
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Triplet energy migration-based photon upconversion by amphiphilic molecular assemblies in aerated water†
Hironori
Kouno
a,
Taku
Ogawa
a,
Shogo
Amemori
a,
Prasenjit
Mahato
a,
Nobuhiro
Yanai
*ab and
Nobuo
Kimizuka
*a
aDepartment of Chemistry and Biochemistry, Graduate School of Engineering, Center for Molecular Systems (CMS), Kyushu University, 744 Moto-oka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: yanai@mail.cstm.kyushu-u.ac.jp; n-kimi@mail.cstm.kyushu-u.ac.jp
bPRESTO, JST, Honcho 4-1-8, Kawaguchi, Saitama 332-0012, Japan
First published on 18th April 2016
Abstract
A molecular self-assembly approach is developed to resolve an outstanding issue in triplet energy migration-based photon upconversion (TEM-UC), that is, air-stable TEM-UC in water. Amphiphilic cationic acceptor (emitter) molecules self-assemble in water via hydrophobic and hydrogen bonding interactions, with which anionic donor (sensitizer) molecules are integrated through electrostatic interactions. Triplet energy is quantitatively transferred from the excited donor to the acceptor, which is followed by effective triplet energy migration among the pre-organized acceptors. It leads to TTA and concomitant UC emission in water. The dense acceptor chromophore arrays with extended hydrogen bonding networks show efficient barrier properties against molecular oxygen, as demonstrated by the stable UC emission even in air-saturated water.
Introduction
Photon upconversion (UC), converting lower-energy photons to higher-energy photons, has attracted considerable attention because of its wide variety of applications ranging from renewable energy production to bioimaging and photodynamic therapy.1–5 Conventional UC mechanisms such as two-photon absorption and multistep excitation of lanthanides, however, require strong excitation light intensity (>W cm−2), thus limiting their applications.6,7 On the other hand, triplet–triplet annihilation (TTA)-based UC with multi-component triplet donor (D, sensitizer) and acceptor (A, emitter) system can operate under a non-coherent, low intensity light source.1,8–15 As summarized in Fig. 1, this mechanism starts with the generation of donor triplets (T1,D) by intersystem crossing (ISC) from the photogenerated singlet state (S1,D), and the succeeding D-to-A triplet–triplet energy transfer (TTET) forms optically dark, metastable acceptor triplets (T1,A). The subsequent diffusion and collision of two excited acceptor triplets generate a higher energy excited singlet state (S1,A) through TTA, from which the upconverted delayed fluorescence is emitted. The TTET and TTA processes occur via an electron exchange mechanism (Dexter energy transfer), which requires the involved molecules to come close within the distance of 1 nm.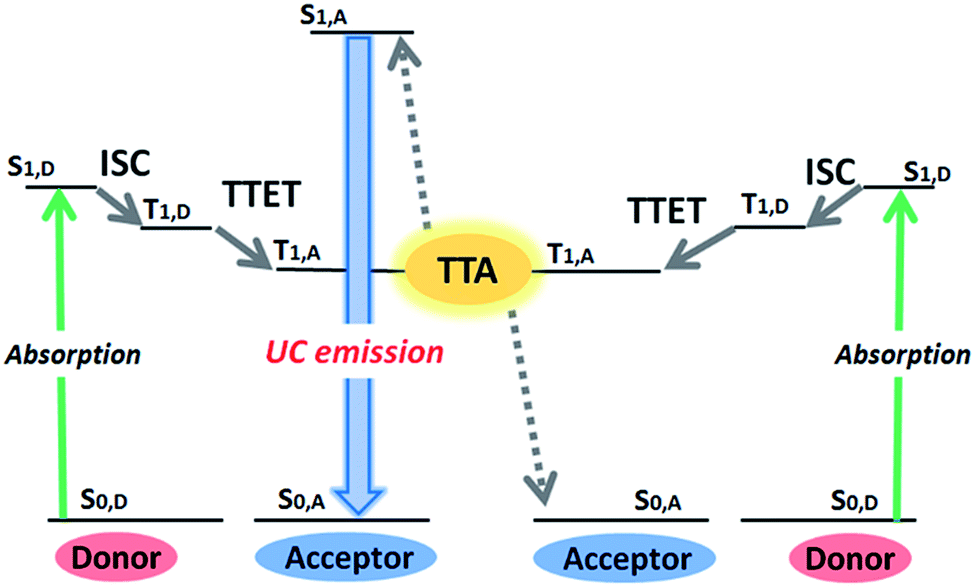 | ||
| Fig. 1 An outline of the TTA-UC process, showing the energy levels involved in the TTA-UC (S = singlet, T = triplet). The TTA-UC utilizes a pair of donor (sensitizer) with high intersystem crossing (ISC) efficiency and acceptor (emitter) with a high fluorescence quantum yield. Green and blue arrows indicate the absorption and emission processes, respectively. TTET: triplet–triplet energy transfer, TTA: triplet–triplet annihilation. | ||
In spite of the growing demand for photocatalytic and biological applications, most studies on TTA-based UC involve investigations in deaerated organic solvents, and examples in air-saturated aqueous phase have been limited because of the massive quenching of excited triplet states by dissolved molecular oxygen. Previous reports have shown that this oxygen quenching can be suppressed by employing specific solid polymers or viscous liquids as matrices to reduce the oxygen concentration.16–21 For example, Kim and coworkers have reported photocatalytic reactions in aerated water by encapsulating UC chromophores in viscous hexadecane/polyisobutylene mixture.17,19,21 Another strategy is to utilize viscous matrices with singlet oxygen scavenging ability.22–26 Li et al. observed UC emission in aerated water by loading UC dyes in nanocapsules containing reductive linoic acid and oleic acid, and applied their nanomaterials to in vivo bioimaging.22 While these approaches allow for air-stable UC emission with reasonably high efficiency, the limited diffusion of large dye molecules in these viscous matrices is forecast to cause potential issues for further advancements.
