
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA room temperature phosphorescence encoding [2]rotaxane molecular shuttle†
Xiang
Ma
 *,
Jing
Zhang
,
Jingjing
Cao
,
Xuyang
Yao
,
Tiantian
Cao
,
Yifan
Gong
,
Chunchang
Zhao
and
He
Tian
*,
Jing
Zhang
,
Jingjing
Cao
,
Xuyang
Yao
,
Tiantian
Cao
,
Yifan
Gong
,
Chunchang
Zhao
and
He
Tian
Key Laboratory for Advanced Materials and Institute of Fine Chemicals, East China University of Science & Technology, Shanghai 200237, P. R. China. E-mail: maxiang@ecust.edu.cn
First published on 29th March 2016
Abstract
A novel bistable molecular shuttle, composed of a Pt(II) porphyrin-containing dibenzo[24]crown-8 macrocycle threaded onto two different recognition sites (secondary dialkylammonium (NH2+) and 4,4′-bipyridinium (Bpym2+) units), and the anthracene (Anth) moiety as one terminal stopper, was synthesized by click chemistry. Its acid–base switchable shuttling could be addressed by both the room temperature phosphorescence (RTP) emission signals of the Pt(II) porphyrin moiety and the fluorescence emission of the Anth unit, as well as their lifetime changes. When the macrocycle was switched to be located on the NH2+ site close to Anth, the Pt(II) porphyrin moiety exhibited strong RTP emission, excited in the Anth band at 370 nm. This was due to the distance-dependent efficient singlet energy transfer between the Anth unit and the porphyrin moiety, followed by intersystem crossing from a singlet to a triplet state in Pt(II) porphyrin, while its RTP emission dramatically decreased when located on the Bpym2+ site far from the Anth unit. When excited in the porphyrin band at 402 nm, the RTP emission lifetimes changed obviously. This is the first rotaxane-type molecular shuttle whose shuttling has been encoded by RTP signals.
Introduction
Rotaxanes,1 as one of the challenging topics of molecular machines, have attracted continued interest and attention. A variety of appealing rotaxanes have been fabricated over the recent years, with various functions or applications in such areas as switches,2 logic gates,3 artificial muscles,4 molecular elevators,5 molecular motors,6 and so forth. The output signals of most rotaxane shuttles were observed by NMR, UV-Vis absorption, cyclic voltammetry, circular dichroism, fluorescence emission, and so forth.7 Room temperature phosphorescence (RTP), with the advantages of longer emission wavelengths and lifetimes, larger ‘Stokes’ shifts, higher signal-to-noise ratios, quick response and low-cost detection etc.,8 is another excellent method which is expected to be employed to address the shuttling and conformation of rotaxane shuttles.9 Up to now, few rotaxane examples employing phosphorescence signal changes as the output have been reported.10Porphyrins are important compounds in relation to electron and energy transfer processes. Stoddart11 and Sauvage12 first introduced the porphyrin moiety into rotaxane systems. Subsequently, several rotaxanes and catenanes containing porphyrins have been investigated.13 Particularly, Pt(II) porphyrins have been reported to have characteristic long wave RTP emissions.14 Therefore, incorporating a Pt(II) porphyrin moiety into a rotaxane might engender new signals, i.e. RTP emission and lifetime changes, to address the shuttling and conformation of rotaxanes.
Herein, we designed and synthesized novel acid–base controlled molecular shuttles15 based on switchable [2]rotaxanes 1 (Scheme 1), with Pt(II) porphyrin-based dibenzo[24]crown-8 as the host macrocycle threaded onto two different recognition sites (secondary dialkylammonium (NH2+) and 4,4′-bipyridinium (Bpym2+) units), and the anthracene (Anth) moiety as one terminal stopper. In this [2]rotaxane, reversible motions of the macrocycle between the two stations could be controlled by the protonation and deprotonation of the NH2+ recognition site, as previously observed in the related system.16 These motions affect the interactions between the porphyrin moiety on the macrocycle and the Anth stopper. Upon protonation of the NH2+ recognition site, the macrocycle is located at said site, and the porphyrin unit nearby could also interact with the Anth unit by energy transfer. Deprotonation of the NH2+ center could engender the displacement of the macrocycle to the Bpym2+ station, and destroy the energy transfer process due to the elongated distance between the Anth and porphyrin units. These switching processes could be investigated using conventional methods, such as 1H NMR spectroscopy and fluorescence spectroscopy. Gratifyingly, after the metallization of the porphyrin core with Pt(II) salt, the shuttling of rotaxane 1-H accompanied by the macrocycle 6 staying between the two recognition sites could also be monitored by obvious RTP signal changes, which makes this the first [2]rotaxane molecular shuttle with switchable RTP output addresses so far.
 | ||
| Scheme 1 Chemical structures of the macrocycle 6, and the [2]rotaxanes 1-H, 1 and 3-H. | ||
Results and discussions
The [2]rotaxane 1-H was synthesized conveniently via a copper(I) catalyzed azide–alkyne ‘click’ cycloaddition. As shown in Scheme 1, treating equal equivalents of 4 and 5 with 2.0 equivalents of macrocycle 6 in the absence of light and oxygen led to the formation of the [2]rotaxane 1-H with a relatively low yield of 13%, which might be due to the relatively lower binding constant of the DB24C8 decorated with Pt(II) porphyrin (macrocycle 6) with the NH2+ center of compound 4 and steric hindrance, besides the obvious product loss during purification by chromatography on silica gel. The triazole ring and the oxygen atom of the dumbbell were used as long linkers not only to increase the distance between the two recognition sites, preventing energy transfer from the anthracene moiety to the 4,4′-bipyridium moiety as much as possible, but also to ensure the stability of the interlocking states by avoiding adverse factors like steric hindrance. Another reference [2]rotaxane 3-H was constructed by replacing macrocycle 6 with porphyrin-free dibenzo[24]crown-8 (DB24C8). The addition of 1.0 equiv. of n-Bu3N16,17 to the solution of [2]rotaxane 1-H results in the deprotonated form of 1. Reprotonation can be performed by the addition of a slight molar excess of CF3COOH.The [2]rotaxane formation and its reversible switching process were monitored by 1H NMR spectroscopy. As shown in Fig. 1, the dumbbell and macrocycle components of 1-H/1 are present in equimolar amounts and are interlocked with each other. The spectrum (Fig. 1c) demonstrates that the macrocycle 6 binds selectively with the NH2+ center, since the characteristic resonances Ha and Hb of the dumbbell 2-H, at δ 5.59 and 4.39 ppm (Fig. 1b), for the four CH2 protons adjacent to the NH2+ group experienced upfield shifts, Ha at δ 5.24 ppm (Δδ = 0.35 ppm) and Hb at 4.22 ppm (Δδ = 0.17 ppm), respectively, upon rotaxane (1-H) formation. Furthermore, the aromatic protons He of the macrocycle 6 were split into two peaks (δ 7.15 and 7.13 ppm) after complexation.
