
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel-catalyzed enantioselective arylation of pyridine†
J. Patrick
Lutz
,
Stephen T.
Chau
and
Abigail G.
Doyle
*
Department of Chemistry, Princeton University, Princeton, New Jersey 08544, USA. E-mail: agdoyle@princeton.edu
First published on 8th March 2016
Abstract
We report an enantioselective Ni-catalyzed cross coupling of arylzinc reagents with pyridinium ions formed in situ from pyridine and a chloroformate. This reaction provides enantioenriched 2-aryl-1,2-dihydropyridine products that can be elaborated to numerous piperidine derivatives with little or no loss in ee. This method is notable for its use of pyridine, a feedstock chemical, to build a versatile, chiral heterocycle in a single synthetic step.
Introduction
Pyridine and its derivatives comprise one of the most important classes of heterocycles in the field of chemistry. For example, a recent analysis showed that piperidine and pyridine are the two most common nitrogen heterocycles in US FDA-approved pharmaceuticals, together found in approximately 12% of these drugs.1 Dihydropyridines are particularly valuable precursors to these motifs and exhibit biological activities in their own right.2,3 While the synthetic and biological utility of 1,4-dihydropyridines is well recognized, a recent review has singled out 1,2-dihydropyridines as a class of compounds that is understudied due to limitations in synthetic methodology.2e In particular, there is a dearth of methods for the enantioselective formation of 1,2-dihydropyridines.The most common methods for the synthesis of 1,2-dihydropyridines involve cyclizations or cycloadditions of linear precursors.4 Regioselective reductions of substituted pyridines to 1,2-dihydropyridines have also been reported.5 However, these strategies have rarely been rendered stereoselective and require the use of prefunctionalized substrates that are accessible only by multi-step synthesis.6–8 A third approach involves the addition of a carbon nucleophile to an activated pyridinium salt, which is typically formed in situ from the corresponding pyridine and an acylating agent. This approach is particularly attractive, as it employs readily available starting materials and provides 2-substituted 1,2-dihydropyridines in a single step. Furthermore, stereoselective variants have been reported. Comins,9 Wanner,10 and Charette11 have pioneered the use of chiral auxiliaries to control the facial selectivity of Grignard additions to pyridinium salts, though a blocking group at the C3 position is often required for high regio- and diastereoselectivity.
By contrast, access to chiral 1,2-dihydropyridines from pyridinium ions in an asymmetric catalytic manner remains largely an unsolved, albeit highly important, problem. Only a few such methods have been disclosed. The first reported example, from Shibasaki, featured the asymmetric addition of cyanide to the 6-position of pyridinium salts derived from nicotinate amides (Scheme 1A).12 Another 1,6-addition was achieved by Nadeau and the process group of Merck Frosst in their rhodium-catalyzed coupling of aryl boronic acids with N-benzyl nicotinate esters.13 These two methods rely on the presence of a carbonyl group conjugated to the pyridinium ring to achieve reactivity, but other reports have demonstrated enantioselective carbon–carbon bond formation in the absence of this structural feature. For example, Feringa and Minnaard reported a copper-catalyzed enantioselective synthesis of 2-alkyl piperidinones from 4-methoxypyridinium salts.14 In 2013, we disclosed a catalytic protocol for enantioselective arylation of this same electrophile class. In this process, a Ni(0) catalyst ligated by a chiral phosphoramidite ligand undergoes oxidative addition to the π system of the pyridinium ion, followed by transmetalation with arylzinc halides and reductive elimination to yield 2-aryl-4-methoxy-1,2-dihydropyridines, which are hydrolyzed to the corresponding piperidinones upon workup with aq. HCl (Scheme 2).15 Numerous arylzinc nucleophiles were successful coupling partners; nevertheless, 4-methoxypyridinium salt was the only competent electrophile under the reported reaction conditions.
