
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free disproportionation of formic acid mediated by organoboranes†
Clément
Chauvier
,
Pierre
Thuéry
and
Thibault
Cantat
*
NIMBE, CEA, CNRS, Université Paris-Saclay, Gif-sur-Yvette, France. E-mail: thibault.cantat@cea.fr; Fax: +33 1 6908 6640
First published on 19th May 2016
Abstract
In the presence of dialkylboranes, formic acid can be converted to formaldehyde and methanol derivatives without the need for an external reductant. This reactivity, in which formates serve as the sole carbon and hydride sources, represents the first example of the disproportionation of formate anions under metal-free conditions. Capitalizing on both experimental and computational (DFT) mechanistic considerations, the role of transient borohydride is highlighted in the reduction of formates and this reactivity was further exemplified in the methylation of TMP (2,2,6,6-tetramethylpiperidine) and in the transfer hydroboration reactions for the reduction of aldehydes.
Introduction
Formic acid is an attractive reductant in organic chemistry because it is a benign and liquid surrogate for molecular hydrogen. In fact, the use of HCO2H in transfer hydrogenation has been successfully applied to a variety of organic substrates,1 including carbonyl groups and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C systems, and the development of catalysts for these transformations is still under active study.2 The reasons behind this success originate in the kinetic and thermodynamic properties of HCO2H. Indeed, formic acid presents a redox potential similar to H2E0(CO2/HCO2H = −0.61 V vs. E0(H2O/H2 = −0.41 V at pH = 7 vs. NHE) but the C–H bond has a Bond Dissociation Energy (BDE) which is about 16 kcal mol−1 weaker3 than the H–H bond and it is thus easier to activate at a metal center. More recently, the high hydrogen content of formic acid (4.4%) has been recognized as a means to store H2 in liquid form, under ambient conditions.4 This concept relies on the selective decomposition of HCO2H to H2 and CO2 and a plethora of molecular catalysts have been designed to catalyze this important transformation.5 While catalysts based on noble metals are the most efficient systems, the state-of-the-art is currently shifting towards earth abundant metal catalysts, based on iron6 or aluminum complexes.7 In 2015, our group reported the first organic catalysts for the dehydrogenation of formic acid, using dialkyboranes.8
C systems, and the development of catalysts for these transformations is still under active study.2 The reasons behind this success originate in the kinetic and thermodynamic properties of HCO2H. Indeed, formic acid presents a redox potential similar to H2E0(CO2/HCO2H = −0.61 V vs. E0(H2O/H2 = −0.41 V at pH = 7 vs. NHE) but the C–H bond has a Bond Dissociation Energy (BDE) which is about 16 kcal mol−1 weaker3 than the H–H bond and it is thus easier to activate at a metal center. More recently, the high hydrogen content of formic acid (4.4%) has been recognized as a means to store H2 in liquid form, under ambient conditions.4 This concept relies on the selective decomposition of HCO2H to H2 and CO2 and a plethora of molecular catalysts have been designed to catalyze this important transformation.5 While catalysts based on noble metals are the most efficient systems, the state-of-the-art is currently shifting towards earth abundant metal catalysts, based on iron6 or aluminum complexes.7 In 2015, our group reported the first organic catalysts for the dehydrogenation of formic acid, using dialkyboranes.8
Beyond formic acid, methanol can store 12.1 wt% hydrogen and this high energy chemical (4900 vs. 2104 W h L−1 for HCOOH) represents an excellent energy vector. Formic acid has been recently proposed as a sustainable intermediate in the production of methanol.9 This strategy relies on the catalytic electroreduction of CO2 to HCO2H, a technically and economically viable process which is under pilot development.10 HCO2H can then serve as a C–H bond shuttle to yield methanol, assuming that efficient systems can promote the disproportionation of formic acid (Scheme 1). Nevertheless, the disproportionation of HCO2H is a difficult transformation because the dehydrogenation of formic acid is favored over the reduction of a formate group. The first catalysts for the disproportionation of formic acid were only unveiled in 2013 (ref. 11) by Goldberg, Miller et al. who showed that iridium(III) complexes could convert formic acid to methanol with a maximum yield of 2.6%. H2 is in fact the main product during the iridium-catalyzed decomposition of HCO2H. In 2014, our group reported ruthenium(II) catalysts for this transformation9 and the selective conversion of HCO2H to 50% methanol was achieved. While the ruthenium catalysts still represent the state of the art in this transformation, Parkin et al. described the first catalysts without noble metals, using molybdenum(II) complexes.12 With all these systems, H2 is produced as a competing product in at least 50% yield.
