DOI:
10.1039/C6SC00771F
(Edge Article)
Chem. Sci., 2016,
7, 4867-4874
Flavoenzyme CrmK-mediated substrate recycling in caerulomycin biosynthesis†
Received
19th February 2016
, Accepted 11th April 2016
First published on 13th April 2016
Abstract
Substrate salvage or recycling is common and important for primary metabolism in cells but is rare in secondary metabolism. Herein we report flavoenzyme CrmK-mediated shunt product recycling in the biosynthesis of caerulomycin A (CRM A 1), a 2,2′-bipyridine-containing natural product that is under development as a potent novel immunosuppressive agent. We demonstrated that the alcohol oxidase CrmK, belonging to the family of bicovalent FAD-binding flavoproteins, catalyzed the conversion of an alcohol into a carboxylate via an aldehyde. The CrmK-mediated reactions were not en route to 1 biosynthesis but played an unexpectedly important role by recycling shunt products back to the main pathway of 1. Crystal structures and site-directed mutagenesis studies uncovered key residues for FAD-binding, substrate binding and catalytic activities, enabling the proposal for the CrmK catalytic mechanism. This study provides the first biochemical and structural evidence for flavoenzyme-mediated substrate recycling in secondary metabolism.
Introduction
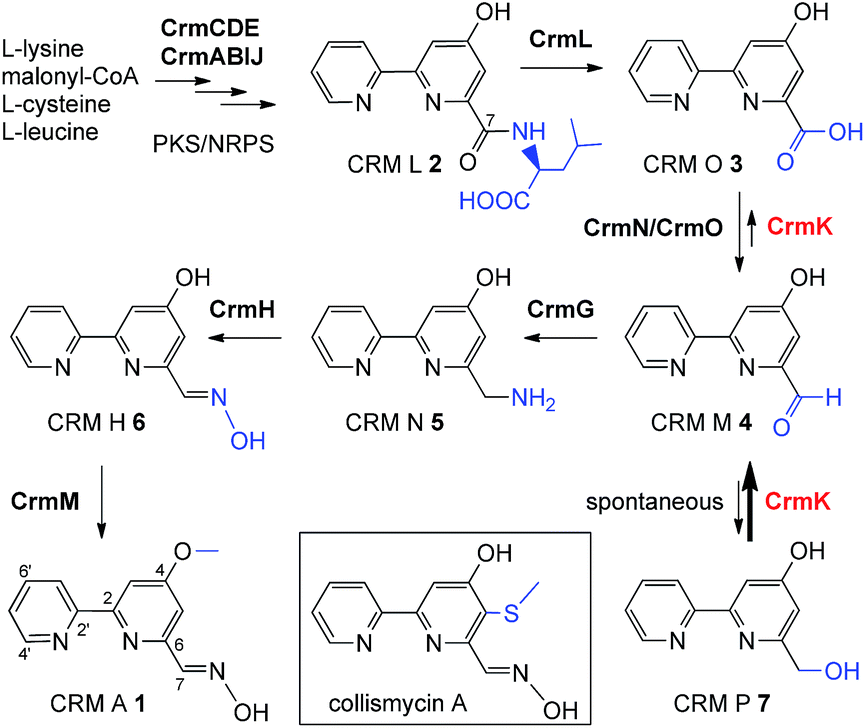
The substrate salvage or recycling pathway is important and common in primary metabolism, such as for nucleosides,1 uracil,2 coenzyme B1,3 tetrahydrobiopterin,4 methionine,5 thiamine,6 and sphingolipids.7,8 The existence of a salvage pathway guarantees the preservation of cell integrity,9 and allows the recycling of metabolites derived from normal cellular catabolism, in compensation to the general de novo biosynthetic pathway.10 However, there are rare examples of the salvage pathway in secondary metabolism.11–13 In this study, we provided evidence of flavoenzyme CrmK-mediated substrate recycling in caerulomycin biosynthesis. Caerulomycins (CRMs) feature a 2,2′-bipyridine core structure and are produced by terrestrial Streptomyces caeruleus14 and marine-derived Actinoalloteichus cyanogriseus WH1-2216-6.15,16 CRMs are of considerable medicinal interest due to their diverse bioactivities.17 Particularly, CRM A 1 (Fig. 1) has shown remarkable promise in immunosuppression of both T- and B-cells, being ten times more potent than the known immunosuppressant cyclosporin A,18 and is currently under development as a novel immunosuppressive agent for the treatment of autoimmunity by Nostrum Pharmaceuticals.19,20
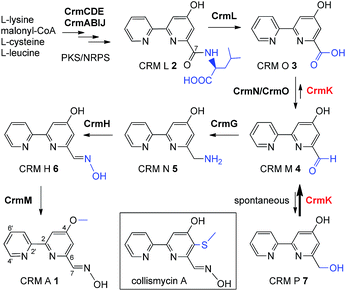 |
| | Fig. 1 The proposed biosynthetic pathway for CRM A 1 and chemical structure for collismycin A. | |
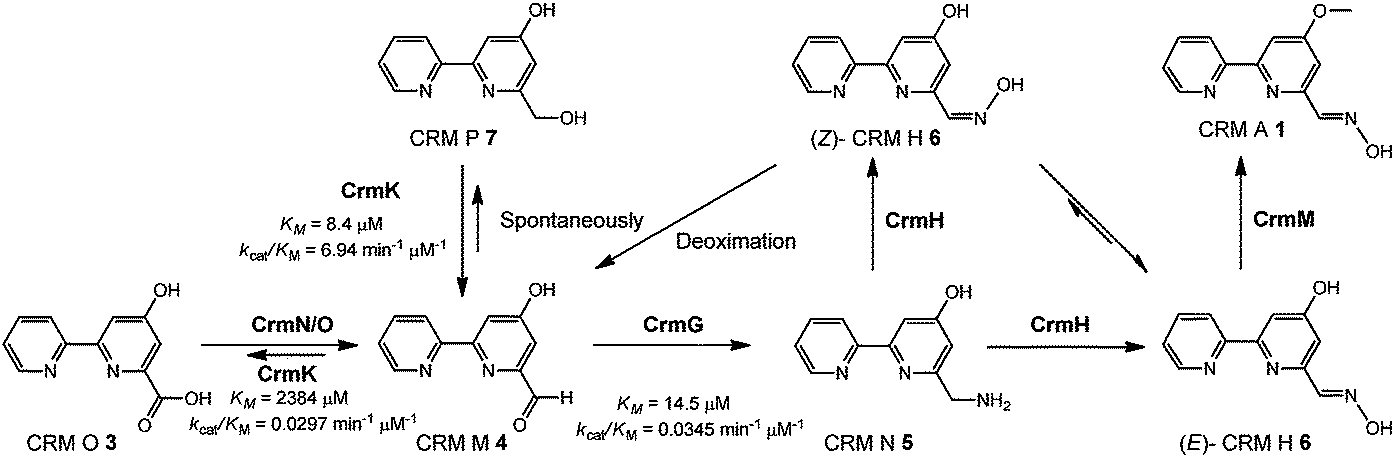
Recently, biosynthetic studies on CRM A 1 and its closely-related analogue collismycin A (Fig. 1) have revealed a common origin of a hybrid polyketide synthase (PKS)/non-ribosomal peptide synthetase (NRPS) system for bipyridine core formation with unclear mechanisms.21–24 The tailoring steps for collismycin biosynthesis have been demonstrated by characterizing a number of collismycin analogues from gene-disrupted mutants.23–25 In parallel, the CRM A 1 biosynthetic pathway has been well established by biochemically elucidating four biosynthetic enzymes, including an unusual amidohydrolase CrmL,21 an aminotransferase CrmG,26 a two-component monooxygenase CrmH,27 and a methyltransferase CrmM (Fig. 1).28 Hence, the conversion of CRM O 3 to CRM M 4 (Fig. 1) remains the only step en route to CRM A 1 without experimental evidence, and was speculated to be catalyzed by a pair of GirC/GirD-like29 dehydrogenase components CrmN/CrmO.21
Interestingly, a flavoenzyme-encoding gene crmK was found to flank the 1 biosynthetic gene cluster, and has been demonstrated to be inessential for CRM A 1 biosynthesis, given the production of 1 in the ΔcrmK mutant.21 In this study, our biochemical characterizations demonstrate that CrmK plays an unexpectedly important role in 1 biosynthesis by catalyzing the conversion of an alcohol shunt product 7 to an aldehyde 4 and then to a carboxylate 3, thus recycling or salvaging the shunt product 7 into the main pathway of 1 (Fig. 1). Moreover, the crystal structures of CrmK in apo form and in complex with the substrate CRM P 7 and site-directed mutagenesis studies provide insights into the catalytic mechanisms and the molecular basis of CrmK in shunt product recycling.