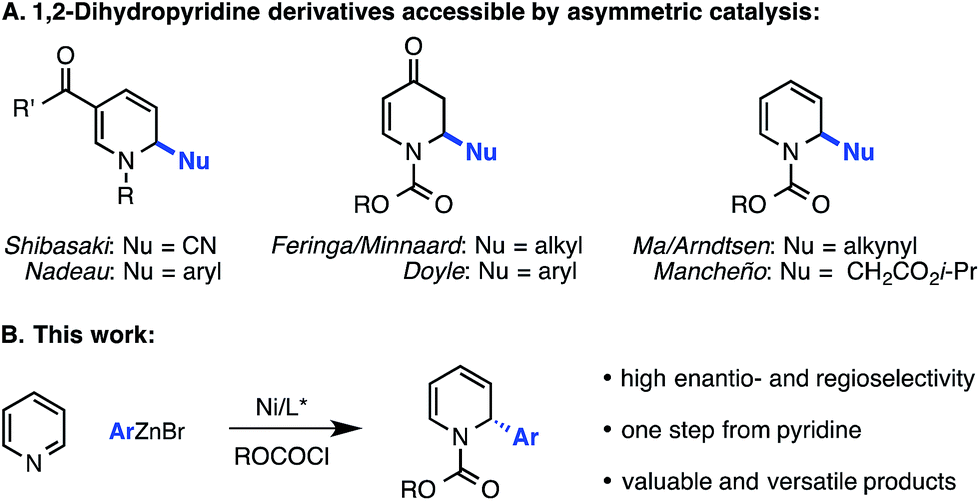 | ||
| Scheme 1 (A) Classes of 1,2-dihydropyridine and piperidinone accessible by asymmetric addition of a nucleophile to a pyridinium salt. (B) Asymmetric arylation of pyridine. | ||
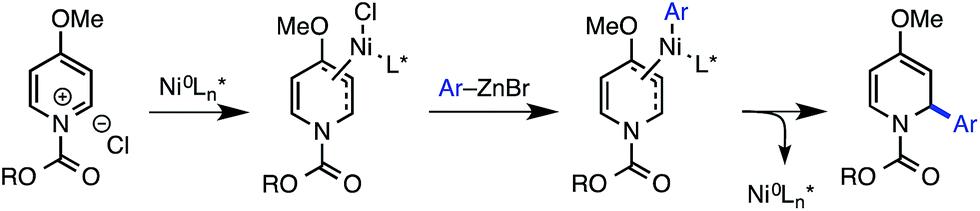 | ||
| Scheme 2 Proposed mechanism for the Ni-catalyzed arylation of 4-methoxypyridinium ions. | ||
In light of the potential utility of simple 2-aryl-1,2-dihydropyridines, we sought to expand this activation strategy to achieve the enantioselective arylation of unsubstituted pyridinium ions formed in situ directly from pyridine and an acylating agent.16,17 Pyridine is a stable, abundant and inexpensive feedstock chemical, rendering it an ideal prochiral starting material for organic synthesis. Despite this, pyridine has seen very limited use as a substrate for metal-catalyzed reactions, and a number of potential challenges were evident for the development of the proposed methodology. First, because pyridine is an excellent ligand, it might simply poison the nickel catalyst. While this obstacle was surmountable with 4-methoxypyridine, pyridine is acylated less efficiently than 4-methoxypyridine, so a greater amount of free pyridine would be expected to be present in the reaction mixture.18 Another principal concern was whether the catalyst could enforce high regioselectivity for 1,2-addition in the absence of a blocking group installed at the C4 position of the ring. Indeed, only two nucleophile classes have succumbed to coupling with unsubstituted pyridinium ions in an asymmetric fashion: copper acetylides,19 and most recently, silyl ketene acetals, though poor regioselectivity for the 1,2-dihydropyridine over the 1,4-dihydropyridine was observed in the latter case.20 Here we report the successful translation of our Ni-catalyzed iminium activation mode to the development of a highly regio- and enantioselective catalytic arylation of pyridinium salts derived from pyridine itself (Scheme 1B). Furthermore, we show that the enantioenriched dihydropyridine products of this new reaction can serve as highly valuable precursors to numerous complex piperidine derivatives.
Results and discussion
Optimization studies
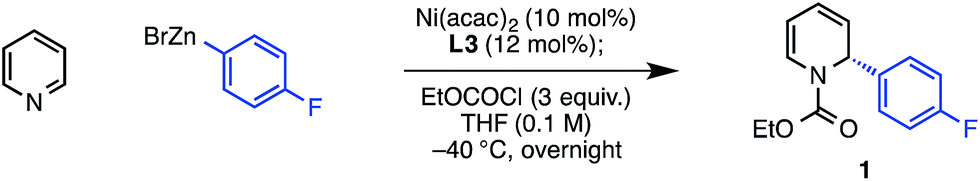
The previously described reaction conditions, which used PhOCOCl as an acylating agent, provided only trace product when using pyridine in place of 4-methoxypyridine. However, we found that EtOCOCl gave measurable amounts of the desired dihydropyridine product 1, albeit initially in only 14% NMR yield and 42% ee.21 Evaluation of precatalysts showed that both Ni(0) and Ni(II) sources were effective (Table 1, entries 2 and 3), with Ni(acac)2 providing the best balance of yield and enantioselectivity (entry 1). The enantioselectivity was not strongly affected by the Ni loading, though below 10 mol% Ni, the reaction yield diminished (entry 4). In the absence of Ni, there is a substantial racemic background reaction, which proceeds to a greater extent than that for 4-methoxypyridine under the original conditions (entry 5).15 We also found that concentration has a significant effect on reaction efficiency and selectivity, with higher yields and ee's observed at higher concentration (entry 6). Since equilibrium favors the pyridinium salt at higher reaction concentrations, these results may be explained by diminished catalyst poisoning by free pyridine in solution.|
|
|||
|---|---|---|---|
| Entry | Conditionsa | Yieldb (%) | eec (%) |
| a Reactions run on a 0.5 mmol scale with 3 equiv. 4-F-PhZnBr. b Isolated yield. c Determined using chiral HPLC analysis after Pd/C-catalyzed hydrogenation to the piperidine. | |||
| 1 | Standard conditions | 64 | 86 |
| 2 | Ni(cod)2 instead of Ni(acac)2 | 71 | 76 |
| 3 | NiBr2·diglyme instead of Ni(acac)2 | 58 | 80 |
| 4 | 5 mol% Ni(acac)2, 6 mol% L3 | 38 | 87 |
| 5 | No Ni or ligand | 72 | 0 |
| 6 | 0.02 M instead of 0.1 M | 56 | 74 |
| 7 | Ligand L1 instead of L3 | 64 | 74 |
| 8 | Ligand L2 instead of L3 | 18 | 7 |
| 9 | Ligand L4 instead of L3 | 24 | 0 |
| 10 | −78 °C → rt instead of −40 °C | 63 | 85 |
| 11 | ArZnBr derived from ArMgBr | 43 | 20 |
| 12 | +3 equiv. MgBr2 | 13 | 0 |
|
|||
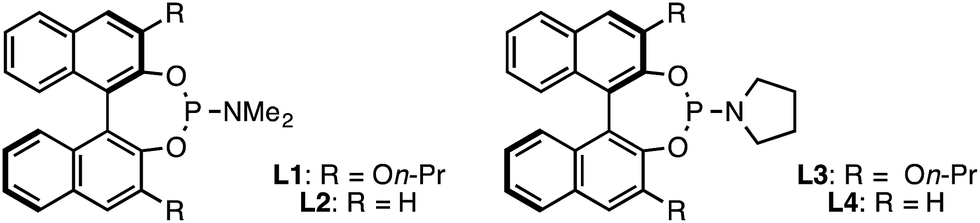
With the modifications described above, the original BINOL-derived phosphoramidite ligand L1 provided 1 with reasonable yield and moderate ee (entry 7). When we investigated the use of (R)-Monophos, L2, as a ligand, we found that 3,3′-substitution was critical for high enantioselectivity, showing a dramatic decrease from 74% ee to only 7% ee (entry 8). In an attempt to identify a more selective ligand, we evaluated several Monophos derivatives possessing differing 3,3′-substitution, but we were unable to identify a 3,3′-substituent that gave higher ee than On-Pr. Consequently, we prepared a number of phosphoramidites that retained the 3,3′-On-Pr substitution but that differed in the identity of the N-substituent.22 Pyrrolidine-derived ligand L3 was optimal, providing 1 in 64% yield and 86% ee (entry 1).
In the course of our optimization studies, we also found that the method of preparation of the arylzinc reagent had a strong influence on the outcome of the reaction. When 4-fluorophenylzinc bromide was prepared by transmetalation of the corresponding aryllithium reagent with ZnBr2, the reaction proceeded smoothly and with high ee (entry 1). However, if the zinc reagent was instead prepared by transmetalation between ZnBr2 and a Grignard reagent, the yield and enantioselectivity fell off dramatically (entry 11). We believe that this effect is due to the presence of Mg(II) salts, as the reaction also fails when exogenous MgBr2 is added to a reaction with an organolithium-derived zinc reagent (entry 12).
