaction conditions (see ESI†) revealed that Ru-(R)-Xyl-SDP/(S,S)-DPEN (R,S,S)-11a efficiently catalyzed the hydrogenation of 5 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) mixture of iPrOH and tBuOH, providing chiral alcohol (−)-6 (cis, trans > 99%) in 95% isolated yield as a 3:1 mixture of the methyl ester (96% ee) and the isopropyl ester (95% ee). Similarly, chiral alcohol (+)-6, which has the opposite configuration, was obtained by using Ru-(S)-Xyl-SDP/(R,R)-DPEN (S,R,R)-11a) as the catalyst (95% yield, (+)-6a, 96% ee, (+)-6b, 95% ee, (+)-6a/(+)-6b = 3:1).
:1 (v/v) mixture of iPrOH and tBuOH, providing chiral alcohol (−)-6 (cis, trans > 99%) in 95% isolated yield as a 3:1 mixture of the methyl ester (96% ee) and the isopropyl ester (95% ee). Similarly, chiral alcohol (+)-6, which has the opposite configuration, was obtained by using Ru-(S)-Xyl-SDP/(R,R)-DPEN (S,R,R)-11a) as the catalyst (95% yield, (+)-6a, 96% ee, (+)-6b, 95% ee, (+)-6a/(+)-6b = 3:1).
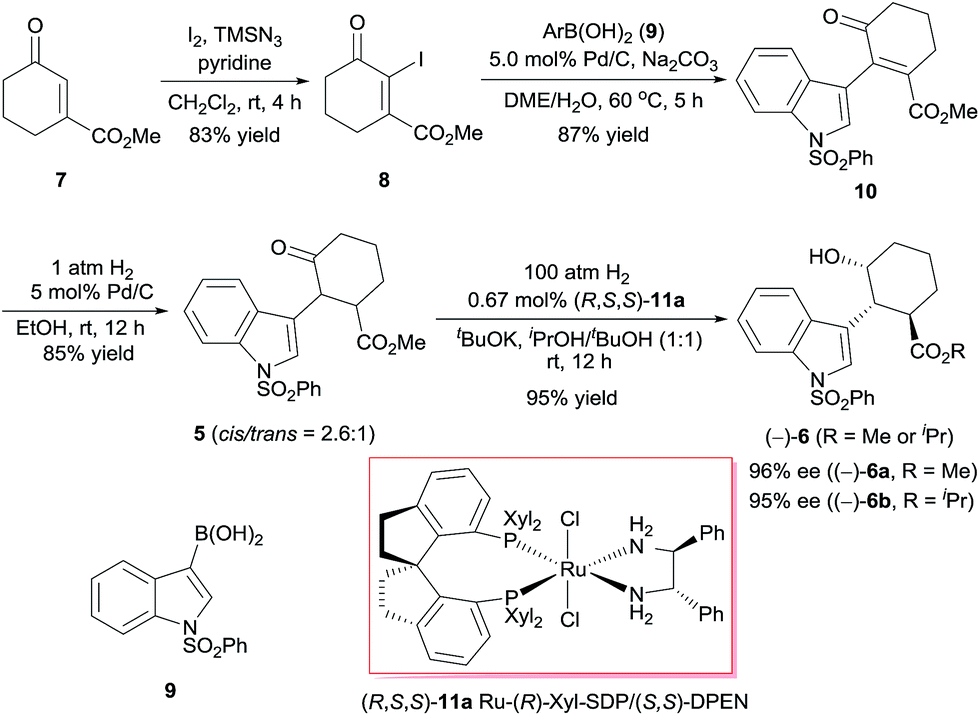 | ||
| Scheme 2 Asymmetric synthesis of chiral alcohol (−)-6. | ||
Reaction of chiral alcohol (−)-6 (as a mixture of methyl and isopropyl esters) with MeLi (6 equiv.) in the presence of cerium chloride12 yielded alcohol (−)-12 in 90% yield (Scheme 3). X-ray diffraction analysis of a crystal of (−)-12 showed its absolute configuration to be 1R,2R,3R. Oxidation of (−)-12 with pyridinium chlorochromate (PCC), followed by dehydration with Burgess's reagent,13 produced ketone (+)-14 in 71% yield over two steps. By means of the same process, ketone (−)-14 was synthesized from alcohol (+)-6 with the 1S,2S,3S configuration. Thus, we obtained ketones (+)-14 and (−)-14, which are key intermediates for the synthesis of hapalindole-type alkaloids, in 37.5% overall yield.
