
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA N,N′-dioxide/Mg(OTf)2 complex catalyzed enantioselective α-addition of isocyanides to alkylidene malonates†
Weiwei
Luo
a,
Xiao
Yuan
a,
Lili
Lin
a,
Pengfei
Zhou
a,
Xiaohua
Liu
*a and
Xiaoming
Feng
*ab
aKey Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: liuxh@scu.edu.cn; xmfeng@scu.edu.cn; Fax: +86 28 85418249; Tel: +86 28 85418249
bCollaborative Innovation Center of Chemical Science and Engineering, Tianjin, China
First published on 22nd April 2016
Abstract
A highly efficient catalytic asymmetric α-addition of isocyanides to alkylidene malonates was accomplished. The process was based on the utilization of a chiral N,N′-dioxide/MgII catalyst, delivering a variety of 2-alkyl-5-aminooxazoles in up to 99% yield and 96% ee under mild reaction conditions. Besides, a chiral imide and dipeptide could be easily obtained by ring-opening of the oxazole product, both of which are important structural motifs for many biologically active compounds. Based on the experimental investigations and previous work, a possible transition state model was proposed.
Introduction
Optically active heterocyclic compounds containing an oxazole motif appear extensively in natural products, pharmaceuticals, and synthetic intermediates.1 Because of the importance of these compounds, versatile approaches have been reported towards nonracemic oxazole derivatives. Early successful examples, such as cyclodehydration reactions and metal-catalyzed cross-coupling reactions were limited to the use of stoichiometric quantities of chiral precursors.2 In contrast, direct catalytic asymmetric synthesis of these compounds is less developed. Until now, only two methods, an asymmetric hetero-ene reaction of 5-methyleneoxazolines with carbonyls3 and α-addition of isocyanides with carbonyls or imines,4,5 have been reported. In relation to the latter, α-addition is a simple but very efficient route to obtain 5-aminooxazoles.α-Additions of isocyanides with both electrophiles and nucleophiles6 have found wide application in organic synthesis since early studies on the Passerini7 and Ugi reactions.8 Although various diastereoselective methods using chiral substrates and/or chiral auxiliaries have been developed in recent decades,9–11 the development of the enantioselective α-addition of isocyanides still remains challenging.4,5,12 The groups of Wang and Zhu,4a–c and Shibasaki4d as well as Zhong4e have made significant contributions to the catalytic enantioselective α-addition of isocyanides to aldehydes, thus affording the desired 2-(1-hydroxyalkyl)-5-aminooxazoles. Recently, Wang and Zhu described an enantioselective α-addition of isocyanides to imines, providing a series of 2-(1-aminoalkyl)-5-aminooxazoles in moderate to good enantioselectivities (Scheme 1a).5 To the best of our knowledge, the reaction of isocyanides with unactivated alkenes remains elusive, which might be due to the low reactivity of the alkenes or the complicated regiochemistry of isocyanides.13,14 Furthermore, ring-chain isomerization of the α-isocyanoacetamides inevitably provided byproducts, C-2 unsubstituted 5-aminooxazoles, in the presence of a Lewis acid (Scheme 1b).15 To further expand the scope in terms of the reaction partners and to complement the established methods for synthesizing enantioenriched oxazole derivatives, we describe herein an efficient asymmetric α-addition of isocyanides to activated alkenes catalyzed by a chiral N,N′-dioxide/MgII complex,3,16 delivering 2-alkyl-5-aminooxazoles in good yields with high enantioselectivities.
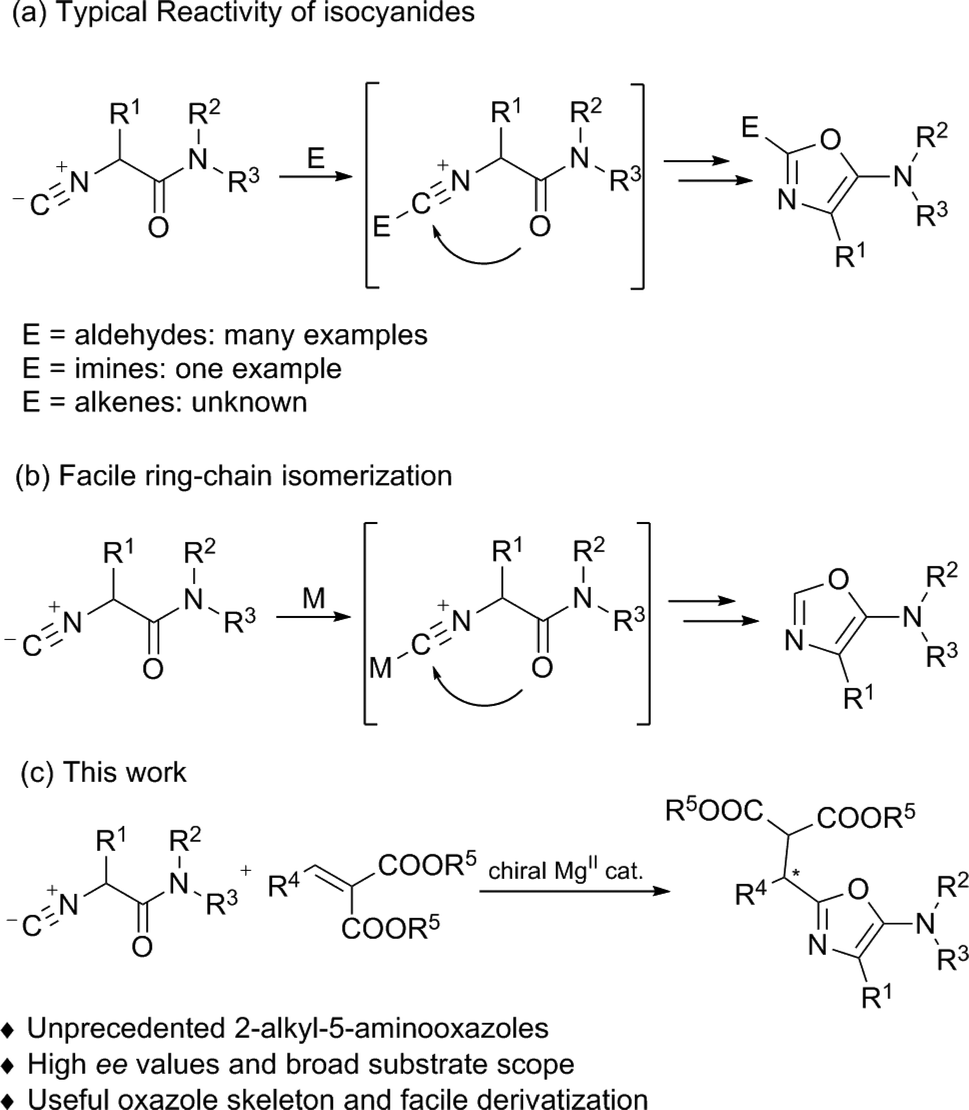 | ||
| Scheme 1 Construction of an oxazole ring using α-isocyanoacetamide. | ||
Results and discussion
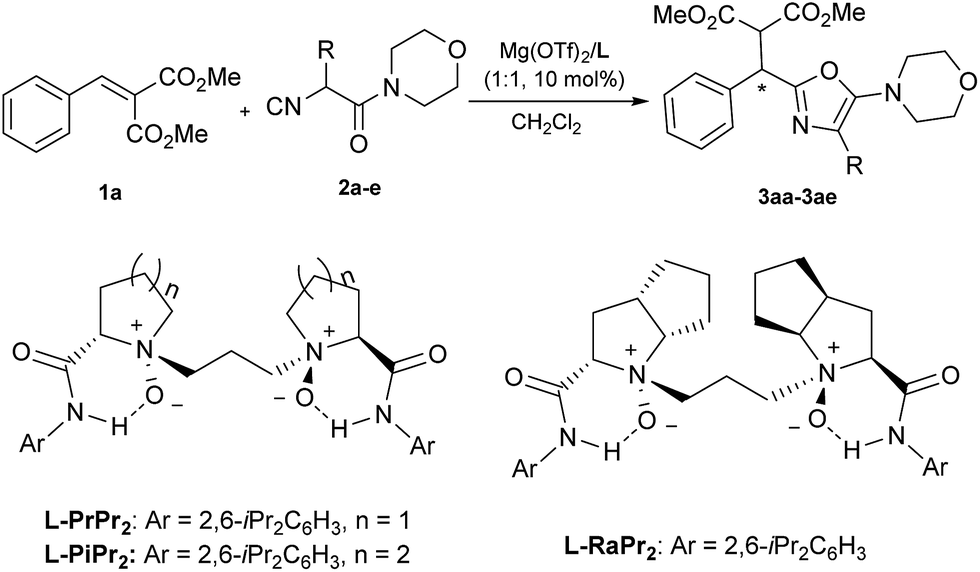
Our investigation began with the addition of a DL-phenylalanine derived α-isocyanoacetamide (2a) to methyl 2-benzylidenemalonate (1a), as a model reaction used to optimize the reaction conditions. Initially, various chiral N,N′-dioxide ligands complexing with Mg(OTf)2 were evaluated (Table 1, entries 1–3). The results suggest that L-ramipril derived L-RaPr2 exhibited superior reactivity compared with L-proline derived L-PrPr2 and L-pipecolic acid derived L-PiPr2, and the desired product 3aa was obtained in 99% yield with 82% ee (entry 3 vs. entries 1 and 2). Decreasing the reaction temperature to 0 °C resulted in a dramatic loss of reactivity but a slight improvement of the enantioselectivity (63% yield and 86% ee; entry 4). In order to improve the reactivity, the structure of the α-isocyanoacetamide was then examined (entries 5–8). With an increase of the steric hindrance of the α-substituent on the isocyanoacetamide, a positive effect was observed (entry 5). Efficient product formation was also observed by applying a sterically demanding DL-tert-leucine derived α-isocyanoacetamide (2e; entry 8), furnishing the corresponding 5-aminooxazole 3ae in 75% yield with 92% ee. Importantly, the yield improved noticeably to 91% with retention of the enantioselectivity when the reaction was carried out with a slight excess amount of the metal salt (entry 9). We also found that the catalytic system was insensitive to both atmospheric oxygen and moisture, making the catalytic system practical. Therefore, the optimized conditions were determined as Mg(OTf)2/L-RaPr2 as the catalyst in CH2Cl2 at 0 °C for 72 h.|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Ligand | R | T (°C) | t (h) | Yieldb (%) | eec (%) |
a Unless specified otherwise, reactions were performed with Mg(OTf)2/L (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 10 mol%), 1a (0.1 mmol) and 2 (0.15 mmol) in 1.0 mL CH2Cl2.
b Isolated yield.
c Determined using HPLC analysis with a chiral stationary phase.
d Reaction was carried out with Mg(OTf)2/L-RaPr2 (1.2:1, 10 mol%). :1, 10 mol%), 1a (0.1 mmol) and 2 (0.15 mmol) in 1.0 mL CH2Cl2.
b Isolated yield.
c Determined using HPLC analysis with a chiral stationary phase.
d Reaction was carried out with Mg(OTf)2/L-RaPr2 (1.2:1, 10 mol%).
|
||||||
| 1 | L-PrPr2 | Bn (2a) | 30 | 24 | 72 | 76 |
| 2 | L-PiPr2 | Bn (2a) | 30 | 24 | 93 | 70 |
| 3 | L-RaPr2 | Bn (2a) | 30 | 24 | 99 | 82 |
| 4 | L-RaPr2 | Bn (2a) | 0 | 48 | 63 | 86 |
| 5 | L-RaPr2 | Ph (2b) | 0 | 48 | 86 | 87 |
| 6 | L-RaPr2 | Me (2c) | 0 | 72 | 61 | 86 |
| 7 | L-RaPr2 | iPr (2d) | 0 | 72 | 91 | 89 |
| 8 | L-RaPr2 | tBu (2e) | 0 | 72 | 75 | 92 |
| 9d | L-RaPr2 | tBu (2e) | 0 | 72 | 91 | 92 |
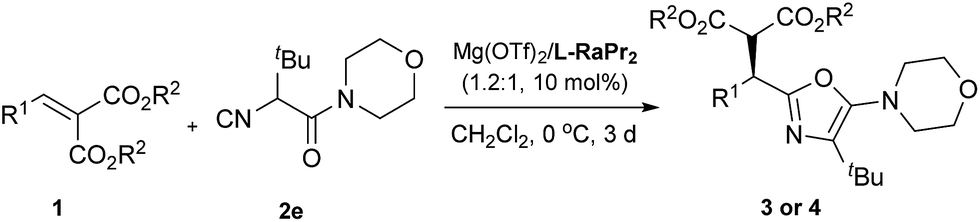
Having established the optimized conditions, we next investigated the scope of the alkylidene malonates. As shown in Table 2, by reaction with α-isocyanoacetamide 2e, a wide range of alkylidene malonates were transformed into the corresponding 2-alkyl-5-aminooxazoles smoothly. Generally, the reactivities and enantioselectivities gradually decreased with increased steric hindrance of the ester group (Table 2, entries 1–3). An o-fluoro group on the phenyl ring in 1d decreased the reactivity and caused a reaction time of 7 days to be required to achieve 66% yield and 80% ee, which might be due to both the electronic nature and steric encumbrance at the reaction site (entry 4). Notably, electron-withdrawing meta substituents on the phenyl ring such as fluorine, chlorine or bromine have no significant influence on the reactivity or enantioselectivity (entries 5–7). Meanwhile, electron-donating meta substituents are also well tolerated (entries 8–10). Substituents at the para position are equally well tolerated and both electron-donating and electron-withdrawing groups provided the corresponding products in high yields and enantioselectivities (entries 11–21). It is noteworthy that having a m-methoxy or p-methoxy group in 1 decreased the reactivity and also caused a longer reaction time to be required to achieve high conversion (entries 9 and 19), results we attribute to the decreased electrophilicity of alkylidene malonates with an electron-donating group on the phenyl ring. Multisubstituted and fused-ring-substituted alkylidene malonates also proceeded well, providing the corresponding products in up to 90% yield and 92% ee (entries 22 and 23). An ortho-substituted heteroaromatic substrate proved detrimental to both the reactivity and selectivity, while a meta substituted one led to a satisfactory result (entries 24 and 25). Aliphatic substrates can also be employed but these gave moderate enantioselectivities (entries 26 and 27).
|
|
|||||
|---|---|---|---|---|---|
| Entrya | R1 | R2 | 3 or 4 | Yieldb (%) | eec (%) |
|
a Unless specified otherwise, reactions were performed with Mg(OTf)2/L-RaPr2 (1.2:1, 10 mol%), 1 (0.1 mmol) and 2e (0.15 mmol) in 1.0 mL CH2Cl2 at 0 °C for 3 days.
b Isolated yield.
c Determined using HPLC analysis with a chiral stationary phase.
d The reaction was carried out over 7 days.
e The absolute configuration of 3ae was determined using X-ray analysis.
|
|||||
| 1 | C6H5 | Me | 3ae | 91 | 92 (R)e |
| 2 | C6H5 | Et | 3be | 71 | 91 |
| 3 | C6H5 | iPr | 3ce | 41 | 82 |
| 4d | 2-FC6H4 | Me | 3de | 66 | 80 |
| 5 | 3-FC6H4 | Me | 3ee | 92 | 91 |
| 6 | 3-ClC6H4 | Me | 3fe | 77 | 91 |
| 7 | 3-BrC6H4 | Me | 3ge | 96 | 91 |
| 8 | 3-MeC6H4 | Me | 3he | 66 | 94 |
| 9d | 3-MeOC6H4 | Me | 3ie | 81 | 90 |
| 10 | 3-PhOC6H4 | Me | 3je | 84 | 88 |
| 11 | 4-FC6H4 | Me | 3ke | 86 | 93 |
| 12 | 4-ClC6H4 | Me | 3le | 96 | 94 |
| 13 | 4-BrC6H4 | Me | 3me | 93 | 94 |
| 14 | 4-F3CC6H4 | Me | 3ne | 86 | 92 |
| 15 | 4-NCC6H4 | Me | 3oe | 98 | 94 |
| 16 | 4-O2NC6H4 | Me | 3pe | 91 | 94 |
| 17 | 4-MeC6H4 | Me | 3qe | 83 | 94 |
| 18 | 4-PhC6H4 | Me | 3re | 98 | 91 |
| 19d | 4-MeOC6H4 | Me | 3se | 87 | 96 |
| 20 | 4-PhOC6H4 | Me | 3te | 64 | 92 |
| 21 | 4-BnOC6H4 | Me | 3ue | 64 | 92 |
| 22 | 3,4-Cl2C6H3 | Me | 3ve | 90 | 92 |
| 23 | 2-Naphthyl | Me | 3we | 81 | 90 |
| 24d | 2-Thienyl | Me | 3xe | 28 | 85 |
| 25d | 3-Furyl | Me | 3yw | 76 | 89 |
| 26 | c-Hexyl | Me | 3ze | 81 | 86 |
| 27 | Me | Me | 4ae | 90 | 72 |
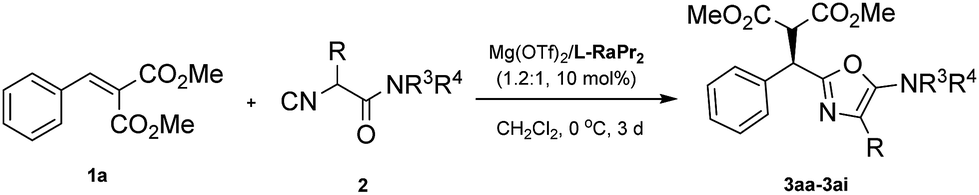
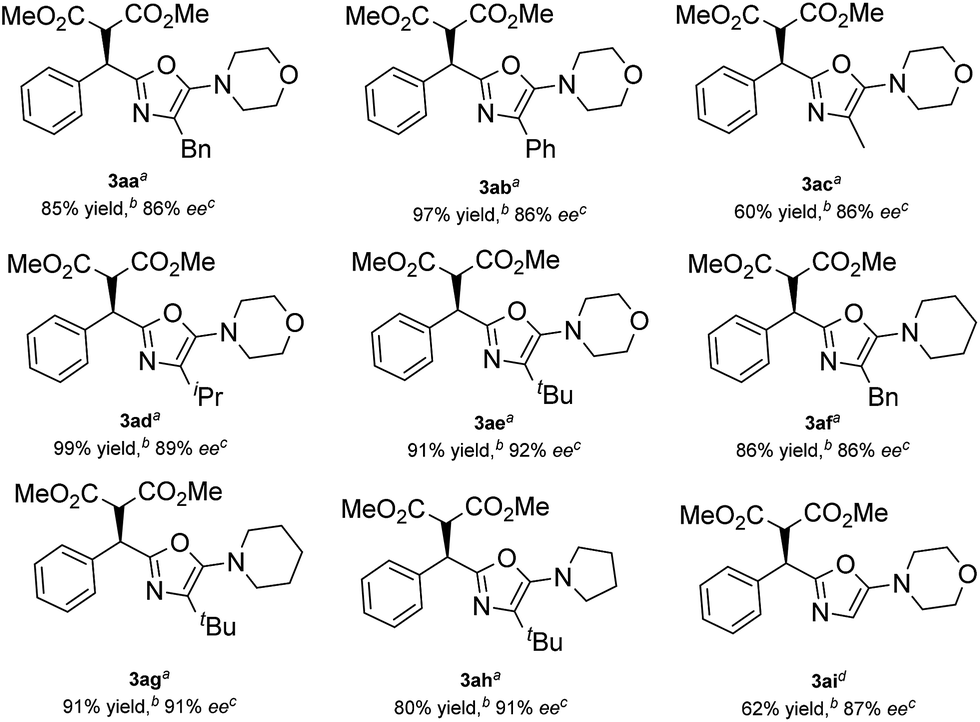
Then, various α-substituted isocyanides were examined (Table 3). Thankfully, isocyanides 2a–2e with different alkyl or phenyl substituents were applicable, giving the corresponding products 3aa–3ae in 60–99% yield and 86–92% ee. The 2-alkyl-5-aminooxazoles generated from piperidine 2f, 2g, and pyrrolidine 2h were also formed in high yield and stereoselectivity. Additionally, a moderate yield (62%) and good enantioselectivity (87% ee) were observed when a glycine derived α-isocyanoacetamide 2i was applied. The absolute configuration of compound 3ae was established unambiguously to be R using single-crystal X-ray structure analysis,17 and the 5-aminooxazoles exhibited a similar (+) Cotton effect in their CD (circular dichroism) spectra (for details, see the ESI†).
To show the prospect of using this methodology in synthesis, a gram scale synthesis of 3ae was performed. Under the optimal conditions, 4.5 mmol of α-isocyanoacetamide 2e reacted well with 3.0 mmol of methyl 2-benzylidenemalonate, providing 0.90 g (70% yield) of the corresponding 5-aminooxazole 3ae with an ee of 91% (Scheme 2a). The enantiopure product could be obtained using a simple recrystallization, with a yield of 67%. Next, simple derivatizations of the product were conducted (Scheme 2b). The product 3ae could be efficiently converted into a useful 1,3-diol 5 through reduction with LiAlH4 (90% yield, 95% ee). In addition, 5-aminooxazole 3ae was readily hydrolyzed in the presence of trifluoroacetic acid,18 and dipeptide 6 could be obtained in 99% yield with the enantioselectivity maintained (1.8:1 d.r., 99% ee). These derivatives are important structural motifs for the synthesis of many biologically active compounds. Next, in the presence of ceric ammonium nitrate, the oxazole ring was opened up, providing an imide product 7 in 51% yield with 98% ee.19 An initial decarboxylation of 7 gave the succinate derivative 8 in 47% yield as a racemic mixture, caused by subjecting the racemization-prone α-substituted carbonyl compound 7 to high temperature. Considering that optically active 2-substituted succinic acid derivatives are useful motifs in numerous biologically active compounds and natural products,20 we turned our attention to accessing these compounds. To our delight, monoester product 9 was obtained in excellent yield with the enantioselectivity maintained (98% yield, 94% ee). Subsequent opening of the oxazole ring gave the target chiral succinate derivative 10 in 70% yield with 94% ee. In the meantime, monoester oxazole 9 underwent a facile reduction with LiAlH4 to provide 11 in 47% yield.
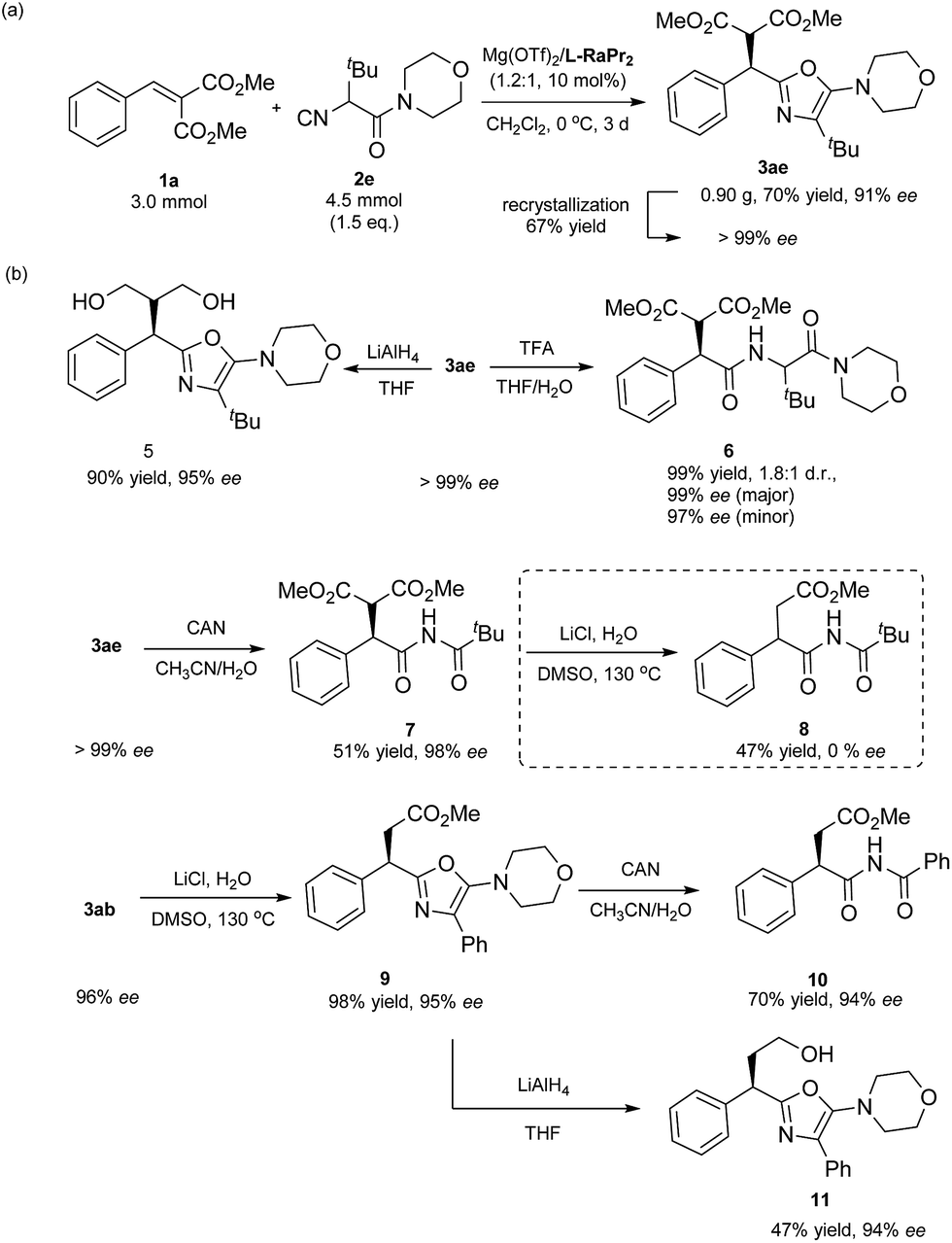 | ||
| Scheme 2 (a) Gram-scale version of the reaction. (b) Synthetic utility. | ||
During the course of this study, a 5-aminooxazole by-product was formed in some cases, and to verify the identity of this by-product an authentic sample was obtained (12; see Scheme 3a).15 In order to understand the reaction profile, 12 was applied in a reaction with alkylidene malonate 1a under the standard reaction conditions. However, no desired product 3ae was obtained after three days, suggesting that a Friedel–Crafts pathway isn't involved in the formation of the final product. Moreover, deuterium labeling studies were carried out (Scheme 3b). While the use of deuteriated isocyanide led to low deuterium labeling of the product, the use of a small amount of D2O resulted in significant deuterium labeling of the product. This interesting observation suggests that the proton transfer is facilitated by a trace amount of water (T3 to product in Fig. 1).
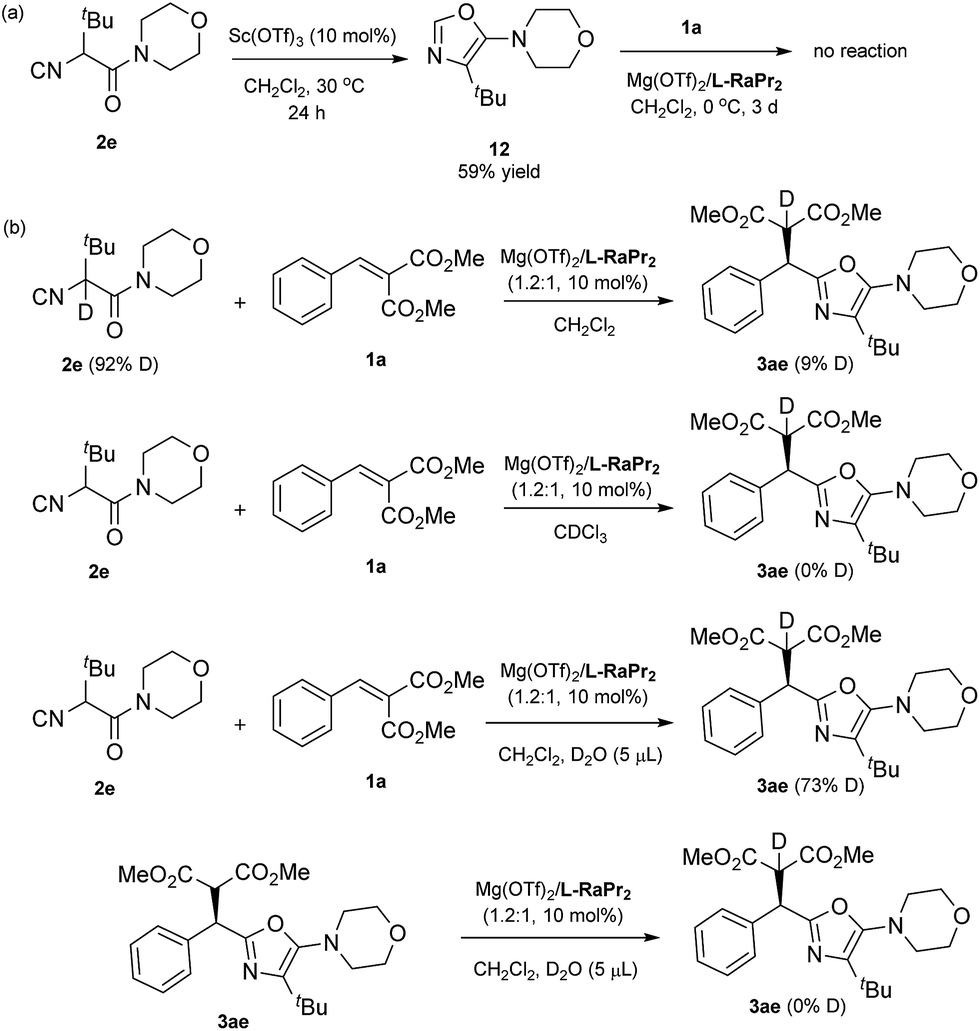 | ||
| Scheme 3 Control experiments. | ||
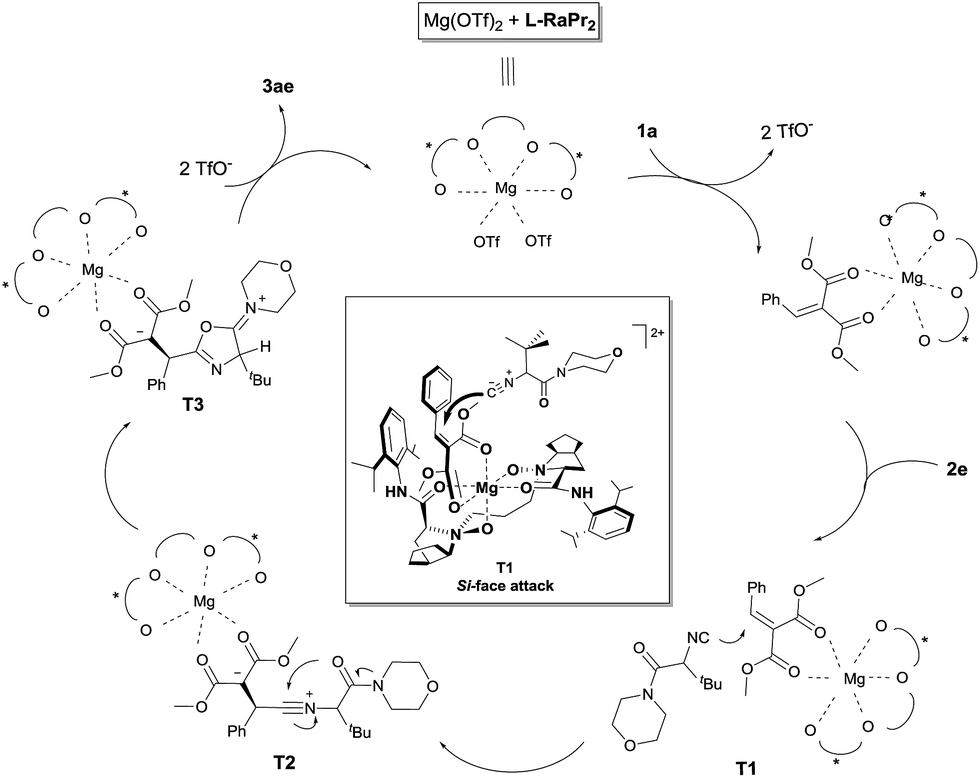 | ||
| Fig. 1 Proposed catalytic cycle. | ||
The HRMS spectrum of a mixture of Mg(OTf)2/L-RaPr2 and methyl 2-benzylidenemalonate 1a (1:1:1) confirmed coordination of the substrate to the catalyst. A peak at m/z 1093.5035 was detected and corresponds to the complex [Mg2+ + L-RaPr2 + 1a + OTf−]+ (cal. 1093.5034). Based on the above results and our previous work,16b,g a possible reaction mechanism and a transition-state model were proposed and are shown in Fig. 1. At first, the N-oxide and amide oxygen atoms of L-RaPr2 coordinate to Mg2+ in a tetradentate manner to form two six-membered chelate rings. Methyl 2-benzylidenemalonate 1a could be activated by coordination to the magnesium atom in a bidentate fashion, and the Re face of the substrate would be shielded by the neighboring 2,6-diisopropylphenyl group of the ligand. So, nucleophilic addition of the divalent carbon atom of isonitrile 2e onto the Si face of the substrate would afford the nitrilium intermediate T2, which could undergo cyclization to afford T3. Finally, deprotonation involving the proton on C4 of T3 would provide the R-configured product, which is in accord with the X-ray crystal structure of 3ae.
Conclusions
In summary, we have developed a chiral N,N′-dioxide/MgII catalyst system to realize the asymmetric α-addition of isocyanides to alkylidene malonates. A range of 2-alkyl-5-aminooxazoles were obtained in moderate to excellent yields (up to 99%) with excellent ee values (up to 96%). This represents the first example of an enantioselective α-addition of isocyanides to activated alkenes and may lay the foundation for the development of the long-awaited enantioselective α-addition to simple alkenes. Further studies on applying this catalyst system to other related reactions are underway.Acknowledgements
The study was funded by the National Natural Science Foundation of China (Nos. 21372162, 21432006, and 21321061).Notes and references
-
(a) P. Wipf, Chem. Rev., 1995, 95, 2115 CrossRef CAS
; (b) D. C. Palmer and S. Venkatraman, in The Chemistry of Heterocyclic compounds, Oxazoles: Synthesis Reactions, and Spectroscopy: Part A., ed. D. C. Palmer, Wiley, Hoboken, NJ, 2003, vol. 60, pp. 138–149 CrossRef
-
(a) P. Y. Coqueron, C. Didier and M. A. Ciufolini, Angew. Chem., Int. Ed., 2003, 42, 1411 CrossRef CAS PubMed
-
(a) W. W. Luo, J. N. Zhao, C. K. Yin, X. H. Liu, L. L. Lin and X. M. Feng, Chem. Commun., 2014, 50, 7524 RSC
-
(a) S.-X. Wang, M.-X. Wang, D.-X. Wang and J. Zhu, Eur. J. Org. Chem., 2007, 4076 CrossRef CAS
-
(a) T. Yue, M.-X. Wang, D.-X. Wang, G. Masson and J. Zhu, Angew. Chem., Int. Ed., 2009, 48, 6717 CrossRef CAS PubMed
-
(a) A. Dömling, Chem. Rev., 2006, 106, 17 CrossRef PubMed
-
(a) M. Passerini, Gazz. Chim. Ital., 1921, 51, 126 CAS
-
(a) A. Dömling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168 CrossRef
-
(a) S. Marcaccini, D. Miguel, T. Torroba and M. García-Valverde, J. Org. Chem., 2003, 68, 3315 CrossRef CAS PubMed
-
(a) H. Kunz and W. Pfrengle, J. Am. Chem. Soc., 1988, 110, 651 CrossRef CAS
-
L. Banfi, A. Basso, G. Guanti and L. Banfi, Asymmetric Isocyanide-based MCRs, in Multicomponent reactions, ed. J. Zhu and H. Bienaymé, Wiley-VCH, Weinheim, 2005, pp. 1–32 Search PubMed
-
(a) S. E. Denmark and Y. Fan, J. Am. Chem. Soc., 2003, 125, 7825 CrossRef CAS PubMed
- For examples of α-carbanion attack, see:
(a) H. Li, J. Song, X. Liu and L. Deng, J. Am. Chem. Soc., 2005, 127, 8948 CrossRef CAS PubMed
- For examples of isocyano group attack, see:
(a) T. Saegusa, Y. Ito, S. Tomita, H. Kinoshita and N. Taka-Ishi, Tetrahedron, 1971, 27, 27 CrossRef CAS
- Y. Odabachian, Q. Wang and J. Zhu, Chem.–Eur. J., 2013, 19, 12229 CrossRef CAS
-
(a) X. H. Liu, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2011, 44, 574 CrossRef CAS PubMed
- CCDC 1416058† (3ae).
-
(a) B. H. Lipshutz, B. E. Huff, K. E. McCarthy, T. A. Miller, S. M. J. Mukarram, T. J. Siahaan, W. D. Vaccaro, H. Webb and A. M. Falick, J. Am. Chem. Soc., 1990, 112, 7032 CrossRef CAS
- D. A. Evans, P. Nagorny and R. Xu, Org. Lett., 2006, 8, 5669 CrossRef CAS PubMed
- Y.-C. Chung, D. Janmanchi and H.-L. Wu, Org. Lett., 2012, 14, 2766 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1416058. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc00689b |
| This journal is © The Royal Society of Chemistry 2016 |