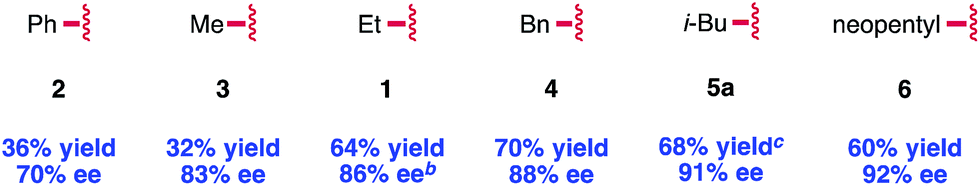
Our early observations regarding the difference in reactivity between EtOCOCl and PhOCOCl led us to investigate the identity of the acylating agent more thoroughly in an attempt to further increase the enantioselectivity of the reaction (Table 2). In doing so, we found that more sterically demanding chloroformates tended to give higher enantioselectivity: methyl chloroformate provided compound 3 in 83% ee, while neopentyl chloroformate provided 6 in 92% ee. Noting that there was only a slight difference in enantioselectivity between isobutyl and neopentyl chloroformate, we elected to use the less expensive reagent, i-BuOCOCl, as the acylating agent in our investigation of the reaction scope. Notably, we found that these optimized conditions provided 1,2-adduct 5a with high regioselectivity; none of the 1,4-adduct was observed by NMR.22
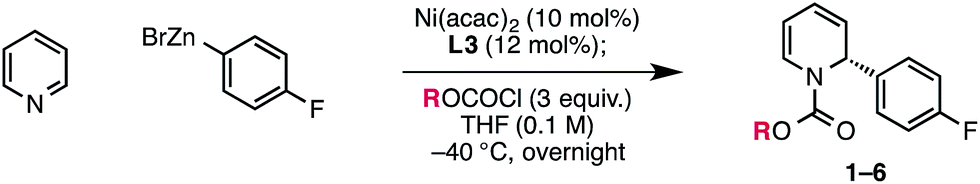
Scope elucidation
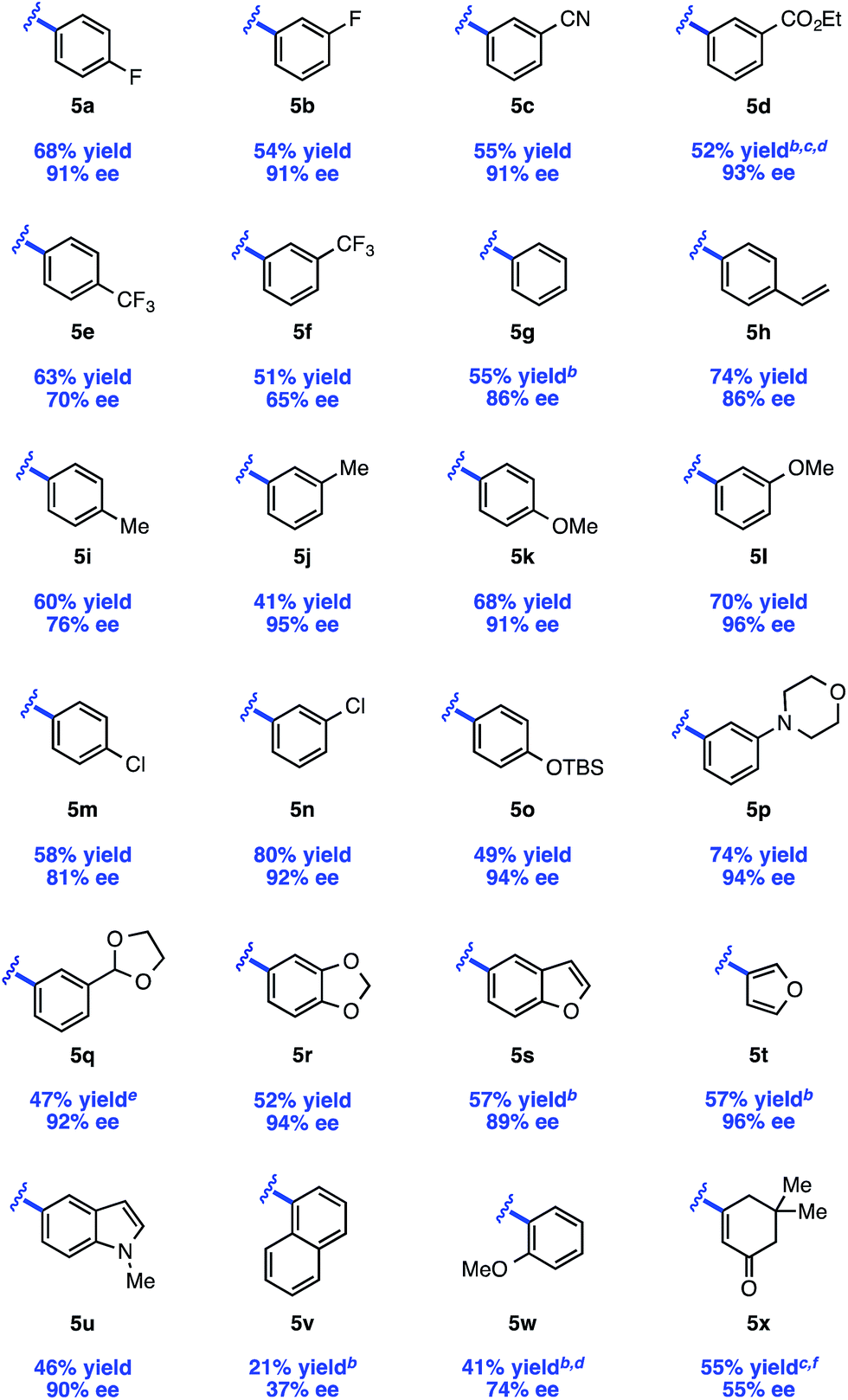
The scope of the reaction with respect to the arylzinc nucleophile was explored (Table 3). π-Electron-withdrawing groups are well tolerated (5c, 5d), though strong σ-withdrawing substituents lead to depressed enantioselectivity (5e, 5f). Neutral arylzinc bromides give acceptable levels of selectivity (5g–j), as do electron-rich nucleophiles (5k, 5l); the latter is notable as electron-rich nucleophiles deliver low enantioselectivity due to a facile background reaction in our previously described arylation of 4-methoxypyridinium ions. Importantly, many functional groups are compatible with the nickel chemistry: nucleophiles containing nitriles (5c), esters (5d), silyl-protected alcohols (5o), and tertiary amines (5p) all afford >90% ee in the arylation. Furthermore, Cl-substituted nucleophiles perform well, providing a handle for subsequent functionalization at these positions, though the meta-Cl nucleophile (5n) gave higher enantioselectivity than the para-Cl nucleophile (5m). This trend was observed in a few other cases as well (5ivs.5j, 5kvs.5l).|
|
|---|
| a Isolated yields and ee's are the average of two runs on a 0.5 mmol scale with 3 equiv. RZnBr. b Reaction was run with 20 mol% Ni(acac)2 and 24 mol% L3. c Reaction was run with RZnI. d Yield and ee determined after Pd/C-catalyzed hydrogenation to the piperidine. e Reaction was run with 1.25 equiv. i-BuOCOCl. f Reaction was run from −78 °C → rt. |
|
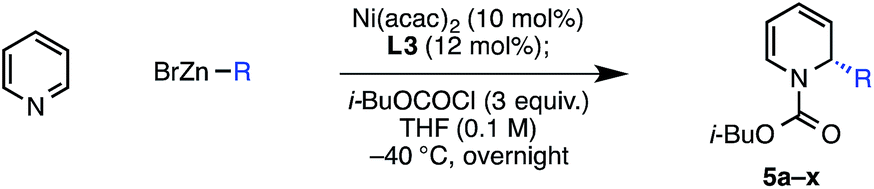