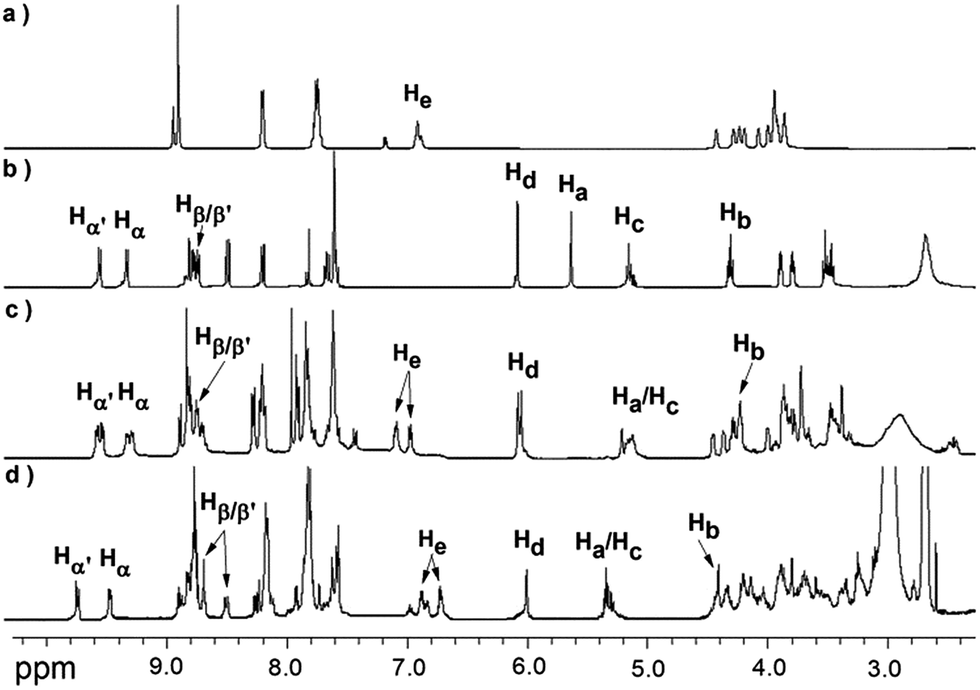 | ||
| Fig. 1 Partial 1H NMR spectra (500 MHz in CD3COCD3 at 298 K) of: (a) the macrocycle 6; (b) the dumbbell compound 2-H; (c) the [2]rotaxane 1-H; and (d) the deprotonated [2]rotaxane 1. | ||
After the addition of n-Bu3N, the 1H NMR spectrum of 1 shows dramatic chemical shift changes with respect to that of 1-H. The resonances (δ 5.24 and 4.22 ppm) of the CH2–NH2+–CH2 protons disappeared and new signals (δ 5.38 and 4.40 ppm, Fig. 1d), assigned to CH2–NH–CH2 protons, appeared. Moreover, the macrocycle 6 glides towards the Bpym2+ unit, resulting in the obvious changes of the proton resonance values of Bpym2+ (δ [Δδ] for Hα′, Hα, Hβ′ and Hβ (ppm): 9.56 [+0.22], 9.28 [+0.18], 8.70 [−0.10] and 8.51 [−0.28], respectively). Upon addition of CF3COOH, the original 1H NMR spectrum was almost regenerated, demonstrating that the switching process was fully reversible via de-/reprotonation (Fig. S5†).
The photophysical and photochemical properties of the shuttling processes of rotaxane 1-H/1 were thoroughly investigated (Table S1†). Pt(II) porphyrin-based dibenzo[24]crown-8 6 displayed relatively weak fluorescence emission bands at 650 and 715 nm in air equilibrated CH3CN (Fig. S7†). Notably, no RTP was exhibited even in a deaerated organic solution, but strong RTP signals were observed in the medium containing the anionic surfactant sodium dodecyl sulfate (SDS) and Na2SO3 for removing oxygen.18 The characteristic RTP emission peaks of 6 were exhibited at 667 and 736 nm (inset, Fig. S7†) when excited in the porphyrin band at 402 nm, and experienced a redshift compared with the fluorescence emission peak (Fig. S7†). Upon forming the [2]rotaxane 1-H, the fluorescence emission was almost quenched,19 while its RTP emission showed a slight increase due to the movement restriction caused by the macrocycle 6 in the rotaxane system when excited at 402 nm (Fig. S7 and S8†).
The switching of the [2]rotaxanes between 1 and 1-H was accompanied by obvious RTP signal changes. As shown in Fig. 2, both RTP spectra of the [2]rotaxanes 1-H and 1 showed intense RTP emissions in the region above 600 nm, upon excitation in the porphyrin band at 402 nm, which were characteristic of the Pt(II) porphyrin moieties. Deprotonation induced very small changes in this band, and the intensity decrease of the deprotonated [2]rotaxane 1 could be due to the quenching effect of the Bpym2+ unit through charge transfer. When the excitation was performed in the Anth band at 370 nm, the RTP spectra exhibited dramatic changes (emission intensities increased by more than 1200%, Fig. 2). Such a porphyrin-based phosphorescence band was still present in 1-H, but with obvious decreases in intensity of 1. The RTP lifetime of 1-H excited at 370 nm was found to be about 57.1 μs (Fig. S12†). The reversibility of the switching process of the rotaxanes 1-H/1 was proven to be favourable via testing the RTP intensity change upon addition of n-Bu3N and CF3COOH alternately (Fig. 2, inset). Moreover, Table 1 lists the RTP lifetimes of all of the Pt(II) porphyrin-containing species. The [2]rotaxane 1 exhibited a shorter RTP lifetime than 1-H, which resulted from the electron transfer to the Bpym2+ moiety in [2]rotaxane 1 upon deprotonation.