An alternate strategy for aqueous UC is to take advantage of fast triplet energy migration (TEM) in condensed chromophore assemblies.12,15,27–32 In essence, TEM-UC is the chemistry challenge of how to pre-organize donor and acceptor molecules for effective donor-to-acceptor TTET and inter-acceptor TEM.32 TEM-UC works even in the absence of molecular diffusion, and therefore it has the potential to solve the inevitable issues associated with molecular-diffusion-based UC. While air-stable TEM-UC has recently been achieved in solvent-free liquids and self-assembled acceptors in organic media,12,15,28,29 oxygen-tolerant TEM-UC has not been realized in aqueous media.
In this work, we show the first example of an air-stable aqueous TEM-UC system (Fig. 2). Ordered aqueous molecular self-assemblies such as bilayer membranes have been shown to give regular chromophore alignment and exert efficient singlet energy transfer characteristics.33,34 We made the assumption that such densely organized molecular assemblies with extended molecular networks such as hydrogen bonding may prevent the intrusion of molecular oxygen from the bulk water into the hydrophobic interior of the molecular self-assemblies.35
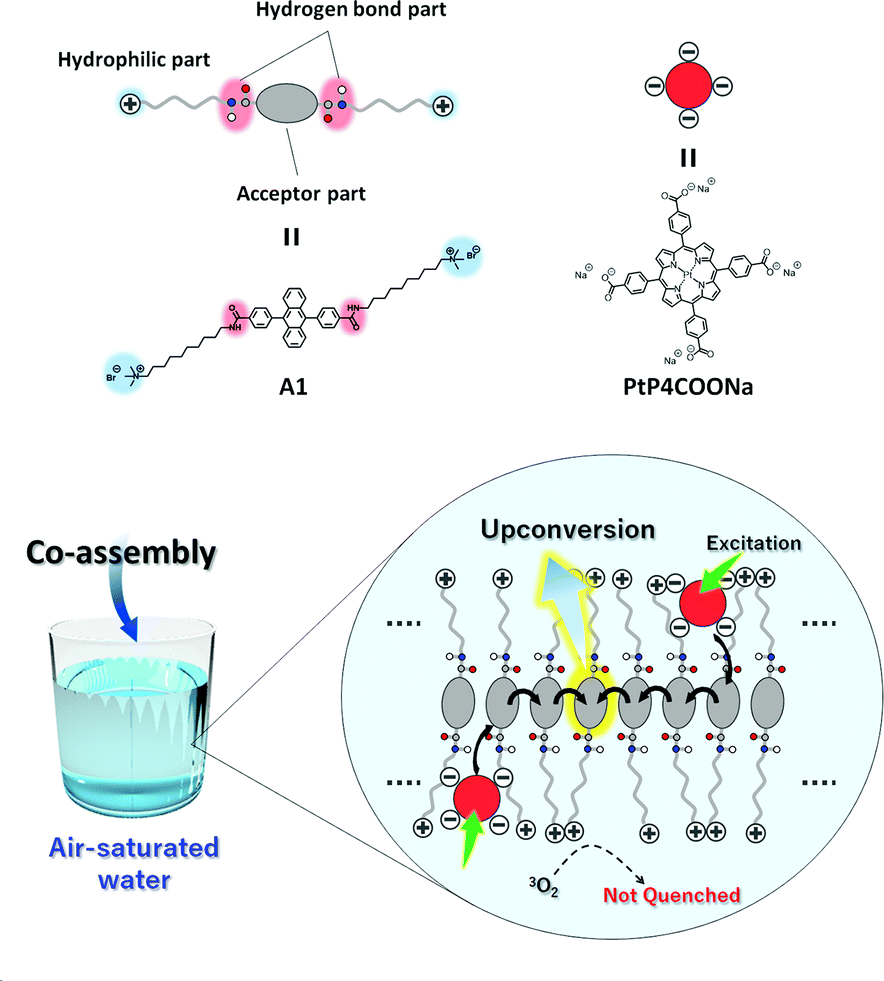 | ||
| Fig. 2 Schematic illustration of the aqueous TEM-UC system. Amphiphilic acceptor A1 self-assembles in water to form monolayer membranes which are stabilized by hydrophobic and hydrogen bonding interactions. Anionic donor PtP4COONa co-assembles with the cationic acceptor membrane A1 in water. Photoexcitation of the donor is followed by ISC, donor-to-acceptor TTET, TEM among the acceptor arrays, interacceptor TTA, and upconverted photoluminescence. Oxygen quenching is effectively avoided by the developed intermolecular hydrogen bonding networks in the acceptor monolayer membranes. | ||
As a proof of concept, we designed a novel amphiphilic acceptor A1 based on the prototypical acceptor 9,10-diphenylanthracene (DPA). Quaternary ammonium groups were introduced at both ends of the DPA chromophore, by reference to the molecular design of aqueous monolayer membranes.36–38 The DPA unit is modified with amide groups since the formation of dense hydrogen bonding networks would be beneficial for blocking oxygen.28,35 The long alkyl chain spacer groups locate the amide groups in the hydrophobic interior of the molecular membrane structure, which would facilitate the formation of hydrogen bond networks in water.39 While the typical donor Pt(II) octaethylporphyrin (PtOEP) was too hydrophobic to be stably dispersed in the aqueous self-assemblies of A1, anionic donor PtP4COONa co-assembles with cationic A1, to which triplet energy was efficiently transferred (Fig. 2). These donor–acceptor co-assemblies showed an efficient TEM-UC emission in deaerated aqueous dispersions, which was largely preserved even in the air-saturated aqueous systems. Control experiments in a water/DMF mixed solvent and with another acceptor A2 that does not have amide groups showed no UC emission, indicating the important roles that dense molecular packing and intermolecular networks play in blocking oxygen molecules.
