
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical reduction of cationic Li+@C60 to neutral Li+@C60˙−: isolation and characterisation of endohedral [60]fulleride†
Hiroshi
Ueno‡
a,
Shinobu
Aoyagi
b,
Yu
Yamazaki
a,
Kei
Ohkubo
cd,
Naohiko
Ikuma
 a,
Hiroshi
Okada
e,
Tatsuhisa
Kato
f,
Yutaka
Matsuo
eg,
Shunichi
Fukuzumi
dh and
Ken
Kokubo
*a
a,
Hiroshi
Okada
e,
Tatsuhisa
Kato
f,
Yutaka
Matsuo
eg,
Shunichi
Fukuzumi
dh and
Ken
Kokubo
*a
aDivision of Applied Chemistry, Graduate School of Engineering, Osaka University, Suita, Osaka 565-0871, Japan. E-mail: kokubo@chem.eng.osaka-u.ac.jp
bDepartment of Information and Basic Science, Nagoya City University, Mizuho-ku, Nagoya 467-8501, Japan
cDepartment of Material and Life Science, Graduate School of Engineering, Osaka University, ALCA and SENTAN, Japan Science and Technology (JST), Suita, Osaka 565-0871, Japan
dDepartment of Chemistry and Nano Science, Ewha Womans University, Seoul 120-750, Korea
eDepartment of Mechanical Engineering, School of Engineering, The University of Tokyo, Bunkyo-ku, Tokyo 113-8656, Japan
fDepartment of Interdisciplinary Environment, Graduate School of Human and Environmental Studies, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan
gHefei National Laboratory for Physical Sciences at Microscale, University of Science and Technology of China, Hefei, Anhui 230026, China
hFaculty of Science and Engineering, Meijo University, ALCA and SENTAN, Japan Science and Technology Agency (JST), Nagoya, Aichi 468-0073, Japan
First published on 20th June 2016
Abstract
Lithium-encapsulated [60]fullerene Li@C60, namely, lithium-ion-encapsulated [60]fullerene radical anion Li+@C60˙−, was synthesised by electrochemical reduction of lithium-ion-encapsulated [60]fullerene trifluoromethanesulfonylimide salt [Li+@C60](TFSI−). The product was fully characterised by UV-vis-NIR absorption and ESR spectroscopy as well as single-crystal X-ray analysis for the co-crystal with nickel octaethylporphyrin. In solution Li@C60 exists as a monomer form dominantly, while in the crystal state it forms a dimer (Li@C60–Li@C60) through coupling of the C60 radical anion cage. These structural features were supported by DFT calculations at the M06-2X/6-31G(d) level of theory.
Introduction
Insertion of a metal atom into the fullerene cage is one of the most attractive methods to tune the electronic properties of a spherically π-conjugated fullerene carbon cage without changing the exterior framework. Ever since the resulting products, called “endohedral metallofullerenes (EMFs)”, were reported1 many studies on various EMFs have been conducted because of their unique electronic structure as a so-called “superatom”; formally defined by an electron transfer from the inner metal atom to the outer fullerene cage (Fig. 1a).2,3 Among EMFs, which have various carbon-cage sizes, the C60-based EMF has attracted special attention owing to its highly symmetrical structure and expected unique electronic properties.4 However, the studies on metallo[60]fullerenes,4,5 even the relatively well-studied alkali metal-encapsulated ones,6 are somewhat stagnant due to the difficulty of their isolation and the lack of structural evidence.7 Although some of us have reported the isolation of a lithium-ion-encapsulated [60]fullerene (Li+@C60) as a SbCl6− salt [Li+@C60](SbCl6−) in 2010,8 the isolated Li+@C60 is regarded as an “ion-encapsulated fullerene” which is a new endohedral fullerene family,9 but is not categorised as part of general EMFs (Fig. 1b).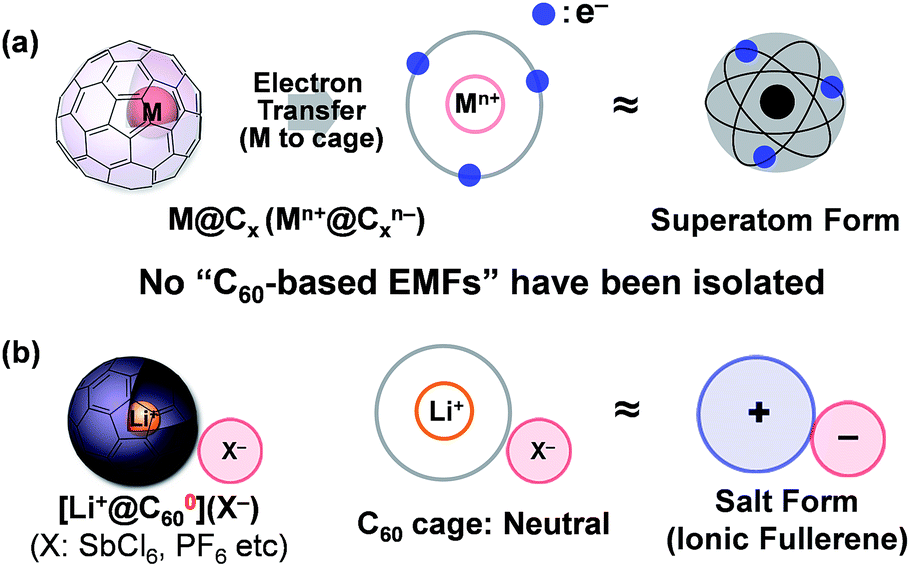 | ||
| Fig. 1 Electronic state of (a) a general endohedral metallofullerene M@Cx, and (b) a lithium-ion-encapsulated fullerene [Li+@C60](X−). The described electrons are derived from charge transfer from the encapsulated metal to the fullerene cage. | ||
Previously, we found that Li@C60, namely, Li+@C60˙−, was generated by the electrochemical reduction of [Li+@C60](PF6−) in o-dichlorobenzene (o-DCB).10 However, only a mixture of the product and the starting Li+@C60 salt was obtained. Purification of the product Li+@C60˙− has been a major challenge to achieve the first isolation of M@C60. The ion-pair form of the [60]fullerene anion, named the fulleride, stabilised by an external counter metal or organic cations is an intriguing material because of their superconductivity11 and magnetism.12 The Li+@C60˙− can be deemed as an “endohedral [60]fulleride” as well as the simplest superatom. Thus, application of this material very promising in the organic electronics and materials chemistry fields.