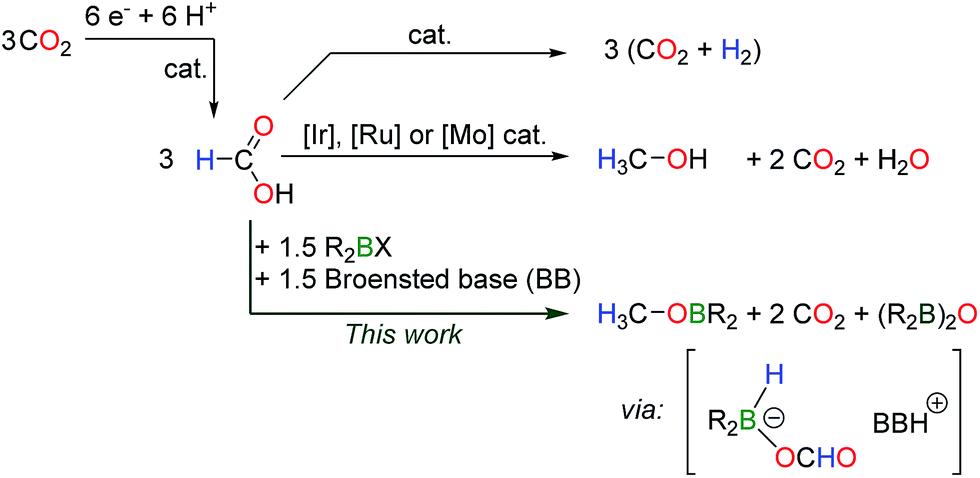 | ||
| Scheme 1 Disproportionation of formic acid to the methanol level and the competing dehydrogenation. | ||
Reasoning that on the one hand dialkylboranes are catalysts for the dehydrogenation of formic acid8 and that on the other hand hydroboranes can reduce CO2 to methoxyboranes, in the presence of a catalytic amount of an organic base,13 we have sought metal-free systems able to mediate the disproportionation of formic acid. Herein, we demonstrate that stoichiometric amounts of dialkyborane derivatives can convert formates to formaldehyde and methanol derivatives, for the first time. Mechanistic insights derived from the experimental results and DFT calculations highlight the role of transient borohydride in these transformations.
Results and discussion
9-Iodo-9-borabicyclo[3.3.1]nonane (BBN–I) was shown to promote the catalytic decarboxylation and dehydrogenation of formic acid and its reaction chemistry was thus explored in the presence of formate anions.8 Adding 2 molar equivalents of formic acid and triethylamine to an acetonitrile solution of BBN–I affords bis(formoxy)borate [HNEt3+, 1−] along with HNEt3I, at room temperature (RT) (eqn (1)).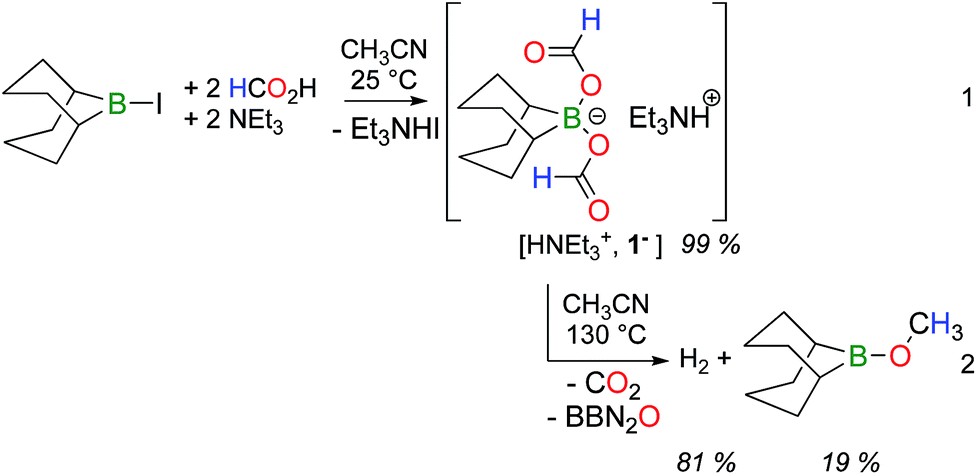
Heating this mixture at 130 °C leads to the complete decomposition of the formate ligands in 1− and the concomitant formation of CO2 was observed by GC and 13C NMR (eqn (2)). As expected, H2 is also observed in the gas phase. Notably, a singlet at 3.71 ppm is also observed in the 1H NMR spectrum, coupled with a weak resonance at 53.9 ppm that was attributed to a primary carbon in the 13C NMR spectrum. To verify this chemical behavior, the same chemical sequence was reproduced with H13CO2H. At RT, in acetonitrile, the protons of the formate in 13C-labelled 1− display a doublet (δ = 8.43 ppm, 1JC–H = 202 Hz) coupled to a singlet at 168.1 ppm in the 13C NMR spectrum. After 3 h at 130 °C, the 1H NMR spectrum of the crude mixture indicates the formation of H2 gas and 13CH3OBBN (1H NMR: δ = 3.71 ppm, 1JC–H = 143 Hz; 13C NMR: δ = 53.9 ppm). After 20 h, 13CO2 and 13CH3OBBN were the only 13C-enriched products observed in solution. After the removal of the volatiles under a reduced pressure, the resulting solid was dissolved in d8-toluene to afford a homogeneous solution comprising methoxyborane (δ(11B) = 57 ppm) and diboroxane (BBN)2O (δ(11B) = 59 ppm) as the sole boron-containing products. These results clearly demonstrate that formate anions can disproportionate to methoxides in the coordination sphere of boron, with the associated release of CO2 and diboroxane. The parallel formation of H2 and CH3OBBN shows that the disproportionation and dehydrogenation of formic acid are competing in the presence of the dialkylborane, although stoichiometric amounts of protons are present in the reaction mixture (in the form of HNEt3+) with respect to the formates. Under catalytic conditions (using 5 mol% [HNEt3+, 1−]), the dehydrogenation of HCO2H was observed after 24 h at 130 °C, together with only a catalytic quantity of CH3OBBN (<5 mol%).