Results and discussion
In vivo characterization of the crmK gene
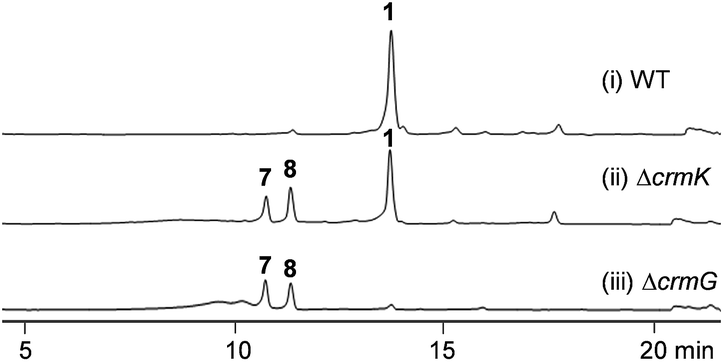
CrmK belongs to a growing family of alcohol oxidase flavoproteins, which share a conserved bicovalent flavin adenine dinucleotide (FAD) binding domain and function to convert an alcohol to an aldehyde or a ketone.30,31 CrmK resembles a number of flavin-dependent oxidoreductases in natural product biosynthesis, such as TrdL32 (or TamL33), Dbv29,34 SpnJ,35 AknOx,36 GilR,37,38 and Pac11.39 These flavoenzymes contain invariant histidine and cysteine residues that are responsible for bicovalent binding of the cofactor FAD (Fig. S1†). HPLC analysis showed that the ΔcrmK mutant accumulated CRM P 7 and CRM F 8 (Fig. 2 and 3A), two products observed in the ΔcrmG mutant,26 along with the major product 1. The yield of 1 in the ΔcrmK mutant was 52% of the yield of 1 in the wild type (WT) strain. In contrast, the total yield of CRMs (1, 7 and 8) was comparable (91%) to the yield of 1 in the WT (Fig. 2). Given that the ΔcrmK mutant could produce two alcohol-containing shunt products CRM P 7 and CRM F 8, we reason that CrmK might act as an alcohol oxidase to convert the alcohols CRM P 7 and CRM F 8 to their aldehyde derivatives (Fig. 3A).
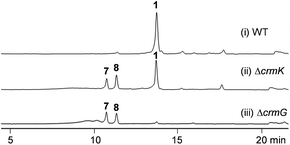 |
| | Fig. 2 HPLC analysis of metabolite profiles of (i) wild type strain (WT), (ii) the ΔcrmK mutant, and (iii) the ΔcrmG mutant with UV detection at 313 nm. | |
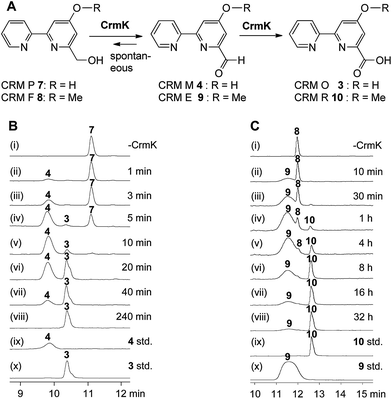 |
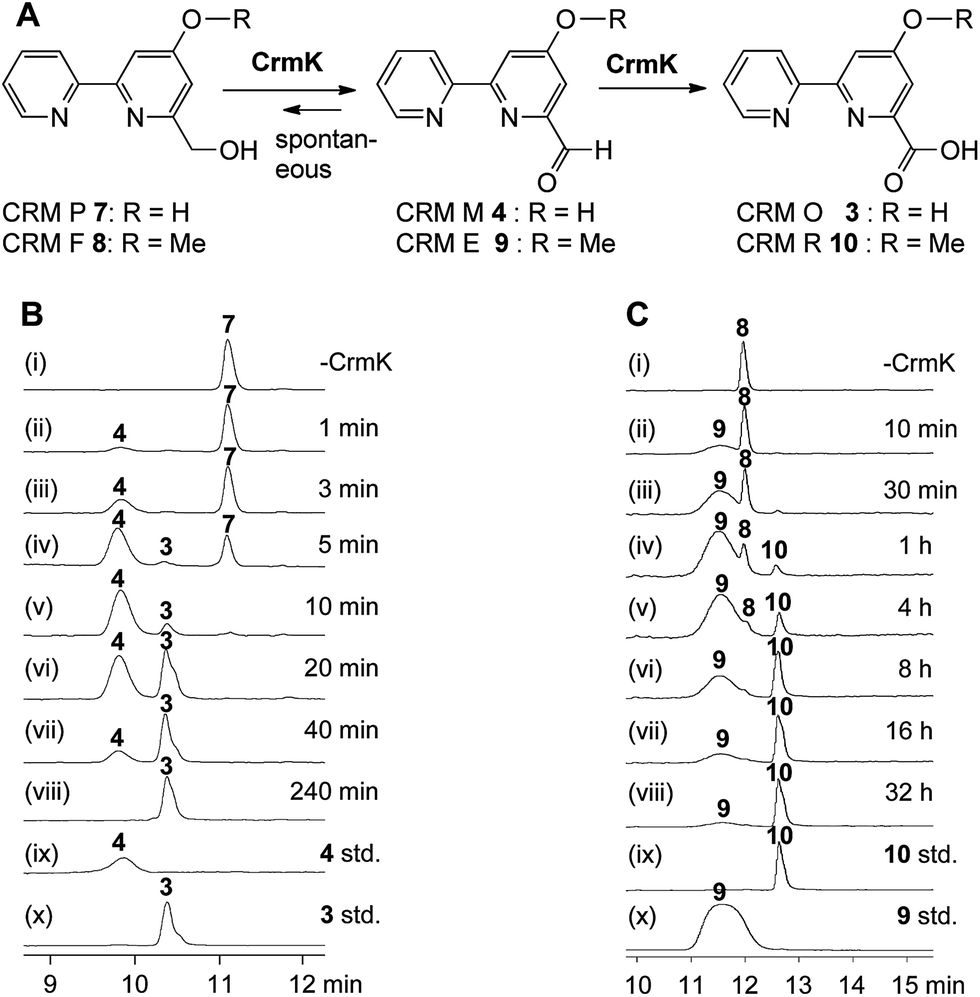
| | Fig. 3
In vitro characterization of CrmK-catalyzed reactions. (A) Scheme for CrmK-catalyzed reactions. (B) HPLC analysis of a time course assay of CrmK with CRM P 7. (i) No CrmK as a control; (ii) 1 min; (iii) 3 min; (iv) 5 min; (v) 10 min; (vi) 20 min; (vii) 40 min; (viii) 240 min; (ix) standard 4; (x) standard 3. (C) HPLC analysis of a time course assay of CrmK with CRM F 8. (i) No CrmK as a control; (ii) 10 min; (iii) 30 min; (iv) 1 h; (v) 4 h; (vi) 8 h; (vii) 16 h; (viii) 32 h; (ix) standard 10; (x) standard 9. A standard CrmK assay was performed in Tris–HCl buffer (50 mM, pH 8.0) comprised of 200 μM 7 and 240 nM CrmK (or 200 μM 8 and 2.4 μM CrmK) at 28 °C. | |