Results and discussion
The novel acceptor A1 was synthesized and fully characterized. Its purity was confirmed by 1H-NMR measurements and elemental analysis (ESI†). The self-assembly behaviour of A1 in water was studied using transmission electron microscopy, dynamic light scattering (DLS), IR spectroscopy and temperature-dependent absorption spectroscopy. Transmission electron microscopy conducted for the aqueous dispersion of A1 exhibited round nanostructures with an average diameter of ca. 100 nm (Fig. 3a). This was consistent with the DLS profile of A1 in water ([A1] = 1 mM), which indicated the formation of nanoassemblies with an average size of 110 nm (Fig. 3b). The IR spectrum of A1 cast film showed a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration at 1631 cm−1 and N–H stretching band at 3319 cm−1, which indicate the formation of intermolecular hydrogen bonding (Fig. S1, ESI†).31,40,41 Importantly, these IR bands were maintained for the aqueous dispersion of A1 (Fig. S1, ESI†). These results clearly confirm that the acceptor amphiphile A1 forms supramolecular nanoassemblies in water which are stabilized by hydrogen bonding and hydrophobic interactions.
O stretching vibration at 1631 cm−1 and N–H stretching band at 3319 cm−1, which indicate the formation of intermolecular hydrogen bonding (Fig. S1, ESI†).31,40,41 Importantly, these IR bands were maintained for the aqueous dispersion of A1 (Fig. S1, ESI†). These results clearly confirm that the acceptor amphiphile A1 forms supramolecular nanoassemblies in water which are stabilized by hydrogen bonding and hydrophobic interactions.
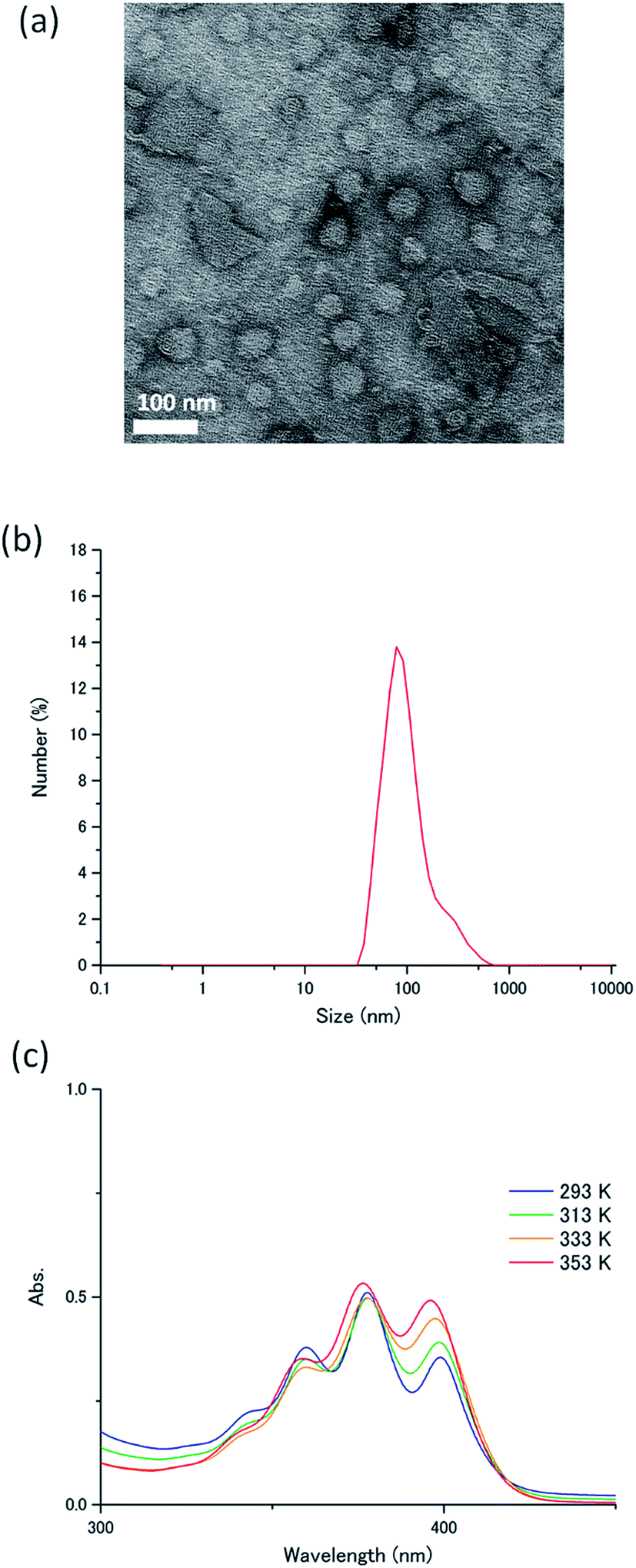 | ||
| Fig. 3 (a) Transmission electron microscopy image of A1. The sample was post stained with uranyl acetate. (b) DLS profile of A1 in water ([A1] = 1 mM). (c) Temperature-dependent absorption spectra of A1 in water ([A1] = 1 mM). | ||
The absorption spectrum of A1 in water ([A1] = 1 mM) exhibited peaks of 1La transitions at 360, 377.5 and 399 nm at 293 K, corresponding to transition dipoles polarized along the short axis of the anthracene moiety.42 Upon heating the aqueous dispersion to 363 K, these bands showed small blue shifts to 358.5, 376, and 395.5 nm (Fig. 3c). The observed small spectral shifts indicate the presence of weak interactions between the transition moments, probably because of the steric effects caused by the conformationally twisted benzene groups attached to the 9,10-positions of the anthracene chromophore. The fluorescence spectrum of A1 in water shows a band at around 440 nm, which doesn't show large overlaps with the donor absorption spectra (Fig. S2, ESI†). A high absolute fluorescence quantum yield of 53% was observed for A1 in water ([A1] = 1 mM).
While the acceptor DPA units are densely arrayed in the A1 assembly, the conformationally twisted phenyl rings attached to the anthracene moiety seem to suppress the strong interchromophore interactions and concomitant quenching of the fluorescence, as observed in absorption and emission behaviours.