Several heteroaromatic nucleophiles are amenable to the reaction, providing access to compounds derived from benzofuran (5s), furan (5t), and N-methylindole (5u) in ≥89% ee. Nucleophiles substituted at the ortho position generally perform poorly in terms of reaction efficiency and selectivity (5v). However, we found that a nucleophile containing coordinating functionality at the ortho position, 2-methoxyphenylzinc bromide, delivers a moderate 74% ee (5w). Finally, an alkenylzinc iodide provides 5x in 55% ee. In this case, no reaction was observed at the typical reaction temperature of −40 °C, possibly suggesting a slower transmetalation for this class of alkenyl nucleophile.23
While the isolated yields are generally moderate, we believe that the use of pyridine as limiting reagent renders this method practical. Furthermore, upon scaling up the reaction, we observed an increase in reaction efficiency with no influence on selectivity. Under the conditions shown in Table 3, a gram-scale reaction with 4-fluorophenylzinc bromide provided 5a in 80% yield and 90% ee (average of two runs).24 Notably, these gram-scale reactions were set up on the benchtop using Schlenk technique, obviating the need for a glovebox.
Product derivatization
With the scope of the reaction established, we sought to illustrate the utility of the 1,2-dihydropyridine products as valuable building blocks for further organic synthesis. To do so, we explored a number of derivatization reactions, each of which was performed with only minimal optimization (Scheme 3). Importantly, these reactions proceeded with essentially no erosion of ee. The 1,2-dihydropyridine 5a can be selectively reduced with triethylsilane to give tetrahydropyridine 7,20 or with Lindlar's catalyst and H2 to give the regioisomeric tetrahydropyridine 8.4d These compounds are valuable synthetic building blocks in their own right. For example, tetrahydropyridine 8 was further functionalized by iodination with NIS followed by Suzuki cross coupling to provide disubstituted tetrahydropyridine 9. Compound 7 was dihydroxylated with OsO4 with >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr (10), demonstrating that the stereocenter set in the pyridinium arylation can provide high substrate control for a diastereoselective reaction.
:1 dr (10), demonstrating that the stereocenter set in the pyridinium arylation can provide high substrate control for a diastereoselective reaction.