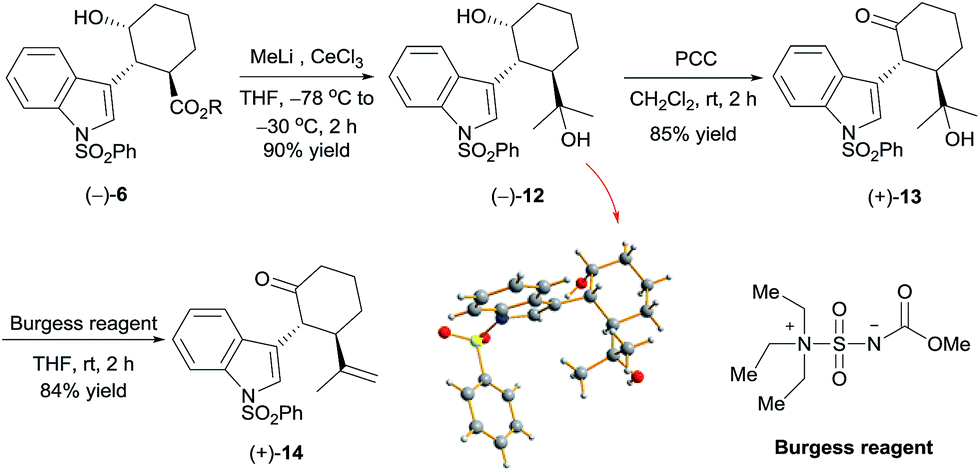 | ||
| Scheme 3 Asymmetric synthesis of cycloketone (+)-14. | ||
Having in hand the optically active ketones (+)-14 and (−)-14, which contain the chiral core structure of the hapalindole family of alkaloids, we turned our efforts to the total synthesis of specific hapalindole alkaloids. The enantioselective total synthesis of (+)-hapalindole Q (1)14 from optically active (+)-14 is outlined in Scheme 4. Selective α-methylation of (+)-14 with methyl iodide in the presence of lithium hexamethyldisilazide (LiHMDS)15 yielded ketone (+)-15 in 75% yield with 6:1 diastereoselectivity. An aldol reaction of (+)-15 with acetaldehyde in the presence of LiHMDS and zinc bromide16 afforded alcohol (+)-16 in 71% yield with 10:1 diastereoselectivity. Subsequently, intersecting with the syntheses described by Vaillancourt and Albizati5a and Baran and Richter,5d (+)-16 was dehydrated with Martin's sulfurane to afford ketone (+)-17 (85% yield). Thus, we constructed the chiral quaternary stereocenter at the C12-position by a sequence involving the introduction of a methyl group and then a vinyl group. Note that all the reactions used for the synthesis of (+)-17 could be performed on a gram scale.