We herein report the electrochemical reduction and complete isolation of Li+@C60˙− endohedral [60]fulleride by utilizing highly soluble Li+@C60 salt. We found that Li+@C60˙− forms Li@C60–Li@C60 dimer in a co-crystal with nickel octaethylporphyrin (NiOEP), which was revealed by X-ray structure analysis and theoretical calculations. This is the first report of the isolation and unambiguous characterisation of a metal-encapsulated [60]fullerene consisted of only lithium and carbon atoms with a 100% encapsulation ratio.
Results and discussion
One of the major advantages of ionic [Li+@C60](X−) is that the solubility of the [Li+@C60](X−) salt can be modified by exchanging the counter anion (X−) whereas the solubility of the other fullerenes could not be tuned without chemical functionalisation of the fullerene cage.13 To prepare the target Li@C60, we focused on the difference in solubility between the starting [Li+@C60](X−) salt and the product Li@C60. After several attempts, lithium-ion-encapsulated fullerene bis(trifluoromethanesulfonyl)imide salt, [Li+@C60](TFSI−) was selected as the starting ionic fullerene because of its good solubility in CH2Cl2, which is a poor solvent for the product. [Li+@C60](TFSI−) salt was prepared by anion exchange from commercially available [Li+@C60](PF6−) salt according to our previously reported procedure.13 The electrochemical reaction was carried out under Ar atmosphere as depicted in Fig. 2. A CH2Cl2 solution of the starting compound was placed in an H-shaped cell, cooled to 253 K, and electrolysed using a Pt electrode at a constant current (0.5 μA) for 3 days. The purple solution gradually became colourless because of deposition of the reduced fullerene-based product on the surface of the cathode. In this process, due to sufficient ionic conductivity of the [Li+@C60](TFSI−) solution (see Fig. S1 in the ESI†), the electrochemical reaction could be carried out without any supporting electrolyte.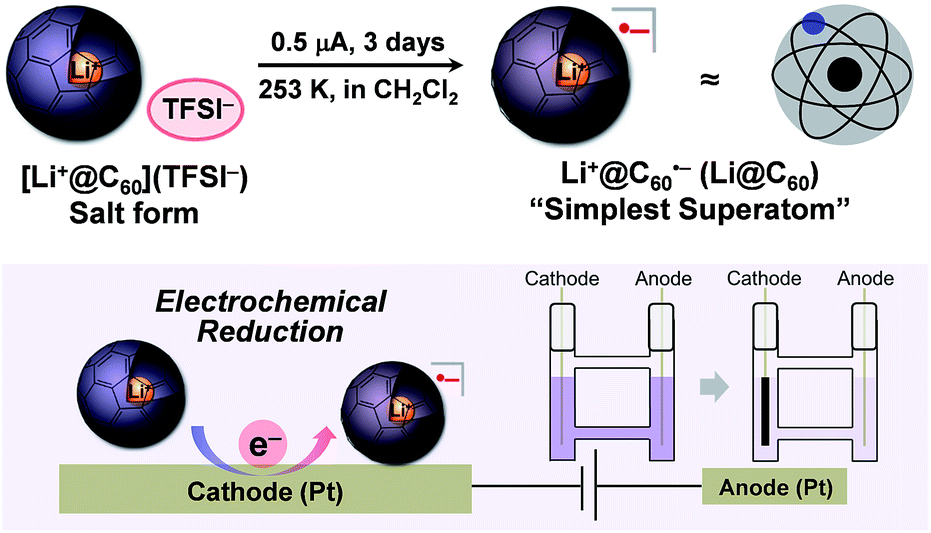 | ||
| Fig. 2 Schematic image of the electrochemical one electron reduction of [Li+@C60](TFSI−) to Li@C60. | ||
The obtained product was dissolved in o-DCB to be characterised by UV-vis-NIR and ESR, and NMR spectroscopy in the solution phase. The UV-vis-NIR spectrum of the product showed characteristic absorption at 1035 nm, which was assignable to the lithium-ion-encapsulated fullerene monovalent radical anion by a TD-DFT calculation (Fig. 3a and also see Fig. S2 in ESI†).10,14,15 The ESR spectrum of the solution is shown in Fig. 3b. The observed g value (2.0010) was nearly identical to the reported value for the empty C60 radical anion and our previous results.10,16 The 7Li and 13C NMR showed no signal due to the paramagnetic relaxation. The 19F NMR spectrum of the product also showed no signals, indicating elimination of TFSI−. These results suggest the absence of the starting Li+@C60 salt in the product and the complete isolation of neutral Li@C60.