The dense molecular alignment with hydrogen bond networks formed in A1 assemblies effectively prevent the quenching of the UC emission by oxygen in water. The photoluminescence spectra of A1 and PtOEP in water ([A1] = 1 mM, [PtOEP] = 1 μM) were measured after deaeration by repeated freeze–pump–thaw cycles. Under excitation at 532 nm, an UC emission was clearly observed at around 440 nm (Fig. S3a, ESI†), and the spectral shape matches well with the fluorescence spectrum of A1 (Fig. S2, ESI†). Very interestingly, this UC emission was found to be largely preserved also in the air-saturated aqueous dispersion (Fig. S3a, ESI†). To understand the role of the intermolecular network structure, we carried out control experiments by synthesizing another new DPA-based amphiphilic acceptor A2 that does not possess amide groups (Fig. S4, ESI†). The A2 molecules also form self-assembled nanostructures in water, as evidenced by transmission electron microscopy, DLS and temperature-dependent absorption measurements (Fig. S4, ESI†). The A2–PtOEP also showed an UC emission in the deaerated aqueous dispersion ([A2] = 1 mM, [PtOEP] = 1 μM), however, this UC emission entirely disappeared in the aerated specimens (Fig. S3b, ESI†). These results indicate that the dense molecular alignment promoted by the developed intermolecular hydrogen bonding networks is crucial to avoid oxygen quenching.
We should note that the UC emission of the A1–PtOEP system was weak even in the deaerated dispersion, which is accompanied by the phosphorescence of PtOEP at 650 nm. Upon incubating the aqueous A1–PtOEP dispersion for 24 h, precipitation of the red PtOEP aggregates was observed. Apparently, the hydrophobic donor PtOEP is not co-dispersed stably and tends to segregate, resulting in poor donor-to-acceptor TTET efficiency.
Meanwhile, by employing the anionic donor PtP4COONa (Fig. 2), the compatibility with the cationic acceptor A1 was improved. When PtP4COONa was mixed with A1 in deaerated dispersion at room temperature ([A1] = 1 mM, [PtP4COONa] = 1 μM), only donor phosphorescence was observed with almost no UC emission, reflecting that the rigid molecular assembly of A1 did not accommodate donor molecules. To co-assemble the donor and acceptor, a mixed aqueous dispersion of A1–PtP4COONa was heated in a microwave at 393 K for 30 min and incubated at room temperature under stirring for 48 h. The obtained aqueous dispersion was deaerated by repeated freeze–pump–thaw cycles. Interestingly, the obtained dispersion showed a clear UC emission at 440 nm (Fig. 4a). Almost no donor phosphorescence at 670 nm indicates the integration of donor molecules in acceptor assemblies, facilitating efficient donor-to-acceptor TTET. The heating procedure may have disassembled the acceptor nanostructure but it ensured the electrostatic co-assembly between the acceptor and donor.