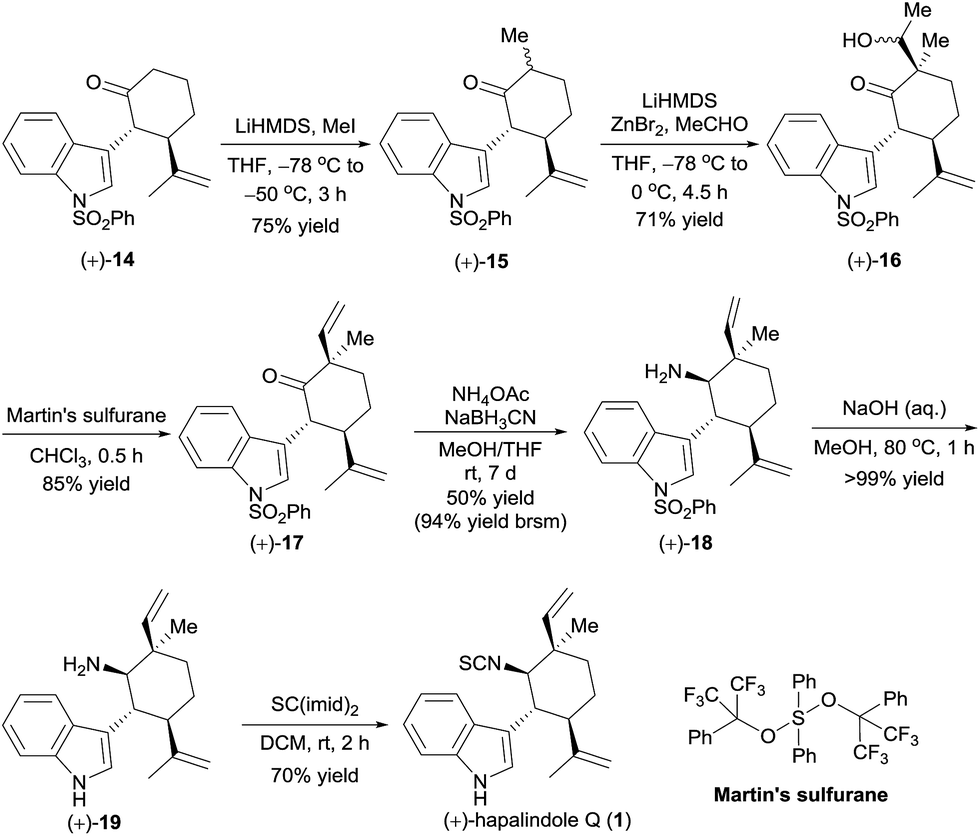 | ||
| Scheme 4 Total synthesis of (+)-hapalindole Q (1). | ||
Ketone (+)-17 was transformed to amine (+)-18 as a 6:1 mixture of diastereomers in 50% yield by a reductive amination with NH4OAc in the presence of NaBH3CN; unreacted ketone (+)-17 was recovered in 47% yield, so the yield of (+)-18 was 94% based on recovered starting material. Removal of the phenylsulfonyl protecting group of (+)-18 by hydrolysis with aqueous methanolic sodium hydroxide, followed by formation of an isothiocyanate by reaction with CS(imid)2 provided (+)-hapalindole Q (1) in 70% yield over two steps. The NMR spectroscopic data and the optical rotation ([α]25D +27.8 (c 1.1 CH2Cl2), lit. [α]25D +24.1 (c 1.1, CH2Cl2)14) of our synthetic sample were comparable to those reported for the natural product.