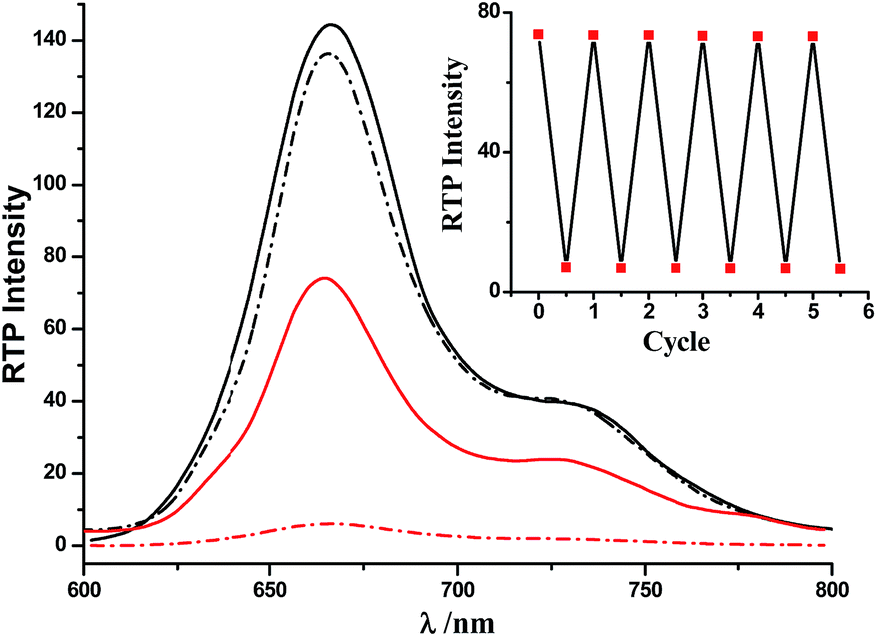 | ||
| Fig. 2 RTP emission spectra of 1-H (solid line) and 1 (dashed line). Excitation was performed in the Anth band at 370 nm (red, the RTP lifetime of 1-H was found to be about 57.1 μs, Fig. S12†) and the Pt(II) porphyrin band at 402 nm (black, for the lifetimes see Table 1). 4.0 × 10−6 mol L−1. Inset: RTP intensity changes at 667 nm after addition of n-Bu3N and CF3COOH to 1-H and 1, over six cycles (λex = 370 nm). | ||
| Compound | 6 | 1-H | 1 |
| λ ex/λem (nm) | 408/667 | 403/665 | 403/666 |
| I RTP | 169.18 | 144.40 | 136.30 |
| τ (μs) | 54.5 | 48.6 | 42.9 |
Electrochemical measurements of 1-H, 1 and the reference [2]rotaxanes 3-H and 3 using the cyclic voltammetry technique were carried out to confirm the possible electron transfer from the Pt(II) porphyrin unit to the bipyridinium moiety in [2]rotaxane 1 (Fig. 3). For the DB24C8-based [2]rotaxanes 3-H and 3, it was clear that the reduction waves of the bipyridinium unit were displaced to more negative positions as a result of being engaged in a charge transfer interaction with the electron donating macrocycle.20 However, for 1-H and 1, the positions of the reduction waves of the bipyridinium unit were almost unaltered although it was proven by NMR that the macrocycle also moved towards the bipyridinium unit when 1-H was turned into 1. For unperturbed bipyridinium reduction potentials, there are few electrochemical experiments in the literature, although several similar systems containing Zn-porphyrin have been reported.20b A probable reason for the unaltered reduction potentials of bipyridinium was the electron transfer from the Pt(II) porphyrin unit to the bipyridinium moiety, since the presence of an electron-donor moiety usually leads to a less negative potential value for the first and second reductions of the bipyridium unit.20a
 | ||
| Fig. 3 Cyclic voltammograms of (a) 1-H and 1 (after the addition of tributylamine); and (b) DB24C8-based [2]rotaxanes 3-H and 3 (after the addition of tributylamine) in acetonitrile at 298 K. | ||
Addition of the base (n-Bu3N) made the RTP emission band of [2]rotaxane 1 almost quenched (Fig. 2). This behaviour cannot be simply assigned to the electron transfer to the Bpym2+ unit; it should be due to the interaction between the Anth unit and the nearby porphyrin moiety of the macrocycle. As in the control experiment, both the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture solutions of the macrocycle 6 with dumbbell 2-H and of 6 with 2 (after the addition of n-Bu3N) showed no obvious RTP emission bands when excited in the Anth band by light at 370 nm (Fig. S17 and S18†). Upon deprotonation, the intensity of the fluorescence emission band of the Anth unit of the dumbbell 2, shown in Fig. 4, decreases. This is attributed to the electron transfer interaction between the Anth moiety and the amino group.20 Conversely, the intensity of the Anth bands for the [2]rotaxane 1 experiences a dramatic increase with respect to that of 1-H. Consequently, both the increase of the porphyrin based RTP emission and the decrease of the Anth based fluorescence emission for 1-H compared with those of its deprotonated species should be attributed to the efficient singlet energy transfer process between the Anth unit and the porphyrin moiety, followed by intersystem crossing from a singlet to triplet state in Pt(II) porphyrin (Fig. 5).
:1 mixture solutions of the macrocycle 6 with dumbbell 2-H and of 6 with 2 (after the addition of n-Bu3N) showed no obvious RTP emission bands when excited in the Anth band by light at 370 nm (Fig. S17 and S18†). Upon deprotonation, the intensity of the fluorescence emission band of the Anth unit of the dumbbell 2, shown in Fig. 4, decreases. This is attributed to the electron transfer interaction between the Anth moiety and the amino group.20 Conversely, the intensity of the Anth bands for the [2]rotaxane 1 experiences a dramatic increase with respect to that of 1-H. Consequently, both the increase of the porphyrin based RTP emission and the decrease of the Anth based fluorescence emission for 1-H compared with those of its deprotonated species should be attributed to the efficient singlet energy transfer process between the Anth unit and the porphyrin moiety, followed by intersystem crossing from a singlet to triplet state in Pt(II) porphyrin (Fig. 5).