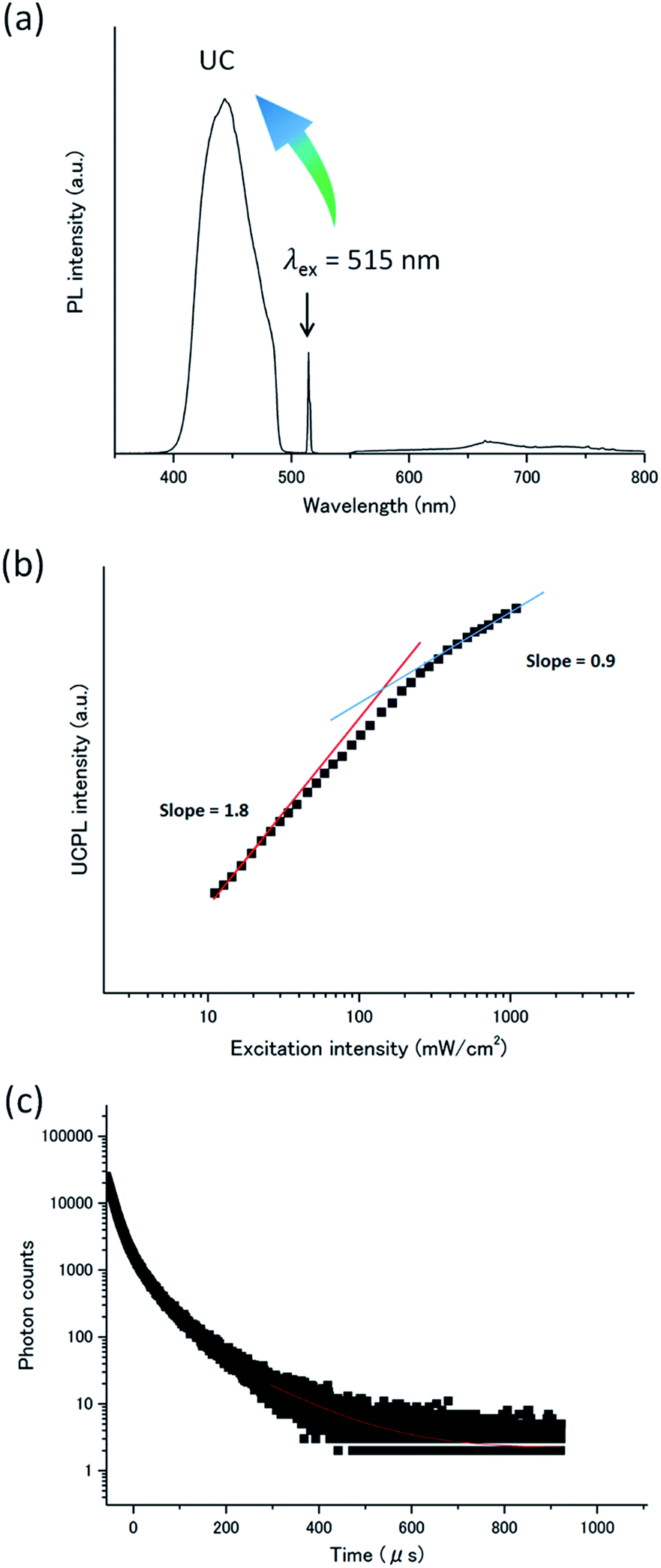 | ||
| Fig. 4 (a) Photoluminescence spectrum of A1–PtP4COONa in deaerated water ([A1] = 1 mM, [PtP4COONa] = 1 μM, λex = 515 nm, excitation intensity = 275 mW cm−2). Scattered incident laser light was removed by 490 nm short-pass filter (for UC emission) and by 550 nm long-pass filter (for phosphorescence). (b) UC emission intensity at 440 nm of A1–PtP4COONa in deaerated water as a function of the excitation intensity ([A1] = 1 mM, [PtP4COONa] = 1 μM, λex = 515 nm). The solid red and blue lines are fitting results with slopes of 1.8 and 0.9 in the low and high power regimes, respectively. (c) UC emission decay at 440 nm of A1–PtP4COONa in deaerated water under 531 nm pulsed excitation. The red fitting curve in the tail part of the decay was obtained by considering the relationship of IUC(t) ∝ exp(−t/τUC) = exp(−2t/τA,T), where τUC is UC emission lifetime and τA,T is acceptor triplet lifetime. | ||
The TTA-based UC mechanism was confirmed by the excitation intensity dependence and lifetime of the UC emission. The TTA-based UC emission intensity generally shows a quadratic and first-order dependence on the incident intensity in the low- and high-excitation intensity ranges, respectively. A double logarithmic plot for the UC emission intensity of the A1–PtP4COONa aqueous dispersion as a function of incident light intensity showed a transition from a slope of ca. 2 to 1, providing unequivocal evidence for TTA-based UC (Fig. 4b). The Ith value of 130 mW cm−2 is experimentally determined as the intersection point. The TTA-based mechanism was further supported by the UC emission decay in microsecond time scale characteristics to UC via a long-lived triplet state (Fig. 4c). It has been well documented that the TTA-based delayed fluorescence is not a single exponential decay, but its tail part obeys single exponential behavior.3,43 The acceptor triplet lifetime τA,T was obtained as 238 μs by considering the known relationship of IUC(t) ∝ exp(−2t/τA,T).3,43
The triplet diffusion constant for the acceptor arrays DT was estimated by using the following relationship,44
| Ith = (αΦET8πDTa0)−1(τA,T)−2, | (1) |
The remarkable air-stability of the aqueous TEM-UC emission was demonstrated by the UC quantum yield and time-dependent UC emission measurements. The UC quantum yield was determined by using an aqueous solution of rhodamine B as a standard. Because the TTA-UC process converts two photons to one photon, the theoretical maximum of the UC quantum yield ΦUC is 50%.10,13,45 Meanwhile, in many reports this value is multiplied with 2 to set the maximum conversion efficiency as 100%. To avoid the confusion between these different definitions, the UC quantum yield is written as Φ′UC (= 2ΦUC) when the maximum efficiency is standardized to be 100%. A high Φ′UC value of 13% was obtained for A1–PtP4COONa in deaerated water (Fig. S5, ESI†). Note that the fluorescence lifetime of A1 was not almost affected by the donor PtP4COONa, indicating the absence of acceptor-to-donor singlet–singlet back energy transfer (Fig. S6, ESI†). Significantly, even in the presence of dissolved oxygen, more than half of the Φ′UC value was maintained (7.0%). This quantum yield is comparable to those of previous highly efficient molecular-diffusion-based UC systems in aerated conditions.21,22 The time-dependent UC emission of A1–PtP4COONa in the air-saturated aqueous dispersion maintained peak intensity right after starting the laser irradiation for more than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 seconds (Fig. 5). It has been reported that the UC emission becomes stronger with time when oxygen molecules are consumed by photochemical reactions.5,28 The absence of such a gradual increase in emission intensity indicates the intrinsic oxygen barrier ability of the aqueous molecular assembly system.