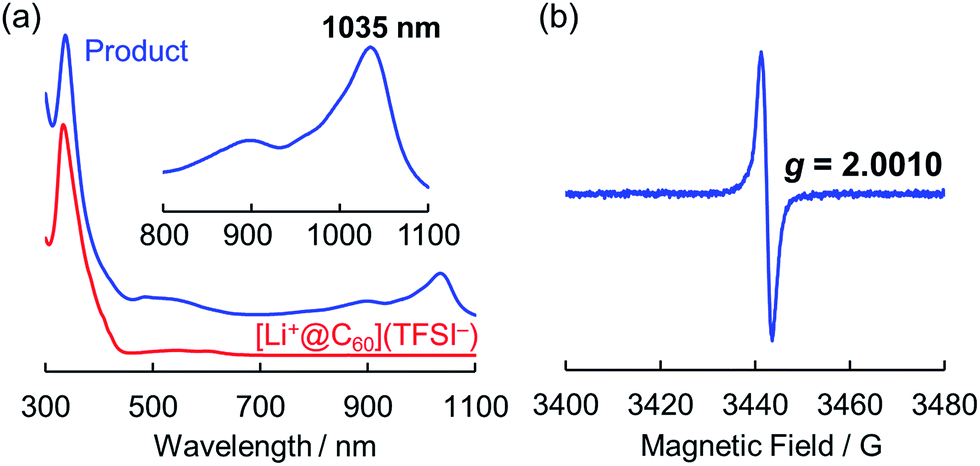 | ||
| Fig. 3 (a) UV-vis-NIR spectra of product (blue line) and starting [Li+@C60](TFSI−) (red line) measured in o-DCB. (b) ESR spectrum of the product measured at 77 K in frozen o-DCB. | ||
Conclusive structural evidence was obtained by X-ray structure analysis for a co-crystal of the product with NiOEP (OEP2− = octaethylporphyrin dianion). The appropriate single crystal was obtained by electrochemical reaction in the presence of NiOEP, which is usually used as a cocrystallising agent for endohedral fullerenes.17 Because of the much lower LUMO level of Li+@C60 than that of NiOEP, only the Li+@C60 was electrochemically reduced on the surface of the anode.18 As shown in Fig. 4, the dimerised structure of Li+@C60˙− (Li+@C60−–Li+@C60−) connected by a single C–C bond was determined in the co-crystal. The dimer could be formed by the coupling of the spin centres of Li+@C60˙−s, and this is not surprising because similar dimerization has been reported in alkali-metal doped fullerides,19 a chemically reduced penta-arylated [60]fullerene derivative,20 an open-shell EMF derivative,21 and an anionic C60−–C60− dimer in the ionic charge-transfer complex of C60 with decamethylchromocene.22 The equilibrium state of a C60 radical anion and its dimer in the solution phase and dimerization of the radicals in the solid phase has been already well-known,23 and thus, in our case as well, the observed structure was attributed to an ion-pair form, Li+@C60˙−, in the solution phase.