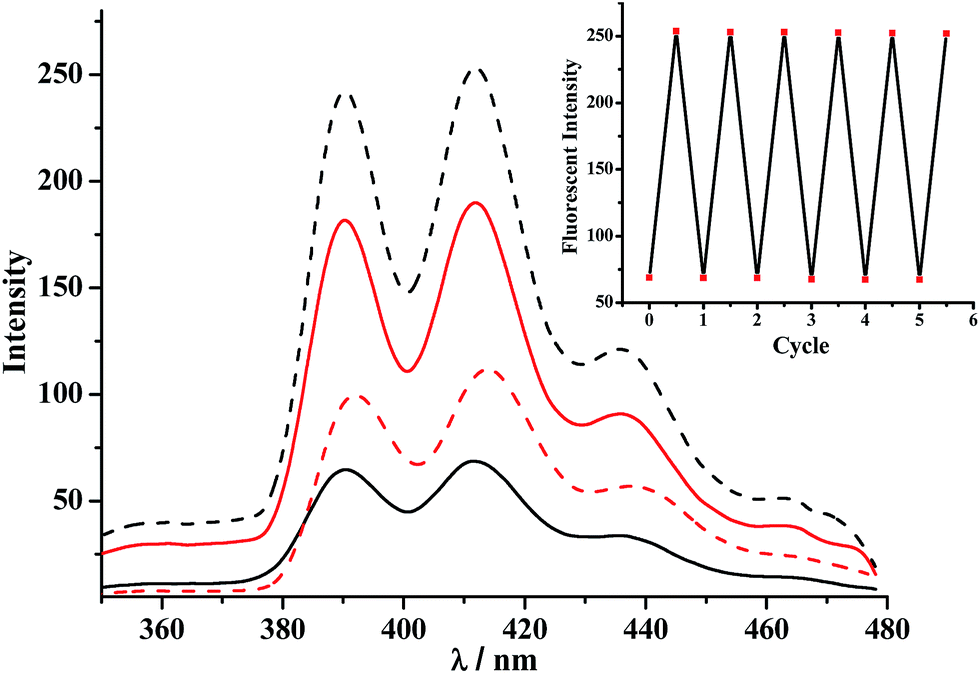 | ||
| Fig. 4 Fluorescence spectra of the [2]rotaxanes 1-H/1 (black, solid/dash lines) and the dumbbell 2-H/2 (red, solid/dash lines). Excitation was performed in the Anth band at 370 nm, 1.0 × 10−5 mol L−1. Inset: fluorescence intensity changes at 412 nm after addition of n-Bu3N and CF3COOH to 1-H and 1, over six cycles (λex = 370 nm). | ||
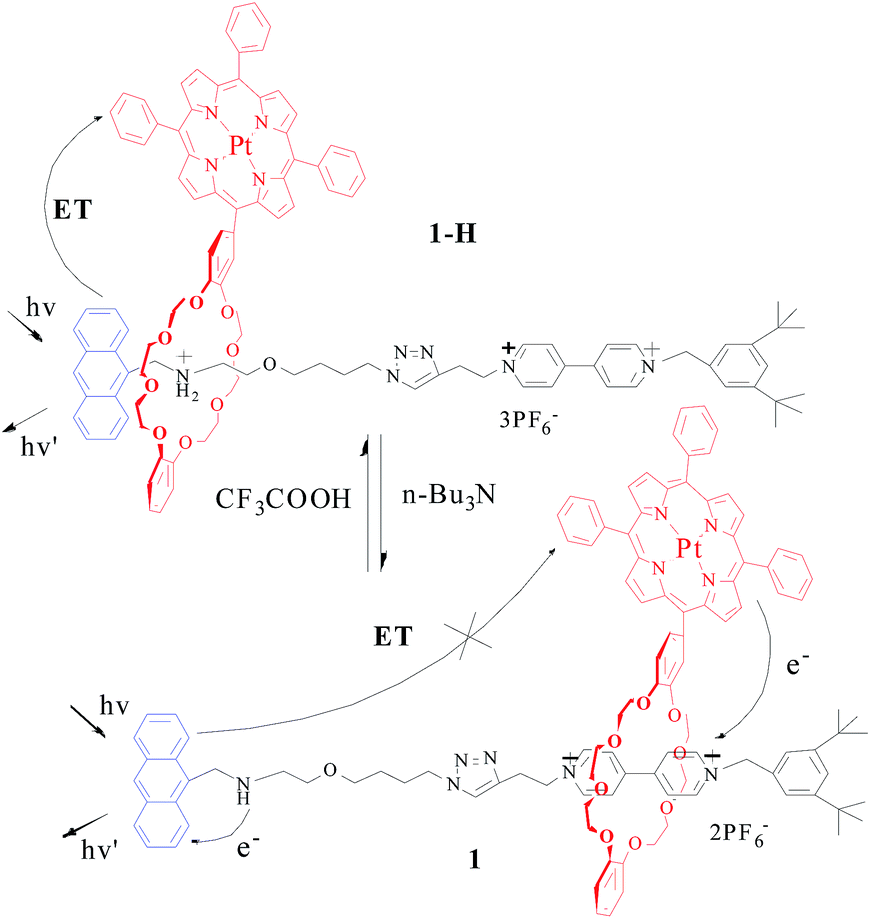 | ||
| Fig. 5 Photoinduced charge and energy transfer processes in [2]rotaxanes 1-H/1 (singlet energy transfer process between the Anth and porphyrin moieties followed by intersystem crossing from a singlet to triplet state in Pt(II) porphyrin). | ||
The quantum yields for the various RTP emission processes were also investigated, as shown in Table 2. The maximum absorption peak of the Pt(II) porphyrin-containing macrocycle 6 is at about 402 nm while the absorption at around 370 nm is very weak (Fig. S6†). Thus the quantum yields of 6, 6 + base and [2]rotaxane 1-H excited at 402 nm were relatively higher than the ones excited at 370 nm. After forming [2]rotaxane, the quantum yield of 1-H (0.16) was slightly increased with respect to that of the host macrocycle 6 due to the efficient singlet energy transfer process between the Anth unit and the porphyrin moiety, followed by intersystem crossing from a singlet to triplet state in Pt(II) porphyrin. While the quantum yields of [2]rotaxane 1 (1-H + base) excited at both 402 nm and 370 nm were too low to quantify, since the 4,4′-bipyridinium (Bpym2+) units in 1 had some quenching effects on the emission of the porphyrin moiety of 6. Also, the distance dependent energy transfer efficiency from the Anth to porphyrin units decreased when the macrocycle 6 was located on the Bpym2+ site far from the Anth moiety.
| Compound | Quantum yields (excited at 370 nm) | Quantum yields (excited at 402 nm) |
|---|---|---|
| 6 | 0.14 | 2.1 |
| 6 + base | 0.10 | 2.2 |
| 1-H | 0.16 | 1.2 |
| 1 (1-H + base) base: tributylamine | N/A | N/A |
The fluorescence lifetime of the excited Anth unit in the [2]rotaxane 1-H was found to be 2.79 ns (Fig. S13†). It was shorter than that of the corresponding dumbbell 2-H (4.13 ns, Fig. S14†), which was attributed to the energy transfer from Anth to Pt(II) porphyrin moieties in 1-H (Fig. S19–S21† and discussion therein). Upon the addition of base, the lifetime of the excited Anth unit in 1 increased compared with 1-H and reached up to 3.79 ns (Fig. S13†), which was due to the break up of the energy transfer process from the Anth to Pt(II) porphyrin units owing to the longer distance between them (Fig. S20†). The emission spectrum showed similarities with that of the original [2]rotaxane 1-H upon addition of an equivalent of CF3COOH to its solution, indicating that the macrocycle was situated on the NH2+ center once again. The fluorescence emission intensity changes of [2]rotaxane 1 (at 412 nm) also showed good reversibility after alternating protonation/deprotonation for several cycles, indicating that its shuttling was totally reversible (Fig. 4, inset).