000 seconds (Fig. 5). It has been reported that the UC emission becomes stronger with time when oxygen molecules are consumed by photochemical reactions.5,28 The absence of such a gradual increase in emission intensity indicates the intrinsic oxygen barrier ability of the aqueous molecular assembly system.
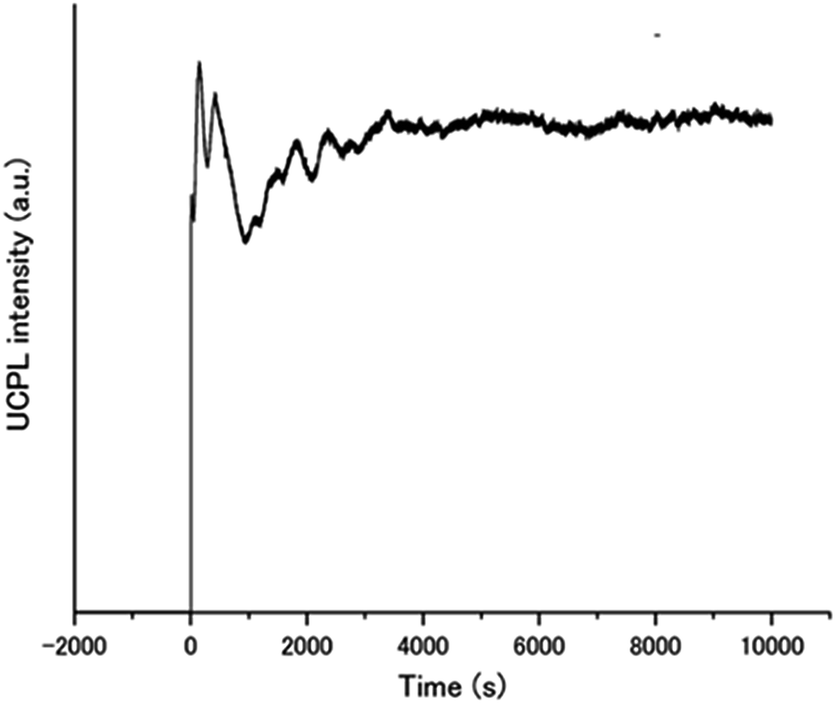 | ||
| Fig. 5 Time dependence of the UC emission intensity of A1–PtP4COONa in air-saturated water at 450 nm upon continuous excitation at 515 nm with a laser light intensity of 5 W cm−2. | ||
To understand this air-stable behaviour, we carried out control experiments by employing a water/DMF (1:1 in volume) mixed solvent instead of pure water for the A1–PtP4COONa pair to weaken the hydrophobic interactions and eventually break the hydrogen bonding networks. In contrast to the temperature-dependent shifts observed in the absorption spectra of A1 by supramolecular assembly/disassembly (Fig. 3c), the absorption peaks of A1 at 357.5, 376, 396 nm did not show any shift between 298 K and 363 K in water/DMF, indicating the absence of interchromophore close-contact assembly in water/DMF (Fig. S7†). We compared the photoluminescence spectra of the A1–PtP4COONa pair in air-saturated water and water/DMF (Fig. 6). Very interestingly, the UC emission, observed in aerated water, was completely quenched in aerated water/DMF. This clear difference supports the principal role of dense molecular alignment of A1 promoted by hydrogen bonding networks in monolayer membranes in avoiding oxygen quenching. Furthermore, the observed air stability of A1–PtP4COONa in pure water is in stark contrast to the control system without hydrogen bond networks, A2–PtP4COONa, for which the UC emission was completely quenched in aerated water similar to the case of A2–PtOEP. These results clearly demonstrate the proper amphiphilic molecular design of acceptor chromophores with dense intermolecular networks such as hydrogen bonds to achieve the air-stable TEM-UC aqueous system.