Biochemical characterization of CrmK
To probe the exact function of CrmK in vitro, the crmK gene was overexpressed in E. coli to yield a soluble N-(His)6-tagged protein (Fig. S2†). Although the purified CrmK protein was yellow in color, no flavin cofactors could be released by simple heat denaturation, consistent with the predicted covalent binding of flavin cofactors. Subsequently, a time course of in vitro CrmK assays revealed that 7 was quickly converted to 4, and 4 began to be slowly converted to 3 after most of 7 was consumed (Fig. 3B). The identities of enzymatic products 3 and 4 were confirmed by LC-HRESIMS (Fig. S3†) and comparison with the standards (Fig. 3B). In a similar manner to the turnover of 7 to 3, CrmK was shown to be capable of catalyzing the conversion of 8 to 10via the intermediate 9 (Fig. 3C), which was confirmed by comparison with the standard 9 (Fig. 3C).26 The product 10 was isolated and purified from 100 mL of a CrmK enzyme reaction, and was confirmed by HRESIMS (C12H10N2O3, m/z [M + H]+ 231.0769) and NMR analyses (Fig. S4†). These experiments demonstrated that CrmK was a dedicated enzyme in CRM A 1 biosynthesis that catalyzed two sequential oxidizing steps to convert an alcohol to a carboxyl through an aldehyde intermediate, and thus to recycle an alcohol shunt product back to the main pathway of CRM A 1 (Fig. 1).
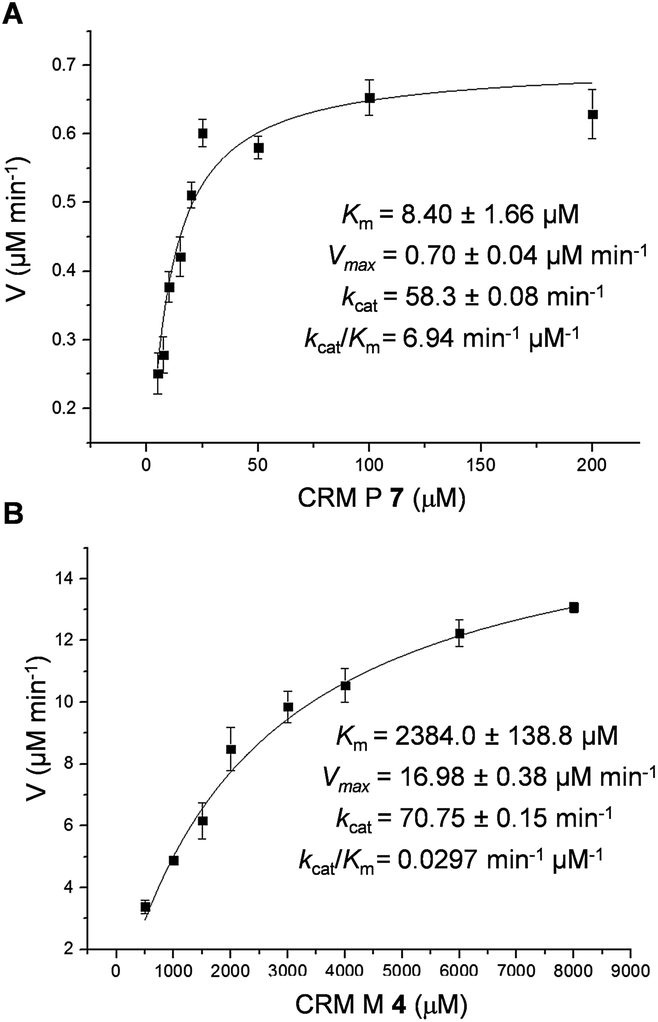
To better understand the CrmK-catalyzed two-step oxidative reactions, kinetic parameters were determined by using the alcohol 7 and the aldehyde 4 as substrates. CrmK displayed much higher binding affinity toward 7 (Km 8.4 μM, Fig. 4A) than 4 (Km 2384.0 μM, Fig. 4B). Comparison of the kcat/Km values revealed that CrmK was almost 230-fold more efficient in catalyzing the first oxidation step from an alcohol 7 to an aldehyde 4 (kcat/Km 6.94 μM−1 min−1) than in catalyzing the second oxidation step from an aldehyde 4 to a carboxylate 3 (kcat/Km 0.0297 μM−1 min−1).