[HNEt3+, 1−] was isolated from the reaction between 9-BBN, HCOOH and triethylamine and its disproportionation to CH3OBBN was investigated to gain further insights into this novel transformation. The thermolysis of [HNEt3+, 1−] is inefficient in THF or benzene and it is best carried out in acetonitrile at 130 °C (entries 1–4 in Table 1). Under these conditions, [HNEt3+, 1−], is completely decomposed to H2 and CH3OBBN after 10.5 h (entry 4 in Table 1). CH3OBBN is formed in 39% yield, meaning that 39% of the C–H bonds present in the formate ligands of 1− are efficiently preserved as C–H bonds in the methoxide ligand (see ESI†). Increasing the reaction temperature to 150 °C only affects the rate of the thermolysis and CH3OBBN is obtained in 33% yield after only 4 h. Importantly, the disproportionation of formates is not specific to the BBN framework and the dicyclohexylborane derivative [HNEt3+, 2−] is also prone to generate the corresponding Cy2BOCH3. Although the latter gave low yields of the methoxyborane at 130 °C, presumably due to the thermal instability of the reaction intermediates, the reaction proceeded smoothly at 120 °C to afford Cy2BOCH3 in 38% yield after 7 h (entry 7 in Table 1). In all cases, free methanol can be ultimately generated by hydrolysis of methoxyborane (see ESI†). The nature of the base used to prepare the starting bis(formoxy)borate also influences the efficiency of the disproportionation. Indeed, the thermolysis of [iPr2EtNH+, 1−] at 130 °C leads to CH3OBBN in 50% yield within only 4.5 h. This improved selectivity demonstrates the positive influence of bulky tertiary amines, such as iPr2EtN, which might arise from the decreased affinity of the amine for the boron center.14
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R2B | BaseH+ | t [h] | T [°C] | Solvent | Yieldb [%] |
| a Reaction conditions: 0.125 mmol 1− or 2−, 0.4 mL solvent. b Yields determined by 1H NMR using mesitylene (10 μL); mean value over at least 2 runs. c Time required to obtain >95% conversion of formate. | ||||||
| 1 | BBN | Et3NH+ | >48 h | 130 | d 8-THF | <5 |
| 2 | BBN | Et3NH+ | >48 h | 130 | C6D6 | <5 |
| 3 | BBN | Et3NH+ | 4 | 150 | CD3CN | 33 |
| 4 | BBN | Et3NH+ | 10.5 | 130 | CD3CN | 39 |
| 5 | BBN | iPr2EtNH+ | 4.5 | 130 | CD3CN | 50 |
| 6 | BCy2 | Et3NH+ | 2.5 | 130 | CD3CN | <5 |
| 7 | BCy2 | Et3NH+ | 7 | 120 | CD3CN | 38 |
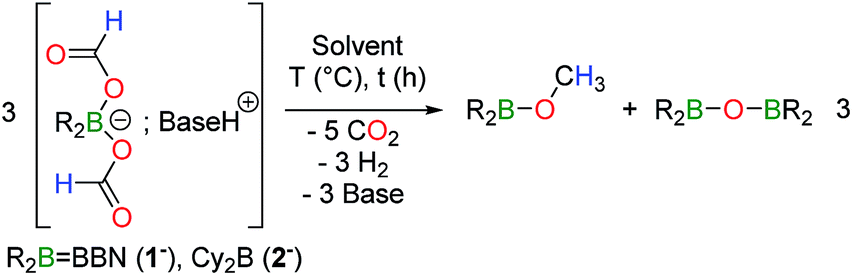
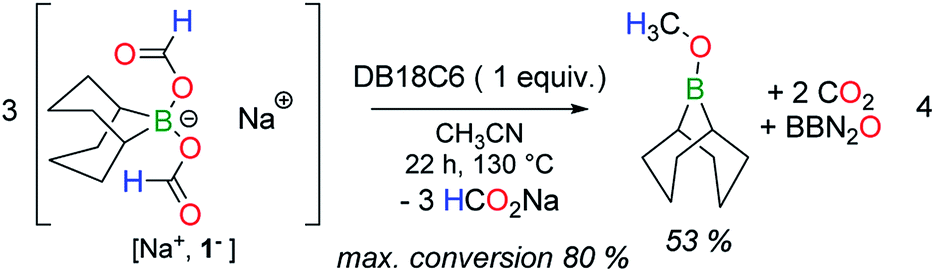
To assess whether H2, released by dehydrogenation, plays any role in disproportionation, the corresponding bis(formoxy)borate [Et3ND+, R2B(OCHO)2−] was synthesized in situ in toluene at RT from HCO2D. As expected, its thermolysis at 130 °C in CH3CN affords gaseous HD (δH = 4.54 ppm, t, 1JHD = 43 Hz), as made evident by 1H NMR. However, no deuterium-containing reduced product (other than HD) was detected by either 1H or 13C NMR (Fig. S17†) spectroscopy at intermediate or full conversion of the formate. In addition, the thermolysis of aprotic [Na+, 1−] in the presence of the crown ether DB18C6 (dibenzo-18-crown-6) also affords CH3OBBN in a 53% yield after 22 h at 130 °C (eqn (4)), thus proving that the disproportionation can proceed without dehydrogenation. In that case, the conversion reached a plateau at ca. 80%, induced by the release of somewhat unreactive HCO2Na under the applied reaction conditions.