Encouraged by the successful synthesis of (+)-hapalindole Q (1), we moved on to the synthesis of (−)-12-epi-hapalindole Q isonitrile (2),17 which had previously been synthesized only in racemic form.4f The C12-epi-quaternary stereocenter was constructed by controlling the stereochemistry of the acylation–methylation sequence (Scheme 5). Specifically, treatment of optically active cycloketone (+)-14 with LiHMDS and trapping with N-acetylimidazole18 yielded diketone (+)-20 in 70% yield. α-Methylation of (+)-20 with MeI in the presence of K2CO3 afforded 91% yield of diketone (+)-21, which has a quaternary carbon center. Selective reduction of the acetyl group, which is less hindered than the ketone in (+)-21, was achieved by reaction with lithium tri-tert-butoxyaluminium hydride (LiAlH(OtBu)3),19 and the resulting alcohol, (+)-22, was dehydrated with Martin's sulfurane to afford ketone (+)-23 in 85% yield (note that the above mentioned reactions could be performed on a gram scale). This sequence resulted in the construction of the C12-epi-quaternary stereocenter with a vinyl group and methyl group arranged in a configuration opposite to that of ketone (+)-17. Next, in analogy to the synthesis of (+)-hapalindole Q (1), reductive amination of ketone (+)-23 gave amines (−)-24 and (−)-25 in 54% and 16% yields, respectively. After dephenylsulfonylation, the amino group of (−)-24 was converted to an isonitrile group by formylation with formic acid and 2-chloro-4,6-dimethyloxy-1,3,5-triazine (CDMT), and dehydration of the resulting formamide with triphosgene5b afforded (−)-12-epi-hapalindole Q isonitrile (2) in 59% yield over three steps. The NMR spectroscopic data were identical to those reported by Braekman17 and Li.4f The absolute configuration of synthetic (−)-12-epi-hapalindole Q isonitrile (2) was assigned as 10R,11R,12S,15R by inference from the X-ray crystal structure of alcohol (−)-12.
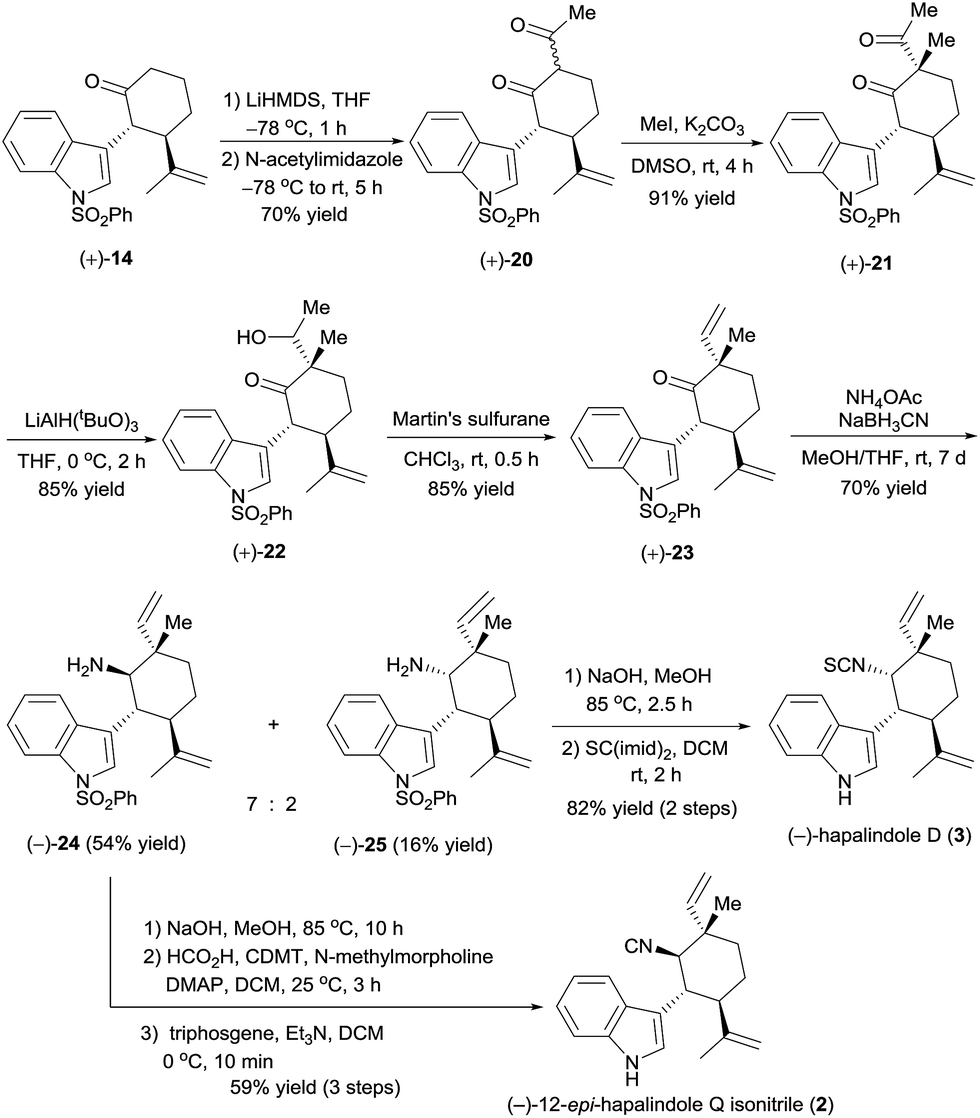 | ||
| Scheme 5 Total synthesis of (−)-12-epi-hapalindole Q isonitrile (2) and (−)-hapalindole D (3). | ||