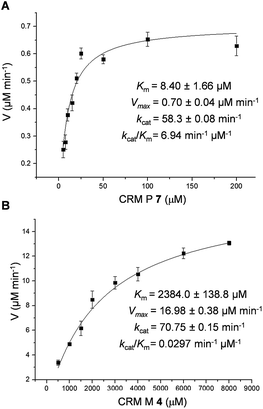 |
| | Fig. 4 Determination of CrmK kinetic parameters for substrate 7 with 5–200 μM 7 (A) and for substrate 4 with 500–8000 μM 4 (B). | |
Overall crystal structure of CrmK
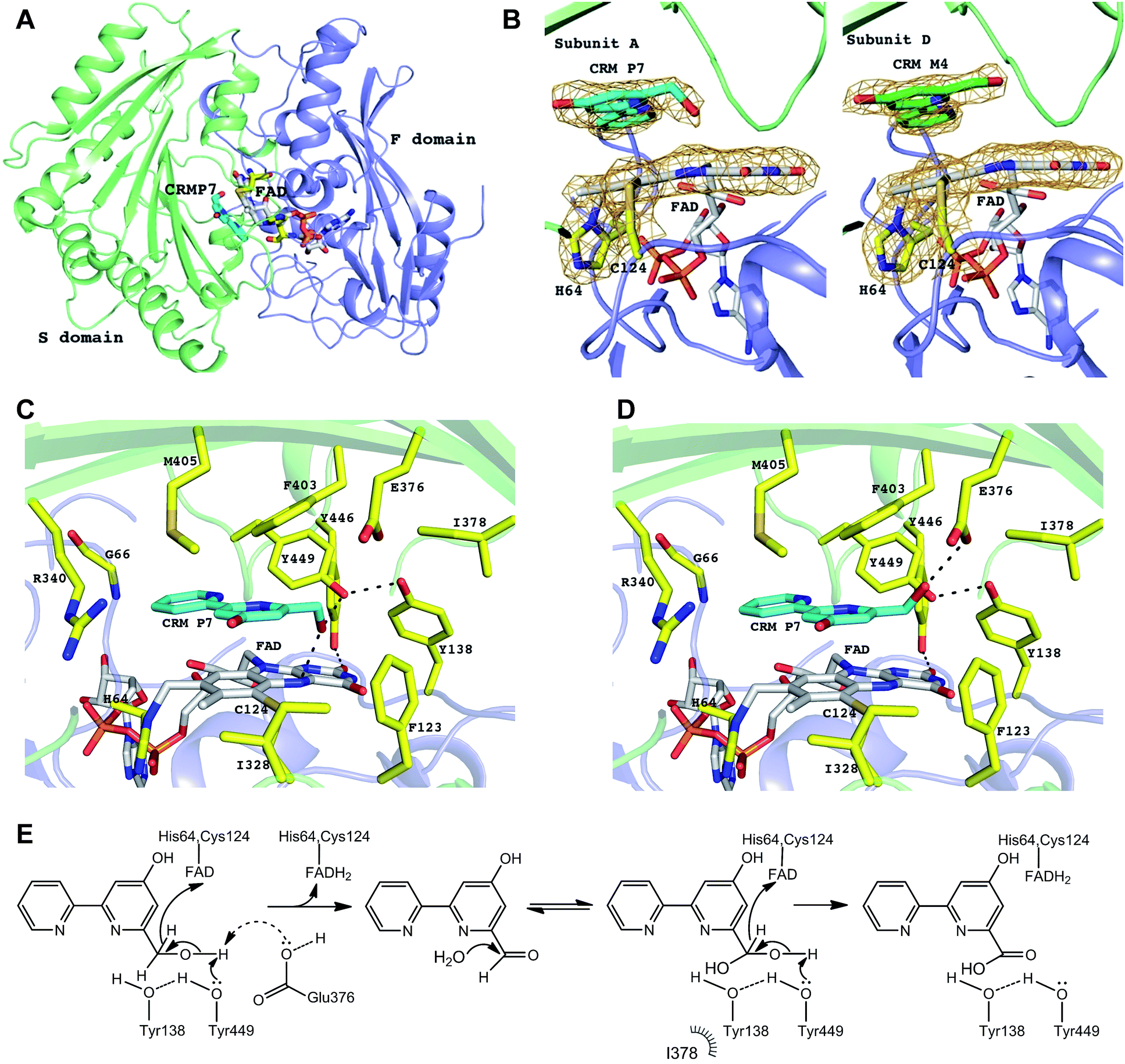
To better understand the enzymatic reaction and substrate binding of CrmK, the crystal structures of CrmK alone and in complex with the substrate CRM P 7 were resolved at 1.84 Å and 2.15 Å (Table 1), respectively. In both crystals, each asymmetric unit contains four subunits of CrmK that form two stable dimers with a dimerization interface of 1695 Å2, comparable to that in the TamL dimer (1680 Å2)33 and more than that in the Dbv29 dimer (∼1300 Å2).34 The closest structural homologs of CrmK include TamL,33 GilR,38 AknOx,36 and Dbv29.34 Each CrmK subunit contains two domains (Fig. 5A): the cofactor FAD binding domain (F) and the substrate binding domain (S).
Table 1 X-ray data collection and refinement statistics (values in parentheses are for the highest-resolution shell)
| Structure |
CrmK-FAD |
CrmK-FAD-CRM P 7 |
| Space group |
P1 |
P1 |
|
a, b, c (Å) |
63.6, 95.7, 98.4 |
63.1, 95.2, 98.0 |
|
α, β, γ (°) |
95.2, 97.0, 104.4 |
94.7, 96.6, 105.2 |
| Wavelength (Å) |
0.9795 |
0.9795 |
| Resolution (Å) |
48.7–1.84 (1.94–1.84) |
45.6–2.15 (2.27–2.15) |
| Observed hkl |
729![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 153 (103972) 153 (103972) |
278363 (40448) |
| Unique hkl |
184445 (26576) |
114678 (16652) |
| Redundancy |
4.0 (3.9) |
2.4 (2.4) |
| Completeness (%) |
95.9 (94.7) |
97.0 (96.1) |
|
R
merge
|
0.118 (0.828) |
0.162 (0.699) |
| CC1/2 |
0.993 (0.498) |
0.950 (0.400) |
|
I/(σI) |
7.5 (2.1) |
5.0 (2.8) |
| Wilson B (Å2) |
22.5 |
23.7 |
|
R
work (# hkl) |
0.161 (175148) |
0.168 (108921) |
|
R
free (# hkl) |
0.194 (9296) |
0.213 (5754) |
|
|
B-factors (# atoms)
|
| Protein |
29.8 (15629) |
35.0 (15577) |
| Solvent |
37.5 (1386) |
37.3 (600) |
| Ligand (FAD) |
19.9 (212) |
23.8 (212) |
| Ligand (7 or 4) |
— |
47.1 (60) |
|
|
Ramachandran
|
| Allowed (%) |
99.9 |
99.9 |
| Generous (%) |
0.1 |
0.1 |
| Disallowed (%) |
0 |
0 |
|
|
rmsd
|
| Bonds (Å) |
0.014 |
0.015 |
| Angles (°) |
1.67 |
1.71 |
| PDB code |
5I1V
|
5I1W
|
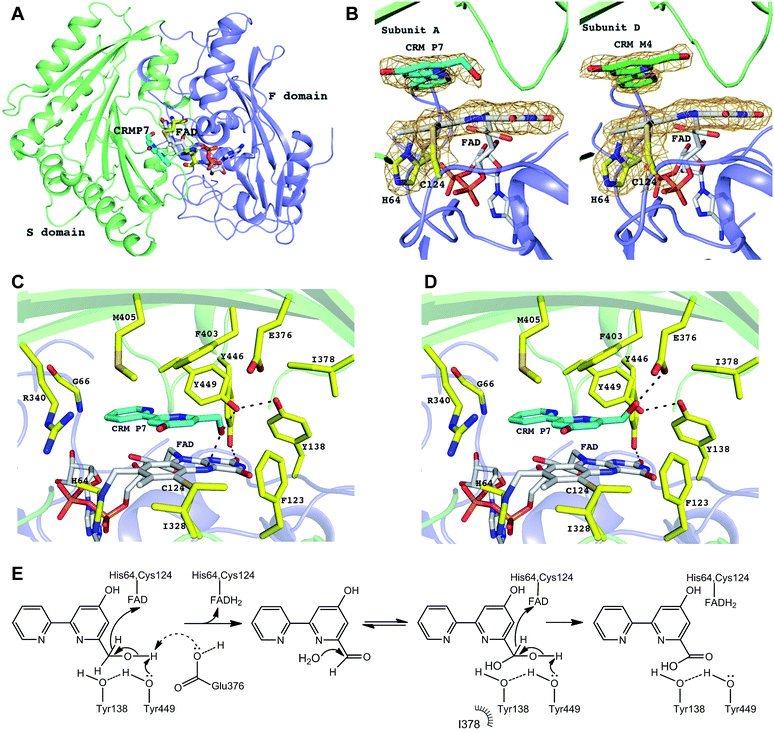 |
| | Fig. 5 CrmK structures and proposed mechanism. (A) Cartoon representation of CrmK subunit. The F domain (colored in blue) and S domain (colored in green) are responsible for the binding of FAD and CRM P 7, respectively. FAD and CRM P 7 molecules are shown in stick mode. (B) FAD is bicovalently bound to CrmK via residues His64 and Cys124. In all the subunits of CrmK, the FAD isoalloxazine ring is not planar with a bend of ∼10°. The substrate molecule CRM P 7 (carbon in cyan) was observed in three (A, B, and C) out of four subunits whereas in the subunit D, the product CRM M 4 (carbon in green) was observed in the binding pocket. The electron density 2Fo-Fc map (colored in orange) is contoured at 1.2σ and, for clarity, it covers only His64, Cys124, the isoalloxazine ring, and the bound ligands. (C) The CrmK active site. The residues lining the active site pocket are shown in stick mode. The H-bonds are indicated by black dashed lines. No water molecules were found to mediate the interactions between CRM P 7 and the protein. (D) E376 as an alternative catalytic base (to abstract a proton from the substrate). The potential conformation of the C7 hydroxyl group of CRM P 7 shown here allows for the formation of an H-bond with E376. (E) The proposed catalytic mechanism of CrmK. | |