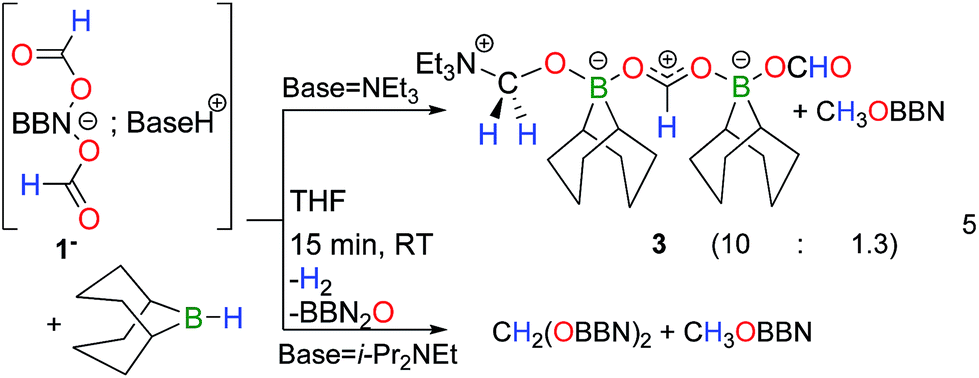
Overall, the disproportionation of formate anions mediated by dialkylboranes proceeds with yields and selectivities (up to 53%), comparable to state-of-the-art metal catalysts.9,11,12 Although stoichiometric amounts of dialkylboranes are required to promote the disproportionation of formates, the formation of methoxyboranes from 1− and 2− represents the first examples of metal-free mediated disproportionations of formates. The mechanism of this transformation was thus investigated both experimentally and computationally in order to track possible reaction intermediates. Monitoring the thermal decomposition of [HNEt3+, 1−] by NMR reveals the formation of a single transient species 3 (<5% yield), characterized by a singlet at 4.40 ppm in the 1H NMR spectrum, which is associated with a 13C resonance at 80.5 ppm. Reasoning that 3 is an acetal compound, its independent synthesis was carried out by reacting hydroborane 9-BBN with [HNEt3+, 1−] (eqn (5)). 3 is obtained as the major reduced product, along with CH3OBBN (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.3 ratio), and the formulation of an acetal compound was confirmed by X-ray diffraction (Fig. 1A). 3 is a zwitterionic adduct of formaldehyde in which the carbon atom is bound to NEt3 and the oxygen atom coordinates a formoxyborane group, namely BBNOCHO. The latter formate ligand coordinates a second equivalent of the Lewis acid BBNOCHO.15
:1.3 ratio), and the formulation of an acetal compound was confirmed by X-ray diffraction (Fig. 1A). 3 is a zwitterionic adduct of formaldehyde in which the carbon atom is bound to NEt3 and the oxygen atom coordinates a formoxyborane group, namely BBNOCHO. The latter formate ligand coordinates a second equivalent of the Lewis acid BBNOCHO.15
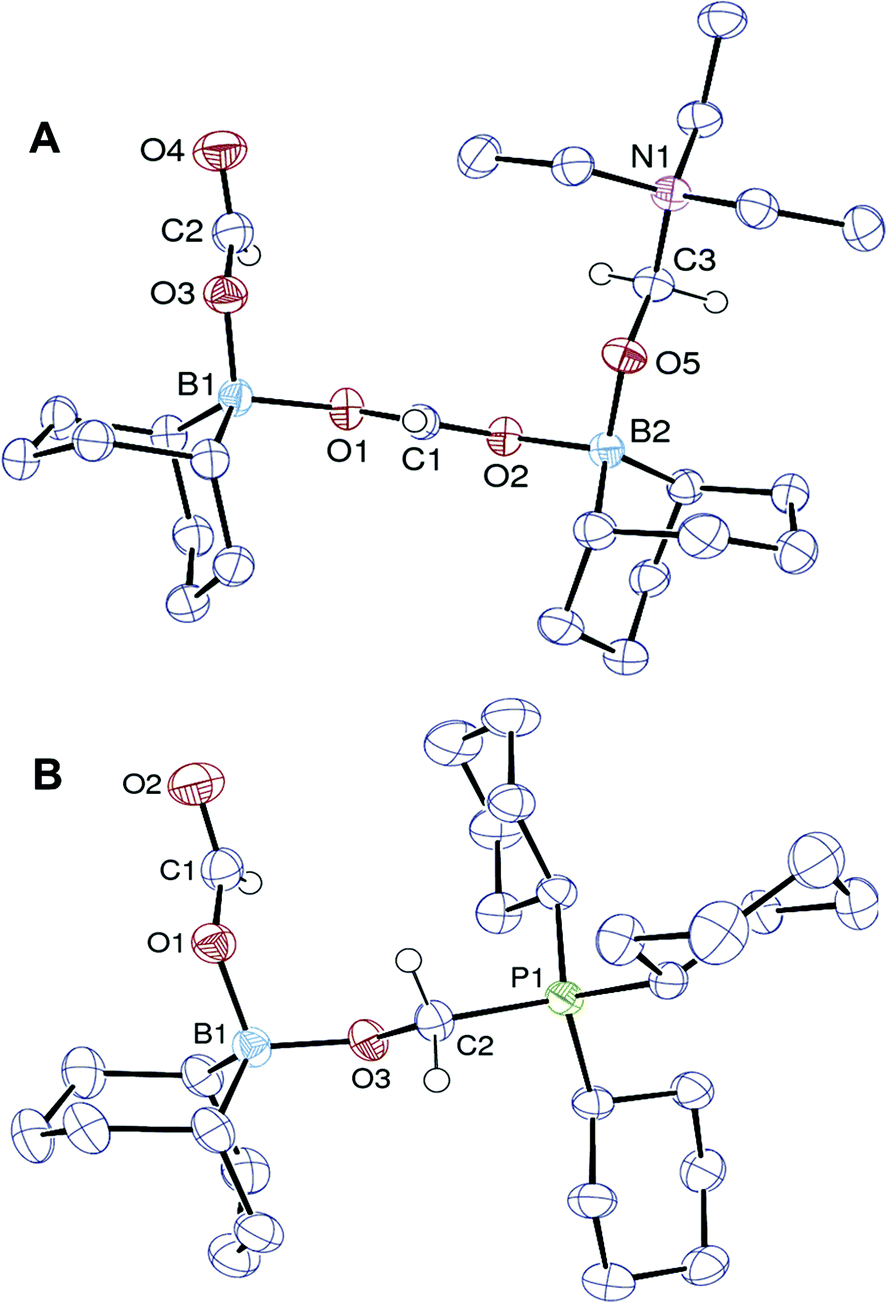 | ||
| Fig. 1 ORTEP views of 3 (A) and 4 (B). The hydrogen atoms of the BBN, ethyl and cyclohexyl groups are omitted for clarity. | ||