In addition, using the minor product of the reductive amination, (−)-25, we synthesized (−)-hapalindole D (3)14 in 82% yield over two steps by dephenylsulfonylation and transformation of the amino group to an isothiocyanate group by using the procedure described for the synthesis of (+)-hapalindole Q (1). By comparing the optical rotation of our synthetic (−)-hapalindole D (3, [α]25D −224 (c 3.1 CH2Cl2)) with the optical rotation of natural hapalindole D ([α]25D +239 (c 3.1 CH2Cl2)),14 we assigned the absolute configuration of natural (+)-hapalindole D to be 10S,11R,12R,15S, which is opposite to that depicted in Scheme 5.
To further demonstrate the utility and generality of this strategy, we selected tetracyclic (+)-12-epi-fischerindole U isothiocyanate (4),20 which was first isolated in 1994 by Moore et al.,21 as another synthetic target. As suggested by the absolute configuration of this molecule, we started the synthesis from ketone (−)-14 (Scheme 6). Ketone (−)-14 was methylated with MeI, and a subsequent aldol reaction with acetaldehyde in the presence of LiHMDS and dehydration using Martin's sulfurane according to the procedure described for the synthesis of (+)-17 (see Scheme 4) afforded ketone (−)-17 in 46% yield over three steps. Ketone (−)-17 was then hydrolyzed in aqueous sodium hydroxide and cyclized in the presence of trimethylsilyl trifluoromethanesulfonate (TMSOTf) to yield ketone (+)-26 in 50% yield over two steps. Finally, intersecting with the synthesis of Baran et al.,5d ketone (+)-26 was converted to (+)-12-epi-fischerindole U isothiocyanate (4) by reductive amination and isothiocyanate formation (34% yield over two steps).
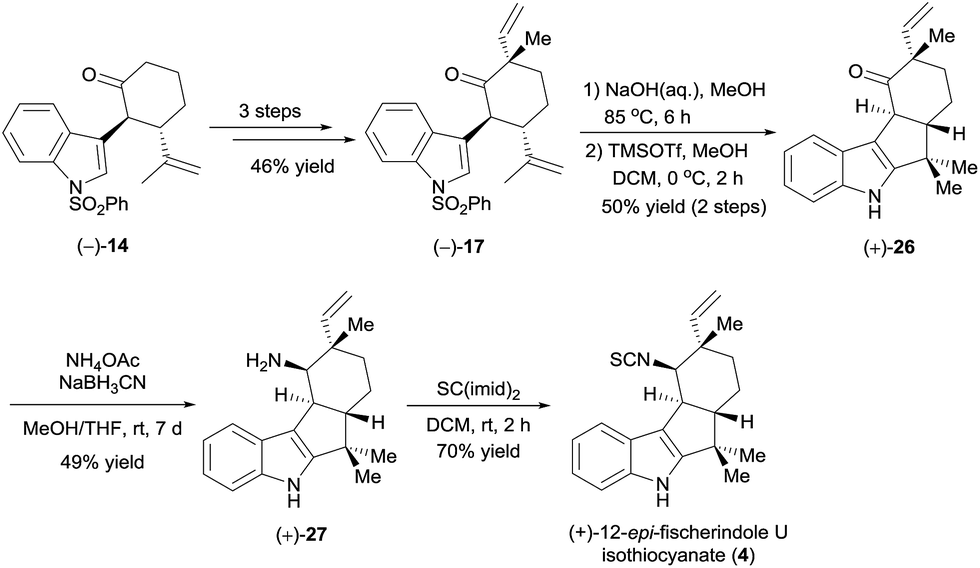 | ||
| Scheme 6 Total synthesis of (+)-12-epi-fischerindole U isothiocyanate (4). | ||
The synthetic (+)-12-epi-fischerindole U isothiocyanate (4) displayed identical spectra and a similar optical rotation ([α]25D +217 (c 0.035, CH2Cl2), lit. [α]25D +231 (c 0.035, CH2Cl2)21) to those reported for the natural product.