FAD-binding site
The electron density maps clearly showed that CrmK is a bicovalently flavinylated enzyme as the FAD isoalloxazine ring forms two covalent bonds via its 8α-methyl group and C6 carbon atom with the Nδ1 atom of His64 and the sulfur atom of Cys124 (Fig. 5B), respectively. Removing either of the covalent linkages by site-directed mutagenesis (e.g. H64A or C124A) leads to severely impaired mutants with only residual activities, and the double mutant (H64A/C124A) completely abolishes CrmK activities (Table 2). Notably, Y446 from the S domain forms a hydrogen bond with the O2 atom of the isoalloxazine moiety of FAD (Fig. 5C). Consequently, Y446F kept only ∼20% activity of wild type CrmK for both steps (Table 2).
Table 2 Relative catalytic efficiency of CrmK mutants compared to CrmK wild type toward CRM P 7 and CRM M 4
| CrmK mutants |
Substrates |
| CRM P 7 |
CRM M 4 |
| H64A |
0.1 |
0.04 |
| C124A |
0.05 |
0.01 |
| H64A/C124A |
0.0 |
0.0 |
| F123A |
0.16 |
0.67 |
| Y138F |
0.54 |
0.11 |
| I328A |
0.67 |
0.32 |
| R340A |
0.08 |
0.11 |
| E376A |
0.35 |
0.56 |
| I378A |
0.67 |
0.03 |
| F403A |
0.10 |
0.08 |
| F403E |
0.11 |
0.05 |
| M405A |
0.47 |
0.24 |
| Y446F |
0.23 |
0.18 |
| Y449F |
0.29 |
0.0 |
| Y449F/Y138F |
0.18 |
0.0 |
| Y449F/F403E |
0.0 |
0.0 |
Substrate-binding site
The substrate-bound structure of CrmK was solved by soaking CrmK crystals with CRM P 7 for a short time (∼3 min). The substrate sits at the active site and is sandwiched between the FAD isoalloxazine ring and the characteristic seven-stranded β-sheet in the S domain (Fig. 5B), which was unambiguously revealed by the electron density in three out of four subunits (A, B, and C) in the CrmK complex. Interestingly, the product CRM M 4 provides a better fit to the electron density in the subunit D (Fig. 5B). In addition to the aromatic stacking with FAD, the 2,2′-bipyridine ring of CRM P 7 forms a number of van der Waals contacts with the surrounding residues (Fig. 5C), such as F403, M405, V342, F123, I328, Y449, the main chain atoms of C65 and G66, and the aliphatic moiety of R340. The importance of these residues was confirmed by site-directed mutagenesis studies. The CrmK mutants F403A, F403E, F123A and R340A led to impaired or largely reduced enzymatic activities toward both oxidative steps (Table 2). Remarkably, the putative catalytic residue Y449 forms the only H-bond (2.6–2.9 Å) between the CRM P 7 and CrmK residues, contributing to anchoring the C7 hydroxyl group of the substrate (Fig. 5C). Interestingly, this hydroxyl group of CRM P 7 is also within the H-bonding distance (2.6–3.0 Å) of the N5 atom on the FAD isoalloxazine ring. The only major conformational change in CrmK upon the substrate binding (compared with the substrate-free form, Fig. S5†) is the swing of the Y449 side chain to enable H-bond (2.6–3.0 Å) formation with the Y138 hydroxyl group. Notably, no water molecules could be identified in the binding pocket to mediate additional H-bond interactions between the substrates and CrmK. Other residues that line the active site pocket also include E376 and I378, which are ∼4.3–5.5 Å away from the ligand (Fig. 5C).