The finding that nucleophilic bases such as NEt3 can stabilize an intermediate formaldehyde adduct suggests the occurrence of a novel and unique transformation, namely the selective disproportionation of formates to CO2 and formaldehyde. We have thus sought a Lewis base able to direct the selectivity towards the formation of formaldehyde surrogates from the disproportionation of boron formates. A phosphonium salt [Cy3PH+, 1−] was synthesized and its thermolysis leads, after 5.5 h at 130 °C, to the formation of H2, CO2, free PCy3 and the formaldehyde adduct 4 as the only reduction product (eqn (6)).
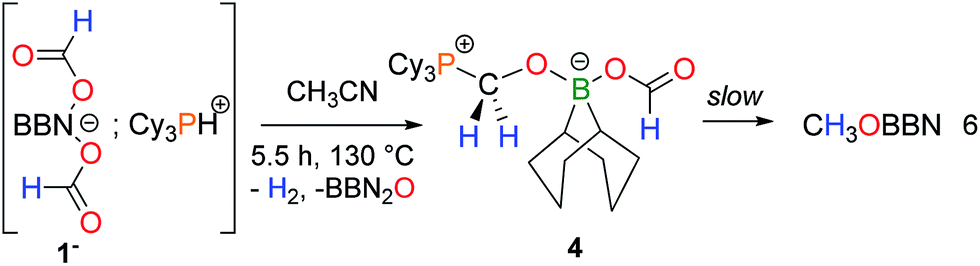
The X-ray analysis of crystals obtained by slowly cooling the reaction mixture reveals that 4 features a formaldehyde unit C-coordinated to the phosphorus Lewis base and O-coordinated to a BBN–OCHO unit (Fig. 1B). It thus resembles the related (tBu3PCH2O)(HC(O)O)B(C8H14) isolated by Stephan and coworkers,16 from the hydroboration of CO2. As expected, 4 exhibits an enhanced stability towards reduction in comparison to 3. Its conversion to CH3OBBN is slow and requires 10 h at 130 °C to proceed (30% yield).17
The disproportionation of 1− to CH2(OBBN)2 and CH3OBBN was investigated using DFT calculations (M06-2X/6-311+G(d,p) level of theory, in acetonitrile (PCM)) so as to determine how a C–H bond from a formate anion can be transferred to a second formate ligand in the boron coordination sphere. The thermodynamic balance for the disproportionation of 1− is given in Scheme 2. From 2 equiv. of 1− the formation of CH2(OBBN)2 is slightly endergonic with ΔG = 10.1 kcal mol−1 and 2 equiv. of HCO2− and 1 equiv. of CO2 are released. Further addition of 1 equiv. of 1− enables the formation of CH3OBBN together with 1 equiv. of HCO2− and CO2 (ΔG = −3.9 kcal mol−1). It is noteworthy that, under the applied reaction conditions (130 °C), 1− also catalyzes the decarboxylation of the free HCO2− anions, thereby shifting the equilibria towards the formation of the products (see ESI†).