Conclusions
In summary, a novel bistable molecular shuttle, based on an acid–base switchable [2]rotaxane in which the Pt(II) porphyrin-containing macrocycle could be stimulated to shuttle back and forth between the two recognition sites, was constructed, whose shuttling process was accompanied by reversible changes both in the RTP emission (both intensities and lifetimes) of the Pt(II) porphyrin moiety and the fluorescence emission of the Anth unit. It should be noticed that this is the first example of a rotaxane-type molecular shuttle whose shuttling could be encoded by RTP signals. The concept and advance in this work, employing RTP signals to address the shuttling of a rotaxane, supply a novel method to encode a molecular shuttle and pave the way for designing molecular machines with multi-responses and multi-functions.Experimental section
Synthesis of 1-H
A solution of the azide 4 (56 mg, 0.11 mmol) and the ring 6 (200 mg, 0.17 mmol) in 10 mL of CHCl3 was stirred for 24 h at room temperature under Ar. Then the BIPY2+ derivative 5 (71 mg, 0.1 mmol) and Cu(MeCN)4PF6 (20 mg, 0.05 mmol) in 10 mL of dry Me2CO were added, and the whole solution was stirred for another 3 days. After the solvent was evaporated, the resulting reddish solid was washed with H2O, air-dried and purified by column chromatography on silica gel (Me2CO to 2% NH4PF6/Me2CO) to obtain the [2]rotaxane 1-H (31 mg, 13%).1H NMR (500 MHz, (CD3)2CO): δH (ppm) 9.56 (d, 2H, 3J = 5.9 Hz), 9.31 (d, 2H, 3J = 5.5 Hz), 8.86 (m, 8H), 8.74 (d, 2H, 3J = 6.2 Hz), 8.50 (d, 2H, 3J = 6.5 Hz), 8.58 (d, 3H, 3J = 9.0 Hz), 8.23 (t, 6H, 3J = 7.3 Hz), 8.11 (d, 2H, 3J = 8.1 Hz), 7.87 (m, 11H), 7.62 (s, 3H), 7.53 (m, 4H), 7.15 (d, 2H, 3J = 6.0 Hz), 7.13 (d, 2H, 3J = 6.0 Hz), 6.09 (s, 2H), 5.59 (s, 2H), 5.24 (t, 2H, 3J = 6.5 Hz), 4.70–3.70 (m, 29H), 3.60 (t, 3H, 3J = 6.0 Hz), 3.39 (m, 4H), 2.64 (s, 2H), 2.15 (s, 2H), 1.32 (s, 18H). 13C NMR (100 MHz, CD3OD): δC (ppm), 153.92, 151.77, 151.50, 147.39, 146.85, 134.40, 133.58, 132.80, 132.24, 131.65, 130.62, 128.85, 128.61, 128.24, 127.72, 126.67, 125.36, 124.96, 124.32, 78.92, 75.18, 71.47, 67.46, 66.56, 62.30, 61.23, 58.31, 56.32, 51.31, 35.94, 31.76, 28.10, 26.02, 23.71, 21.87, 18.38, 14.44. HRMS (ESI): m/z [1-H-PF6]+ calc. for [C112H115N10F12O9P2Pt]+ 2228.8, found 2229.2. MALDI-TOF-MS: m/z [1-H-3PF6]3+ calc. for C112H115N10O9Pt, 1938.85, found 1938.78.Synthesis of 2-H
A solution of the azide 4 (197.6 mg, 0.4 mmol), the BIPY2+ derivative 5 (140 mg, 0.2 mmol), and Cu(MeCN)4PF6 (33.2 mg, 0.1 mmol) in dry Me2CO (10 mL) was stirred for 48 h at room temperature under Ar. After the solvent was evaporated, the resulting reddish solid was washed with H2O, air dried and purified by column chromatography on silica gel (Me2CO to 2% NH4PF6/Me2CO) to obtain the thread 2-H (73 mg, 30%). 1H NMR (500 MHz, (CD3)2CO): δH (ppm) 9.49 (d, 2H, 3J = 5.9 Hz), 9.31 (d, 2H, 3J = 5.5 Hz), 8.74 (d, 2H, 3J = 6.2 Hz), 8.50 (d, 2H, 3J = 6.5 Hz), 8.58 (d, 3H, 3J = 9.0 Hz), 8.11 (d, 2H, 3J = 8.1 Hz), 7.62 (s, 3H), 7.53 (m, 4H), 6.09 (s, 2H), 5.59 (s, 2H), 5.24 (t, 2H, 3J = 6.5 Hz), 4.39 (s, 2H), 3.84 (s, 1H), 3.60 (t, 3H, 3J = 6.0 Hz), 3.39 (m, 4H), 2.64 (s, 2H), 2.15 (s, 2H), 1.32 (s, 18H). 13C NMR (100 MHz, CD3OD): δC (ppm) 151.62, 148.78, 146.05, 145.58, 141.63, 133.50, 130.85, 130.73, 129.13, 127.18, 127.06, 126.46, 125.58, 124.37, 123.58, 123.34, 123.17, 69.86, 65.45, 64.06, 59.96, 49.25, 42.37, 34.79, 31.22, 26.71, 26.07. HRMS (ESI): m/z [2-H-PF6]+ calc. for [C50H61F12OP2]+ 1051.4, found 1051.3.Synthesis of 3-H