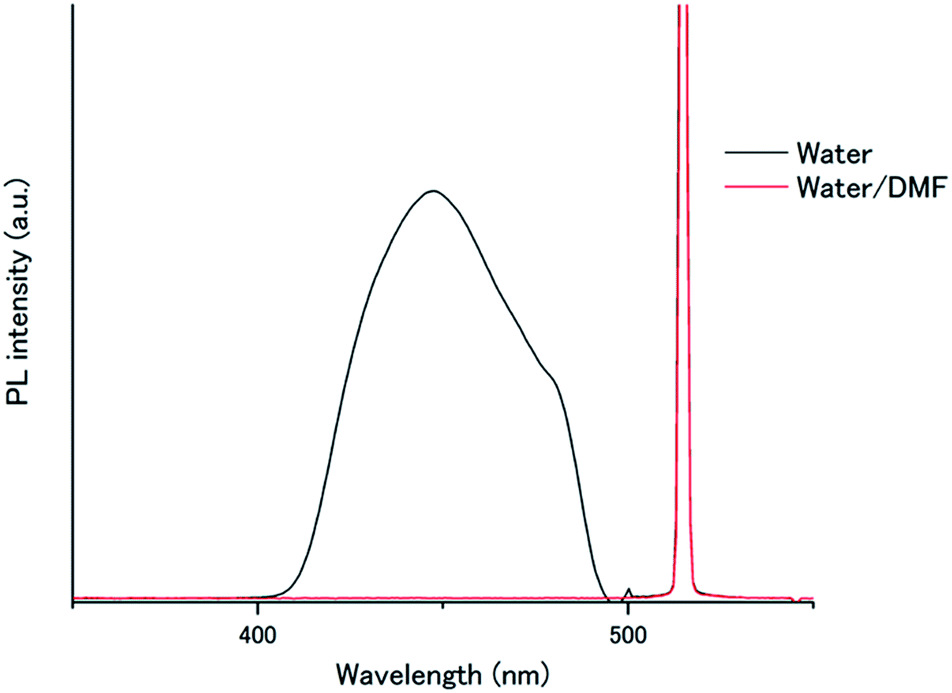 | ||
| Fig. 6 Photoluminescence spectra of A1–PtP4COONa ([A1] = 1 mM, [PtP4COONa] = 1 μM) in aerated water (black) and water/DMF (red) (λex = 515 nm, excitation intensity = 1.3 W cm−2). Scattered incident light was removed by a 490 nm short-pass filter. | ||
Conclusions
A supramolecular TEM-UC system showing an air-stable UC emission in an aqueous environment was developed for the first time. The rational design guideline to achieve this goal is demonstrated; the penetration of oxygen molecules is effectively blocked by the dense intermolecular networks driven by hydrogen bonding interactions in the hydrophobic environment of the acceptor self-assemblies. The stabilized acceptor triplets, sensitized by the co-assembled donor, migrate in the acceptor arrays and annihilate to produce efficient UC emission. The chromophores self-assembled in the hydrophobic interior with developed hydrogen bond networks of aqueous assembly systems thus are less influenced by dissolved molecular oxygen, which provides a new perspective in the design of photochemical or photophysical functions that require the interplay of reactive photo-generated species as observed in natural thylakoid membranes.Acknowledgements
We acknowledge Dr H. Yonemura and Prof. S. Yamada in Kyushu University for the access to the Ar laser. This work was partly supported by a Grants-in-Aid for Scientific Research (S) (25220805), a Grants-in-Aid for Young Scientists (B) (26810036), a Grant-in-Aid for Scientific Research on Innovative Area (16H00844) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and the JSPS-NSF International Collaborations in Chemistry (ICC) program.Notes and references
- S. Baluschev, T. Miteva, V. Yakutkin, G. Nelles, A. Yasuda and G. Wegner, Phys. Rev. Lett., 2006, 97, 143903 CrossRef CAS PubMed.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS.
- A. Monguzzi, F. Bianchi, A. Bianchi, M. Mauri, R. Simonutti, R. Ruffo, R. Tubino and F. Meinardi, Adv. Energy Mater., 2013, 3, 680–686 CrossRef CAS.
- T. F. Schulze and T. W. Schmidt, Energy Environ. Sci., 2015, 8, 103–125 CAS.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395–465 CrossRef CAS PubMed.
- K.-L. Wong, G.-L. Law, W.-M. Kwok, W.-T. Wong and D. L. Phillips, Angew. Chem., Int. Ed., 2005, 44, 3436–3439 CrossRef CAS PubMed.
- M. Haase and H. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 5808–5829 CrossRef CAS PubMed.
- K. Tanaka, K. Inafuku and Y. Chujo, Chem. Commun., 2010, 46, 4378–4380 RSC.
- J. Zhao, S. Ji and H. Guo, RSC Adv., 2011, 1, 937–950 RSC.
- A. Monguzzi, R. Tubino, S. Hoseinkhani, M. Campione and F. Meinardi, Phys. Chem. Chem. Phys., 2012, 14, 4322–4332 RSC.
- Y. C. Simon and C. Weder, J. Mater. Chem., 2012, 22, 20817–20830 RSC.
- P. Duan, N. Yanai and N. Kimizuka, J. Am. Chem. Soc., 2013, 135, 19056–19059 CrossRef CAS PubMed.
- T. W. Schmidt and F. N. Castellano, J. Phys. Chem. Lett., 2014, 5, 4062–4072 CrossRef CAS PubMed.
- S. H. C. Askes, A. Bahreman and S. Bonnet, Angew. Chem., Int. Ed., 2014, 53, 1029–1033 CrossRef CAS PubMed.