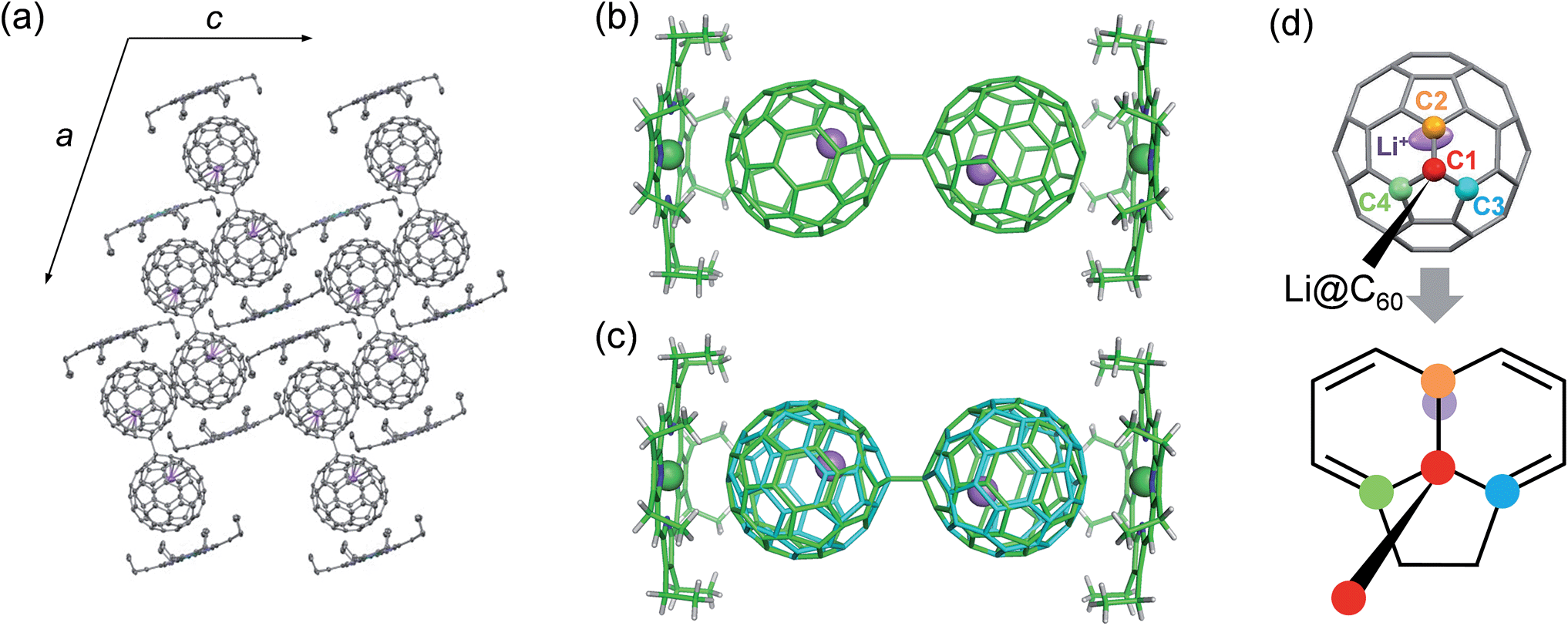 | ||
| Fig. 4 Crystal structure of the Li+@C60˙−–NiOEP co-crystal. (a) The molecular arrangement with thermal ellipsoids at the 50% probability level viewed along the b-axis at 100 K. Hydrogen atoms and dichloromethane solvent molecules are omitted in the Figure. (b) and (c) The Li+@C60−–Li+@C60− dimer coordinated by two NiOEP molecules at 100 and 250 K, respectively. The Li+@C60−–Li+@C60− dimer at 250 K is disordered by the overlapping of two molecules (green and blue) in a mirror relationship. (d) View along the single C–C bond connecting Li@C60s. The four carbon atoms around the inter-fullerene single C–C bond are labeled as C1–C4. The length of each bond is 1.594(5) Å (C1–C1′), 1.528(5) Å (C1–C2), 1.546(6) Å (C1–C3), and 1.558(5) Å (C1–C4), respectively. | ||
The Li+@C60−–Li+@C60− dimer in the co-crystal at 100 K showed the trans conformation (Fig. 4b), which has been proposed by the X-ray powder diffraction study of the dimerised phase of AC60 fullerides and theoretical calculations of C60−–C60− and (C59N)2 as a stable conformation.19,24 The length of a single C–C bond connected to each C60 cage (C1–C1′) was 1.594(5) Å, which was longer than the normal C(sp3)–C(sp3) bond length (1.54 Å) and comparable to an interfullerene single C–C bond in the empty C60−–C60− dimer (1.597(7) Å).22 Although it has been reported that the single C–C bond of the empty C60−–C60− dimer starts to break at 200–220 K in the crystal, our co-crystal did not show cleavage of the interfullerene bond up to 400 K and is stable in air. As shown in Fig. 4c, the Li+@C60−–Li+@C60− dimer showed a disordered structure attributed to a ratchet motion of the C60− cage along the single C–C interfullerene bond with a rotating angle of about 39° at 250 K. The ratchet motion was induced in the high temperature phase through the phase transition around 250 K. The linear temperature dependence of the lattice constants with a small anomaly at the phase transition temperature suggests that the phase transition is of the order–disorder type (see Fig. S4 in ESI†).
The endohedral Li+ was clearly observed, and localised near the carbon atom (C2) nearest to the carbon atom (C1) forming the interfullerene single C–C bond (Fig. 4d). The oxidation state of Li was estimated as +0.8(3) from the electron charge-densities which were obtained from the X-ray diffraction data by using the maximum entropy method (see Fig. S5 in ESI†). The result clearly indicated the formation of a “superatom state” as in reported EMFs, and coincided with the results of ESR spectroscopy. The C1 carbon atom bonded with the C2 carbon atom by a short 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 bond fusing two hexagons, and C3 and C4 carbon atoms bonded by a long 6:5 bond fusing a hexagon and a pentagon. The C1–C2, C1–C3 and C1–C4 bond lengths were 1.528(5), 1.546(6) and 1.558(5) Å, respectively, which were much longer than the 6:6 and 6:5 bond lengths for neutral C60 (1.39 and 1.45 Å, respectively),25 and comparable to the normal C(sp3)–C(sp3) bond length (1.54 Å). These results indicate that the C2 atom bonded to the C1 atom by the shorter 6:6 bond has excess electrons, and the excess electrons attract the encapsulated Li+, causing the localisation of the Li+ near the C2 atom. The Li–C2 distance was 2.20(1) Å at 100 K. The Li–C distance was shorter than that of 2.344(6) Å in a cubic [Li+@C60](PF6−) crystal around 25 K, in which the Li+ equivalently localizes under the centres of two hexagons on the three-fold inversion axis by the electrostatic interaction from the coordinated six PF6− anions.26 The ion-pairing with the short Li–C bond contributes to the stabilisation of the anionic C60 cage bonded by the single C–C bond in the Li+@C60−–Li+@C60−–NiOEP co-crystal.