A solution of the azide 4 (200 mg, 0.4 mmol) and DB24C8 (225 mg, 0.5 mmol) in CHCl3 (30 mL) was stirred for 48 h at room temperature under argon. Then the BIPY2+ derivative 5 (140 mg, 0.2 mmol) and the catalyst Cu(CH3CN)4PF6 (150 mg, 0.4 mmol) were added and the whole solution was stirred for another 2 days. After the solvent was removed under reduced pressure, the resulting reddish solid was washed with H2O, air dried and purified by column chromatography on silica gel (Me2CO to 2% NH4PF6/Me2CO) to afford the DB24C8 based [2]rotaxane 3-H (62 mg, 19%).1H NMR (400 MHz, DMSO-d6): δH (ppm) 9.61 (d, 2H, J = 6.0 Hz), 9.42 (d, 2H, J = 6.0 Hz), 8.77 (s, 1H), 8.57–8.48 (m, 4H), 8.36 (d, 2H, J = 6.0 Hz), 8.17 (d, 2H, J = 8.0 Hz), 7.73 (s, 1H), 7.69–7.54 (m, 6H), 7.46 (s, 1H), 6.75–6.61 (m, 4H), 6.51–6.31 (m, 4H), 5.89 (s, 2H), 5.21 (s, 2H), 5.07 (t, 2H, J = 8.0 Hz), 4.32 (t, 2H, J = 6.4 Hz), 4.11–3.99 (m, 4H), 3.96 (m, 4H), 3.83–3.66 (m, 10H), 3.64–3.41 (m, 12H), 3.16 (d, 2H, J = 4.8 Hz), 1.93–1.78 (m, 2H), 1.59–1.48 (m, 2H), 1.30 (s, 18H). 13C NMR (100 MHz, DMSO-d6): δC (ppm) 151.63, 146.77, 146.54, 145.24, 142.89, 133.78, 130.95, 130.69, 129.13, 127.00, 126.59, 125.54, 124.39, 123.81, 123.41, 123.11, 122.25, 120.50, 111.98, 70.22, 69.89, 69.47, 67.78, 49.14, 46.86, 42.56, 34.80, 31.22, 26.71, 26.14, 25.07, 22.55. HRMS (ESI): m/z [3-H-3PF6]3+ calc. for [C74H93N6O9]3+ 1209.69, found 1209.64.Acknowledgements
We acknowledge the financial support from the Programme of Introducing Talents of Discipline to Universities (B16017), NSFC (21421004, 21476075 & 21272072), the National Basic Research 973 Program and the Fundamental Research Funds for the Central Universities. We also thank Profs D. Qu and Q. Wang for their helpful discussion.Notes and references
- (a) D. Leigh, M. A. F. Morales, E. M. Pérez, J. K. Y. Wong, C. G. Saiz, A. M. Z. Slawin, A. J. Carmichael, D. M. Haddleton, A. M. Brouwer, W. J. Buma, G. W. H. Wurpel, S. León and F. Zerbetto, Angew. Chem., Int. Ed., 2005, 44, 3062 CrossRef CAS PubMed; (b) H. Tian and Q.-C. Wang, Chem. Soc. Rev., 2006, 35, 361 RSC; (c) E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72 CrossRef CAS PubMed; (d) V. Balzani, A. Credi and M. Venturi, Chem. Soc. Rev., 2009, 38, 1542 RSC; (e) X. Ma and H. Tian, Chem. Soc. Rev., 2010, 39, 70 RSC; (f) Z.-J. Zhang, H.-Y. Zhang, H. Wang and Y. Liu, Angew. Chem., Int. Ed., 2011, 50, 10834 CrossRef CAS PubMed; (g) N. D. Suhan, S. J. Loeb and S. H. Eichhorn, J. Am. Chem. Soc., 2013, 135, 400 CrossRef CAS PubMed; (h) C. Ke, R. A. Smaldone, T. Kikuchi, H. Li, A. P. Davis and J. F. Stoddart, Angew. Chem., Int. Ed., 2013, 52, 381 CrossRef CAS PubMed; (i) J. Winn, A. Pinczewska and S. M. Goldup, J. Am. Chem. Soc., 2013, 135, 13318 CrossRef CAS PubMed; (j) Y.-H. Lin, C.-C. Lai, Y.-H. Liu, S.-M. Peng and S.-H. Chiu, Angew. Chem., Int. Ed., 2013, 52, 10231 CrossRef CAS PubMed; (k) M. J. Langton and P. D. Beer, Acc. Chem. Res., 2014, 47, 1935 CrossRef CAS PubMed; (l) S. Chao, C. Romuald, K. F. Marotte, C. Clavel and F. Coutrot, Angew. Chem., Int. Ed., 2014, 27, 6914 CrossRef PubMed; (m) Y.-D. Yang, C.-C. Fan, B. M. Rambo, H.-Y. Gong, L.-J. Xu, J.-F. Xiang and J. L. Sessler, J. Am. Chem. Soc., 2015, 137, 12966 CrossRef CAS PubMed; (n) A. Fernandez, E. M. Pineda, C. A. Muryn, S. Sproules, F. Moro, G. A. Timco, E. J. L. McInnes and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2015, 127, 11008 CrossRef; (o) M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Chem. Rev., 2015, 115, 7398 CrossRef CAS PubMed.
- (a) C. P. Collier, G. Mattersteig, E. W. Wang, Y. Luo, K. Beverly, J. Sampaio, F. M. Raymo, J. F. Stoddart and J. R. Heath, Science, 2000, 289, 1172 CrossRef CAS PubMed; (b) V. Blanco, D. A. Leigh, U. Lewandowska, B. Lewandowski and V. Marcos, J. Am. Chem. Soc., 2014, 136, 15775 CrossRef CAS PubMed; (c) A. Caballero, L. Swan, F. Zapata and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11854 CrossRef CAS PubMed; (d) M. Woźny, J. Pawłowska, A. Osior, P. Świder, R. Bilewicz and B. Korybut-Daszkiewicz, Chem. Sci., 2014, 5, 2836 RSC; (e) P. G. Young, K. Hirose and Y. Tobe, J. Am. Chem. Soc., 2014, 136, 7899 CrossRef CAS PubMed; (f) E. Moulin and N. Giuseppone, Nat. Nanotechnol., 2014, 9, 331 CrossRef CAS PubMed.