Conclusions
In conclusion, we achieved divergent enantioselective total syntheses of four hapalindole-type alkaloids, namely, (+)-hapalindole Q (1, 13 steps, 5.9% (11.0% brsm) overall yield), (−)-12-epi-hapalindole Q isonitrile (2, 15 steps, 5.5% overall yield), (−)-hapalindole D (3, 14 steps, 2.3% overall yield), and (+)-12-epi-fischerindole U isothiocyanate (4, 14 steps, 3.0% overall yield), from commercially available methyl 3-oxocyclohex-1-enecarboxylate. Our synthetic strategy features a ruthenium-catalyzed asymmetric hydrogenation of a ketone via DKR to construct the chiral core structure and a switchable sequence of methylation and acetylation/aldol reaction to install a chiral quaternary stereocenter. We anticipate that this strategy will find applications in the synthesis of other hybrid isoprenoid–indole alkaloids.Acknowledgements
This project was supported by the National Natural Science Foundation of China, the National Basic Research Program of China (973 Program) (No. 2012CB821600), and the “111” project (No. B06005) of the Ministry of Education of China.Notes and references
- V. Bhat, A. Dave, J. A. MacKay and V. H. Rawal, in The Alkaloids: Chemistry and Biology, ed. K. Hans-Joachim, Academic Press, New York, 2014, vol. 73, pp. 65–160 Search PubMed.
- R. E. Moore, C. Cheuk and G. M. Patterson, J. Am. Chem. Soc., 1984, 106, 6456 CrossRef CAS.
- T. J. Maimome, Y. Ishihara and P. S. Baran, Tetrahedron, 2015, 71, 3652 CrossRef PubMed.