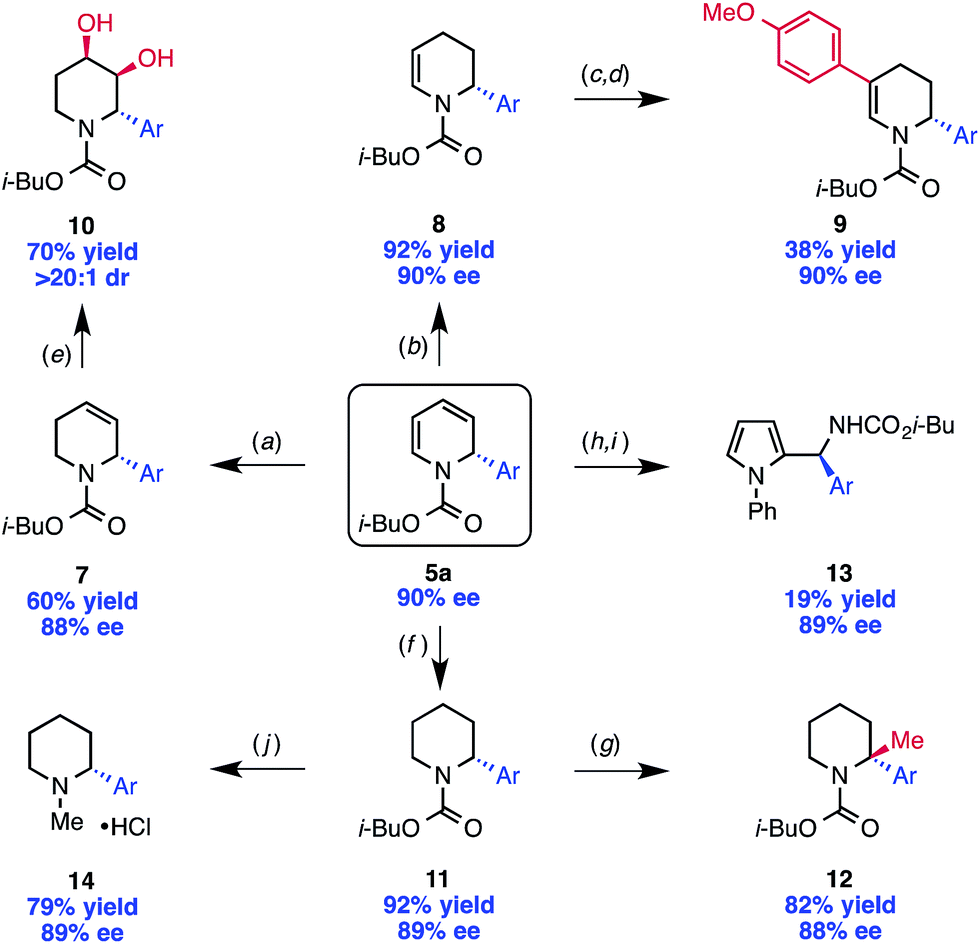 | ||
| Scheme 3 Derivatization of dihydropyridine 5a. Ar = 4-fluorophenyl. Reagents and conditions: (a) Et3SiH (1.05 equiv.), trifluoroacetic acid (20 equiv.), CH2Cl2, −10 °C, 45 s; (b) H2 (1 atm), Lindlar catalyst (3 mol%), MeOH, rt, overnight; (c) N-iodosuccinimide (3 equiv.), DMF, rt, 2 h; (d) Buchwald precatalyst (2 mol%), XPhos (4 mol%), K3PO4 (2 equiv.), 4-MeO-PhB(OH)2 (1.5 equiv.), THF, 80 °C, overnight; (e) OsO4 (4 mol%), N-methylmorpholine-N-oxide (1.2 equiv.), THF:H2O (2:1), rt, 3 h; (f) H2 (100 psi), Pd/C (5 mol%), MeOH, rt, overnight; (g) n-BuLi (1.2 equiv.), THF, −50 °C, 30 min, then MeI (3 equiv.), THF, −78 °C, 2 h; (h) nitrosobenzene (2 equiv.), CH2Cl2, rt, 2 h; (i) CuCl (20 mol%), MeOH, rt, overnight; (j) LiAlH4 (4 equiv.), THF, 50 °C, overnight, then HCl in Et2O. | ||
Full hydrogenation of 5a yields the piperidine product 11, which can be lithiated at low temperature and trapped with an appropriate electrophile with almost no loss in ee, allowing for the formation of a fully substituted, stereodefined carbon (12).25 The diene system of 5a is also amenable to Diels–Alder cycloaddition. An interesting example is the Diels–Alder reaction with nitrosobenzene, which provides an unusual bicycle that can be opened with CuCl to afford the chiral pyrrole derivative 13.26
The carbamate protecting group can be removed from 11 with LiAlH4 to provide N-methyl piperidine derivative 14. For access to the free N–H piperidine 15, we found that the most convenient route was the hydrogenation of the Cbz-protected dihydropyridine 4, which was prepared in only slightly lower ee than 5a (88% vs. 91%, Scheme 4) under our standard reaction conditions.
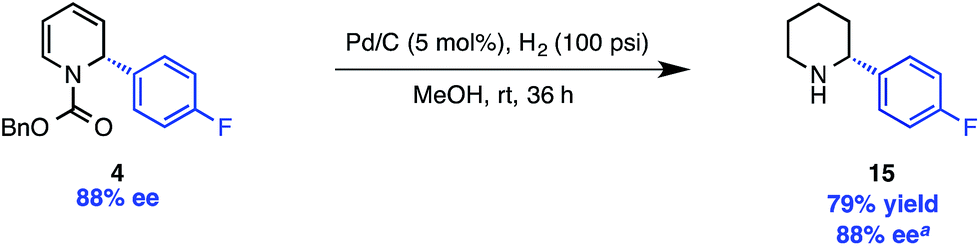 | ||
| Scheme 4 Hydrogenation/deprotection of Cbz-protected dihydropyridine. aDetermined after Boc protection. | ||
Conclusions
In summary, we have developed an enantioselective synthesis of 2-aryl-1,2-dihydropyridines, previously only accessible in a diastereoselective or racemic fashion. The reaction proceeds through a pyridinium salt derived from pyridine and an inexpensive chloroformate, and the products are formed by Ni-catalyzed cross coupling with arylzinc halide nucleophiles. The dihydropyridine products are valuable chiral building blocks, as demonstrated by a number of transformations that proceed with little or no loss in ee, providing a variety of chiral piperidine derivatives. More generally, this methodology is noteworthy for its use of pyridine, a feedstock chemical, as a prochiral substrate in a transition metal-catalyzed asymmetric reaction.Acknowledgements
Financial support from NIGMS (R01 GM100985), Eli Lilly, and Amgen is gratefully acknowledged. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE-1148900 (to J. P. L.). We thank Jake Essman for ligand synthesis and assistance with product derivatization, and Dr István Pelczer and Ken Conover for help with NMR experiments. A. G. D. is a Camille Dreyfus Teacher-Scholar and Arthur C. Cope Scholar.Notes and references
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed.
- For reviews on the synthesis of dihydropyridines, see: (a) U. Eisner and J. Kuthan, Chem. Rev., 1972, 72, 1 CrossRef CAS; (b) D. M. Stout and A. I. Meyers, Chem. Rev., 1982, 82, 223 CrossRef CAS; (c) R. Lavilla, J. Chem. Soc., Perkin Trans. 1, 2002, 1141 RSC; (d) J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charette, Chem. Rev., 2012, 112, 2642 CrossRef CAS PubMed; (e) E. M. P. Silva, P. A. M. M. Varandas and A. M. S. Silva, Synthesis, 2013, 45, 3053 CrossRef CAS.
- For a review on the biological activity of dihydropyridines, see: N. Edraki, A. R. Mehdipour, M. Khoshneviszadeh and R. Miri, Drug Discovery Today, 2009, 14, 1058 CrossRef CAS PubMed.
- Selected examples: (a) T. Koike, Y. Shinohara, M. Tanabe, N. Takeuchi and S. Tobinaga, Chem. Pharm. Bull., 1999, 47, 1246 CrossRef CAS; (b) K. Tanaka, H. Mori, M. Yamamoto and S. Katsumura, J. Org. Chem., 2001, 66, 3099 CrossRef CAS PubMed; (c) T. Harschneck and S. F. Kirsch, J. Org. Chem., 2011, 76, 2145 CrossRef CAS PubMed; (d) B. M. Trost and B. Biannic, Org. Lett., 2015, 17, 1433 CrossRef CAS PubMed.
- Selected examples: (a) T. J. Donohoe, A. J. McRiner and P. Sheldrake, Org. Lett., 2000, 2, 3861 CrossRef CAS PubMed; (b) T. J. Donohoe, A. J. McRiner, M. Helliwell and P. Sheldrake, J. Chem. Soc., Perkin Trans. 1, 2001, 1435 RSC.
- For cyclization reactions to prepare chiral 1,2-dihydropyridines from single-enantiomer compounds, see: (a) T. Shono, Y. Matsumura, O. Onomura and Y. Yamada, Tetrahedron Lett., 1987, 28, 4073 CrossRef CAS; (b) Y. Matsumura, Y. Nakamura, T. Maki and O. Onomura, Tetrahedron Lett., 2000, 41, 7685 CrossRef CAS; (c) D. Monguchi, S. Majumdar and T. Kawabata, Heterocycles, 2006, 68, 2571 CrossRef CAS; (d) M. Motamed, E. M. Bunnelle, S. W. Singaram and R. Sarpong, Org. Lett., 2007, 9, 2167 CrossRef CAS PubMed.
- For a diastereoselective synthesis of 1,2-dihydropyridines, see: K. Tanaka, T. Kobayashi, H. Mori and S. Katsumura, J. Org. Chem., 2004, 69, 5906 CrossRef CAS PubMed.
- For enantioselective syntheses of substituted 1,2-dihydropyridines via [2 + 2 + 2] or [4 + 2] cycloadditions, see: (a) M. Amatore, D. Leboeuf, M. Malacrian, V. Gandon and C. Aubert, J. Am. Chem. Soc., 2013, 135, 4576 CrossRef CAS PubMed; (b) P. Ramaraju, N. A. Mir, D. Singh, V. K. Gupta, R. Kant and I. Kumar, Org. Lett., 2015, 17, 5582 CrossRef CAS PubMed.
- (a) D. L. Comins, R. R. Goehring, S. P. Joseph and S. O'Connor, J. Org. Chem., 1990, 55, 2574 CrossRef CAS; (b) D. L. Comins, H. Hong and J. M. Salvador, J. Org. Chem., 1991, 56, 7197 CrossRef CAS; (c) D. L. Comins, S. P. Joseph and R. R. Goehring, J. Am. Chem. Soc., 1994, 116, 4719 CrossRef CAS; (d) D. L. Comins and L. Guerra-Weltzien, Tetrahedron Lett., 1996, 37, 3807 CrossRef CAS; (e) D. L. Comins, J. Heterocycl. Chem., 1999, 36, 1491 CrossRef CAS.
- (a) C. E. Hoesl, M. Maurus, J. Pabel, K. Polborn and K. Th. Wanner, Tetrahedron, 2002, 58, 6757 CrossRef CAS; (b) C. E. Hoesl, J. Pabel, K. Polborn and K. Th. Wanner, Heterocycles, 2002, 58, 383 CrossRef CAS.
- (a) A. B. Charette, M. Grenon, A. Lemire, M. Pourashraf and J. Martel, J. Am. Chem. Soc., 2001, 123, 11829 CrossRef CAS PubMed; (b) C. Legault and A. B. Charette, J. Am. Chem. Soc., 2003, 125, 6360 CrossRef CAS PubMed; (c) A. Lemire, M. Grenon, M. Pourashraf and A. B. Charette, Org. Lett., 2004, 6, 3517 CrossRef CAS PubMed.
- E. Ichikawa, M. Suzuki, K. Yabu, M. Albert, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2004, 126, 11808 CrossRef CAS PubMed.
- C. Nadeau, S. Aly and K. Belyk, J. Am. Chem. Soc., 2011, 133, 2878 CrossRef PubMed.
- M. Á. Fernández-Ibáñez, B. Maciá, M. G. Pizzuti, A. J. Minnaard and B. L. Feringa, Angew. Chem., Int. Ed., 2009, 48, 9339 CrossRef PubMed.
- S. T. Chau, J. P. Lutz, K. Wu and A. G. Doyle, Angew. Chem., Int. Ed., 2013, 52, 9153 CrossRef CAS PubMed.
- (a) T. J. A. Graham, J. D. Shields and A. G. Doyle, Chem. Sci., 2011, 2, 980 RSC; (b) K. T. Sylvester, K. Wu and A. G. Doyle, J. Am. Chem. Soc., 2012, 134, 16967 CrossRef CAS PubMed; (c) J. D. Shields, D. T. Ahneman, T. J. A. Graham and A. G. Doyle, Org. Lett., 2014, 16, 142 CrossRef CAS PubMed.
- For an example of asymmetric arylation of pyridine N-oxide, see: M. Hussain, T. S.-L. Banchelin, H. Andersson, R. Olsson and F. Almqvist, Org. Lett., 2013, 15, 54 CrossRef CAS PubMed.
- J. Pabel, C. E. Hösl, M. Maurus, M. Ege and K. Th. Wanner, J. Org. Chem., 2000, 65, 9272 CrossRef CAS PubMed.
- (a) Z. Sun, S. Yu, Z. Ding and D. Ma, J. Am. Chem. Soc., 2007, 129, 9300 CrossRef CAS PubMed; (b) D. A. Black, R. E. Beveridge and B. A. Arndtsen, J. Org. Chem., 2008, 73, 1906 CrossRef CAS PubMed.
- O. G. Mancheño, S. Asmus, M. Zurro and T. Fischer, Angew. Chem., Int. Ed., 2015, 54, 8823 CrossRef PubMed.
- Reaction conditions: pyridine (0.05 mmol, 1 equiv.), EtOCOCl (4 equiv.), THF, 0 °C, 10 min, then NiBr2·diglyme (10 mol%), L1 (12 mol%), 4-fluorophenylzinc bromide (4 equiv.), THF (0.01 M), −78 °C → rt, overnight.
- See ESI† for details.
- We have found that other pyridine electrophiles, including pyridinium salts derived from 4-methoxypyridine, 4-methylpyridine, and 3-ethylpyridine, have limited success under the reported reaction conditions. See ESI† for details.
- Similarly, 5j was prepared on gram scale with 96% ee and an improved 58% yield.
- N. S. Sheikh, D. Leonori, G. Barker, J. D. Firth, K. R. Campos, A. J. H. M. Meijer, P. O'Brien and I. Coldham, J. Am. Chem. Soc., 2012, 134, 5300 CrossRef CAS PubMed.
- F. Berti, V. Di Bussolo and M. Pineschi, J. Org. Chem., 2013, 78, 7324 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, and optimization tables. See DOI: 10.1039/c6sc00702c |
| This journal is © The Royal Society of Chemistry 2016 |