- (a) D.-H. Qu, Q.-C. Wang and H. Tian, Angew. Chem., Int. Ed., 2005, 44, 5296 CrossRef CAS PubMed; (b) D.-H. Qu, Q.-C. Wang and H. Tian, Adv. Mater., 2006, 18, 2035 CrossRef CAS; (c) S. Kou, H. N. Lee, D. van Noort, K. M. K. Swamy, S. H. Kim, J. H. Soh, K.-M. Lee, S.-W. Nam, J. Yoon and S. Park, Angew. Chem., Int. Ed., 2008, 47, 872 CrossRef CAS PubMed; (d) F. Lohmann, J. Weigandt, J. Valero and M. Famulok, Angew. Chem., Int. Ed., 2014, 53, 10372 CrossRef CAS PubMed; (e) A. Wolf, E. Moulin, J.-J. Cid, A. Goujon, G. Du, E. Busseron, G. Fuks and N. Giuseppone, Chem. Commun., 2015, 51, 4212 RSC.
- (a) Y. Liu, A. H. Flood, P. A. Bonvallet, S. A. Vigon and J. F. Stoddart, J. Am. Chem. Soc., 2005, 127, 9745 CrossRef CAS PubMed; (b) R. E. Dawson, S. F. Lincoln and C. J. Easton, Chem. Commun., 2008, 3980 RSC; (c) C. Gao, X. Ma, Q. Zhang, Q. Wang, D.-H. Qu and H. Tian, Org. Biomol. Chem., 2011, 9, 1126 RSC; (d) C. J. Bruns and J. F. Stoddart, Nat. Nanotechnol., 2013, 8, 794 CrossRef CAS PubMed; (e) C. J. Bruns and J. F. Stoddart, Acc. Chem. Res., 2014, 47, 2186 CrossRef CAS PubMed; (f) C. J. Bruns, M. Frasconi, J. Iehl, K. J. Hartlieb, S. T. Schneebeli, C. Cheng, S. I. Stupp and J. F. Stoddart, J. Am. Chem. Soc., 2014, 136, 4714 CrossRef CAS PubMed; (g) L. Gao, Z. Zhang, B. Zheng and F. Huang, Polym. Chem., 2014, 5, 5734 RSC.
- (a) J. D. Badjic, V. Balzani, A. Credi, S. Silvi and J. F. Stoddart, Science, 2004, 303, 1845 CrossRef CAS PubMed; (b) J. D. Badjic, C. M. Ronconi, J. F. Stoddart, V. Balzani, S. Silvi and A. Credi, J. Am. Chem. Soc., 2006, 128, 1489 CrossRef CAS PubMed; (c) Z.-J. Zhang, M. Han, H.-Y. Zhang and Y. Liu, Org. Lett., 2013, 15, 1698 CrossRef CAS PubMed.
- (a) M. Schliwa, Molecular Motors, Wiley-VCH, Weinheim, 2003 Search PubMed; (b) G. S. Kottas, L. I. Clarke, D. Horinek and J. Michl, Chem. Rev., 2005, 105, 1281 CrossRef CAS PubMed; (c) S. P. Fletcher, F. Dumur, M. M. Pollard and B. L. Feringa, Science, 2005, 310, 80 CrossRef CAS PubMed; (d) E. R. Kay and D. A. Leigh, Pure Appl. Chem., 2008, 80, 17 CrossRef CAS; (e) S. Lee and W. Lu, Nanotechnology, 2011, 22, 205501 CrossRef PubMed.
- (a) C. A. Stanier, S. J. Alderman, T. D. W. Claridge and H. L. Anderson, Angew. Chem., Int. Ed., 2002, 41, 1769 CrossRef; (b) T. Iijima, S. A. Vignon, H.-R. Tseng, T. Jarrosson, J. K. M. Sanders, F. Marchioni, M. Venturi, E. Apostoli, V. Balzani and J. F. Stoddart, Chem.–Eur. J., 2004, 10, 6375 CrossRef CAS PubMed; (c) A. Altieri, F. G. Gatti, E. R. Kay, D. A. Leigh, D. Martel, F. Paolucci, A. M. Z. Slawin and J. K. Y. Wong, J. Am. Chem. Soc., 2003, 125, 8644 CrossRef CAS PubMed; (d) G. Bottari, D. A. Leigh and E. M. Pérez, J. Am. Chem. Soc., 2003, 125, 13360 CrossRef CAS PubMed; (e) X. Ma, Q.-C. Wang, D.-H. Qu, Y. Xu, F.-Y. Ji and H. Tian, Adv. Funct. Mater., 2007, 17, 829 CrossRef CAS; (f) Q.-C. Wang, D.-H. Qu and H. Tian, Angew. Chem., Int. Ed., 2004, 43, 2661 CrossRef CAS; (g) E. M. Pérez, D. T. F. Dryden, D. A. Leigh, T. Gilberto and Z. Francesco, J. Am. Chem. Soc., 2004, 126, 12210 CrossRef PubMed.
- (a) Y. Wei, S. Shuang, C. Dong, W. Jin, C. Gao, Y. Wei and C. Liu, Trends Appl. Spectrosc., 2004, 5, 195 CAS; (b) I. Sánchez-Barragán, J. M. Costa-Fernández, M. Valledor, J. C. Campo and A. Sanz-Medel, Trends Anal. Chem., 2006, 25, 958 CrossRef.
- (a) R. A. Paynter, S. L. Wellons and J. D. Winefordner, Anal. Chem., 1974, 46, 736 CrossRef CAS; (b) L. J. C. Love, M. Skrilec and J. G. Habarta, Anal. Chem., 1980, 52, 754 CrossRef CAS.
- (a) X. Ma, J. Cao, Q. Wang and H. Tian, Chem. Commun., 2011, 47, 3559 RSC; (b) J. Cao, X. Ma, M. Min, T. Cao, S. Wu and H. Tian, Chem. Commun., 2014, 50, 3224 RSC; (c) H. Masai, J. Terao, S. Makuta, Y. Tachibana, T. Fujihara and Y. Tsuji, J. Am. Chem. Soc., 2014, 136, 14714 CrossRef CAS PubMed; (d) H. Chen, X. Ma, S. Wu and H. Tian, Angew. Chem., Int. Ed., 2014, 53, 14149 CrossRef CAS PubMed.
- P. R. Ashton, M. R. Johnston, J. F. Stoddart, M. S. Tolley and J. W. Wheeler, J. Chem. Soc., Chem. Commun., 1992, 1128 RSC.
- J. C. Chambron, V. Heitz and J. P. Sauvage, J. Chem. Soc., Chem. Commun., 1992, 1131 RSC.