- P. Duan, N. Yanai, H. Nagatomi and N. Kimizuka, J. Am. Chem. Soc., 2015, 137, 1887–1894 CrossRef CAS PubMed.
- J.-H. Kim, F. Deng, F. N. Castellano and J.-H. Kim, Chem. Mater., 2012, 24, 2250–2252 CrossRef CAS.
- J.-H. Kim and J.-H. Kim, J. Am. Chem. Soc., 2012, 134, 17478–17481 CrossRef CAS PubMed.
- Z. Jiang, M. Xu, F. Li and Y. Yu, J. Am. Chem. Soc., 2013, 135, 16446–16453 CrossRef CAS PubMed.
- J.-H. Kim, F. Deng, F. N. Castellano and J.-H. Kim, ACS Photonics, 2014, 1, 382–388 CrossRef CAS.
- A. J. Svagan, D. Busko, Y. Avlasevich, G. Glasser, S. Baluschev and K. Landfester, ACS Nano, 2014, 8, 8198–8207 CrossRef CAS PubMed.
- J.-H. Kim and J.-H. Kim, ACS Photonics, 2015, 2, 633–638 CrossRef CAS.
- Q. Liu, B. Yin, T. Yang, Y. Yang, Z. Shen, P. Yao and F. Li, J. Am. Chem. Soc., 2013, 135, 5029–5037 CrossRef CAS PubMed.
- F. Marsico, A. Turshatov, R. Peköz, Y. Avlasevich, M. Wagner, K. Weber, D. Donadio, K. Landfester, S. Baluschev and F. R. Wurm, J. Am. Chem. Soc., 2014, 136, 11057–11064 CrossRef CAS PubMed.
- S. H. C. Askes, N. L. Mora, R. Harkes, R. I. Koning, B. Koster, T. Schmidt, A. Krosa and S. Bonnet, Chem. Commun., 2015, 51, 9137–9140 RSC.
- O. S. Kwon, H. S. Song, J. Conde, H. Kim, N. Artzi and J.-H. Kim, ACS Nano, 2016, 10, 1512–1521 CrossRef CAS PubMed.
- H. Kim, O. S. Kwon, S. Kim, W. Choi and J.-H. Kim, Energy Environ. Sci., 2016, 9, 1063–1073 CAS.
- S. Baluschev, V. Yakutkin, G. Wegner, B. Minch, T. Miteva, G. Nelles and A. Yasuda, J. Appl. Phys., 2007, 101, 023101 CrossRef.
- T. Ogawa, N. Yanai, A. Monguzzi and N. Kimizuka, Sci. Rep., 2015, 5, 10882 CrossRef CAS PubMed.
- S. Hisamitsu, N. Yanai and N. Kimizuka, Angew. Chem., Int. Ed., 2015, 54, 11550–11554 CrossRef CAS PubMed.
- P. Mahato, A. Monguzzi, N. Yanai, T. Yamada and N. Kimizuka, Nat. Mater., 2015, 14, 924–930 CrossRef CAS PubMed.
- M. Hosoyamada, N. Yanai, T. Ogawa and N. Kimizuka, Chem.–Eur. J., 2016, 22, 2060–2067 CrossRef CAS PubMed.
- N. Yanai and N. Kimizuka, Chem. Commun., 2016, 52, 5354–5370 RSC.
- T. Kunitake, Angew. Chem., Int. Ed., 1992, 31, 709–726 CrossRef.
- T. Nakashima and N. Kimizuka, Adv. Mater., 2002, 14, 1113–1116 CrossRef CAS.
- R. Nishiyabu, C. Aimé, R. Gondo, T. Noguchi and N. Kimizuka, Angew. Chem., Int. Ed., 2009, 48, 9465–9468 CrossRef CAS PubMed.
- Y. Okahata and T. Kunitake, J. Am. Chem. Soc., 1979, 101, 5231–5234 CrossRef CAS.
- M. Shimomura, H. Hashimoto and T. Kunitake, Chem. Lett., 1982, 1285–1288 CrossRef CAS.
- J. H. Fuhrhop and D. Fritsch, Acc. Chem. Res., 1986, 19, 130–137 CrossRef CAS.
- N. Kimizuka, T. Kawasaki, K. Hirata and T. Kunitake, J. Am. Chem. Soc., 1998, 120, 4094–4104 CrossRef CAS.
- Y. Maeda, T. Higuchi and I. Ikeda, Langmuir, 2000, 16, 7503–7509 CrossRef CAS.
- A. Onoda, Y. Yamada, M. Doi, T. Okamura and N. Ueyama, Inorg. Chem., 2001, 40, 516–521 CrossRef CAS PubMed.
- N. Harada, Y. Takuma and H. Uda, J. Am. Chem. Soc., 1978, 100, 4029–4036 CrossRef CAS.
- M. Pepo and C. E. Swenberg, Electronic Processes in Organic Crystals, Oxford University Press, 1982 Search PubMed.
- A. Monguzzi, J. Mezyk, F. Scotognella, R. Tubino and F. Meinardi, Phys. Rev. B, 2008, 78, 195112 CrossRef.
- V. Gray, D. Dzebo, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, Phys. Chem. Chem. Phys., 2014, 16, 10345–10352 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, TTA-based UC mechanism, IR, absorption, emission and UC spectra, structure of A2, UC quantum yield. See DOI: 10.1039/c6sc01047d |
| This journal is © The Royal Society of Chemistry 2016 |