:6 bond fusing two hexagons, and C3 and C4 carbon atoms bonded by a long 6:5 bond fusing a hexagon and a pentagon. The C1–C2, C1–C3 and C1–C4 bond lengths were 1.528(5), 1.546(6) and 1.558(5) Å, respectively, which were much longer than the 6:6 and 6:5 bond lengths for neutral C60 (1.39 and 1.45 Å, respectively),25 and comparable to the normal C(sp3)–C(sp3) bond length (1.54 Å). These results indicate that the C2 atom bonded to the C1 atom by the shorter 6:6 bond has excess electrons, and the excess electrons attract the encapsulated Li+, causing the localisation of the Li+ near the C2 atom. The Li–C2 distance was 2.20(1) Å at 100 K. The Li–C distance was shorter than that of 2.344(6) Å in a cubic [Li+@C60](PF6−) crystal around 25 K, in which the Li+ equivalently localizes under the centres of two hexagons on the three-fold inversion axis by the electrostatic interaction from the coordinated six PF6− anions.26 The ion-pairing with the short Li–C bond contributes to the stabilisation of the anionic C60 cage bonded by the single C–C bond in the Li+@C60−–Li+@C60−–NiOEP co-crystal.
We also performed X-ray diffraction measurements for the powder sample of Li+@C60˙− resulting from electrolysis of [Li+@C60](TFSI−) (see Fig. S6 in ESI†). Unfortunately, the sample had a more complicated crystal structure and/or contained multiple phases, and thus indexing of the powder diffraction pattern with a single phase by using DICVOL27 was unsuccessful. Incidentally, the powder sample also showed a similar ESR signal to that measured in frozen o-DCB (see Fig. S3 in ESI†), which indicated that while the Li@C60 dimer could be formed perfectly in a well-ordered crystal, the Li+@C60˙− monomer remained partially in the disordered solid state.
The observed equilibrium behaviour of the Li+@C60˙− monomer and its dimer was consistent with the results of DFT calculations at M06-2X/6-31G(d) level of theory (Fig. 5). An estimation of the change in Gibbs free energy (ΔG) of dimerization of Li+@C60˙− in o-DCB was made using the SCRF(IEFPCM) methodology. The results revealed that while the formation of singlet dimer was energetically favourable in vacuum (ΔG = −4.35 kcal mol−1), Li+@C60˙− monomer was slightly stable in o-DCB solution (ΔG = +0.29 kcal mol−1), which indicated the higher stability of the Li+@C60˙− monomer as a result of the surrounding solvent molecules in the solution phase.28 We also calculated the ΔG of dimerization of the empty C60 radical anion in o-DCB using the same method, and the value was calculated to be +11.5 kcal mol−1 (see Fig. S7 in ESI†), which was higher than that of Li+@C60˙−. The difference could be explained by the degree of electrostatic repulsion between anionic fullerene spheres relaxed by the unique ion-pair form in the endohedral fullerides. In Li+@C60˙−, the positional relation between an encapsulated Li+ ion and an anionic C60 must be nearly constant because Li+ cannot be released from the anionic cage. The effect of solvation is known as important factor when the electrostatic interaction is considered,29 but the solvation may not be important for such a unique shielded ion. Thus the negative charge on the anionic C60 cage might be cancelled more effectively by the “perfect” ion pairing of the internal Li+ ion and the negative charge on the C60 cage, which makes the electrostatic repulsion force between Li+@C60˙−s weaker. Nevertheless, as we already mentioned, the Li+@C60−– Li+@C60− dimer was not observed in o-DCB solution by any spectroscopic analyses, which indicated that majority of the Li+@C60˙− exists as a monomer at ambient temperature in the solution phase.