- (a) M. J. Blanco, M. C. Jiménez, J. C. Chambron, V. Heitz, M. Linke and J. P. Sauvage, Chem. Soc. Rev., 1999, 28, 293 RSC; (b) H. Sasabe, N. Kihara, Y. Furusho, K. Mizuno, A. Ogawa and T. Takata, Org. Lett., 2004, 6, 3957 CrossRef CAS PubMed; (c) K. Li, D. I. Shuster, D. M. Guldi, M.-A. Herranz and L. Echegoyen, J. Am. Chem. Soc., 2004, 126, 3388 CrossRef CAS PubMed; (d) D. I. Schster, K. Li, D. M. Guldi and J. Ramey, Org. Lett., 2004, 6, 1919 CrossRef PubMed; (e) J.-S. Marois, K. Cantin, A. Desmarais and J.-F. Morin, Org. Lett., 2008, 10, 33 CrossRef CAS PubMed; (f) J. P. Collin, J. Frey, V. Heitz, J. P. Sauvage, C. Tock and L. Allouche, J. Am. Chem. Soc., 2009, 131, 5609 CrossRef CAS PubMed; (g) M. J. Langton, J. D. Matichak, A. L. Thompson and H. L. Anderson, Chem. Sci., 2011, 2, 1897 RSC; (h) Y. Yamada, N. Mihara, S. Shibano, K. Sugimoto and K. Tanaka, J. Am. Chem. Soc., 2013, 135, 11505 CrossRef CAS PubMed.
- (a) D. B. Papkovsky, G. V. Ponomarev, W. Trettnak and P. O'Leary, Anal. Chem., 1995, 67, 4112 CrossRef CAS; (b) P. Hartmann and W. Trettnak, Anal. Chem., 1996, 68, 2615 CrossRef CAS PubMed; (c) A. Mills and A. Lepre, Anal. Chem., 1997, 69, 4653 CrossRef CAS; (d) M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151 CrossRef CAS; (e) C.-M. Che, Y.-J. Hou, M. C. W. Chan, J. H. Guo, Y. Liu and Y. Wang, J. Mater. Chem., 2003, 13, 1362 RSC; (f) T. Mani, M. Tanabe, S. Yamauchi, N. V. Tkachenko and S. A. Vinogradov, J. Phys. Chem. Lett., 2012, 3, 3115 CrossRef CAS PubMed.
- (a) A. M. Brouwer, C. Frochot, F. G. Gatti, D. A. Leigh, L. Mottier, F. Paolucci, S. Roffia and G. W. H. Wurpel, Science, 2001, 291, 2124 CrossRef CAS PubMed; (b) J. Wu, K. C.-F. Leung, D. Benítez, J.-Y. Han, S. J. Cantrill, L. Fang and J. F. Stoddart, Angew. Chem., Int. Ed., 2008, 39, 7470 CrossRef PubMed; (c) S.-Y. Hsueh, C.-T. Kuo, T.-W. Lu, C.-C. Lai, Y.-H. Liu, H.-F. Hsu, S.-M. Peng, C.-H. Chen and S.-H. Chiu, Angew. Chem., Int. Ed., 2010, 48, 9170 CrossRef PubMed; (d) Z. Zhang, C. Han, G. Yu and F. Huang, Chem. Sci., 2012, 3, 3026 RSC; (e) Z. Meng, J.-F. Xiang and C.-F. Chen, Chem. Sci., 2014, 5, 1520 RSC; (f) H. Li, X. Li, A. Hans and D.-H. Qu, Org. Lett., 2014, 16, 4940 CrossRef CAS PubMed.
- (a) M.-V. Martínez-Díaz, N. Spencer and J. F. Stoddart, Angew. Chem., Int. Ed., 1997, 36, 1904 CrossRef; (b) P. R. Ashton, R. Ballardini, V. Balzani, I. Baxter, A. Credi, M. C. T. Fyfe, M. T. Gandolfi, M. Gómez-López, M.-V. Martínez-Díaz, A. Piersanti, N. Spencer, J. F. Stoddart, M. Venturi, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1998, 120, 11932 CrossRef CAS.
- Since the Bpym2+ unit is very sensitive to the nucleophilic base, tributylamine (Bu3N) was chosen as the ideal base, which was strong enough to deprotonate the NH2+ center and behaved as a relatively weak (or unreactive) nucleophile towards Bpym2+ units. See ref. 16 and I. Eléna, C. Alberto, B. Vincenzo, S. Francesco and M. Luigi, Chem.–Eur. J., 1999, 5, 984 CrossRef.
- (a) M. Roza-Fernández, M. J. Valencia-González and M. E. Díaz-Gacría, Anal. Chem., 1997, 69, 2406 CrossRef PubMed; (b) J. A. M. Pulgarín, A. A. Molina and P. F. López, Analyst, 1997, 122, 253 RSC; (c) A. S. Carretero, C. C. Blanco, B. C. Díaz and A. F. Gutiérrez, Analyst, 1998, 123, 1069 RSC; (d) Y.-T. Wang and W.-J. Jin, Spectrochim. Acta, Part A, 2008, 70, 871 CrossRef PubMed.
- (a) H. Onagi and J. Rebek, Jr., Chem. Commun., 2005, 36, 4604 RSC; (b) W.-Y. Wong, K. C.-F. Leung and J. F. Stoddart, Org. Biomol. Chem., 2010, 8, 2332 RSC; (c) H. Zhang, B. Zhou, H. Li, D.-H. Qu and H. Tian, J. Org. Chem., 2013, 78, 2091 CrossRef CAS PubMed; (d) H. Zhang, J. Hu and D.-H. Qu, Org. Lett., 2012, 14, 2334 CrossRef CAS PubMed; (e) C.-S. Kwan, A. S. C. Chan and K. C.-F. Leung, Org. Lett., 2016, 18, 976 CrossRef CAS PubMed.
- (a) P. R. Ashton, R. Ballardini, V. Balzani, M. Bělohradský, M. T. Gandol, D. Philp, L. Prodi, F. M. Raymo, M. V. Reddington, N. Spencer, J. F. Stoddart, M. Venturi and D. J. Williams, J. Am. Chem. Soc., 1996, 118, 4931 CrossRef CAS; (b) M. Fathalla, J. C. Barnes, R. M. Young, K. J. Hartlieb, S. M. Dyar, S. W. Eaton, A. A. Sarjeant, D. T. Co, M. R. Wasielewski and J. F. Stoddart, Chem.–Eur. J., 2014, 20, 14690 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, general procedures and synthesis; additional characterization data of compounds, control experiments and corresponding discussions. See DOI: 10.1039/c6sc00769d |
| This journal is © The Royal Society of Chemistry 2016 |