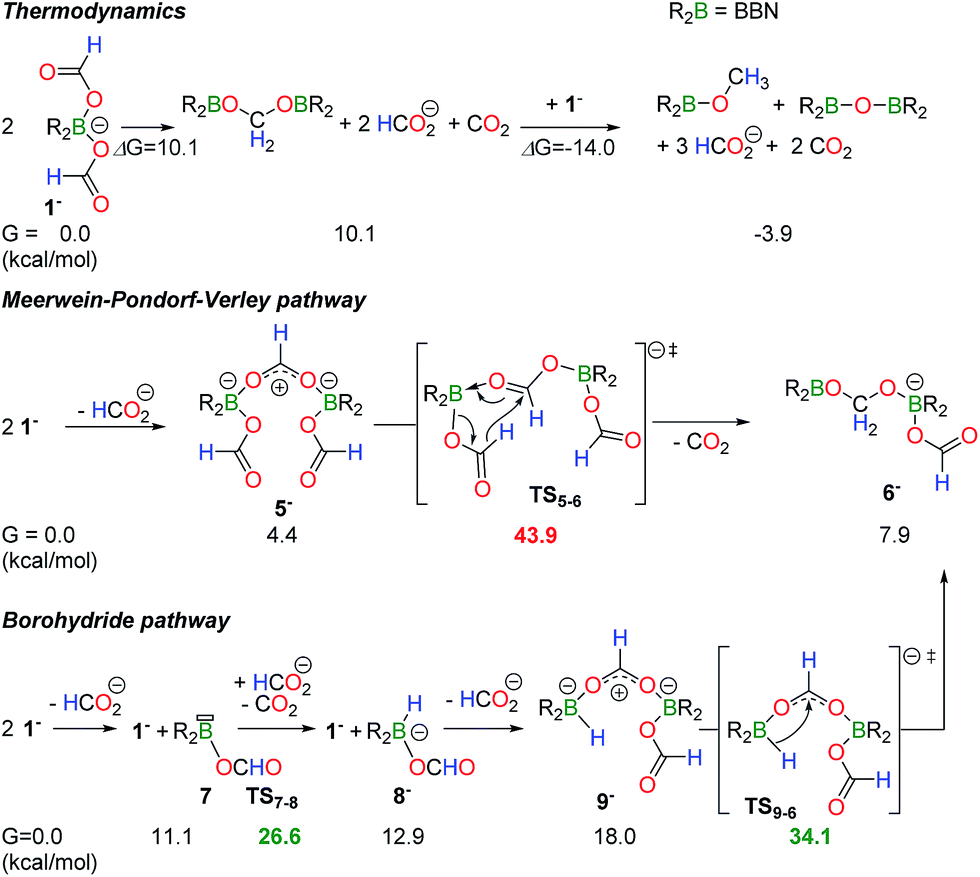 | ||
| Scheme 2 Computed mechanism for the disproportionation of 1− to the formaldehyde level. The full energy surface is provided in Scheme S1.† | ||
Experimental observations show that the disproportionation of 1− to the formaldehyde redox state is rate determining, as CH2(OBBN)2 and 3 do not accumulate, and thus only the mechanism of this step was computed. Two routes can be proposed. First, transfer hydrogenation with aluminum(III) isopropoxide salts is well documented and a mechanism similar to the Meerwein–Pondorf–Verley (MPV) reaction represents a first plausible pathway.18 In a second option, the decarboxylation of a formate ligand could afford a boron hydride, whose B–H functionality reduces a second formate anion. DFT calculations were performed to distinguish the two pathways and the results are summarized in Scheme 2, with the full energy surface being provided in Scheme S1 (ESI†). Starting from 2 equiv. of 1−, the exclusion of one formate ligand yields dimer 5− in a slightly endergonic process (ΔG = +4.4 kcal mol−1). In dimer 5−, one formate ligand is bridging two boron centers and this activation increases the electrophilicity of the carbon atom. In fact, the NBO charge of the carbon atom in the μ2(O,O′)–HCOO− ligand is 0.77 vs. 0.71 in the other two κ1-HCOO− ligands. Hydride transfer from a monodentate formate ligand thus provides intermediate 6−, which further dissociates into CH2(OBBN)2 and a free HCO2− anion. This pathway represents a boron variant of the MPV mechanism. Nevertheless, its occurrence is highly unlikely because the transition state (TS5−6) connecting 5− and 6− is high in energy and it involves an overall barrier of 43.9 kcal mol−1, incompatible with the experimental conditions.
Alternatively, a mechanism relying on the transient formation of a borohydride species is favored. The dissociation of one formate ligand from 1− is slightly endergonic (+11.1 kcal mol−1) but it provides an unsaturated formatoborane (7), able to promote the decarboxylation of HCO2− with a low energy barrier of 15.5 kcal mol−1 by hydride abstraction. This process affords a reductant, BBN(H)(OCOH)− (8−). The displacement of a formate ligand in 1− with 8− provides dimer 9− (+18.0 kcal mol−1) which features a bridging HCOO− ligand, similar to 5− and 3. Hydride transfer from boron to the bidentate formate ligand in 9− yields 6−, with a low energy barrier of 16.1 kcal mol−1. From 6−, the acetal CH2(OBBN)2 (+10.1 kcal mol−1) can be released or trapped as adduct 3 (−8 kcal mol−1) in the presence of NEt3. Overall, this mechanism only involves an energy barrier of 34.1 kcal mol−1, corresponding to the energy span between the starting materials and TS9−6, and it is compatible with thermolysis occurring within several hours at 130 °C.