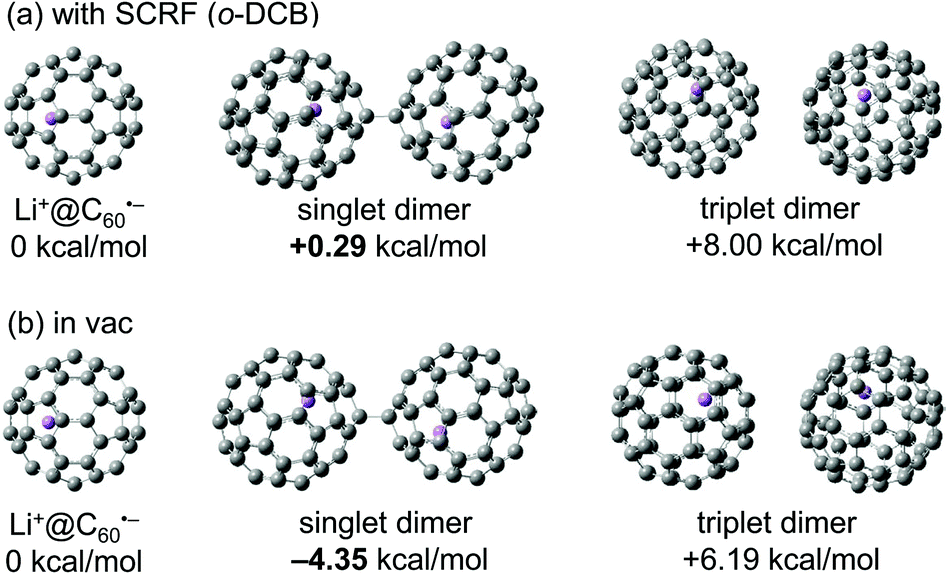 | ||
| Fig. 5 Change in Gibbs free energy of dimerization of Li+@C60˙− calculated by DFT (M06-2X/6-31G(d)) (a) using the SCRF(IEFPCM) methodology and (b) in vacuum. | ||
Li+@C60˙− and its dimer are also interesting in terms of their quasi-atomic/molecular electronic structure. Because the Li+@C60˙−, consisting of encapsulated monocationic Li+ and a nearly spherical anionic C60 cage with a single unpaired-electron, is very similar to the nucleus and orbital of a hydrogen atom, Li+@C60˙− can be considered as the “simplest” superatom. Through the dimerization of Li+@C60˙−, in addition, the superatomic structure converts to the model of a “pseudo homonuclear diatomic molecule”, again like a hydrogen molecule. The unique structure of the superatomic molecular orbital of doped C60 has been predicted only by theoretical calculation.30 Thus, the present Li+@C60˙− will play an important role as a simplest example of a superatom, providing a unique model of the molecule-like orbital structure.
Conclusions
In summary, we have successfully isolated the lithium-ion-encapsulated fullerene radical anion Li+@C60˙−, which can be considered as a general EMF form of lithium-encapsulated fullerene Li@C60, by means of the electrochemical reduction of ionic [Li+@C60](TFSI−). Due to the solubility difference between ionic Li+@C60 and the Li+@C60˙− superatom, only the product was deposited on the surface of the electrode. Thus, we did not need to purify the product by common methods for the purification of metallofullerenes such as the preparative HPLC technique. The product was unambiguously characterised by spectroscopic analyses as well as X-ray structure analysis for the co-crystal with NiOEP. The Li@C60 exists dominantly as a monomer form, while in the crystal state it forms a dimer (Li+@C60−–Li+@C60−) through coupling of the radical centre on the anionic C60 cage. These structural features were supported by DFT calculations at the M06-2X/6-31G(d) level of theory. This is the first report of the isolation and characterization of a [60]fullerene-based metallofullerene. With this detailed structural information and great anticipation for the expected unique properties, utilisation of this new metallofullerene for various applications may be possible.Acknowledgements
This work was partly supported by the Adaptable and Seamless Technology Transfer Program through target-driven R&D, JST, Health Labour Sciences research grants from the MHLW of Japan and the Program for Creating Future Wisdom, Osaka University, selected in 2014 (to K. K.), and the Funding Program for a Grants-in-Aid for Scientific Research (15H05760 to Y. M., 26620154 and 26288037 to K. O.). The X-ray diffraction measurements were performed at SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2014A0100 and 2015A0100). K. K. and Y. Y. also thank to Professor H. Sakurai (Osaka University) for useful discussions.Notes and references
- J. R. Heath, S. C. O’Brien, Q. Zhang, Y. Liu, R. F. Curl, H. W. Kroto, F. K. Tittle and R. E. Smalley, J. Am. Chem. Soc., 1985, 107, 7779 CrossRef CAS.
- (a) H. Shinohara, Rep. Prog. Phys., 2000, 63, 843 CrossRef CAS; (b) Endohedral Fullerenes: A New Family of Carbon Clusters, ed. T. Akasaka and S. Nagase, Kluwer, Dordrecht, 2002 Search PubMed; (c) A. A. Popov, S. Yang and L. Dunsch, Chem. Rev., 2013, 113, 5989 CrossRef CAS PubMed.
- (a) R. D. Johnson, M. S. Vries, J. Salem, D. S. Bethune and C. S. Yannoni, Nature, 1992, 355, 239 CrossRef CAS; (b) S. Nagase and K. Kobayashi, J. Chem. Soc., Chem. Commun., 1994, 1837 RSC.
- (a) Y. Wang, D. Tománek and R. S. Ruoff, Chem. Phys. Lett., 1993, 208, 79 CrossRef CAS; (b) Z. Slanina, S. L. Lee, L. Adamowicz, F. Uhlík and S. Nagase, Int. J. Quantum Chem., 2005, 104, 272 CrossRef CAS; (c) R. F. Sabirianov, W.-N. Mei, J. Lu, Y. Gao, X. C. Zeng, R. D. Bolskar, P. Jeppson, N. Wu, A. Caruso and P. A. Dowben, J. Phys.: Condens. Matter, 2007, 19, 082201 CrossRef.
- (a) Y. Chai, T. Guo, C. M. Jin, R. E. Haufler, L. P. F. Chibante, J. Fure, L. H. Wang, J. M. Alford and R. E. Smalley, J. Phys. Chem., 1991, 95, 7564 CrossRef CAS; (b) Y. Kubozono, H. Maeda, Y. Takabayashi, K. Hiraoka, T. Nakai, S. Kashino, S. Emura, S. Ukita and T. Sogabe, J. Am. Chem. Soc., 1996, 118, 6998 CrossRef CAS; (c) T. Ogawa, T. Sugai and H. Shinohara, J. Am. Chem. Soc., 2000, 122, 3538 CrossRef CAS.
- (a) R. Tellgmann, N. Krawez, S.-H. Lin, I. V. Hertel and E. E. B. Campbell, Nature, 1996, 382, 407 CrossRef CAS; (b) E. E. B. Campbell, R. Tellgmann, N. Krawez and I. V. Hertel, J. Phys. Chem. Solids, 1997, 58, 1763 CrossRef CAS; (c) A. Gromov, W. Krätschmer, N. Krawez, R. Tellgmann and E. E. B. Campbell, Chem. Commun., 1997, 2003 RSC; (d) A. Gromov, D. Ostrovskii, A. Lassesson, M. Jönsson and E. E. B. Campbell, J. Phys. Chem. B, 2003, 107, 11290 CrossRef CAS; (e) M. Zhang, L. B. Harding, S. K. Gray and S. A. Rice, J. Phys. Chem. A, 2008, 112, 5478 CrossRef CAS PubMed; (f) H. Reis, O. Loboda, A. Avramopoulos, M. G. Papadopoulos, B. Kirtman, J. M. Luis and R. Zaleśny, J. Comput. Chem., 2011, 32, 908 CrossRef CAS PubMed.
- Recently, Shinohara and co-workers reported a powerful method to isolate “missing metallofullerenes” by applying suitable chemical functionalisation. For details of such metallofullerenes, see: (a) Z. Wang, S. Aoyagi, H. Omachi, R. Kitaura and H. Shinohara, Angew. Chem., Int. Ed., 2016, 55, 199 CrossRef CAS PubMed; (b) Z. Wang, Y. Nakanishi, S. Noda, H. Niwa, J. Zhang, R. Kitaura and H. Shinohara, Angew. Chem., Int. Ed., 2013, 52, 11770 CrossRef CAS PubMed.
- S. Aoyagi, E. Nishibori, H. Sawa, K. Sugimoto, M. Tanaka, Y. Miyata, R. Kitaura, H. Shinohara, H. Okada, T. Sakai, Y. Ono, K. Kawachi, K. Yokoo, S. Ono, K. Omote, Y. Kasama, S. Ishikawa, T. Komuro and H. Tobita, Nat. Chem., 2010, 2, 678 CrossRef CAS PubMed.
- (a) H. Ueno, T. Nishihara, Y. Segawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 3707 CrossRef CAS PubMed; (b) H. Ueno, H. Kawakami, K. Nakagawa, H. Okada, N. Ikuma, S. Aoyagi, K. Kokubo, Y. Matsuo and T. Oshima, J. Am. Chem. Soc., 2014, 136, 11162 CrossRef CAS PubMed.
- H. Ueno, K. Kokubo, Y. Nakamura, K. Ohkubo, N. Ikuma, H. Moriyama, S. Fukuzumi and T. Oshima, Chem. Commun., 2013, 49, 7376 RSC.
- (a) K. Tanigaki, I. Hirosawa, T. W. Ebbesen, J. Mizuki, Y. Shimakawa, Y. Kubo, J. S. Tsai and S. Kuroshima, Nature, 1992, 356, 419 CrossRef CAS; (b) A. Y. Ganin, Y. Takabayashi, P. Jeglič, D. Arčon, A. Potočnik, P. J. Baker, Y. Ohishi, M. Tanaka, M. J. Rosseinsky and K. Prassides, Nature, 2010, 466, 221 CrossRef CAS PubMed; (c) H. Moriyama, H. Kobayashi, A. Kobayashi and T. Watanabe, J. Am. Chem. Soc., 1993, 115, 1185 CrossRef CAS; (d) H. Moriyama, M. Abe, H. Motoke, T. Watanabe, S. Hayashi and H. Kobayashi, Synth. Met., 1998, 94, 167 CrossRef CAS.
- P. W. Stephens, D. Cox, J. W. Lauher, L. Mihaly, J. B. Wiley, P.-M. Allemand, A. Hirsh, K. Holczer, Q. Li, J. D. Thompson and F. Wudl, Nature, 1992, 355, 331 CrossRef CAS.
- H. Okada and Y. Matsuo, Fullerenes, Nanotubes, Carbon Nanostruct., 2014, 22, 262 CrossRef CAS.
- (a) S. Fukuzumi, K. Ohkubo, Y. Kawashima, D. S. Kim, J. S. Park, A. Jana, V. M. Lynch, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2011, 133, 15938 CrossRef CAS PubMed; (b) K. Ohkubo, Y. Kawashima and S. Fukuzumi, Chem. Commun., 2012, 48, 4314 RSC.
- For the absorption spectrum of empty C60˙−, see: (a) T. Kato, T. Kodama, T. Shida, T. Nakagawa, Y. Matsui, S. Suzuki, H. Shiromaru, K. Yamauchi and Y. Achiba, Chem. Phys. Lett., 1991, 180, 446 CrossRef CAS; (b) T. Kato, T. Kodama, M. Oyama, S. Okazaki, T. Shida, T. Nakagawa, Y. Matsui, S. Suzuki, H. Shiromaru, K. Yamauchi and Y. Achiba, Chem. Phys. Lett., 1991, 186, 35 CrossRef CAS.
- T. Kato, T. Kodama and T. Shida, Chem. Phys. Lett., 1993, 205, 405 CrossRef CAS.
- (a) S. Stevenson, G. Rice, T. Glass, K. Harich, F. Cromer, M. R. Jordan, J. Craft, E. Hadju, R. Bible, M. M. Olmstead, K. Maitra, A. J. Fisher, A. L. Balch and H. C. Dorn, Nature, 1999, 401, 55 CrossRef CAS; (b) H. M. Lee, M. M. Olmstead, T. Suetsuna, H. Shimotani, N. Dragoe, R. J. Cross, K. Kitazawa and A. L. Balch, Chem. Commun., 2002, 1352 RSC.
- J.-H. Fuhrhop, K. M. Kadish and D. G. Davis, J. Am. Chem. Soc., 1973, 95, 5140 CrossRef CAS PubMed.
- G. Oszlányi, G. Bortel, G. Faigel, L. Gránásy, G. M. Bendele, P. W. Stephens and L. Forró, Phys. Rev. B: Condens. Matter, 1996, 54, 11849 CrossRef.
- Y. Matsuo and E. Nakamura, J. Am. Chem. Soc., 2005, 127, 8457 CrossRef CAS PubMed.
- Y. Maeda, S. Sato, K. Inada, H. Nikawa, M. Yamada, N. Mizorogi, T. Hasegawa, T. Tsuchiya, T. Akasaka, T. Kato, Z. Slanina and S. Nagase, Chem.– Eur. J., 2010, 16, 2193 CrossRef CAS PubMed.
- D. V. Konarev, S. S. Khasanov, A. Otsuka and G. Saito, J. Am. Chem. Soc., 2002, 124, 8520 CrossRef CAS PubMed.
- C. A. Reed and R. D. Bolskar, Chem. Rev., 2000, 100, 1075 CrossRef CAS PubMed.
- K. H. Lee, S. S. Park, Y. Suh, T. Yamabe, E. Osawa, H. P. Lüthi, P. Gutta and C. Lee, J. Am. Chem. Soc., 2001, 123, 11085 CrossRef CAS PubMed.
- W. I. F. David, R. M. Ibberson, J. C. Matthewman, K. Prassides, T. J. S. Dennis, J. P. Hare, H. W. Kroto, R. Taylor and D. R. M. Walton, Nature, 1991, 353, 147 CrossRef CAS.
- S. Aoyagi, Y. Sado, E. Nishibori, H. Sawa, H. Okada, H. Tobita, Y. Kasama, R. Kitaura and H. Shinohara, Angew. Chem., Int. Ed., 2012, 51, 3377 CrossRef CAS PubMed.
- A. Boultif and D. Louer, J. Appl. Crystallogr., 2004, 37, 724 CrossRef CAS.
- Another functional (wB97XD) and solvent parameter (SMD) showed a similar trend as shown in Table S2.† Although wB97XD/SMD resulted in an energetically favorable singlet dimer in solution (ΔG = −5.1 kcal mol−1), the dimerization interfered with the solvent as compared with the energy in vacuum (ΔG = −13.5 kcal mol−1).
- Y. Marcus and G. Hefter, Chem. Rev., 2006, 106, 4585 CrossRef CAS PubMed.
- (a) M. Feng, J. Zhao and H. Petek, Science, 2008, 320, 359 CrossRef CAS PubMed; (b) J. Zhao, M. Feng, J. Yang and H. Petek, ACS Nano, 2009, 3, 853 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Ionic conductivity; solid state ESR spectrum; crystallographic table; MEM analysis; and DFT calculations. CCDC 1458690 and 1458691. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc01209d |
| ‡ Present address: Department of Materials, Physics, and Energy Engineering, Nagoya University, Chikusa-ku, Nagoya 464-8603, Japan. |
| This journal is © The Royal Society of Chemistry 2016 |