Proposed catalytic mechanism of CrmK
Most characterized alcohol oxidases catalyze a two-electron oxidation to convert an alcohol to a ketone (e.g. TrdL/TamL,32,33 SpnJ,35 GilR,37,38 and Pac1139). However, a four-electron oxidation process was observed for CrmK catalysis, similar to the glycopeptide hexose oxidase Dbv29, to catalyze the conversion of an alcohol to a carboxylate.34,40 In Dbv29, this conversion was proposed to undergo an aldehyde intermediate (only trappable by chemical derivatization) and a diol species.34,40 In sharp contrast to the Dbv29-catalyzed reaction, an aldehyde 4 was readily available as a CrmK reaction product and was also trapped in one subunit of the CrmK–ligand complex structure, providing solid evidence for the first two-electron oxidation process to convert an alcohol to an aldehyde. Interestingly, the mass unit of a diol intermediate (m/z 219 [M + H]+) was readily detectable by simply dissolving 4 (m/z 201 [M + H]+) in water (Fig. S6†). When 4 was dissolved in 18O-labeled water, a series of mass units were observed to correspond to 18O-labeled aldehyde 4 (m/z 203 [M + H]+), single 18O-labeled diol species (m/z 221 [M + H]+), and double 18O-labeled diol species (m/z 223 [M + H]+). These observations indicated a spontaneous hydration/dehydration equilibrium in water for an interconversion between the aldehyde 4 and the diol species (Fig. S6†). When the CrmK reaction with 7 was performed in Tris–HCl buffer prepared with 18O-labeled water, the final product exhibited a mass unit (m/z 221 [M + H]+) for double 18O-labeled 3 (Fig. S6†). Cumulatively, these data unequivocally demonstrated that CrmK catalyzed two oxidative steps, to convert the alcohol 7 to the carboxylate 3via the aldehyde 4 (or its diol species), mechanistically analogous to the Dbv29 catalysis.34,40
The overall arrangement of active site residues in the CrmK structure in complex with CRM P 7 suggested that Y449, Y138 and E376 are important catalytic residues (Fig. 5C and D). Site-directed mutagenesis studies revealed that the mutants Y449F, Y138F and E376A impaired both oxidative steps (Table 2). Especially, Y449F completely lost the ability to catalyze the second step from an aldehyde to a carboxylate. The double mutant Y138F/Y449F maintains 18% activity for the first step. In sharp contrast, the double mutants of tyrosine residues (Y138/Y449 equivalents) in AknOx and Dbv29 completely lost their catalytic activity,34,36 and the single mutant Y448F in GilR led to a 400-fold reduction in activity.37,38 This unique feature of CrmK catalysis may be attributed to the presence of a second catalytic base E376, alternative to Y449. E376 is unique in CrmK and is not found in its homologous enzymes (Fig. S1†). The E376A mutant keeps only 35% of the CrmK activity, similar to that of Y449F (29%). Structurally, E376 could form an H-bond with the C7 hydroxyl group of 7 that adopts a different orientation from the one observed in the crystal structure (Fig. 5D). Interestingly, AknOx has a dual active site (Y450/Y144 and Y378) catalyzing two different dehydrogenations (Fig. S7†).36 Superimposition of CrmK and AknOx structures reveals that the position of the carboxylate of E376 in the active site of CrmK matches well with the position of the hydroxyl group of Y378 in AknOx (1.2–1.3 Å away, Fig. S8†), thus also indicating the potential role of E376 as a catalytic base in CrmK.
Taken together, a catalytic mechanism for CrmK was proposed (Fig. 5E). The first-step oxidation is likely initiated via proton abstraction from the C7 hydroxyl group by Y449 and/or E376 with concomitant hydride transfer from C7 to the N5 atom of FAD. The proton transfer by Y449 can be aided by Y138, given the H-bond between their hydroxyl groups, similar to the tyrosine pairs in TamL,33 AknOx,36 Dbv29,34 and GilR.38 In Dbv29 catalysis, two well-defined water molecules were found in the vicinity of diol groups that may facilitate the formation of the diol intermediate.34 In CrmK, the equivalent position is occupied by the F123 side chain, thus excluding the presence of water there. The lack of well-defined water molecules in the CrmK active site may suggest that the first half reaction product (CRM M 4) is released to the solvent to become a diol intermediate prior to re-binding to the active site. As such, CRM M 4 became an available product during CrmK catalytic cycles.
Substrate recycling in caerulomycin biosynthesis
In this study, the flavoenzyme CrmK was demonstrated to play an unexpectedly important role in refluxing shunt products back to the main pathway of CRM A 1 (Fig. 6). Our previous characterization of CrmG kinetic parameters revealed that the conversion of CRM M 4 to CRM N 5 (Km 14.5 μM, kcat/Km 0.0345 μM−1 min−1) was a rather slow process,26 owing to an intrinsically complex ping-pong mechanism for amino transfer.41 We have also previously shown that CrmH could catalyze the conversion of CRM N 5 into both E- and Z-configured CRM H 6, while the unstable Z-configured CRM H 6 could be spontaneously transformed into the aldehyde CRM M 4 (Fig. 6).27 These data indicated the possibility of the accumulation of the aldehyde CRM M 4 in cells. However, cells usually could not tolerate the accumulation of aldehydes because aldehydes are reactive electrophiles that form adducts with cellular protein thiols and amines and thus can cause toxicity.42 Thus, aldehydes are detoxified in cells by enzymatic or spontaneous oxidation to acids and/or reduction to alcohols.42 Consistent with this statement,42 alcohols 7 and 8 were accumulated in the ΔcrmG mutant (Fig. 2), and the expected aldehydes 4 and 9 were not found but were confirmed in vitro to be spontaneously converted to 7 and 8, putatively via a Cannizzaro reaction.26 Similar results were also observed in the aminotransferase gene disrupted mutants for studying collismycin A and borrelidin biosynthesis.24,43 It would be a great energy waste for cells to produce alcohols 7 and 8 in 1 biosynthesis. As such, the flavoenzyme CrmK was incorporated into the 1 biosynthetic pathway, to convert 7 and 8 to their corresponding aldehydes, which could be further processed by downstream enzymes to make the final product 1 (Fig. 6). Indeed, CRM P 7 was almost completely converted to 1 when fed to the ΔcrmA (a hybrid PKS/NRPS encoding gene) mutant, in which the production of CRMs was blocked (Fig. S9†).21 CRM F 8 was also converted