- For selected total syntheses of hapalindole-type alkaloids in racemic form, see: (a) H. Muratake and M. Natsume, Tetrahedron, 1990, 46, 6331 CrossRef CAS; (b) H. Muratake, H. Kumagami and M. Natsume, Tetrahedron, 1990, 46, 6351 CrossRef CAS; (c) A. C. Kinsman and M. A. Kerr, Org. Lett., 2001, 3, 3189 CrossRef CAS PubMed; (d) A. Chandra and J. N. Johnston, Angew. Chem., Int. Ed., 2011, 50, 7641 CrossRef CAS PubMed; (e) R. J. Rafferty and R. M. Williams, J. Org. Chem., 2012, 77, 519 CrossRef CAS PubMed; (f) Z. Lu, M. Yang, P. Chen, X. Xiong and A. Li, Angew. Chem., Int. Ed., 2014, 53, 13840 CrossRef CAS PubMed; for selected synthetic studies on hapalindole-type alkaloids, see: (g) H. Muratake and M. Natsume, Tetrahedron, 1990, 46, 6343 CrossRef CAS; (h) P. Harrington and M. A. Kerr, Synlett, 1996, 1047 CrossRef CAS; (i) P. Harrington and M. A. Kerr, Can. J. Chem., 1998, 76, 1256 CrossRef CAS; (j) A. C. Kinsman and M. A. Kerr, Org. Lett., 2000, 2, 3517 CrossRef CAS PubMed; (k) M. A. Brown and M. A. Kerr, Tetrahedron Lett., 2001, 42, 983 CrossRef CAS; (l) J. N. Johnston, M. A. Plotkin, R. Viswanathan and E. N. Prabhakaran, Org. Lett., 2001, 3, 1009 CAS; (m) R. Viswanathan, E. N. Prabhakaran, M. A. Plotkin and J. N. Johnston, J. Am. Chem. Soc., 2003, 125, 163 CrossRef CAS PubMed; (n) R. Viswanathan, D. Mutnick and J. N. Johnston, J. Am. Chem. Soc., 2003, 125, 7266 CrossRef CAS PubMed; (o) A. Chandra, R. Viswanathan and J. N. Johnston, Org. Lett., 2007, 9, 5027 CrossRef CAS PubMed; (p) R. J. Rafferty and R. M. Williams, Tetrahedron Lett., 2011, 52, 2037 CrossRef CAS PubMed; (q) R. J. Rafferty and R. M. Williams, Heterocycles, 2012, 86, 219 CrossRef CAS PubMed.
- (a) V. Vaillancourt and K. F. Albizati, J. Am. Chem. Soc., 1993, 115, 3499 CrossRef CAS; (b) T. Fukuyama and X. Chen, J. Am. Chem. Soc., 1994, 116, 3125 CrossRef CAS; (c) M. Sakagami, H. Muratake and M. Natsume, Chem. Pharm. Bull., 1994, 42, 1393 CrossRef CAS; (d) P. S. Baran and J. M. Richter, J. Am. Chem. Soc., 2004, 126, 7450 CrossRef CAS PubMed; (e) P. S. Baran and J. M. Richter, J. Am. Chem. Soc., 2005, 127, 15394 CrossRef CAS PubMed; (f) P. S. Baran, T. J. Maimone and J. M. Richter, Nature, 2007, 446, 404 CrossRef CAS PubMed; (g) J. M. Richter, Y. Ishihara, T. Masuda, B. W. Whitefield, T. Liamas, A. Pohjakallio and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 17938 CrossRef CAS PubMed; (h) A. D. Huters, K. W. Quasdorf, E. D. Styduhar and N. K. Garg, J. Am. Chem. Soc., 2011, 133, 15797 CrossRef CAS PubMed; (i) K. W. Quasdorf, A. D. Huters, M. W. Lodewyk, D. J. Tantillo and N. K. Garg, J. Am. Chem. Soc., 2012, 134, 1396 CrossRef CAS PubMed; (j) K. M. Allan, K. Kobayashi and V. H. Rawal, J. Am. Chem. Soc., 2012, 134, 1392 CrossRef CAS PubMed; (k) N. A. Weires, E. D. Styduhar, E. L. Baker and N. K. Garg, J. Am. Chem. Soc., 2014, 136, 14710 CrossRef CAS PubMed.
- K. Komine, Y. Nomura, J. Ishihara and S. Hatakeyama, Org. Lett., 2015, 17, 3918 CrossRef CAS PubMed.
- A. C. Kinsman and M. A. Kerr, J. Am. Chem. Soc., 2003, 125, 14120 CrossRef CAS PubMed.