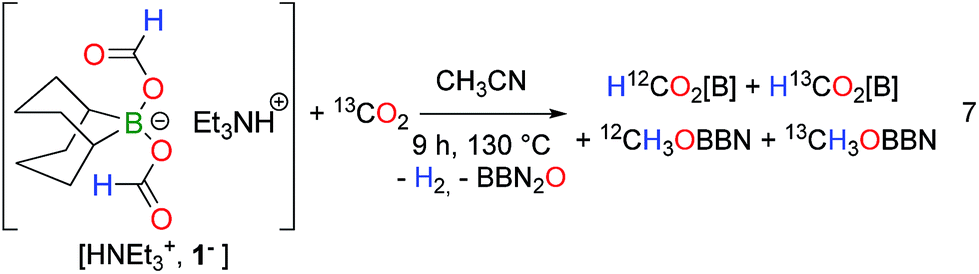
Experimentally, strong evidence for the involvement of a B–H bond was further gained by carrying out the thermolysis of [HNEt3+, (H12C(O)O)2BBN−] under 13CO2 atmosphere (1 atm) (eqn (7)). After 3 h at 130 °C, a mixture of H12C(O)O[B] and H13C(O)O[B] along with 12CH3OBBN was observed and, after 9 h, the additional formation of 13CH3OBBN became evident (Fig. S14†). This result stresses the presence of an equilibrium between CO2 and HCOO− and, while such a process is unlikely for an MPV pathway, it rather points to the involvement of an intermediate borohydride species BBN(H) (OCHO)− (8−). This proposal is also consistent with the observation that HB(C6F5)3− (obtained by H2 splitting with a Frustrated Lewis Pair) and BH4− are able to reduce CO2 to the methanol and formate levels respectively.19
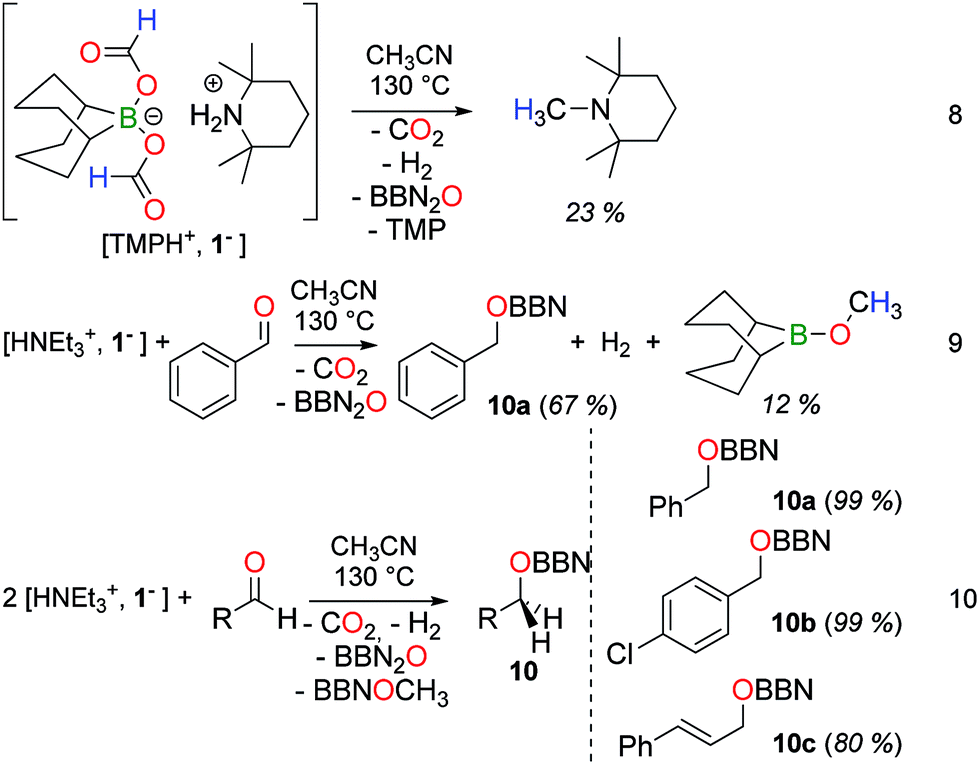
The thermal decarboxylation of a formate anion in 1− provides a reductant (8−) and we have computed that 8− has a hydride donor ability similar to BBNH2− (34 vs. 33 kcal mol−1 respectively).208− (and 1−) is thus a potent reductant and its reductive properties were further investigated, in the presence of various oxidants (eqn (9) and (10)). Building on the disproportionation of formates in 1− to CH3OBBN, the methylation of a secondary amine was attempted. The tetramethylpiperidinium salt [TMPH+, 1−] was first prepared and decomposed quantitatively within 17 h at 130 °C in acetonitrile. Interestingly, CH3OBBN does not form under these conditions and N-methyl-tetramethylpiperidine was obtained instead in 23% yield, along with H2, CO2, BBN2O and free TMP (eqn (8)). In this reaction, the formate ligands in 1− serve both as a H and C source for the formation of the N–CH3 linkage.21
Interestingly, when the thermolysis of [HNEt3+, 1−] is carried out in the presence of 1 molar equiv. of benzaldehyde, PhCH2OBBN (10a) is obtained in a 67% yield, further confirming that bis(formoxy)borate 1− is able to deliver a useful reducing agent (eqn (9)). The parallel formation of CH3OBBN (12% yield) shows that the disproportionation of formates is a surprisingly facile process from 1−. This result represents the first example of reduction of a carbonyl substrate by transfer hydrogenation from formic acid, under metal-free conditions. It can also be described as a transfer hydroboration.22 In fact, the reduction of various aldehydes can be efficiently carried out with 2 molar equivalents of [HNEt3+, 1−]. Under these conditions, benzaldehyde is reduced to 10a in 99% yield and borylethers 10b and 10c were obtained respectively in 99 and 80% yield from the corresponding aldehydes (eqn (10)).