to 1 by the ΔcrmA mutant, but in a ratio of less than 5% (Fig. S9†), indicating that the tailoring enzymes (e.g. CrmG26 and CrmH27) could not efficiently process the derivatives of 8. It should be also noted that, on the basis of kinetic parameters of CrmK and CrmG, the aldehyde CRM M 4 would be captured by the aminotransferase CrmG (the Km value of the aldehyde 4 for CrmG26 is 160-fold lower than that for the second step oxidation by CrmK). That means, under physiological conditions, the second step oxidation of 4 (Km 2384 μM) to 3 by CrmK (kcat/Km 0.0297 μM−1 min−1) is unlikely to happen (Fig. 6). By this way, nature has developed a smart way in 1 biosynthesis to avoid wasting energy by kinetically controlling the flow of substrates, both in recycling or salvaging shunt products (from 7 to 4), and in preventing over-oxidation (from 4 to 3). Elegantly, CrmK has evolved to have two independent catalytic residues (Y449 and E376) to enforce the recycling of 7 to 4. Similarly, in collismycin A biosynthesis, the ΔclmD2 (crmK homologue) mutant accumulated alcohol-containing products, indicating a similar recycling role of ClmD2 in the collismycin pathway.24 The homology model of ClmD2 constructed based on the structure of CrmK reveals the existence of a less bulky Val residue (V343, equivalent to I328 in CrmK) to better accommodate the extra –SCH3 group on collismycin DH, the natural substrate of ClmD2 (Fig. S10†).24
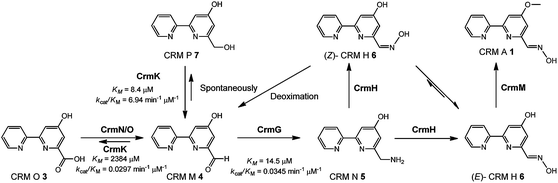 |
| | Fig. 6 A model for the flavoenzyme CrmK-mediated substrate recycling in CRM A 1 biosynthesis with fine-tuned kinetic control. | |
Natural product biosynthetic gene clusters (BGCs) are well known to often carry additional genes for resistance,44–46 transport,47 regulation,48,49 or carrying out natural protection group chemistry.21,50,51 Now the functional characterization of CrmK in caerulomycin biosynthesis adds a recycling mechanism that may operate in secondary metabolism. The majority of antibiotic BGCs carry genes with unknown or unassigned functions, and this work indicates that a salvage/recycling role could be a hypothesis to test for some of such genes. The existence of a “saving pathway” was reported for the biosynthesis of deoxysugars in neomycin11 and landomycins,12 putatively via a short sugar-activation pathway that was previously found for L-fucose.52 The dTDP-glucose synthase-like protein LanZ2 was speculated to be involved in this short sugar-activation pathway but further genetic and biochemical evidence was required to fully address its function.13 With the exponential growth in characterization of BGCs for secondary metabolites, it is expected that more and more enzymes with a salvaging/recycling function could be found in the future and explored for the yield improvement of secondary metabolites.
Conclusions
The substrate salvage pathway is common and plays an important role in controlling primary metabolism. However, rare examples of salvage pathways have been reported in secondary metabolism. In this study, we provided biochemical and structural insights into the flavoenzyme CrmK in CRM A 1 biosynthesis, and set an unusual example of a substrate recycling/salvaging pathway for secondary metabolites.
Acknowledgements
This work is supported in part by NSFC (31470204, 31290233, 31125001), CAS (XDA11030403), the Administration of Ocean and Fisheries of Guangdong Province (GD2012-D01-002), and by a NSERC discovery grant (436202) to R. Shi. We are grateful to the analytical facility at SCSIO for recording spectroscopic data. The X-ray crystallographic studies were performed at the Canadian Light Source, which is supported by the Canada Foundation for Innovation, Natural Sciences and Engineering Research Council of Canada, the University of Saskatchewan, the Government of Saskatchewan, Western Economic Diversification Canada, the National Research Council Canada, and the Canadian Institutes of Health Research. We thank the anonymous reviewers for their insightful comments.
References
- F. Gazzaniga, R. Stebbins, S. Z. Chang, M. A. McPeek and C. Brenner, Microbiol. Mol. Biol. Rev., 2009, 73, 529–541 CrossRef CAS PubMed.
- F. Arsene-Ploetze, H. Nicoloff, B. Kammerer, J. Martinussen and F. Bringel, J. Bacteriol., 2006, 188, 4777–4786 CrossRef CAS PubMed.
- J. D. Woodson and J. C. Escalante-Semerena, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 3591–3596 CrossRef CAS PubMed.
- C. A. Nichol, C. L. Lee, M. P. Edelstein, J. Y. Chao and D. S. Duch, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 1546–1550 CrossRef CAS.
- A. Sekowska and A. Danchin, BMC Microbiol., 2002, 2, 8 CrossRef PubMed.
- R. Karunakaran, K. Ebert, S. Harvey, M. E. Leonard, V. Ramachandran and P. S. Poole, J. Bacteriol., 2006, 188, 6661–6668 CrossRef CAS PubMed.
- G. Tettamanti, R. Bassi, P. Viani and L. Riboni, Biochimie, 2003, 85, 423–437 CrossRef CAS PubMed.
- K. Kitatani, J. Idkowiak-Baldys and Y. A. Hannun, Cell. Signalling, 2008, 20, 1010–1018 CrossRef CAS PubMed.
- L. Popolo, T. Gualtieri and E. Ragni, Med. Mycol., 2001, 39, 111–121 CrossRef CAS PubMed.
- A. Rongvaux, F. Andris, F. Van Gool and O. Leo, BioEssays, 2003, 25, 683–690 CrossRef CAS PubMed.
- S. K. Goda and M. Akhtar, J. Antibiot., 1992, 45, 984–994 CrossRef CAS PubMed.
- J. Rohr, S. E. Wohlert, C. Oelkers, A. Kirschning and M. Ries, Chem. Commun., 1997, 973–974 RSC.
- A. Luzhetskyy, M. Fedoryshyn, C. Durr, T. Taguchi, V. Novikov and A. Bechthold, Chem. Biol., 2005, 12, 725–729 CrossRef CAS PubMed.
- A. Funk and P. V. Divekar, Can. J. Microbiol., 1959, 5, 317–321 CrossRef CAS PubMed.
- P. Fu, S. Wang, K. Hong, X. Li, P. Liu, Y. Wang and W. Zhu, J. Nat. Prod., 2011, 74, 1751–1756 CrossRef CAS PubMed.
- P. Fu, P. Liu, X. Li, Y. Wang, S. Wang, K. Hong and W. Zhu, Org. Lett., 2011, 13, 5948–5951 CrossRef CAS PubMed.
- R. K. Gurram, W. Kujur, S. K. Maurya and J. N. Agrewala, J. Biol. Chem., 2014, 289, 17515–17528 CrossRef CAS PubMed.
-
A. K. Singla, J. N. Agrewala, R. M. Vohra and R. S. Jolly, US pat., 20070078167, 2007.
- A. K. Singla, R. K. Gurram, A. Chauhan, N. Khatri, R. M. Vohra, R. S. Jolly and J. N. Agrewala, Transplantation, 2014, 97, e57–59 CrossRef PubMed.
- S. Kaur, G. Srivastava, A. N. Sharma and R. S. Jolly, Br. J. Pharmacol., 2015, 172, 2286–2299 CrossRef CAS PubMed.
- Y. Zhu, P. Fu, Q. Lin, G. Zhang, H. Zhang, S. Li, J. Ju, W. Zhu and C. Zhang, Org. Lett., 2012, 14, 2666–2669 CrossRef CAS PubMed.
- X. Qu, B. Pang, Z. Zhang, M. Chen, Z. Wu, Q. Zhao, Q. Zhang, Y. Wang, Y. Liu and W. Liu, J. Am. Chem. Soc., 2012, 134, 9038–9041 CrossRef CAS PubMed.
- I. Garcia, N. M. Vior, A. F. Brana, J. Gonzalez-Sabin, J. Rohr, F. Moris, C. Mendez and J. A. Salas, Chem. Biol., 2012, 19, 399–413 CrossRef CAS PubMed.
- I. Garcia, N. M. Vior, J. Gonzalez-Sabin, A. F. Brana, J. Rohr, F. Moris, C. Mendez and J. A. Salas, Chem. Biol., 2013, 20, 1022–1032 CrossRef CAS PubMed.
- C. Sialer, I. Garcia, J. Gonzalez-Sabin, A. F. Brana, C. Mendez, F. Moris and J. A. Salas, Bioorg. Med. Chem. Lett., 2013, 23, 5707–5709 CrossRef CAS PubMed.
- Y. Zhu, J. Xu, X. Mei, Z. Feng, L. Zhang, Q. Zhang, G. Zhang, W. Zhu, J. Liu and C. Zhang, ACS Chem. Biol., 2016, 11, 943–952 CrossRef CAS PubMed.
- Y. Zhu, Q. Zhang, S. Li, Q. Lin, P. Fu, G. Zhang, H. Zhang, R. Shi, W. Zhu and C. Zhang, J. Am. Chem. Soc., 2013, 135, 18750–18753 CrossRef CAS PubMed.
- P. Fu, Y. Zhu, X. Mei, Y. Wang, H. Jia, C. Zhang and W. Zhu, Org. Lett., 2014, 16, 4264–4267 CrossRef CAS PubMed.
- H. Suzuki, Y. Ohnishi and S. Horinouchi, J. Antibiot., 2007, 60, 380–387 CrossRef CAS PubMed.
- C. T. Walsh and T. A. Wencewicz, Nat. Prod. Rep., 2013, 30, 175–200 RSC.
- N. G. Leferink, D. P. Heuts, M. W. Fraaije and W. J. van Berkel, Arch. Biochem. Biophys., 2008, 474, 292–301 CrossRef CAS PubMed.
- X. Mo, H. Huang, J. Ma, Z. Wang, B. Wang, S. Zhang, C. Zhang and J. Ju, Org. Lett., 2011, 13, 2212–2215 CrossRef CAS PubMed.
- J. C. Carlson, S. Y. Li, S. S. Gunatilleke, Y. Anzai, D. A. Burr, L. M. Podust and D. H. Sherman, Nat. Chem., 2011, 3, 628–633 CrossRef CAS PubMed.
- Y. C. Liu, Y. S. Li, S. Y. Lyu, L. J. Hsu, Y. H. Chen, Y. T. Huang, H. C. Chan, C. J. Huang, G. H. Chen, C. C. Chou, M. D. Tsai and T. L. Li, Nat. Chem. Biol., 2011, 7, 304–309 CrossRef CAS PubMed.
- H. J. Kim, R. Pongdee, Q. Wu, L. Hong and H. W. Liu, J. Am. Chem. Soc., 2007, 129, 14582–14584 CrossRef CAS PubMed.
- I. Alexeev, A. Sultana, P. Mantsala, J. Niemi and G. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6170–6175 CrossRef CAS PubMed.
- M. K. Kharel, P. Pahari, H. Lian and J. Rohr, ChemBioChem, 2009, 10, 1305–1308 CrossRef CAS PubMed.
- N. Noinaj, M. A. Bosserman, M. A. Schickli, G. Piszczek, M. K. Kharel, P. Pahari, S. K. Buchanan and J. Rohr, J. Biol. Chem., 2011, 286, 23533–23543 CrossRef CAS PubMed.
- A. E. Ragab, S. Gruschow, D. R. Tromans and R. J. Goss, J. Am. Chem. Soc., 2011, 133, 15288–15291 CrossRef CAS PubMed.
- Y. S. Li, J. Y. Ho, C. C. Huang, S. Y. Lyu, C. Y. Lee, Y. T. Huang, C. J. Wu, H. C. Chan, C. J. Huang, N. S. Hsu, M. D. Tsai and T. L. Li, J. Am. Chem. Soc., 2007, 129, 13384–13385 CrossRef CAS PubMed.
- D. Schiroli and A. Peracchi, Biochim. Biophys. Acta, Proteins Proteomics, 2015, 1854, 1200–1211 CrossRef CAS PubMed.
- P. J. O'Brien, A. G. Siraki and N. Shangari, Crit. Rev. Toxicol., 2005, 35, 609–662 CrossRef.
- C. Olano, S. J. Moss, A. F. Brana, R. M. Sheridan, V. Math, A. J. Weston, C. Mendez, P. F. Leadlay, B. Wilkinson and J. A. Salas, Mol. Microbiol., 2004, 52, 1745–1756 CrossRef CAS PubMed.
- J. M. Blair, M. A. Webber, A. J. Baylay, D. O. Ogbolu and L. J. Piddock, Nat. Rev. Microbiol., 2014, 13, 42–51 CrossRef PubMed.
- J. B. Biggins, K. C. Onwueme and J. S. Thorson, Science, 2003, 301, 1537–1541 CrossRef CAS PubMed.
- H. Xu, W. Huang, Q. L. He, Z. X. Zhao, F. Zhang, R. Wang, J. Kang and G. L. Tang, Angew. Chem., Int. Ed., 2012, 51, 10532–10536 CrossRef CAS PubMed.
- J. F. Martin, J. Casqueiro and P. Liras, Curr. Opin. Microbiol., 2005, 8, 282–293 CrossRef CAS PubMed.
- J. F. Martin and P. Liras, Curr. Opin. Microbiol., 2010, 13, 263–273 CrossRef CAS PubMed.
- A. A. Brakhage, Nat. Rev. Microbiol., 2012, 11, 21–32 CrossRef PubMed.
- Y. Shinohara, F. Kudo and T. Eguchi, J. Am. Chem. Soc., 2011, 133, 18134–18137 CrossRef CAS PubMed.
- H. Hong, M. Samborskyy, F. Lindner and P. F. Leadlay, Angew. Chem., Int. Ed., 2016, 55, 1118–1123 CrossRef CAS PubMed.
- M. L. Reitman, I. S. Trowbridge and S. Kornfeld, J. Biol. Chem., 1980, 255, 9900–9906 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: The experimental procedures, materials, and characterization of compounds. See DOI: 10.1039/c6sc00771f |
| ‡ Y. Zhu and M.-È. Picard contributed equally to this work. |
| § Equal Correspondence to C. Z. for biochemistry or R. S. for structural biology. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Weiming
Zhu
c,
Rong
Shi§
*b and
Changsheng
Zhang§
*a
b,
Weiming
Zhu
c,
Rong
Shi§
*b and
Changsheng
Zhang§
*a