- (a) S.-F. Zhu and Q.-L. Zhou, in Privileged Chiral Ligands and Catalysts, ed. Q.-L. Zhou, Wiley-VCH Verlag GmbH, Weinheim, 2011, pp. 137–170 Search PubMed; (b) J.-H. Xie and Q.-L. Zhou, Aldrichimica Acta, 2015, 48, 33 Search PubMed; for papers, see: (c) J.-Q. Chen, J.-H. Xie, D.-H. Bao, S. Liu and Q.-L. Zhou, Org. Lett., 2012, 14, 2714 CrossRef CAS PubMed; (d) C. Liu, J.-H. Xie, Y.-L. Li, J.-Q. Chen and Q.-L. Zhou, Angew. Chem., Int. Ed., 2013, 52, 593 CrossRef CAS PubMed; (e) G. Li, J.-H. Xie, J. Hou, S.-F. Zhu and Q.-L. Zhou, Adv. Synth. Catal., 2013, 355, 1597 CrossRef CAS; (f) L.-J. Cheng, J.-H. Xie, Y. Chen, L.-X. Wang and Q.-L. Zhou, Org. Lett., 2013, 15, 764 CrossRef CAS PubMed.
- C.-K. Sha and S.-J. Huang, Tetrahedron Lett., 1995, 36, 6927 CrossRef CAS.
- F.-X. Felpin, J. Org. Chem., 2005, 70, 8575 CrossRef CAS PubMed.
- (a) J.-H. Xie, L.-X. Wang, S.-F. Zhu, Y. Fu, B.-M. Fan, H.-F. Duan and Q.-L. Zhou, J. Am. Chem. Soc., 2003, 125, 4404 CrossRef CAS PubMed; (b) J.-H. Xie and Q.-L. Zhou, Acta Chim. Sin., 2014, 72, 778 CrossRef CAS.
- T. Imamoto, N. Takiyama, K. Nakamura, T. Hatajima and Y. Kamiya, J. Am. Chem. Soc., 1989, 111, 4392 CrossRef CAS.
- Y. Ueda, K. Iwahashi, K. Iguchi and H. Ito, Synthesis, 2011, 1532 CAS.
- R. E. Moore, C. Cheuk, X.-Q. G. Yang, G. M. L. Patterson, R. Bonjouklian, T. A. Smitka, J. S. Mynderse, R. S. Foster, N. D. Jones, J. K. Swartzendruber and J. B. Deeter, J. Org. Chem., 1987, 52, 1036 CrossRef CAS.
- O. L. Epstein and J. K. Cha, Angew. Chem., Int. Ed., 2005, 44, 121 CrossRef CAS PubMed.
- K. Tanino, K. Onuki, K. Asano, M. Miyashita, T. Nakamura, Y. Takahashi and I. Kuwajima, J. Am. Chem. Soc., 2003, 125, 1498 CrossRef CAS PubMed.
- D. Klein, D. Daloze, J. C. Braekman, L. Hoffmann and V. Demoulin, J. Nat. Prod., 1995, 58, 1781 CrossRef CAS.
- T. Kaiho, K. Sannohe, S. Kajiya, T. Suzuki, K. Otsuka, T. Ito, J. Kamiya and M. Maruyama, J. Med. Chem., 1989, 32, 351 CrossRef CAS PubMed.
- (a) E. W. Colvin, J. Martin, W. Parker, R. A. Raphael, B. Shroot and M. Doyle, J. Chem. Soc., Perkin Trans. 1, 1972, 860 RSC; (b) D. R. Bhowmik and R. V. Venkateswaran, Tetrahedron Lett., 1999, 40, 7431 CrossRef CAS.
- Baran et al. have recently reported the enantioselective total synthesis of (−)-epi-fischerindole U isothiocyanate from (R)-carvone, the enantiomer of the natural (+)-12-epi-fischerindole U isothiocyanate (4). See ref. 5d and g.
- K. Stratmann, R. E. Moore, R. Bonjouklian, J. B. Deeter, G. M. L. Patterson, S. Shaffer, C. D. Smith and T. A. Smitka, J. Am. Chem. Soc., 1994, 116, 9935 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1453314. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc00686h |
| ‡ Y. L. and L.-J. C. contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |