
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheranostic metal–organic framework core–shell composites for magnetic resonance imaging and drug delivery†
Huai-Xin
Zhao‡
b,
Quan
Zou‡
a,
Shao-Kai
Sun
a,
Chunshui
Yu
ac,
Xuejun
Zhang
a,
Rui-Jun
Li
a and
Yan-Yan
Fu
*a
aSchool of Medical Imaging, Tianjin Medical University, Tianjin 300203, China. E-mail: fuyanyan365@163.com
bCollege of Chemistry, Research Center for Analytical Sciences, State Key Laboratory of Medicinal Chemical Biology (Nankai University), Tianjin Key Laboratory of Molecular Recognition and Biosensing, and Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Nankai University, 94 Weijin Road, Tianjin 300071, China
cDepartment of Radiology, Tianjin Key Laboratory of Functional Imaging, Tianjin Medical University General Hospital, Tianjin 300052, China
First published on 26th April 2016
Abstract
Metal–organic frameworks (MOFs) have shown great potential in designing theranostic probes for cancer diagnosis and therapy due to their unique properties, including versatile structures and composition, tunable particle and pore size, enormous porosity, high surface area, and intrinsic biodegradability. In this study, we demonstrate novel MOF-based theranostic Fe3O4@UiO-66 core–shell composites constructed by in situ growth of a UiO-66 MOF shell on a Fe3O4 core for simultaneous drug delivery and magnetic resonance (MR) imaging. In the composites, the UiO-66 shell is devoted for encapsulating the drug, whereas the Fe3O4 core serves as a MR contrast agent. The Fe3O4@UiO-66 core–shell composites show good biocompatibility, high drug loading capacity, sustained drug release, and outstanding MR imaging capability, as well as effective chemotherapeutic efficacy, demonstrating the feasibility of designing theranostic Fe3O4@UiO-66 core–shell composites for cancer diagnosis and therapy.
Introduction
Metal–organic frameworks (MOFs) are an emerging class of organic–inorganic hybrid porous materials built from metal ion or cluster nodes and organic linkers, which have already been explored for a variety of applications, including separation, catalysis, and gas storage.1,2 In recent years, MOFs have been scaled down to nanometer sizes to form nanoscale MOFs and focused on their preliminary biomedical applications in drug delivery and bioimaging.3–6MOFs have been successfully employed as drug delivery vehicles owing to their unique properties suitable for drug loading and release.7 Different from most of the existing pure organic and inorganic carrier materials, MOFs have exceptionally high surface area and enormous porosity, which are favorable for entrapment of large amounts of drugs, tunable pore size and hydrophilic–hydrophobic cavities to host a variety of drugs with different physico-chemical properties, controllable host–guest interactions and intrinsic biodegradability. Since Ferey's group8 first reported the ability of iron-based MOFs as drug carriers to encapsulate ibuprofen molecules at unprecedented levels and deliver the drug continuously, with no burst effect, many researchers have developed other MOFs, such as UMCM-1, ZIF-8, MIL-53, MOF-74, Gd-MOF, UiO-66, Cu-BTC, and chiral MOFs, for drug delivery.9–18
Paramagnetic metal ions-containing MOFs are also promising as contrast agents for magnetic resonance (MR) imaging.19,20 Compared with clinical small molecule contrast agents, the framework construction features ensure that MOFs not only possess large amounts of paramagnetic metal centers, but also exhibit enhanced per-metal-based relaxivity. Lin and co-workers first demonstrated the potential of Gd-based MOFs as MR contrast agents.21 The Gd-based MOFs show excellent longitudinal relaxivity; however, leaching of the free Gd3+ ions causes nephrogenic systemic fibrosis,22 which precludes their clinical applications. Given that Mn2+ and Fe3+ ions are also known as potent paramagnetic metal ions, with much lower toxicity than Gd3+ ions, low toxic Mn-based MOFs and non-toxic iron-carboxylate MOFs, have been developed for T1/T2-weighted MR contrast enhancement.23–25 The biocompatibility of Mn/Fe-MOFs-based MR contrast agents has been improved, but the moderate relaxivity of Fe/Mn-MOFs still limits the imaging sensitivity, which hinders their practical applications. To circumvent the problem and make full use of the extraordinary drug encapsulation capacity of MOFs, the incorporation of nanoparticles with unique magnetic properties into MOFs is indeed an effective strategy.
Iron oxide nanoparticles are prevalent in MR imaging due to their ability to significantly shorten transverse relaxation time and excellent biocompatibility.26 Moreover, various formulations of iron oxide nanoparticles, such as ferumoxsil, ferrixan and ferumoxide, have already been approved by the FDA as T2-MR contrast agents for clinical use. Recently, Fe3O4-based core–shell nanoparticles, such as Fe3O4@Au, Fe3O4@CuInS2, Fe3O4@PDA, Fe3O4@PPy, Fe3O4@Cu2−xS, have also drawn considerable attention owing to their superior contrast effect in MR imaging.27–33 For these reasons, the employment of Fe3O4 as magnetic nanoparticles to design multifunctional MOF-based composites with high relaxivity, large drug payload and good biocompatibility is feasible. Recently, two groups have developed multifunctional Fe3O4@PAA/AuNCs/ZIF-8 NPs34 and RITC-Fe3O4@IRMOF-3/FA NPs35 by coating Fe3O4 with two different Zn-based MOFs for imaging and drug delivery, which demonstrate the practicability of using MOF-based composites in biomedicine. However, the poor moisture stability of IRMOF-3 and the toxicity of Zn-based MOFs, originating from the ion channel/DNA damage caused by the competition of Zn2+ with Fe2+ and Ca2+, hinder their practical application in biomedicine.36,37 Furthermore, the synthesis procedures usually include multiple steps and the capability of MOF-based composites for imaging and drug delivery, as well as the toxicity should be further systematically investigated. Thus, further development of novel MOF-based theranostic agents via a simple method is necessary and of great interest.
Herein, we report our initial effort to simply incorporate Fe3O4 nanoparticles into the MOF UiO-66 to fabricate novel theranostic MOF core–shell composites (Fe3O4@UiO-66) for in vitro and in vivo MR imaging and drug delivery (Scheme 1). UiO-66, as one class of zirconium-based MOFs, is constructed with Zr(IV) and 2-amino-1,4-benzenedicarboxylate (NH2–H2BDC) ligands and has received great attention in drug delivery due to its excellent chemical and solvent stability.12,15 The coordination of a Zr(IV)-cluster and linear ligands forms a cubic rigid 3D porous structure comprising octahedral cavities with a diameter of 1.1 nm and tetrahedral cavities with a diameter of 0.6 nm.38 The presence of Zr–O clusters, numerous open cavities, metal sites and amphiphilic character make UiO-66 advantageous for capturing and releasing anticancer drugs such as doxorubicin (DOX) based on the strong coordination interactions between the hydroxyl groups in DOX and Zr(IV) centers in UiO-66. In addition, the toxicity of UiO-66 has been demonstrated to be relatively low.37 Thus, UiO-66, which not only exhibits exceptional chemical and solvent stability, but also possesses good biocompatibility, was selected to fabricate the shell over the Fe3O4 nanoparticles for DOX delivery. Fe3O4@UiO-66 core–shell composites were synthesized via a facile in situ growth method based on the controllable growth of a UiO-66 shell on a carboxylate-terminated Fe3O4 core. The formed Fe3O4@UiO-66 composites simultaneously possessed the T2-MR contrast properties of the Fe3O4 cores and the drug delivery ability originating from the MOF shells. As a result, the Fe3O4@UiO-66 composites show excellent stability, high drug loading capacity, low cytotoxicity, negligible in vivo toxicity and obvious MR signal attenuation effect. Furthermore, the DOX encapsulated composites show a sustained drug release and exhibit long-lasting and efficient anticancer therapeutic efficacy. All the results demonstrate that the developed Fe3O4@UiO-66 core–shell composites possess great potential as a novel theranostic agent for MR imaging and drug delivery in biomedicine.
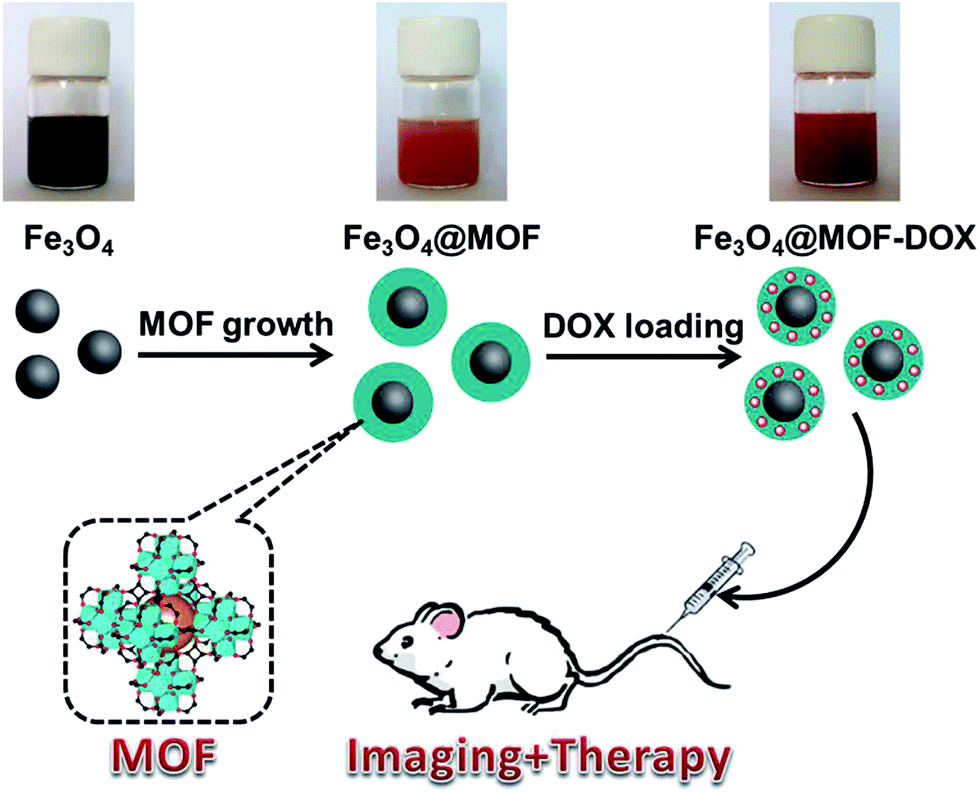 | ||
| Scheme 1 Schematic of the fabrication of Fe3O4@MOF core–shell composites for imaging and therapy. | ||
Results and discussion
Synthesis and characterization of Fe3O4 and Fe3O4@UiO-66
Fe3O4 nanoparticles were synthesized through a hydrothermal method according to the literature.39 The as-synthesized Fe3O4 nanoparticles were spherical with a diameter of 150 nm (Fig. 1A) and in the cubic phase (JCPDS: 19-0629) (Fig. 1E). The Fe3O4@UiO-66 composites were then synthesized by an in situ self-assembly of UiO-66 on the surface of Fe3O4 to obtain the core–shell theranostic agent. In a typical process, Fe3O4 nanoparticles were directly dispersed into the synthetic precursor of UiO-66 composed of ZrCl4, NH2–H2BDC and DMF, and the reaction was accomplished through a simple hydrothermal procedure. The developed method avoids time-consuming layer-by-layer MOF growth and further modification of core particles, which is much simpler than most of the previous methods of synthesizing MOF-based core–shell composites.40,41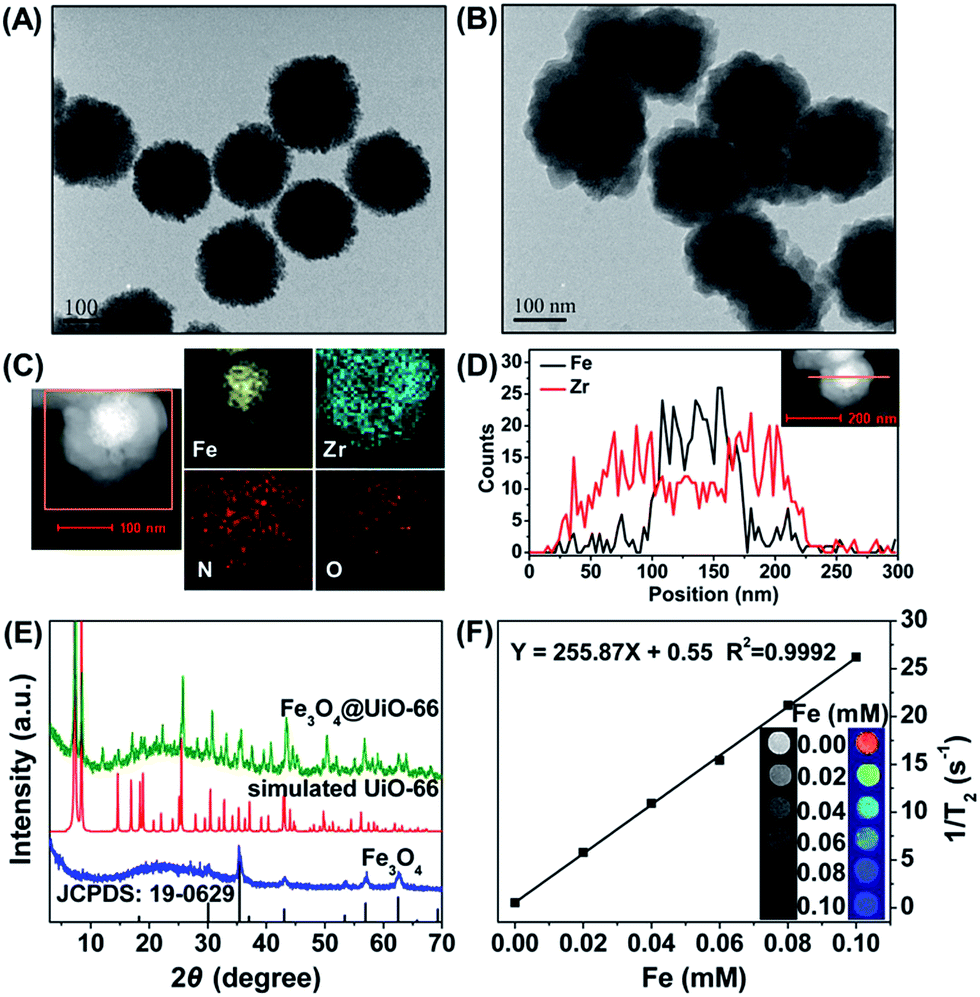 | ||
| Fig. 1 (A and B) TEM images of the synthesized Fe3O4 (A) and the Fe3O4@UiO-66 core–shell composites (B). (C) HAADF-STEM image and the corresponding elemental mapping of one core–shell Fe3O4@UiO-66 composite. (D) HAADF-STEM image of Fe3O4@UiO-66 composites and the corresponding EDS line scan. (E) XRD patterns of synthesized Fe3O4@UiO-66, Fe3O4, simulated UiO-66 and the JCPDS file of Fe3O4. (F) T2-weighted MR images and transverse relaxivity of Fe3O4@UiO-66 at different Fe concentrations. | ||
To control the morphology of the core–shell composites, the concentrations of the precursors of UiO-66 were optimized. After mixing with different concentrations of UiO-66 precursor solutions, the Fe3O4@UiO-66 composites with different UiO-66 shell thickness (5, 25, 50 nm) were obtained (Fig. S1, ESI†). The Fe3O4@UiO-66 composites show uniform core–shell morphology with a 25 nm thickness of the UiO-66 shell when the ratio of Fe3O4 and UiO-66 precursor reached 25 mg in 37.5 mg ZrCl4 (29 mg NH2–H2BDC/18 mL DMF) (Fig. 1B). Either the UiO-66 shell was too thick or no shell was formed if the ratio was higher or lower than the optimized ratio. Thus, the Fe3O4@UiO-66 core–shell composites synthesized under the optimized condition were used for the following experiments. Furthermore, high angle annular dark field scanning transmission electron microscopy (HAADF-STEM) and elemental mapping analysis were used to verify the core–shell structure of the Fe3O4@UiO-66 composite (Fig. 1C and D).
A clear contrast between the core and shell was obtained wherein the core appeared dark, whereas the shell appeared bright. Moreover, the EDS (energy dispersive spectrometry) line scanning data and elemental mapping analysis reveal that the Fe and Zr elements were distributed in the core and shell, respectively. All the results demonstrated the successful formation of a core–shell structure for the Fe3O4@UiO-66 composite.
The simultaneous existence of the characteristic peaks of Fe3O4 and UiO-66 in the X-ray diffraction (XRD) pattern of Fe3O4@UiO-66 indicated the successful formation of a UiO-66 shell on the surface of Fe3O4 nanoparticles without altering their crystallinity (Fig. 1E). The characteristic peaks of UiO-66 at 1570 cm−1, 1435 cm−1, and 1386 cm−1 and Fe3O4 at 1651 cm−1 and 595 cm−1 in the Fourier transform infrared (FT-IR) spectrum of Fe3O4@UiO-66 indicated the formation of the Fe3O4@UiO-66 composites (Fig. S2, ESI†). The two peaks at 3416 cm−1 and 3373 cm−1 in the spectrum of Fe3O4@UiO-66, belonged to the asymmetric and symmetric stretching absorptions of the primary amine groups in the NH2–H2BDC ligands, respectively, which further verified the successful growth of the UiO-66 shell on the Fe3O4 core. The contents of Fe and Zr elements in the prepared Fe3O4@UiO-66 were 30.6% and 0.9%, respectively. Thermogravimetric analysis results showed that the Fe3O4@UiO-66 composites decomposed at 340 °C, which is a similar breakdown temperature to that of the synthesized UiO-66 nanocrystals (Fig. S3, ESI†). The hydrodynamic size and surface zeta potential of Fe3O4@UiO-66 composites in phosphate buffer solution (PBS) were 241.5 ± 28.5 nm (polydispersity index = 0.340) and −25.7 ± 5.2 mV, respectively (Fig. S4, ESI†). The results indicate that the composites had good dispersity and colloidal stability, which ensured their practical application in drug delivery and MR imaging.
The drug loading capacity of Fe3O4@UiO-66 composites depends on the porosity of the UiO-66 shells. N2 adsorption isotherms were used to characterize the surface area and porosity of Fe3O4@UiO-66 (Fig. 2A). The total pore volume (Vtotal) and BET surface area (SBET) of the synthesized Fe3O4@UiO-66 were calculated to be 0.21 cm3 g−1 and 149.75 m2 g−1, respectively, which are large for drug loading yet much lower than those of the pure UiO-66 due to the inner nonporous Fe3O4 core. In addition, the Fe3O4@UiO-66 composites possessed an inter-particle mesopore at 3.5 nm (Table S1, ESI†), which makes them efficient for drug delivery.
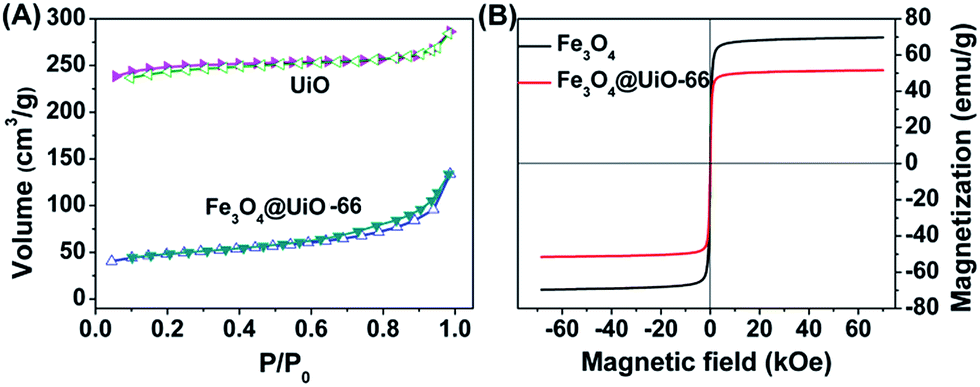 | ||
| Fig. 2 (A) N2 adsorption–desorption isotherms of UiO-66 and Fe3O4@UiO-66. (B) Magnetization hysteresis curves of Fe3O4 and Fe3O4@UiO-66. | ||
Superparamagnetism is essential to MR imaging. Thus, the magnetic properties of the Fe3O4@UiO-66 composites were investigated in a magnetic field range from −70 to +70 kOe at room temperature (Fig. 2B). The saturation magnetization (Ms) of the Fe3O4@UiO-66 was 51.58 emu g−1, smaller than that of Fe3O4 nanoparticles (69.69 emu g−1) due to the encapsulation by the UiO-66 layer. The high Ms value and the existing hysteresis loop without significant coercivity and remanence in the magnetization hysteresis curves demonstrated the strong superparamagnetic character of the as-synthesized Fe3O4@UiO-66 composites.42 Furthermore, the Fe3O4@UiO-66 composites could be quickly collected using the magnet (Fig. S5, ESI†).
The strong superparamagnetism of the Fe3O4@UiO-66 composites ensured their ability to act as a T2 contrast agent for MR imaging. T2-weighted MR images of Fe3O4@UiO-66 showed an obvious concentration-dependent darkening effect with a high transverse relaxivity (r2) of 255.87 mM−1 s−1 (Fig. 1F), and the r2 values decreased as the thickness of the UiO-66 shell increased due to the reduced ratio of Fe3O4 to UiO-66 in the composite (Table S2 and Fig. S6, ESI†). Furthermore, the r2 value of the prepared Fe3O4@UiO-66 (1396 mg−1 mL s−1) was much higher than that of Fe3O4@PAA/AuNCs/ZIF-8 NPs (53.79 mL mg−1 s−1)34 and several clinical Fe-based T2-weighted contrast agents such as ferumoxsil (72 mM−1 s−1), ferumoxide (98.3 mM−1 s−1) and Resovist (150 mM−1 s−1).43 These results show the great potential of Fe3O4@UiO-66 in T2-MR imaging.
Drug loading/release of Fe3O4@UiO-66 composites
The stability of the nanocarrier in a wide pH range from basic to acidic is essential for drug loading/release. Thus, we investigated the stability of the as-synthesized Fe3O4@UiO-66 composites at different pH values (4.0, 5.0, 6.0, 7.4, 8.0). No significant changes in the XRD pattern (Fig. 3A) or the morphology (Fig. S7, ESI†) of the Fe3O4@UiO-66 composites at these pH values were observed. The results indicated that the composites had super stability.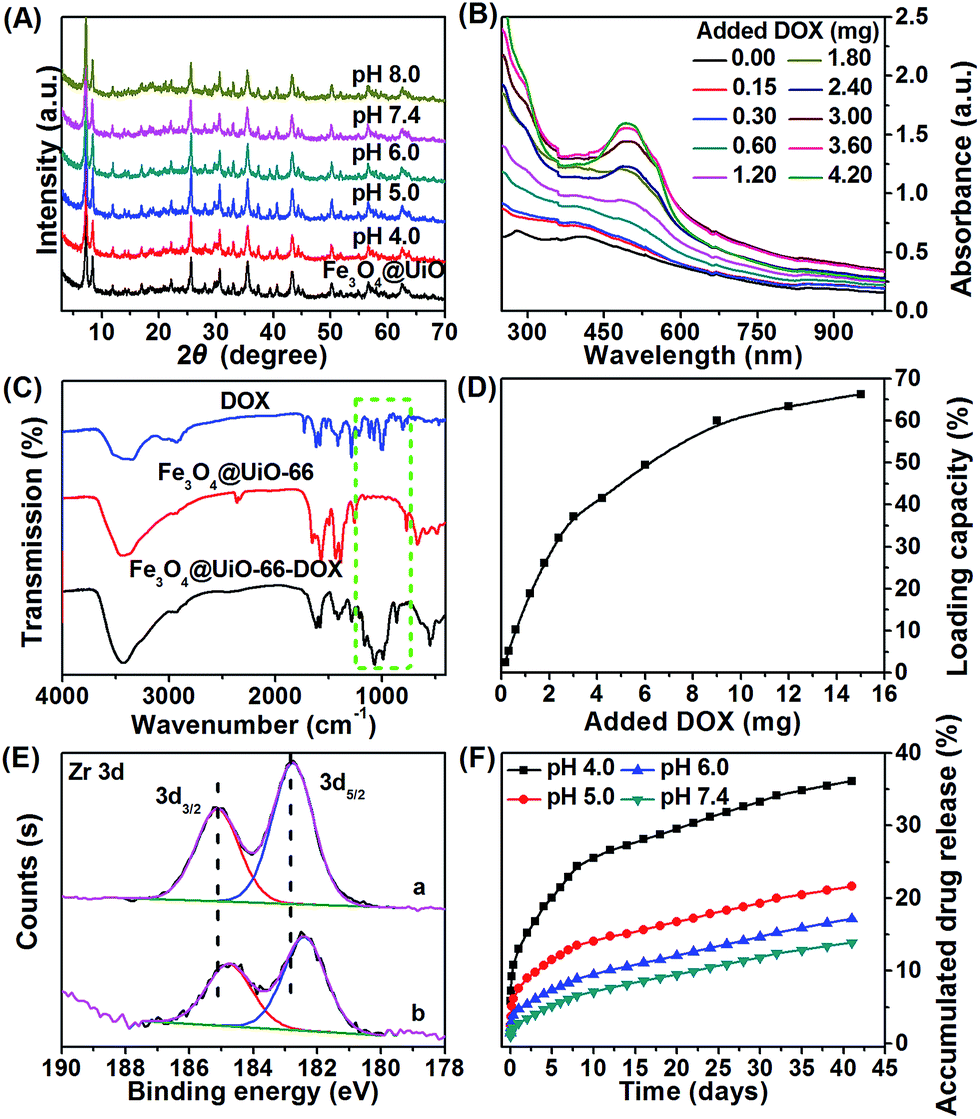 | ||
| Fig. 3 (A) XRD patterns of Fe3O4@UiO-66 after immersion in different pH solutions for one week. (B) UV-Vis-NIR spectra of Fe3O4@UiO-66-DOX obtained at various DOX added contents after removal of excess free drug molecules. (C) FT-IR spectra of DOX, Fe3O4@UiO-66 and Fe3O4@UiO-66-DOX. (D) Drug loading capacity of Fe3O4@UiO-66 at different DOX added contents. (E) XPS spectra of Zr 3d for Fe3O4@UiO-66 (a) and Fe3O4@UiO-66-DOX (b). (F) Accumulated drug release (ADR) from Fe3O4@UiO-66-DOX composites in buffers with four different pH values. | ||
To evaluate the drug loading capacity of the Fe3O4@UiO-66 composites, a commonly used anticancer drug, DOX, was mixed with Fe3O4@UiO-66 in PBS at pH 8.0 for 24 h. After removing the excess unloaded DOX, the drug loaded composites Fe3O4@UiO-66-DOX were obtained. UV-Vis-NIR spectra of Fe3O4@UiO-66 before and after encapsulation with DOX were measured. The appearance of the characteristic peak of DOX at 500 nm in the UV-Vis-NIR spectra of Fe3O4@UiO-66-DOX and the increase of the characteristic peak with the added DOX confirmed the successful loading of DOX into the Fe3O4@UiO-66 composites (Fig. 3B). Moreover, the presence of characteristic absorption bands (indicated by green dashed rectangle) of DOX in the spectra of Fe3O4@UiO-66-DOX also indicated the incorporation of DOX molecules into Fe3O4@UiO-66 (Fig. 3C).
The drug loading capacity increased from 2.5 to 66.3 wt% as the amount of DOX increased from 0.15 to 15 mg (Fig. 3D). Furthermore, the drug loading capacity increased with the thickness of the UiO-66 shell (Table S2, ESI†). The DOX loading capacity of Fe3O4@UiO-66 could reach 66.3 wt% with an ultrahigh loading content of 2.0 mg DOX per mg composites, which is almost the highest DOX payload among the MOFs carriers.25,44–46 The impressive result was meaningful for clinical applications, because the administration of high dosages could be realized using a small amount of composites.
The high drug loading capacity of Fe3O4@UiO-66 composites was probably attributed to the large surface area of the UiO-66 shell and the interactions between DOX and UiO-66. A remarkable fluorescence quenching of DOX along with color change from brown to wine for the Fe3O4@UiO-66-DOX dispersion confirmed the strong interaction between DOX and Fe3O4@UiO-66 (Fig. S5 and S8, ESI†). Potential interactions include π–π stacking between the aromatic anthracycline of DOX and the aromatic pore walls of UiO-66, hydrogen bonding between the oxygen atoms of DOX and the amino groups in UiO-66, van der Waals forces, electrostatic interactions, and coordination bonding.47 Among the above interactions, stable coordination bonding between the deprotonated hydroxyls in DOX and the numerous Zr sites in the UiO-66 framework played the leading role in drug loading, as confirmed by ultraviolet-visible (UV-Vis) spectroscopy and X-ray photoelectron spectroscopy (XPS) (Fig. 3E and S9, ESI†). Red shift of the UV-Vis spectrum of Fe3O4@UiO-66-DOX in comparison with that of DOX after DOX loading into Fe3O4@UiO-66 indicates the formation of a DOX–Zr complex in the Fe3O4@UiO-66 framework, which is also supported by the similar spectral modification of DOX with free Zr(IV) ions (Fig. S9†). XPS results showed that the binding energy of Zr 3d shifted from 182.76 and 185.12 eV for Fe3O4@UiO-66 to 182.40 and 184.76 eV for Fe3O4@UiO-66-DOX (Fig. 3E). The binding energy of Zr 3d shifting to lower levels after DOX encapsulation could be attributed to electron transfer due to binding of DOX to active Zr sites.48–50 The formation of coordination bonds between DOX and metal centres (Zr, Fe, Zn) has also been described previously.44,51,52 Owing to the strong coordination bonding of DOX–Zr, the Fe3O4@UiO-66 showed a remarkable and efficient DOX payload.
The DOX release behavior of the Fe3O4@UiO-66-DOX composites was investigated at different pH values (4.0, 5.0, 6.0, and 7.4). The time-dependent accumulated drug release (ADR) curves showed a slow and sustained release pattern without any burst effect (Fig. 3F). The release rate would generate a stable drug concentration and provide sufficient time for the Fe3O4@UiO-66-DOX to accumulate at the tumor site. As shown in Fig. 3F, about 36.1% and 21.6% of the DOX were released in 41 days at pH 4.0 and 5.0, respectively, whereas 17.1% and 13.8% of DOX were released at pH 6.0 and 7.4, indicating the sensitivity of Fe3O4@UiO-66-DOX to acidic tumor microenvironments. The pH-responsive DOX release was controlled by the drug–matrix interactions under acidic conditions. At acidic pH, the amino group in DOX was easily protonated, giving DOX a positive charge. Moreover, the surface zeta potential of Fe3O4@UiO-66 became less negative in acidic conditions (−9.8 ± 1.1 mV at pH 4.0 and −25.7 ± 5.2 mV at pH 7.4). Thus, the electrostatic interactions between Fe3O4@UiO-66 and DOX were weakened, which promoted the drug release in acidic conditions.53 Moreover, the breakage of the coordination bonds between the protonated hydroxyls in DOX and Zr sites under acidic conditions would accelerate the DOX release. In addition to the pH dependent DOX release, the tendency of endogenous phosphate salts in the endosomes to coordinate to the Zr sites also facilitates the release of DOX from the Fe3O4@UiO-66 composites.52,54 These results indicate that Fe3O4@UiO-66 nanocarriers could deliver the drug to tumor tissue sustainably and effectively, which would be beneficial for the diminishing of toxic side effects and decreasing of patient discomfort.
In vitro cytotoxicity
In vitro cell viabilities of different concentrations of Fe3O4@UiO-66, Fe3O4@UiO-66-DOX and free DOX on HeLa cells were evaluated by MTT assay to study the bio-toxicity of Fe3O4@UiO-66 and the therapeutic effect of Fe3O4@UiO-66-DOX. The HeLa cells treated with Fe3O4@UiO-66 showed no obvious toxicity (nearly 100% cell viability), even at a concentration up to 500 mg L−1, indicating the good biocompatibility of the synthesized Fe3O4@UiO-66 composites (Fig. 4A). In contrast, the Fe3O4@UiO-66-DOX exhibited significant increase in anticancer activity against HeLa cells with the increase of the loaded DOX concentration (Fig. 4B). Nearly 60% of HeLa cells were killed after incubation with Fe3O4@UiO-66-DOX, even at a low DOX loading concentration of 20 mg L−1 and the Fe3O4@UiO-66-DOX had similar cell toxicity to that of free DOX. In addition, the Fe3O4@UiO-66-DOX showed greater lethality against HeLa cells after incubating for a longer time (48 h), indicating the long-term and sustained DOX release from the Fe3O4@UiO-66 composites.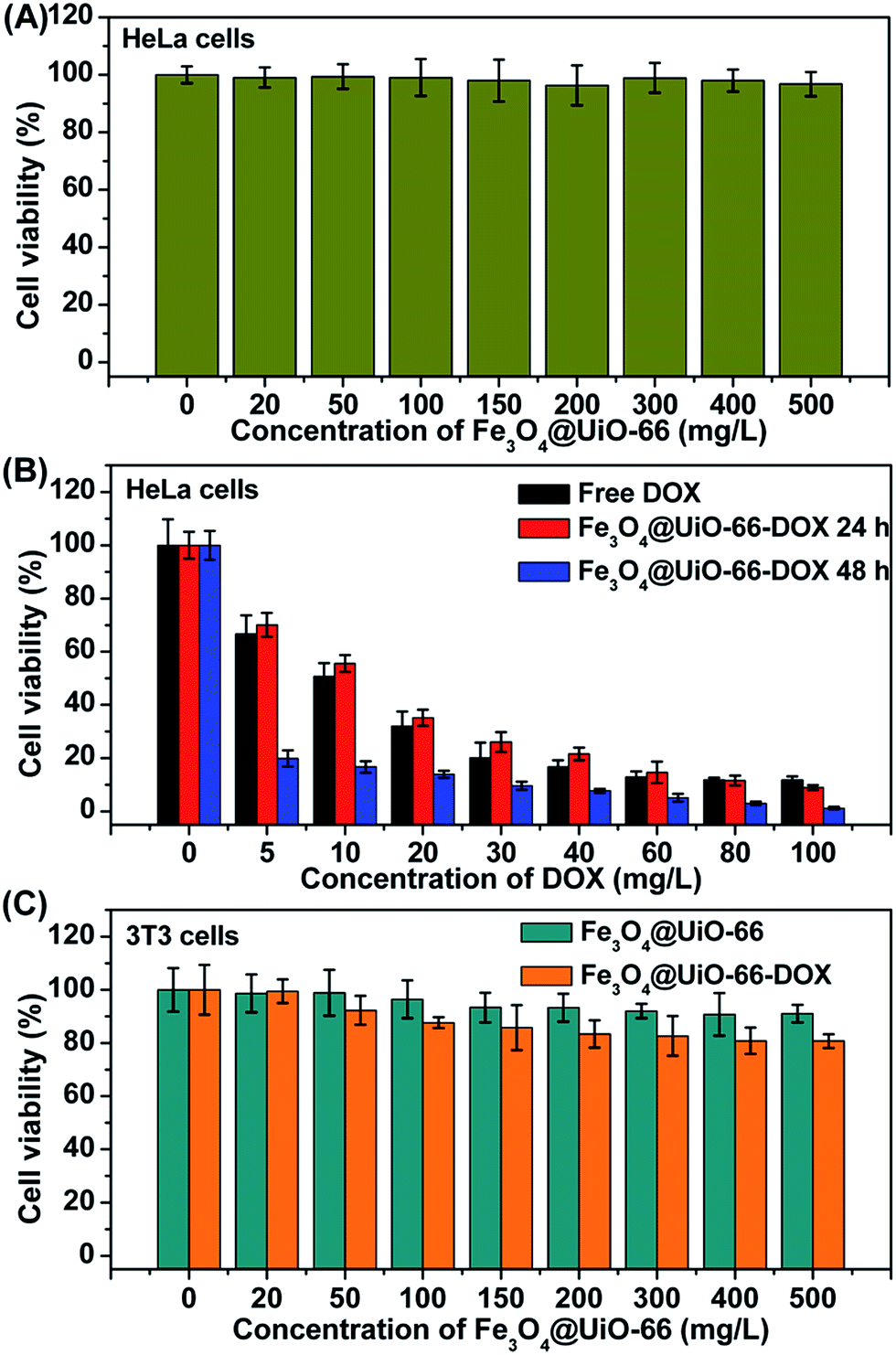 | ||
| Fig. 4 (A) Cell viability of HeLa cells after incubation with different concentrations of Fe3O4@UiO-66. (B) Cell viability of HeLa cells after incubation with free DOX and Fe3O4@UiO-66-DOX for 24 h or 48 h at the same concentration of DOX. (C) Cell viability of 3T3 cells after incubation with Fe3O4@UiO-66 and Fe3O4@UiO-66-DOX for 24 h at the same concentration of Fe3O4@UiO-66. | ||
To demonstrate the low toxic and side effects of DOX loaded Fe3O4@UiO-66 on normal cells, we further evaluated the biocompatibility of different concentrations of Fe3O4@UiO-66 and Fe3O4@UiO-66-DOX on 3T3 cells by MTT assay (Fig. 4C). As expected, the 3T3 cells treated with Fe3O4@UiO-66 showed high viability, further indicating the good biocompatibility of the Fe3O4@UiO-66 composites. Furthermore, the DOX loaded Fe3O4@UiO-66 also showed negligible toxicity on 3T3 cells, demonstrating the low side effect of Fe3O4@UiO-66-DOX on normal cells. Although Fe3O4@UiO-66-DOX could be phagocytized by both normal cells and cancer cells, the tremendous difference in cell viability between the HeLa cells and the 3T3 cells may be attributed to the different DOX release rates determined by the cellular microenvironments. The fast reproduction of cancer cells makes the cellular microenvironment acidic, which facilitates DOX release from Fe3O4@UiO-66-DOX, and thus leads to high toxicity to cancer cells. These results demonstrate the availability of the Fe3O4@UiO-66 composites as drug carriers for cancer cell killing.
In vitro and in vivo MR imaging
The high transverse relaxivity of the synthesized Fe3O4@UiO-66 core–shell composites gives them potential as a contrast agent for cancer diagnosis. The T2-weighted MR images of HeLa cells incubated with different concentrations of Fe3O4@UiO-66 composites (0, 25, 50, 100, 150 and 200 mg L−1) became much darker with increasing of the concentration of Fe3O4@UiO-66 due to the dose-dependent cellular uptake. The results demonstrate the capability of Fe3O4@UiO-66 composites as a T2-weighted MR contrast agent for in vitro MR imaging (Fig. 5A).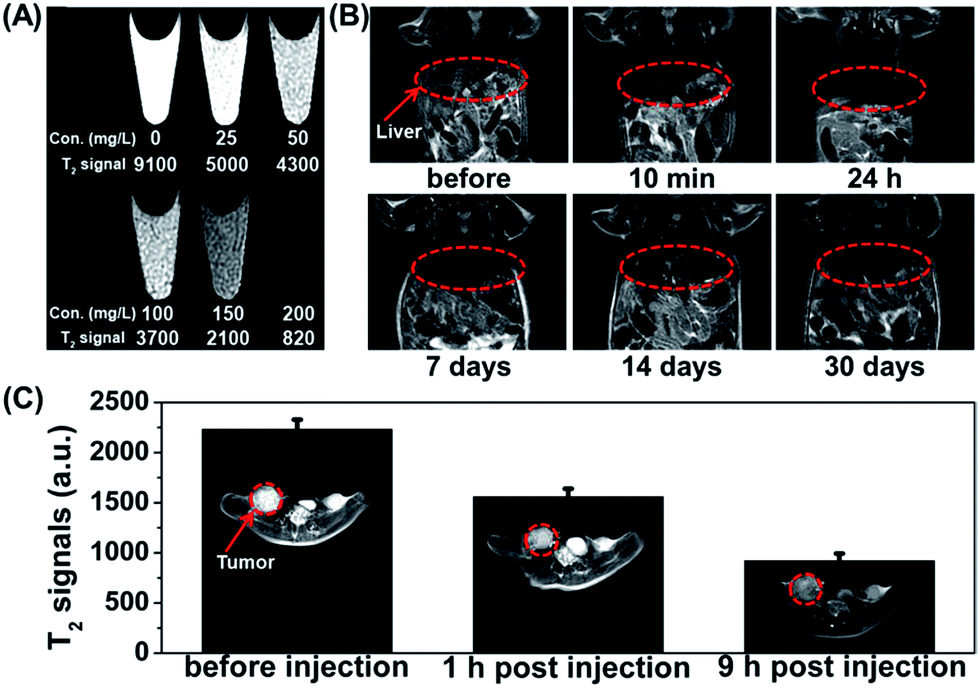 | ||
| Fig. 5 (A) MR images of HeLa cells after incubation with different concentrations (0, 25, 50, 100, 150 and 200 mg L−1) of Fe3O4@UiO-66 for 24 h. (B) T2-weighted MR images of the Kunming mouse before and after intravenous injection of Fe3O4@UiO-66 at different time points (liver region marked by red cycles). (C) T2-weighted MR images and T2-MR signals of tumor on HeLa-tumor bearing mice before injection, 1 h and 9 h post injection of Fe3O4@UiO-66 intravenously (tumor region marked by red cycles). | ||
The feasibility of the Fe3O4@UiO-66 composites for in vivo MR imaging was also tested. A significant darkening effect was observed in the liver region of the Kunming mouse at 10 min post-injection of Fe3O4@UiO-66 composites (400 μL, 5 mg mL−1, 24 mg Fe per kg), indicating the ability of Fe3O4@UiO-66 to enhance in vivo T2-weighted images (Fig. 5B). The dramatic T2 signal intensity decrease is probably due to the phagocytosis of the Fe3O4@UiO-66 composites by the liver macrophage cells in reticuloendothelial systems (RES).55 Considering the excellent MR imaging capability of Fe3O4@UiO-66 composites, MR imaging of HeLa tumor-bearing mice was then carried out. After being intravenously injected with Fe3O4@UiO-66 composites, remarkable darkening effect was observed in the tumor area just 1 h post-injection and the MR image became even darker at 9 h post-injection (Fig. 5C). The quantified T2-weighted MR signals in the tumor also showed a gradual decrease over 9 h post-injection, demonstrating the accumulation of Fe3O4@UiO-66 composites in the tumor, which is probably due to the enhanced permeability and retention (EPR) effect of tumors.
Biodistribution and toxicology studies
The potential toxic effects of nanomaterials are a major concern for their biomedical applications. Thus, the metabolism, biodistribution and long-term toxicity of the as-synthesized Fe3O4@UiO-66 core–shell composites were systematically investigated. For metabolism study, the T2-weighted images of the Kunming mouse injected with the composites were collected after 1, 7, 14 and 30 days of injection. Strong darkening effect after injection and signal recovery at 30 days post-injection were observed in the liver region, but no time-dependent darkening effect was observed in the urinary bladder, indicating that Fe3O4@UiO-66 was not excreted from the kidneys but from the liver (Fig. 5B). For biodistribution study, the major organs (heart, liver, spleen, lung, kidney) of the mice treated with Fe3O4@UiO-66 for 1, 7 and 30 days were collected, weighed and solubilized by aqua regia for AAS measurement of Fe and ICP-MS determination of Zr concentrations, respectively. As expected and consistent with the T2-weighted MR images, high levels of Fe and Zr contents mainly accumulated in mononuclear phagocyte systems such as the spleen and liver (Fig. 6A and S10, ESI†). The Fe levels in all measured organs constantly decreased as the post-injection time prolonged and nearly dropped back to the normal levels after 30 days, except for the liver, in which the Fe content was slightly higher than the control. It was noteworthy that Zr was not detected in the heart, lung or kidney due to the absence of Zr in the organism and no accumulation of Fe3O4@UiO-66 in these organs. In addition, the Zr levels in the liver and spleen could be gradually metabolized over time, which was consistent with the biodistribution of Fe.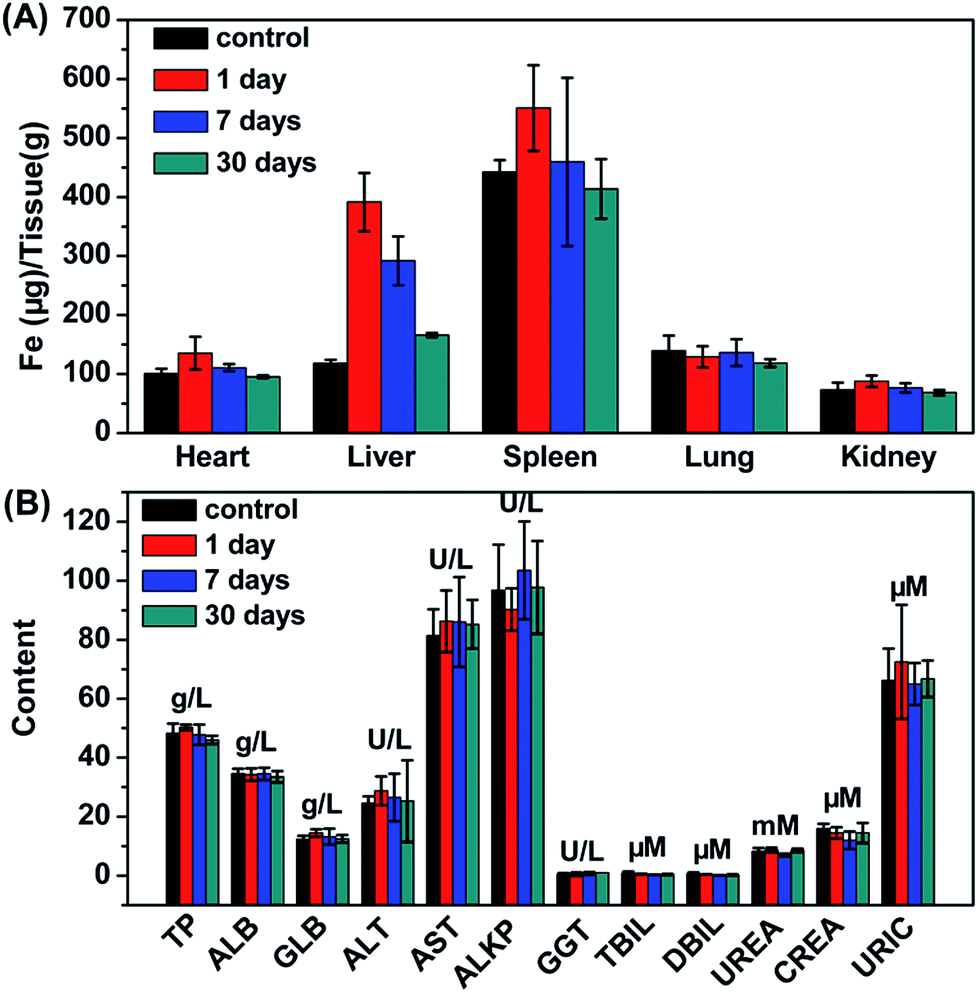 | ||
| Fig. 6 (A) Time-dependent biodistribution of Fe in various organs of mice. (B) Blood biochemistry of mice treated with Fe3O4@UiO-66 at dose of 24 mg kg−1 measured at 1, 7 and 30 days post-injection. | ||
For in vivo long-term toxicity, the body weight and the blood analysis were evaluated. The body weights of the control group and the experimental group treated with Fe3O4@UiO-66 maintained similar increases over 30 days, and no death or significant body weight drop were observed in the experimental group, illustrating that the injection of the Fe3O4@UiO-66 composites did not perceivably interfere with the growth of the mice (Fig. S11, ESI†). Blood analysis parameters, including liver function markers (total protein, TP; albumin, ALB; globulin GLB; alanine aminotransferase, ALT; aspartate aminotransferase, AST; alkaline phosphatase, ALKP; gamma glutamyl transaminase, GGT; total bilirubin, TBIL; direct bilirubin, DBIL) and kidney function markers (urea, UREA; creatinine, CREA; uric acid URIC), at different time points post-injection of Fe3O4@UiO-66 appeared to be normal compared with those in the control group, indicating that no obvious liver or kidney disorders were induced by the injection of Fe3O4@UiO-66 (Fig. 6B). All these results demonstrated that the as-synthesized Fe3O4@UiO-66 was a relatively safe theranostic agent for biomedical applications.
In vivo antitumor efficacy
Inspired by the biocompatibility of Fe3O4@UiO-66 and in vitro anticancer effect of the DOX-loaded Fe3O4@UiO-66, the in vivo chemotherapy performance of Fe3O4@UiO-66-DOX was investigated using the HeLa tumor-bearing nude mice. As shown in Fig. 7A, the tumors in the mice treated with PBS grew quickly. In contrast, the tumor growth on mice treated with Fe3O4@UiO-66-DOX was effectively inhibited due to the preferential DOX accumulation and release at the tumor site via the EPR effect. Furthermore, the images and sizes of the tumor blocks isolated 30 days after being treated showed that the mice treated with Fe3O4@UiO-66-DOX had the smallest tumor size (Fig. 7B and C). All these results make a proof of concept that Fe3O4@UiO-66 can efficiently carry and release drugs into tumors, leading to effective antitumor efficacy.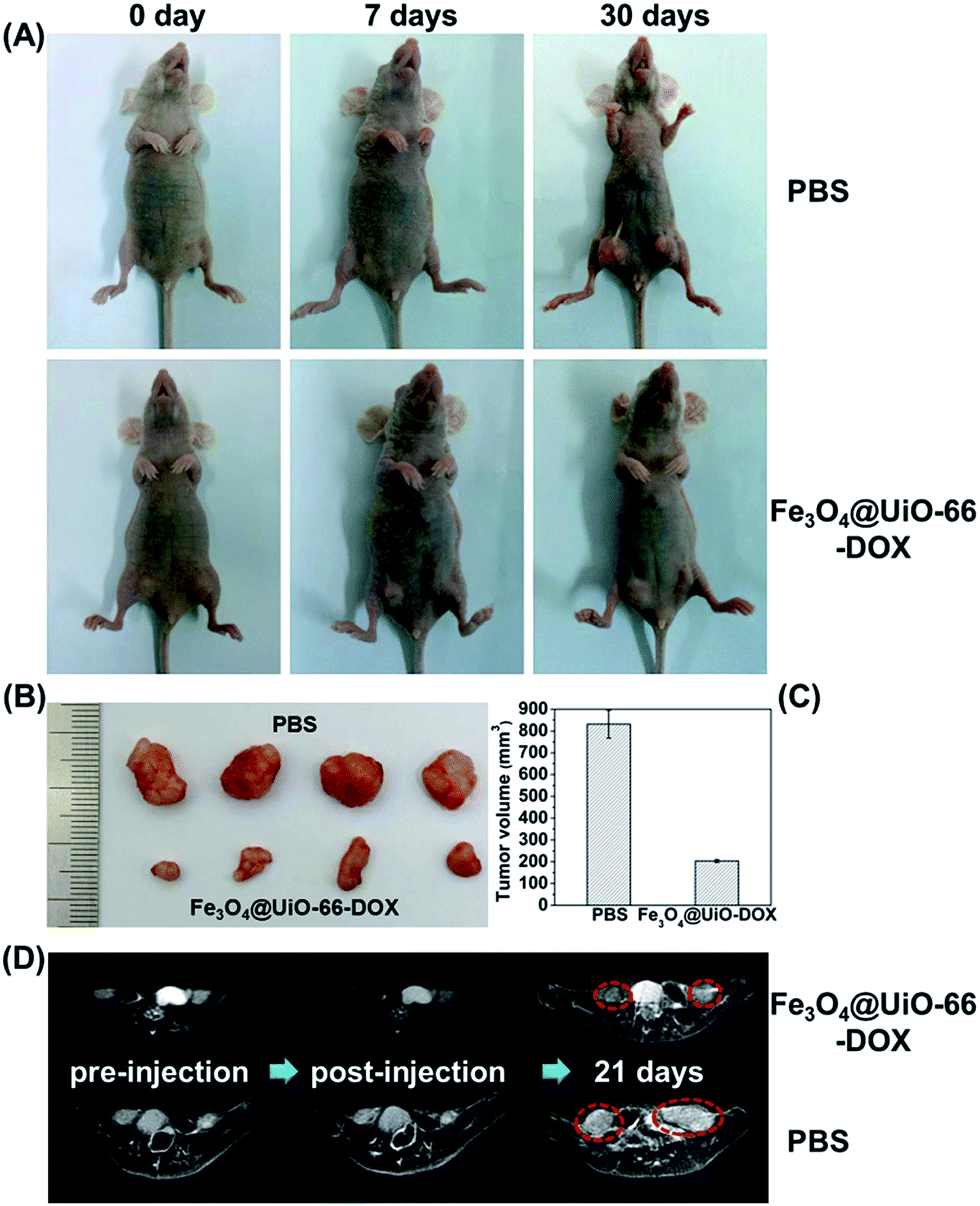 | ||
| Fig. 7 In vivo antitumor efficacy. (A) Representative images of mice before and after intravenous injection with PBS or Fe3O4@UiO-66-DOX after 7 and 30 days. (B) Images and (C) volumes of tumor blocks collected from PBS and Fe3O4@UiO-66-DOX treated groups of mice on day 30. (D) MR images of HeLa-tumor bearing mice treated with PBS or Fe3O4@UiO-66-DOX collected at different time points (tumor region marked by red cycles). | ||
Although visualized tumor size could be observed from the images of the mice and tumor blocks, adopting non-invasive MR imaging to monitor tumor development is necessary to realize precision medicine. Thus, monitoring the tumor change of the mice after being treated with Fe3O4@UiO-66-DOX via MR imaging was carried out to accurately evaluate the antitumor efficacy. The MR images of HeLa-tumor bearing mice injected with Fe3O4@UiO-66-DOX (200 μL, 5 mg mL−1) showed obvious darkening effect in the tumor region, indicating that the Fe3O4@UiO-66-DOX could passively accumulate in the tumor at 7 days post-injection (Fig. 7D and S12, ESI†). Continuous monitoring of tumor development by MR imaging was performed and efficient tumor growth inhibition in the mice treated with Fe3O4@UiO-66-DOX was observed through the obtained MR images at 21 days post-injection. In contrast, the tumors of the mice treated with PBS grew rapidly, as revealed by MR images. All the results not only further convincingly demonstrated the efficient antitumor efficacy of Fe3O4@UiO-66-DOX in terms of MR imaging, but also indicated the great potential of Fe3O4@UiO-66 in imaging-guided therapy.
Conclusions
In conclusion, we developed a novel Fe3O4@UiO-66 theranostic agent by in situ growth of a UiO-66 MOF shell on a Fe3O4 core. The obtained Fe3O4@UiO-66 core–shell composites can serve as nanocarriers and contrast agents for simultaneous drug delivery and T2-weighted MR imaging. The exceptionally high drug loading capacity (∼63 wt%, 2.0 mg DOX per mg composites), and sustained and effective drug release make Fe3O4@UiO-66 an excellent drug delivery carrier. Moreover, high transverse relaxivity (255.87 mM−1 s−1) revealed that Fe3O4@UiO-66 has the ability to act as a contrast agent for MR imaging. The cytotoxicity assay, biodistribution and in vivo toxicology studies demonstrated that the Fe3O4@UiO-66 composites possess low toxicity and good biocompatibility, which inspired us to explore their antitumor efficiency and MR imaging capability in vitro and in vivo. High cancer cell mortality, remarkable tumor size inhibition and significant darkening effect were obtained after treatment with Fe3O4@UiO-66 or Fe3O4@UiO-66-DOX in vitro and in vivo. All the results indicate that the presented novel multifunctional MOF-based composites should be very promising in cancer therapy and diagnosis due to their effective drug delivery and MR imaging.Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grants 21505099, 21435001, 21405112) and the China Postdoctoral Science Foundation (Grants 2015M571270, 2014M550146).Notes and references
- D. Zhao, D. J. Timmons, D. Yuan and H.-C. Zhou, Acc. Chem. Res., 2010, 44, 123 CrossRef PubMed.
- A. Carne, C. Carbonell, I. Imaz and D. Maspoch, Chem. Soc. Rev., 2011, 40, 291 RSC.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232 CrossRef CAS PubMed.
- A. C. McKinlay, R. E. Morris, P. Horcajada, G. Ferey, R. Gref, P. Couvreur and C. Serre, Angew. Chem., Int. Ed., 2010, 49, 6260 CrossRef CAS PubMed.
- S. Keskin and S. Kızılel, Ind. Eng. Chem. Res., 2011, 50, 1799 CrossRef CAS.
- C. Wang, D. Liu and W. Lin, J. Am. Chem. Soc., 2013, 135, 13222 CrossRef CAS PubMed.
- J. Della Rocca, D. Liu and W. Lin, Acc. Chem. Res., 2011, 44, 957 CrossRef CAS PubMed.
- P. Horcajada, C. Serre, M. Vallet-Regi, M. Sebban, F. Taulelle and G. Ferey, Angew. Chem., Int. Ed., 2006, 45, 5974 CrossRef CAS PubMed.
- L.-L. Tan, H. Li, Y.-C. Qiu, D.-X. Chen, X. Wang, R.-Y. Pan, Y. Wang, S. X.-A. Zhang, B. Wang and Y.-W. Yang, Chem. Sci., 2015, 6, 1640 RSC.
- J. Zhuang, C.-H. Kuo, L.-Y. Chou, D.-Y. Liu, E. Weerapana and C.-K. Tsung, ACS Nano, 2014, 8, 2812 CrossRef CAS PubMed.
- Y. Wu, M. Zhou, S. Li, Z. Li, J. Li, B. Wu, G. Li, F. Li and X. Guan, Small, 2014, 10, 2927 CrossRef CAS PubMed.
- D. Cunha, M. Ben Yahia, S. Hall, S. R. Miller, H. Chevreau, E. Elkaim, G. Maurin, P. Horcajada and C. Serre, Chem. Mater., 2013, 25, 2767 CrossRef CAS.