Conclusions
In conclusion, we have shown that formic acid can undergo disproportionation reactions, using stoichiometric quantities of dialkylborane reagents. In the coordination sphere of boron, formate ligands were decarboxylated to provide borohydride intermediates, which were successfully utilized to promote the disproportionation of formates to formaldehyde and methanol scaffolds and the reduction of aldehydes under metal-free conditions, for the first time. Current work in our laboratory is devoted to facilitating the generation of borohydrides from formic acid and increasing the hydride donor ability of the resulting B–H group.Acknowledgements
For financial support of this work, we acknowledge CEA, CNRS, the CHARMMMAT Laboratory of Excellence and the European Research Council (ERC Starting Grant Agreement no. 336467). T. C. thanks the Foundation Louis D. – Institut de France for its support.Notes and references
-
(a) A. Fujii, S. Hashiguchi, N. Uematsu, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 2521–2522 CrossRef CAS
; (b) N. Uematsu, A. Fujii, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 4916–4917 CrossRef CAS
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed
-
(a) C. Fellay, P. J. Dyson and G. Laurenczy, Angew. Chem., Int. Ed., 2008, 47, 3966–3968 CrossRef CAS PubMed
-
(a) T. C. Johnson, D. J. Morris and M. Wills, Chem. Soc. Rev., 2010, 39, 81–88 RSC
-
(a) A. Boddien, B. Loges, F. Gärtner, C. Torborg, K. Fumino, H. Junge, R. Ludwig and M. Beller, J. Am. Chem. Soc., 2010, 132, 8924–8934 CrossRef CAS PubMed
- T. W. Myers and L. A. Berben, Chem. Sci., 2014, 5, 2771–2777 RSC
- C. Chauvier, A. Tlili, C. Das Neves Gomes, P. Thuery and T. Cantat, Chem. Sci., 2015, 6, 2938–2942 RSC
- S. Savourey, G. Lefevre, J. C. Berthet, P. Thuery, C. Genre and T. Cantat, Angew. Chem., Int. Ed., 2014, 53, 10466–10470 CrossRef CAS PubMed
- A. S. Agarwal, Y. Zhai, D. Hill and N. Sridhar, ChemSusChem, 2011, 4, 1301–1310 CrossRef CAS PubMed
- A. J. Miller, D. M. Heinekey, J. M. Mayer and K. I. Goldberg, Angew. Chem., Int. Ed., 2013, 52, 3981–3984 CrossRef CAS PubMed
- M. C. Neary and G. Parkin, Chem. Sci., 2015, 6, 1859–1865 RSC
-
(a) M.-A. Courtemanche, M.-A. Légaré, L. Maron and F.-G. Fontaine, J. Am. Chem. Soc., 2013, 135, 9326–9329 CrossRef CAS PubMed
- H. C. Brown and S. U. Kulkarni, Inorg. Chem., 1977, 16, 3090–3094 CrossRef CAS
- In contrast, the combination of [iPr2EtNH+, 1−] with 9-BBN exclusively leads to H2C(OBBN)2 and CH3OBBN, without involvement of any zwitterionic formaldehyde adduct.
- T. Wang and D. W. Stephan, Chem. Commun., 2014, 50, 7007–7010 RSC
- Analogously, the thermolysis of the adduct 3 eventually affords CH3OBBN albeit much faster than 4.
- R. Cohen, C. R. Graves, S. T. Nguyen, J. M. Martin and M. A. Ratner, J. Am. Chem. Soc., 2004, 126, 14796–14803 CrossRef CAS PubMed
-
(a) A. E. Ashley, A. L. Thompson and D. O'Hare, Angew. Chem., Int. Ed., 2009, 48, 9839–9843 CrossRef CAS PubMed
- Z. M. Heiden and A. P. Lathem, Organometallics, 2015, 34, 1818–1827 CrossRef CAS
- S. Savourey, G. Lefevre, J. C. Berthet and T. Cantat, Chem. Commun., 2014, 50, 14033–14036 RSC
- A similar concept has recently emerged for transfer hydrosilylation. See: M. Oestreich, Angew. Chem., Int. Ed., 2016, 55, 494–499 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational procedures and physical properties of compounds. CCDC 1450409–1450413 and 1454182. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc01410k |
| This journal is © The Royal Society of Chemistry 2016 |