- Q. Hu, J. Yu, M. Liu, A. Liu, Z. Dou and Y. Yang, J. Med. Chem., 2014, 57, 5679 CrossRef CAS PubMed.
- T. Kundu, S. Mitra, P. Patra, A. Goswami, D. Díaz Díaz and R. Banerjee, Chem.–Eur. J., 2014, 20, 10514 CrossRef CAS PubMed.
- X. Zhu, J. Gu, Y. Wang, B. Li, Y. Li, W. Zhao and J. Shi, Chem. Commun., 2014, 50, 8779 RSC.
- C. He, K. Lu, D. Liu and W. Lin, J. Am. Chem. Soc., 2014, 136, 5181 CrossRef CAS PubMed.
- F. Ke, Y.-P. Yuan, L.-G. Qiu, Y.-H. Shen, A.-J. Xie, J.-F. Zhu, X.-Y. Tian and L.-D. Zhang, J. Mater. Chem., 2011, 21, 3843 RSC.
- C.-Y. Sun, C. Qin, C.-G. Wang, Z.-M. Su, S. Wang, X.-L. Wang, G.-S. Yang, K.-Z. Shao, Y.-Q. Lan and E.-B. Wang, Adv. Mater., 2011, 23, 5629 CrossRef CAS PubMed.
- J. Della Rocca and W. Lin, Eur. J. Inorg. Chem., 2010, 2010, 3725 CrossRef.
- D. Liu, K. Lu, C. Poon and W. Lin, Inorg. Chem., 2014, 53, 1916 CrossRef CAS PubMed.
- W. J. Rieter, K. M. L. Taylor, H. An, W. Lin and W. Lin, J. Am. Chem. Soc., 2006, 128, 9024 CrossRef CAS PubMed.
- D. R. Broome, M. S. Girguis, P. W. Baron, A. C. Cottrell, I. Kjellin and G. A. Kirk, Am. J. Roentgenol., 2007, 188, 586 CrossRef.
- K. M. L. Taylor, W. J. Rieter and W. Lin, J. Am. Chem. Soc., 2008, 130, 14358 CrossRef CAS PubMed.
- K. M. L. Taylor-Pashow, J. D. Rocca, Z. Xie, S. Tran and W. Lin, J. Am. Chem. Soc., 2009, 131, 14261 CrossRef CAS PubMed.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J.-S. Chang, Y. K. Hwang, V. Marsaud, P.-N. Bories, L. Cynober, S. Gil, G. Ferey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172 CrossRef CAS PubMed.
- N. Lee and T. Hyeon, Chem. Soc. Rev., 2012, 41, 2575 RSC.
- S. V. Salihov, Y. A. Ivanenkov, S. P. Krechetov, M. S. Veselov, N. V. Sviridenkova, A. G. Savchenko, N. L. Klyachko, Y. I. Golovin, N. V. Chufarova, E. K. Beloglazkina and A. G. Majouga, J. Magn. Magn. Mater., 2015, 394, 173 CrossRef CAS.
- H. Cai, K. Li, J. Li, S. Wen, Q. Chen, M. Shen, L. Zheng, G. Zhang and X. Shi, Small, 2015, 11, 4584 CrossRef CAS PubMed.
- J. Li., Y. Hu, J. Yang, P. Wei, W. Sun, M. Shen, G. Zhang and X. Shi, Biomaterials, 2015, 38, 10 CrossRef CAS PubMed.
- J. Shen, Y. Li, Y. Zhu, X. Yang, X. Yao, J. Li, G. Huang and C. Li, J. Mater. Chem. B, 2015, 3, 2873 RSC.
- L.-S. Lin, Z.-X. Cong, J.-B. Cao, K.-M. Ke, Q.-L. Peng, J. Gao, H.-H. Yang, G. Liu and X. Chen, ACS Nano, 2014, 8, 3876 CrossRef CAS PubMed.
- C. Wang, H. Xu, C. Liang, Y. Liu, Z. Li, G. Yang, H. Cheng, Y. Li and Z. Liu, ACS Nano, 2013, 7, 6782 CrossRef CAS PubMed.
- Q. Tian, J. Hu, Y. Zhu, R. Zou, Z. Chen, S. Yang, R. Li, Q. Su, Y. Han and X. Liu, J. Am. Chem. Soc., 2013, 135, 8571 CrossRef CAS PubMed.
- R. Bian, T. Wang, L. Zhang, L. Li and C. Wang, Biomater. Sci., 2015, 3, 1270 RSC.
- A. Ray Chowdhuri, D. Bhattacharya and S. K. Sahu, Dalton Trans., 2016, 45, 2963 RSC.
- P. Guo, D. Dutta, A. G. Wong-Foy, D. W. Gidley and A. J. Matzger, J. Am. Chem. Soc., 2015, 137, 2651 CrossRef CAS PubMed.
- C. Tamames-Tabar, D. Cunha, E. Imbuluzqueta, F. Ragon, C. Serre, M. J. Blanco-Prieto and P. Horcajada, J. Mater. Chem. B, 2014, 2, 262 RSC.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850 CrossRef PubMed.
- F. Xu, C. Cheng, D.-X. Chen and H. Gu, ChemPhysChem, 2012, 13, 336 CrossRef CAS PubMed.
- F. Ke, L.-G. Qiu, Y.-P. Yuan, X. Jiang and J.-F. Zhu, J. Mater. Chem., 2012, 22, 9497 RSC.
- N. T. Zhang, B. J. Zhu, F. M. Peng, X. Y. Yu, Y. Jia, J. Wang, L. T. Kong, Z. Jin, T. Luo and J. H. Liu, Chem. Commun., 2014, 50, 7686 RSC.
- Z. Xiong, Y. Ji, C. Fang, Q. Zhang, L. Zhang, M. Ye, W. Zhang and H. Zou, Chem.–Eur. J., 2014, 20, 7389 CrossRef CAS PubMed.
- Y.-X. J. Wang, Quant. Imaging Med. Surg., 2011, 1, 35 Search PubMed.
- H. Zheng, Y. Zhang, L. Liu, W. Wan, P. Guo, A. M. Nyström and X. Zou, J. Am. Chem. Soc., 2016, 138, 962 CrossRef CAS PubMed.
- X. G. Wang, Z. Y. Dong, H. Cheng, S. S. Wan, W. H. Chen, M. Z. Zou, J. W. Huo, H. X. Deng and X. Z. Zhang, Nanoscale, 2015, 7, 16061 RSC.
- R. Chen, J. F. Zhang, Y. Wang, X. F. Chen, J. A. Zapien and C. S. Lee, Nanoscale, 2015, 7, 17299 RSC.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232 CrossRef CAS PubMed.
- L. Shen, W. Wu, R. Liang, R. Lin and L. Wu, Nanoscale, 2013, 5, 9374 RSC.
- J. Yang, Y. Dai, X. Zhu, Z. Wang, Y. Li, Q. Zhuang, J. Shi and J. Gu, J. Mater. Chem. A, 2015, 3, 7445 CAS.
- Y.-M. Zheng, L. Yu and J. P. Chen, J. Colloid Interface Sci., 2012, 367, 362 CrossRef CAS PubMed.
- L. N. Nagy, J. Mihaly, A. Polyak, B. Debreczeni, B. Csaszar, I. C. Szigyarto, A. Wacha, Z. Czegeny, E. Jakab, S. Klebert, E. Drotar, G. Dabasi, A. Bota, L. Balogh and E. Kiss, J. Mater. Chem. B, 2015, 3, 7529 RSC.
- R. Anand, F. Borghi, F. Manoli, I. Manet, V. Agostoni, P. Reschiglian, R. Gref and S. Monti, J. Phys. Chem. B, 2014, 118, 8532 CrossRef CAS PubMed.
- F. Gao, L. Li, T. Liu, N. Hao, H. Liu, L. Tan, H. Li, X. Huang, B. Peng, C. Yan, L. Yang, X. Wu, D. Chen and F. Tang, Nanoscale, 2012, 4, 3365 RSC.
- C. He, K. Lu, D. Liu and W. Lin, J. Am. Chem. Soc., 2014, 136, 5181 CrossRef CAS PubMed.
- C. He, Y. Hu, L. Yin, C. Tang and C. Yin, Biomaterials, 2010, 31, 3657 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, experimental details and characterization. See DOI: 10.1039/c6sc01359g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |