
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
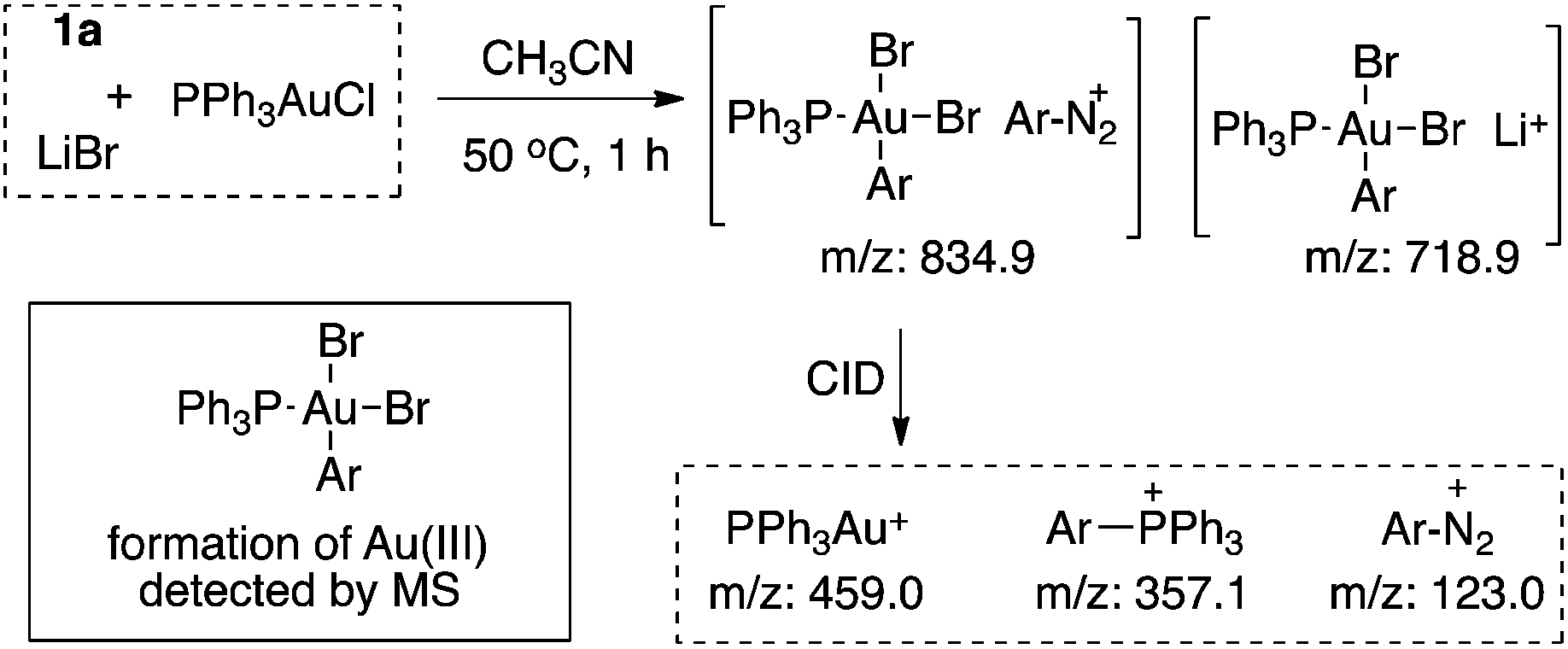
Nucleophile promoted gold redox catalysis with diazonium salts: C–Br, C–S and C–P bond formation through catalytic Sandmeyer coupling†
Haihui
Peng
a,
Rong
Cai
b,
Chang
Xu
c,
Hao
Chen
*c and
Xiaodong
Shi
*a
aDepartment of Chemistry, University of South Florida, Tampa, FL 33620, USA. E-mail: xmshi@usf.edu
bC. Eugene Bennett Department of Chemistry, West Virginia University, Morgantown, WV 26506, USA
cCenter for Intelligent Chemical Instrumentation, Department of Chemistry and Biochemistry, Edison Biotechnology Institute, Ohio University, Athens, OH 45701, USA
First published on 10th June 2016
Abstract
Gold-catalyzed C-heteroatom (C–X) coupling reactions are evaluated without using sacrificial oxidants. Vital to the success of this methodology is the nucleophile-assisted activation of aryldiazonium salts, which could be an effective oxidant for converting Au(I) to Au(III) even without the addition of an assisting ligand or photocatalyst. By accelerating the reaction kinetics to outcompete C–C homo-coupling or diazonium dediazoniation, gold-catalyzed Sandmeyer reactions were achieved with different nucleophiles, forming C–Br, C–S and C–P bonds in high yields and selectivities.
Homogeneous gold catalysis has been well developed for the activation of C–C multiple bond in the past two decades.1 However, compared with Pd(0), a d10 isoelectronic counterpart, traditional redox chemistry with Au(I) is relatively rare due to the higher oxidation potential between Au(I) and Au(III).2 To maximize the potential of gold catalysis, extensive effort has been put into the development of this new branch of gold chemistry.3 Typically, strong external oxidants, such as Selectfluor and hypervalent iodine, are usually required to access catalytically active Au(III) intermediates. The need for strong oxidants made gold redox chemistry less attractive, especially for the synthesis of complex molecules. One of the most significant improvements in gold redox chemistry is dual photoredox and gold catalysis, first reported by Glorius' and Toste's groups (Scheme 1).4 In their studies, a photocatalyst was used to promote gold redox oxidation under mild conditions. More recently, Hashmi and coworkers further extended this chemistry to photosensitizer-free conditions, achieving alkyne 1,2-difunctionalization with only a gold catalyst under visible-light.5 In this study, a gold(III) intermediate was successfully isolated, which supported a gold redox catalytic mechanism under photo-initiated conditions. Herein, we report the investigation of nucleophile promoted diazonium activation for promoting gold(I) oxidation. Through mechanistic investigation using NMR and electrospray ionization mass spectrometry (ESI-MS), nucleophile was identified as a critical factor in promoting this gold redox chemistry. In addition, through suppressing the undesired C–C homocoupling (via trans-metallation and reductive elimination), catalytic Sandmeyer coupling was achieved and C–X bonds (X = Br, S and P) were formed in good to excellent yields.6 Under these new conditions, no strong oxidants or photocatalysts are required to promote gold oxidation, which will potentially open new avenues for future developments in gold redox chemistry.
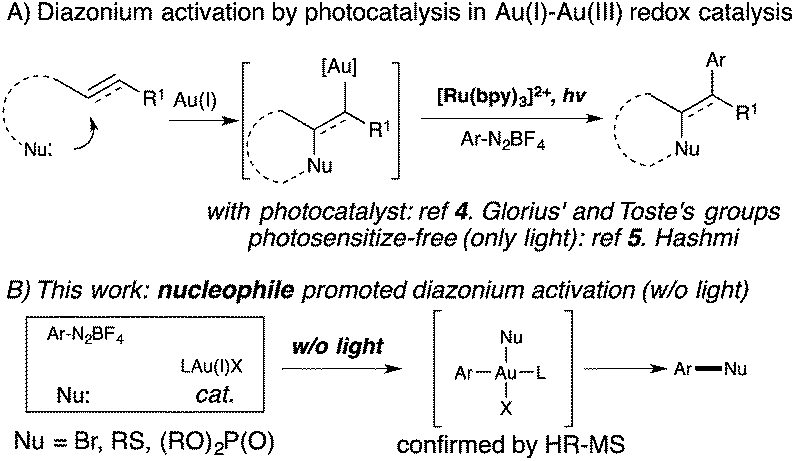 | ||
| Scheme 1 Gold redox catalysis. | ||
The high-oxidation potential between Au(I) and Au(III) has been a major concern that has hindered the development of gold redox catalysis for a long time. Thus, achieving gold oxidation under mild conditions is crucial. Our group recently reported the gold catalyzed C–C coupling reaction between alkynes and aryldiazonium salts.7 Based on that study, diazonium activation can be achieved with the help of a 2,2′-bipyridine (bpy) ligand even without light. Although visible-light conditions are extremely mild and readily accessible, understanding the function of bpy ligand will certainly help the elucidation of reaction mechanism, which will be beneficial for the further development of gold redox chemistry under mild conditions.
Notably, Shin and coworkers have reported the detection of an Au(III) intermediate (using XPS) through mixing PPh3AuCl and an aryl diazonium salt in MeOH/CH3CN (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 60 °C.8 To explore the role of bpy ligands, we monitored the reaction of diazonium salt 1a (p-F-C6H4N2BF4) and PPh3AuCl using 31P NMR. Interestingly, when mixing 1a and PPh3AuCl in CH3CN, no reaction was observed, even under long exposure to light at 50 °C (Fig. 1a). In contrast, with MeOH/CH3CN (9:1) as the solvent, phosphonium salt 2a was detected (22.5 ppm, Fig. 1b), though in a low yield (23% based on NMR). Interestingly, with the addition of 1.0 equiv. of bpy, 2a was formed at a much faster rate and PPh3AuCl was totally consumed within an hour (Fig. 1c and d).9
:1) at 60 °C.8 To explore the role of bpy ligands, we monitored the reaction of diazonium salt 1a (p-F-C6H4N2BF4) and PPh3AuCl using 31P NMR. Interestingly, when mixing 1a and PPh3AuCl in CH3CN, no reaction was observed, even under long exposure to light at 50 °C (Fig. 1a). In contrast, with MeOH/CH3CN (9:1) as the solvent, phosphonium salt 2a was detected (22.5 ppm, Fig. 1b), though in a low yield (23% based on NMR). Interestingly, with the addition of 1.0 equiv. of bpy, 2a was formed at a much faster rate and PPh3AuCl was totally consumed within an hour (Fig. 1c and d).9
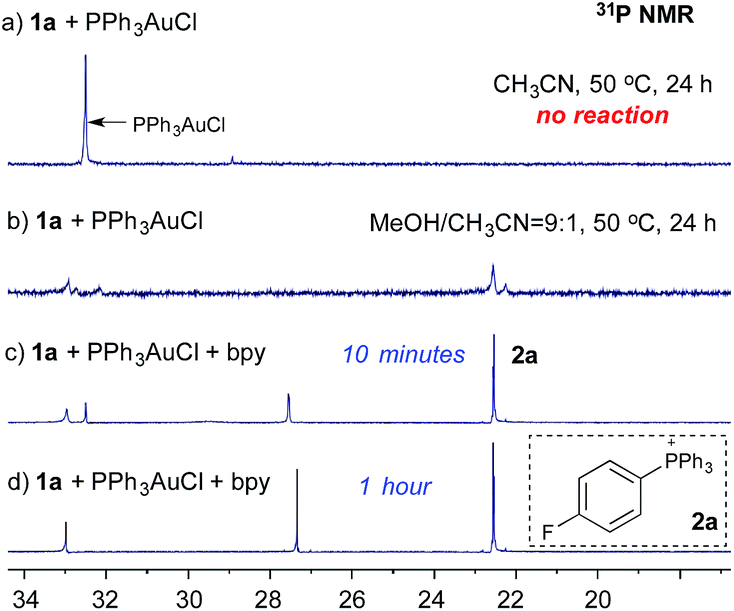 | ||
| Fig. 1 Monitoring the reaction of 1a and PPh3AuCl with 31P NMR. | ||
The formation of phosphonium salt 2a strongly suggested that an Au(III) intermediate is formed during the reaction of PPh3AuCl and diazonium salts with assistance from bpy. Thus, it is likely that the combination of bpy and a diazonium salt is the actual oxidant for the oxidation of PPh3AuCl. Notably, it has been reported in the literature that pyridine can promote diazonium activation through nucleophilic addition.10 Thus, a similar function of bpy is expected as a nucleophile in assisting diazonium activation, which accounts for the observed gold oxidation even without photoinitiation. ESI-MS was used to explore the reaction intermediates. As expected, treating PPh3AuCl/ArN2+/bpy (m/z = 745.12) gave the clear formation of a [PPh3Au(Ar)bpy]+ cation which was also supported by further collision induced dissociation (CID) studies (MS/MS, see details in the ESI†). This result confirmed the gold oxidation by diazonium salts with the assistance of a bpy ligand. Encouraged by this discovery of nucleophilic ligand assisted diazonium activation, we wondered whether it was possible to further extend this gold redox chemistry into challenging C–X bond coupling. Our hypothesis was to explore appropriate anionic nucleophiles to achieve both diazonium activation (for gold activation) and coupling (through reductive elimination) under these mild gold redox conditions with no need for additional photosensitizers (Scheme 2).11
 | ||
| Scheme 2 Proposed Ar–Nu coupling with gold-redox catalysis. | ||
It is well known that the conversion of ArN2+ to ArCl or ArBr can be achieved through standard Sandmeyer conditions using a stoichiometric amount of CuX.12 Successful examples of catalytic Sandmeyer reactions are rare. More importantly, CuX could not promote effective C–S and C–P bond formation through a coupling mechanism. Compared with C–C bond coupling, the formation of a C–X bond from a coupling reaction is thermodynamically less favorable. Thus, there were only a few successful examples reported where this important transformation was achieved catalytically.13 Therefore, the proposed gold-catalyzed coupling is attractive not only due to the mechanistic novelty (no need for a strong oxidant or photo-activation), but also because of its potential synthetic applications (the formation of challenging C–X bonds under catalytic conditions).
In the NMR studies shown in Fig. 1, only a trace amount of aryl chloride was observed, although a stoichiometric amount of PPh3AuCl was used. One possibility is that the reductive elimination of Ar–Cl from Au(III) is unfavorable under gold redox conditions. In fact, Toste group recently confirmed the reductive elimination rate as I > Br > Cl through careful evaluation of different Au(III)–X bond dissociation energies.14 To explore the proposed catalytic C–X bond formation using gold redox chemistry, we started our investigation with the C–Br bond. To our great satisfaction, an excellent yield of aryl bromide 5a was achieved using the gold catalyst under mild conditions (3% PPh3AuCl, 81% in 5 h). Results from some alternative conditions are shown in Table 1.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Variations from above conditions | Time | Conv. (%) | 5a (%) | 3a (%) | 4a (%) |
| a Reaction conditions: 1 (0.1 mmol), NaBr (0.4 mmol), cat. Au (5 mol%) in acetonitrile (ACN), 50 °C. b 19F NMR yield with benzotrifluoride as the internal standard. | ||||||
| 1 | None | 5 h | 100 | 83 | 7 | <5 |
| 2 | Blue LED, No Ph3PAuCl | 12 h | 50 | <10 | Trace | 33 |
| 3 | LiBr instead of NaBr | 12 h | 100 | 78 | 8 | <5 |
| 4 | Acetone instead of ACN | 5 h | 100 | 11 | 37 | <5 |
| 5 | Ph3PAuNTf2 instead of Ph3PAuCl | 5 h | 100 | 68 | 10 | 7 |
| 6 | Ph3PAuNTf2 and 20 mol% bpy | 12 h | 100 | 63 | 8 | 15 |
| 7 | 3 mol% Ph3PAuCl | 5 h | 100 | 81 | 7 | <5 |
| 8 | 1 mol% Ph3PAuCl | 5 h | 100 | 63 | 13 | 9 |
| 9 | No light (darkness) | 5 h | 100 | 76 | 8 | <5 |
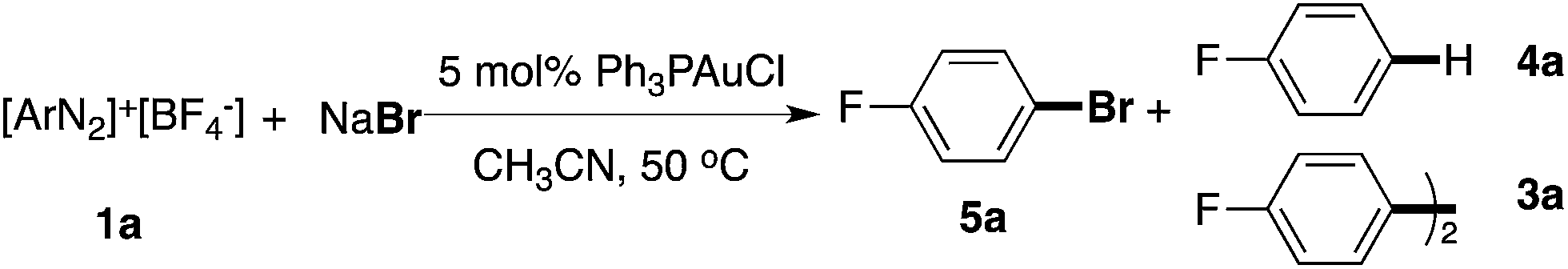
Firstly, the bpy ligand is not required in this reaction, which suggests that Br− could act as an activation factor for the diazonium salts. In fact, reacting a diazonium salt with I− gave the formation of aryl iodide even without any catalyst.15 Less than 10% Ar–Br was observed without the gold catalyst (entry 2). Switching the solvent to acetone gave a significantly increased yield of the homo-coupling product 3a, which suggested either a different reductive elimination reaction rate (relative to transmetallation) or an alternative radical reaction path. Lowering the catalyst loading to 1% led to a reduced yield of 5a (entry 8, 63%) due to the increased aryl homo-coupling and diazonium decomposition (formation of ArH, 4a). The cationic gold(I) catalyst PPh3AuNTf2 also promoted the reaction, though with lower yields (entries 5 and 6), which is similar to the performance of the Ph3PAuBr catalyst. Importantly, a similar reaction yield was observed while conducting the reaction under dark conditions (entry 9), confirming the reaction as nucleophile-promoted activation rather than light-promoted diazonium decomposition. Overall, to the best of our knowledge, this is the first example of a catalytic Sandmeyer reaction using only a gold catalyst (no photo-activation). With this new optimal condition, various substrates were tested. The reaction substrate scope is shown in Table 2.
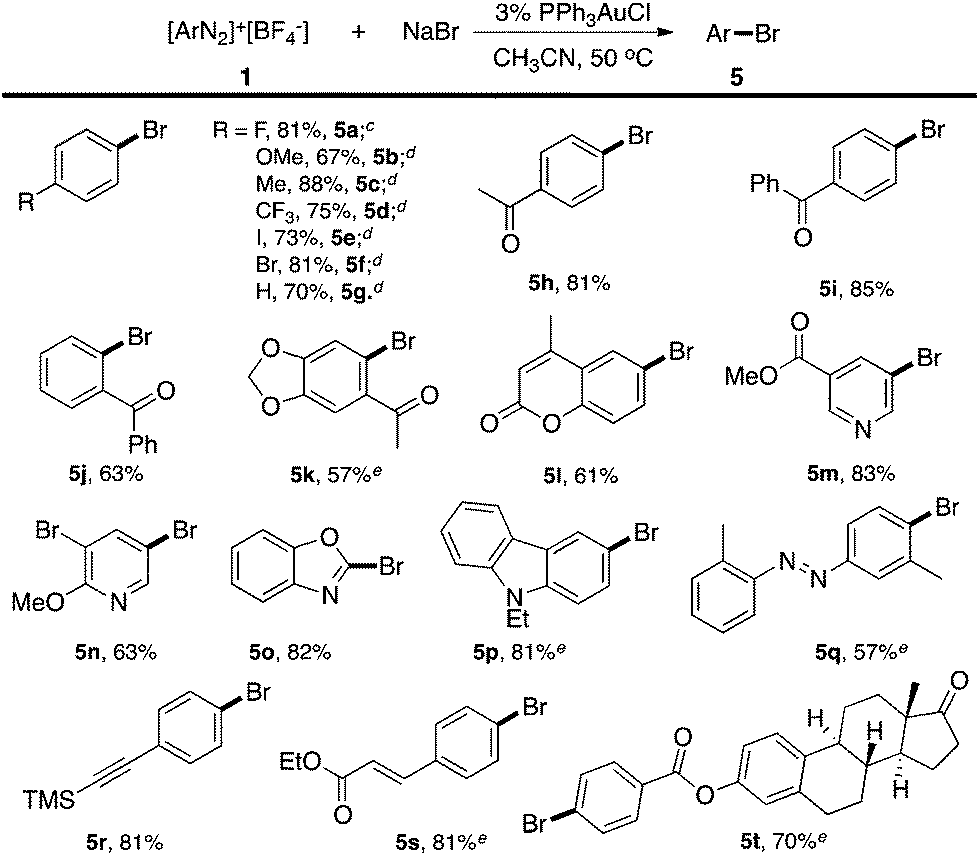
Excellent substrate compatibility was found. Diazonium salts with EWGs and EDGs all furnished the products in good yields (5a–5g). Notably, aryl iodide is also compatible in this catalytic system (5e), highlighting the orthogonal reactivity of the Au catalyst over Pd, Cu, and Ni (for which oxidative addition can occur). Carbonyl groups (5h, 5i and 5j), a benzodioxole (5k) and an azobenzene (5q) were well tolerated in this reaction. Hetero-aromatic diazonium salts, such as pyridines (5m and 5n) and indoles (5p) also worked well in this reaction. Moreover, this reaction proceeded with high efficiency and selectivity for an α,β-unsaturated ester (5s) and p-acetylide aryl diazonium (5r) to give the corresponding products. To further evaluate the synthetic utility and generality of this reaction, we tested a coumarin derivative (5l) and estrone derivative (5t) under the reaction conditions. The desired products were achieved with good yields, highlighting the good potential of this catalytic system for complex molecular synthesis.
ESI-MS studies were performed to explore the reaction mechanism. As shown in Fig. 2, a bisbromide-aryl-gold(III) intermediate was observed with MS under the standard reaction conditions. Through collision induced dissociation (CID) studies (MS/MS), the composition of this intermediate was confirmed (see details in the ESI†). This result provided strong evidence for the formation of an Au(III) intermediate as proposed.
 | ||
| Fig. 2 Evidence of an Au(III) intermediate from ESI-MS. | ||
Encouraged by the success of the gold catalyzed C–Br bond formation, we turned our attention to the synthesis of more challenging C–S and C–P bonds. Unlike the C–Br bond, which can be alternatively prepared using a stoichiometric amount of CuBr, sulfur and phosphine are invalid nucleophiles under Sandmeyer conditions due to the strong coordination of sulfur or phosphine with the Cu cation (completely quenched metal reactivity).
Thiols (RSH) are good nucleophiles in general and can react with arenediazonium salts through an SNAr mechanism with the assistance of a base, especially for acidic thiophenols.16 However, as demonstrated above, one major side reaction of the diazonium decomposition is dediazoniation (the formation of Ar–H). This side reaction was more prevalent when using proton-containing nucleophiles (NuH). For example, as shown in Table 3, the reaction of cysteine derivative 6a with diazonium salt 1a gave only the dediazoniation product 4a in 23% yield. The addition of base (2 equiv. of Na2CO3) did help the formation of the desired thioether 7a (37% yield), however, a significant amount of the dediazoniation by-product 4a was obtained (55%). The application of a stoichiometric amount of Cu(OAc)2 did not help the reaction at all.
|
|
|||||
|---|---|---|---|---|---|
| Catalyst (mol%) | Base (equiv.) | Time | Conv. (%) | 7a (%) | 4a (%) |
| a Reaction conditions: 1a (0.2 mmol), 6a (0.1 mmol), cat. (5 mol%), Na2CO3 (0.2 mmol) in acetonitrile (ACN), rt. b 19F NMR yield with benzotrifluoride as the internal standard. | |||||
| None | None | 24 h | 30 | 0 | 23 |
| None | Na2CO3 (2) | 10 h | 100 | 37 | 55 |
| Cu(OAc)2 (100) | Na2CO3 (2) | 10 h | 100 | 31 | 65 |
| PPh3AuCl (5) | None | 10 h | 55 | 49 | 38 |
| PPh3AuCl (5) | Na2CO3 (2) | 3 h | 100 | 87 | 8 |
| PPh3AuCl (3) | Na2CO3 (2) | 3 h | 100 | 86 | 7 |
| PPh3AuCl (1) | Na2CO3 (2) | 7 h | 100 | 53 | 30 |
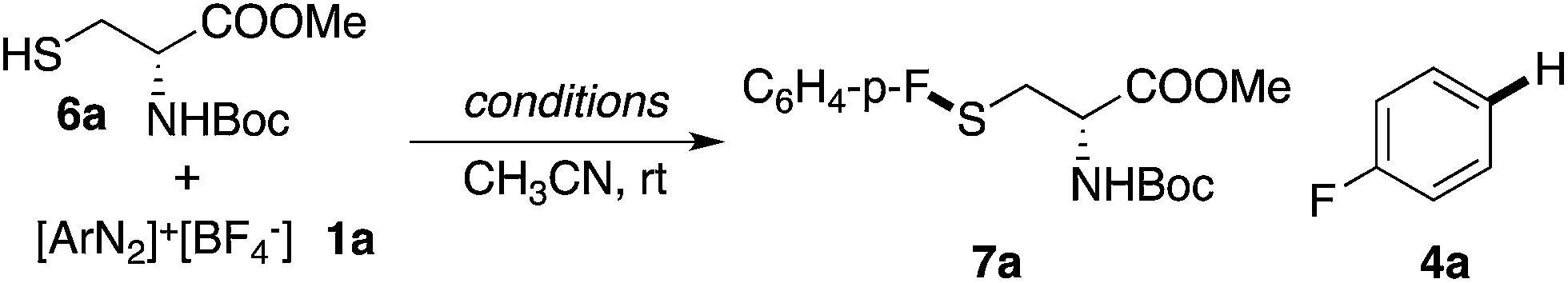
Interestingly, with PPh3AuCl as the catalyst, the desired thioether 7a was obtained even without a base (49% yield). These results suggest that with the help of a thiol nucleophile, PPh3AuCl can be an effective catalyst for diazonium decomposition, forming Au(III) even at room temperature. With the aid of a base, this challenging C–S coupling was achieved in 86% yield with only a 3 mol% gold catalyst loading. Based on the reaction kinetics, the C–S bond formation was dramatically improved with the gold catalyst.17 The reaction scope is shown in Table 4.
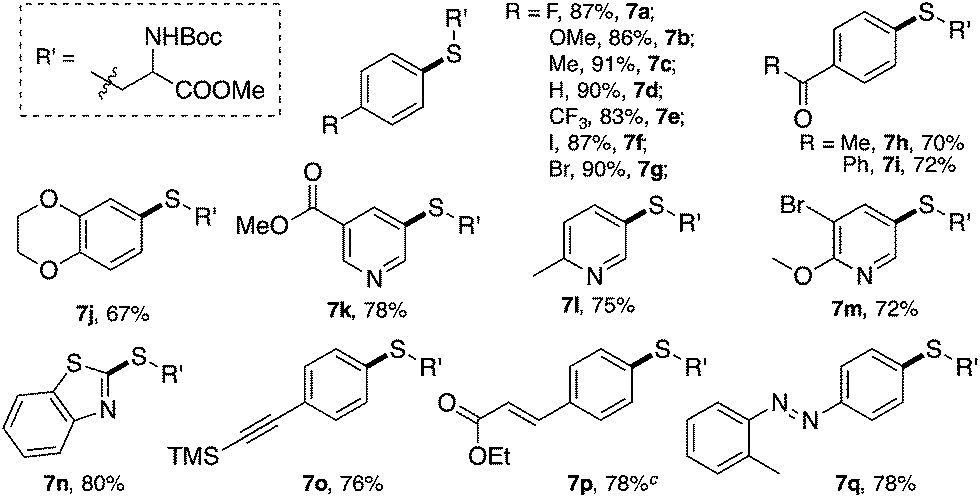
Various cysteine derivatives were successfully prepared in good yields. Both electron-rich (7b, 7c and 7j) and electron-deficient (7a and 7e–7i) diazonium salts were suitable for this transformation with excellent yields. A diazonium salt with an iodide substituent was also tolerated in this reaction (7f), which could be a potential synthetic handle for further functionalization. Heterocycles, including various substituted pyridines (7k–7m) and benzothiazole (7n), gave the desired products efficiently using this catalytic system. An acetylide (7o) and α,β-unsaturated ester (7p) also reacted with good yields. Notably, sulfur containing molecules, as an important class of compounds for both chemical and biological research, are challenging to construct through traditional cross-coupling strategies because of the potential coordination between sulfur and transition metal catalysts.18 This new catalytic system thus provides an efficient strategy to achieve bioactive amino acids.
Our last attempt is to explore the possibility of C–P bond formation using gold redox catalysis. Compared with the C–S bond, C–P bond formation is more challenging as H-phosphonate is much less nucleophilic and it could also be a potential reductant for diazonium salts.19 Thus, the C–P bond formation with diazonium salts cannot be achieved through either SNAr or Cu-promoted Sandmeyer reactions. Recently, Toste and coworkers reported the application of a photocatalyst in gold-catalyzed oxidative coupling to achieve this C–P bond formation.20 Based on the results discussed above, we wondered whether this nucleophile-promoted gold redox catalysis could be used to achieve this C–P bond formation.
As shown in Table 5, no desired arylphosphonate (8a) was obtained using base and/or copper acetate. Impressively, 8a was formed even with solely PPh3AuCl, though in a low yield (25%). The addition of Na2CO3 did not improve the cross-coupling but promoted Ar–H formation. The combination of PPh3AuNTf2 and bpy in the presence of Na2CO3 (previously reported C–C bond coupling conditions) also failed to increase the yield of the desired C–P coupling product. Considering that a nucleophilic ligand is crucial in this gold redox catalysis, we turned our attention to other pyridine derivatives. Through a comprehensive screening, 3-Cl-pyridine was identified as the optimal nucleophile (see detailed screenings in the ESI†), giving the desired C–P bond coupling product 8a in 83% isolated yield. Using PPh3AuNTf2 as the catalyst led to a lower yield of 8a due to the increased yield of the side reactions. Notably, without a gold catalyst, diaza compound 9a was formed as the major product at room temperature whereas no desired coupling product 8a was detected.22 At 50 °C, the reaction was very messy and 8a was not detected at all, which suggested that 8a was not formed from the decomposition of diaza compound 9a. The reaction substrate scope is shown in Table 6.
|
|
||||||
|---|---|---|---|---|---|---|
| Catalyst (mol%) | Additives (equiv.) | Time | Conv. (%) | 8a (%) | 4a (%) | 9a (%) |
|
a Reaction conditions: 1a (0.2 mmol), HP(O)(OEt)2 (0.1 mmol), cat. (5 mol%), base (0.2 mmol) in acetonitrile (ACN), 50 °C.
b
19F NMR yield with benzotrifluoride as the internal standard.
c ACN:EtOH = 6:1.
d Room temperature.21
|
||||||
| None | None | 10 h | 50 | 0 | 31 | 0 |
| None | Na2CO3 (2) | 10 h | 100 | 0 | 70 | 0 |
| Cu(OAc)2 (100) | Na2CO3 (2) | 10 h | 100 | 0 | 75 | 0 |
| PPh3AuCl (5) | None | 10 h | 50 | 25 | 13 | 0 |
| PPh3AuCl (5) | Na2CO3 (2) | 10 h | 100 | 11 | 38 | 0 |
| PPh3AuNTf2 (5) | bpy (0.2), Na2CO3 (2) | 10 h | 100 | <5 | 53 | 11 |
| PPh3AuCl (5)c | 3-Cl-py (2) | 3 h | 100 | 83 | 7 | 0 |
| PPh3AuNTf2 (5)c | 3-Cl-py (2) | 3 h | 100 | 70 | 15 | 0 |
| Noned | 3-Cl-py (2) | 10 h | 69 | 0 | 5 | 44 |
| None | 3-Cl-py (2) | 10 h | >90 | 0 | 25 | 4 |
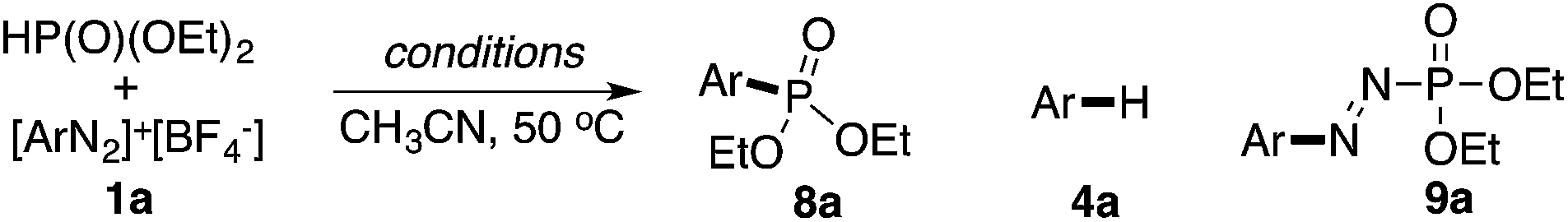
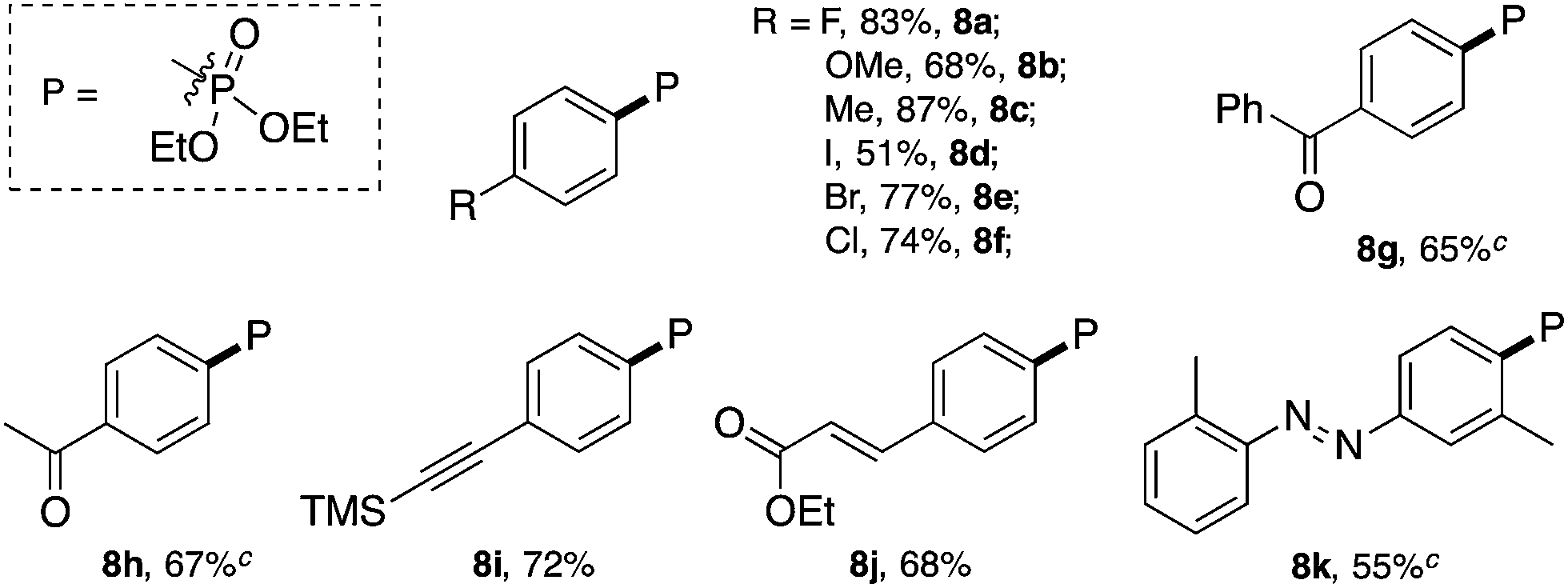
Similar to the C–Br and C–S coupling, a broad substrate scope is observed for the C–P bond formation reactions. Aryl phosphonates with electron rich (8b and 8c) and electron deficient (8a and 8d–8h) substituents could all be generated with good yields. Halogen substituent groups (8a, 8e and 8f) were all tolerated. An alkyne (8i), α,β-unsaturated ester (8j) and azobenzene (8k) also gave good results, suggesting the great synthetic potential of this methodology.
Conclusions
In summary, we reported C–Br, C–S, and C–P bond formation through gold redox catalysis. We demonstrated that nucleophiles play a crucial role in the Au(I) promoted diazonium decomposition. With this strategy, various C–X couplings could be achieved with excellent yields and a broad substrate scope simply using LAuCl (no need for an external oxidant). These results not only provide a new practical strategy to achieve challenging C–X bond couplings, but also, more importantly, reveal some new mechanistic insight regarding gold redox catalysis, which will likely further enrich the pedigree of gold catalysis.Acknowledgements
We are grateful for financial support from the NSF (CHE-1619590) and NSFC (21228204). CX and HC thank support from NSF career Award (CHE-1149367) and NSF IDBR (CHE-1455554).Notes and references
- Selected recent reviews on Au catalysis: (a) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180 CrossRef CAS PubMed; (b) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410 CrossRef PubMed; (c) D. Gorin and F. D. Toste, Nature, 2007, 446, 395 CrossRef CAS PubMed; (d) A. Arcadi, Chem. Rev., 2008, 108, 3266 CrossRef CAS PubMed; (e) Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239 CrossRef CAS PubMed; (f) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351 CrossRef CAS PubMed; (g) R. A. Widenhoefer, Chem.–Eur. J., 2008, 14, 5382 CrossRef CAS PubMed; (h) A. Fürstner, Chem. Soc. Rev., 2009, 38, 3208 RSC; (i) N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994 CrossRef CAS PubMed; (j) M. Bandani, Chem. Soc. Rev., 2011, 40, 1358 RSC; (k) M. N. Hopkinson, A. D. Gee and V. Gouverneur, Chem.–Eur. J., 2011, 17, 8248 CrossRef CAS PubMed; (l) A. Corma, A. Leyva-Pérez and M. Sabater, Chem. Rev., 2011, 111, 1657 CrossRef CAS PubMed; (m) T. de Haro and C. Nevado, Synthesis, 2011, 2530 CAS; (n) L. Liu and G. B. Hammond, Chem. Soc. Rev., 2012, 41, 3129 RSC; (o) M. Rudolph and A. S. K. Hashmi, Chem. Soc. Rev., 2012, 41, 2448 RSC; (p) L. Zhang, Acc. Chem. Res., 2014, 47, 877 CrossRef CAS PubMed; (q) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028 CrossRef CAS PubMed; (r) D. Pflästerer and A. S. K. Hashmi, Chem. Soc. Rev., 2016, 45, 1331 RSC.
- E 0 (AuI/AuIII) = +1.40 V. See: CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, Boca Raton, FL, 84th edn, 2004 CrossRef CAS. For a stable example, including X-ray crystal structure analysis, of a dicoordinated gold(I) center not undergoing oxidative addition with an aryl iodide that could intramolecularly react through a six-membered transition state, see: A. S. K. Hashmi, C. Lothschütz, R. Döpp, M. Ackermann, J. D. B. Becker, M. Rudolph, C. Scholz and F. Rominger, Adv. Synth. Catal., 2012, 354, 133 CrossRef CAS.
- For selected examples, see: (a) A. S. K. Hashmi, T. D. Ramamurthi and F. Rominger, J. Organomet. Chem., 2009, 694, 592 CrossRef CAS; (b) G. Zhang, Y. Peng, L. Cui and L. Zhang, Angew. Chem., Int. Ed., 2009, 48, 3112 CrossRef CAS PubMed; (c) M. N. Hopkinson, J. E. Ross, G. T. Giuffredi, A. D. Gee and V. Gouverneur, Org. Lett., 2010, 12, 4904 CrossRef CAS PubMed; (d) G. Zhang, L. Cui, Y. Wang and L. Zhang, J. Am. Chem. Soc., 2010, 132, 1474 CrossRef CAS PubMed; (e) A. D. Melhado, W. E. Brenzovich, A. D. Lackner and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 8885 CrossRef CAS PubMed; (f) T. de Haro and C. Nevado, J. Am. Chem. Soc., 2010, 132, 1512 CrossRef CAS PubMed; (g) A. Leyva-Põrez, A. Domõnech, S. I. Al-Resayes and A. Corma, ACS Catal., 2012, 2, 121 CrossRef; (h) S. Shi, T. Wang, W. Yang, M. Rudolph and A. S. K. Hashmi, Chem.–Eur. J., 2013, 19, 6576 CrossRef CAS PubMed; (i) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Science, 2012, 337, 1644 CrossRef CAS PubMed; (j) W. J. Wolf, M. S. Winston and F. D. Toste, Nat. Chem., 2014, 6, 159 CrossRef CAS PubMed; (k) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, J. Am. Chem. Soc., 2014, 136, 254 CrossRef CAS PubMed; (l) M. Joost, A. Zeineddine, L. Estõvez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 14654 CrossRef CAS PubMed; (m) Q. Wu, C. Du, Y. Huang, X. Liu, Z. Long, F. Song and J. You, Chem. Sci., 2015, 6, 288 RSC; (n) M. Joost, L. Estõvez, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 5236 CrossRef CAS PubMed; (o) M. D. Levin, S. Kim and F. D. Toste, ACS Cent. Sci., 2016, 2, 293 CrossRef CAS PubMed.
- (a) B. Sahoo, M. N. Hopkinson and F. Glorius, J. Am. Chem. Soc., 2013, 135, 5505 CrossRef CAS PubMed; (b) X.-Z. Shu, M. Zhang, Y. He, H. Frei and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5844 CrossRef CAS PubMed; (c) M. Wieteck, Y. Tokimizu, M. Rudolph, F. Rominger, H. Ohno, N. Fujii and A. S. K. Hashmi, Chem.–Eur. J., 2014, 20, 16331 CrossRef CAS PubMed; (d) J. Xie, S. Shi, T. Zhang, N. Mehrkens, M. Rudolph and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2015, 54, 6046 CrossRef CAS PubMed; (e) A. Tlahuext-Aca, M. N. Hopkinson, B. Sahoo and F. Glorius, Chem. Sci., 2016, 7, 89 RSC; (f) S. Kim, J. Rojas-Martin and F. D. Toste, Chem. Sci., 2016, 7, 85 RSC; (g) J. Xie, T. Zhang, F. Chen, N. Mehrkens, F. Rominger, M. Rudolph and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 2934 CrossRef CAS PubMed; (h) S. Hosseyni, L. Wojtas, M. Li and X. Shi, J. Am. Chem. Soc., 2016, 138, 3994 CrossRef CAS PubMed.
- (a) L. Huang, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 4808 CrossRef CAS PubMed; (b) L. Huang, F. Rominger, M. Rudolph and A. S. K. Hashmi, Chem. Commun., 2016, 52, 6435 RSC.
- The monitored quantitative rate constant kobs for sp2 C–C coupling is (1.5 ± 0.1) × 10−4 S−1 (−52 °C), see ref. 3h; the homo-coupling product was generated from transmetallation between Au(III) and Au(I) intermediate followed by reductive elimination, see ref. 3f.
- R. Cai, M. Lu, E. Y. Aguilera, Y. Xi, N. G. Akhmedov, J. L. Peterson, H. Chen and X. Shi, Angew. Chem., Int. Ed., 2015, 54, 8772 CrossRef CAS PubMed.
- D. V. Patil, H. Yun and S. Shin, Adv. Synth. Catal., 2015, 357, 2622 CrossRef CAS.
- The structure with 27.7 ppm in 31P NMR is not clear right now. According to the literature, the 31P NMR signal of the Au(III) intermediate is around 27 ppm (ref. 4b and 5).
- A pyridine promoted diazonium decomposition through the formation of an aryl radical was proposed: T. Sakakura, M. Hara and M. Tanaka, J. Chem. Soc., Chem. Commun., 1985, 1545 RSC
. - Another possible mechanism is that the nucleophiles are reacting with LAuCl to generate more electron-rich LAuNu, which would be more susceptible to oxidation by diazonium salts.
- For reviews on diazonium salts in cross-coupling, see: (a) S. Bräse, Acc. Chem. Res., 2004, 37, 805 CrossRef PubMed; (b) A. Roglans, A. Pla-Quintana and M. Moreno-Mañas, Chem. Rev., 2006, 106, 4622 CrossRef CAS PubMed; (c) F. Mo, G. Dong, Y. Zhang and J. Wang, Org. Biomol. Chem., 2013, 11, 1582 RSC. For selected examples using diazonium salts, see: (d) C. Molinaro, J. Mowat, F. Gosselin, P. D. O'Shea, J.-F. Marcoux, R. Angelaud and I. W. Davies, J. Org. Chem., 2007, 72, 1856 CrossRef CAS PubMed; (e) I. P. Beletskaya, A. S. Sigeev, A. S. Peregudov and P. V. Petrovskii, Synthesis, 2007, 2534 CrossRef CAS; (f) G. Danoun, B. Bayarmagnai, M. F. Grünberg and L. J. Gooβen, Angew. Chem., Int. Ed., 2013, 52, 7972 CrossRef CAS PubMed; (g) J. Dai, C. Fang, B. Xiao, J. Yi, J. Xu, Z. Liu, X. Lu, L. Liu and Y. Fu, J. Am. Chem. Soc., 2013, 135, 8436 CrossRef CAS PubMed; (h) X. Wang, Y. Xu, F. Mo, G. Ji, D. Qiu, J. Feng, Y. Ye, S. Zhang, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2013, 135, 10330 CrossRef CAS PubMed; (i) C. Matheis, V. Wagner and L. J. Gooβen, Chem.–Eur. J., 2016, 22, 79 CrossRef CAS PubMed.
- For selected examples, see: (a) X. Shen, A. M. Hyde and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 14076 CrossRef CAS PubMed; (b) J. Pan, X. Wang, Y. Zhang and S. L. Buchwald, Org. Lett., 2011, 13, 4974 CrossRef CAS PubMed; (c) Y. Imazaki, E. Shirakawa, R. Ueno and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 14760 CrossRef CAS PubMed; (d) M. Feller, Y. Diskin-Posner, G. Leitus, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2013, 135, 11040 CrossRef CAS PubMed; (e) C. M. Le, P. J. C. Menzies, D. A. Petrone and M. Lautens, Angew. Chem., Int. Ed., 2015, 54, 254 CrossRef CAS PubMed; (f) C. Chen, L. Hou, M. Cheng, J. Su and X. Tong, Angew. Chem., Int. Ed., 2015, 54, 3092 CrossRef CAS PubMed.
- M. S. Winston, W. J. Wolf and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 7921 CrossRef CAS PubMed.
- See recent example: A. Hubbard, T. Okazaki and K. K. Laali, J. Org. Chem., 2008, 73, 316 CrossRef CAS PubMed.
- (a) P. C. B. Page, R. D. Wilkes and D. Reynolds, in Comprehensive Organic Functional Group Transformations, ed. A. R. Katritzky, O. Meth-Cohn and C. W. Rees, Elsevier, Oxford, 1995, p. 113 Search PubMed; For selected recent examples, see: (b) M. Majek and A. J. V. Wangelin, Chem. Commun., 2013, 49, 5507 RSC; (c) M. Barbero, I. Degani, N. Diulgheroff, S. Dughera, R. Fochi and M. Migliaccio, J. Org. Chem., 2000, 65, 5600 CrossRef CAS PubMed; (d) X. Wang, G. D. Cuny and T. Noël, Angew. Chem., Int. Ed., 2013, 52, 7860 CrossRef CAS PubMed.
- Without a catalyst, C–S coupling product 7a has nearly the same kinetics as the by-product 4a; with the gold catalyst, the formation of 7a is accelerated with less than 10% 4a formed. See the detailed kinetics study in the ESI.†.
- For selected examples, see: (a) P. Johannesson, G. Lindeberg, A. Johannesson, G. V. Nikiforovich, A. Gogoli, B. Synergren, M. Le Greves, F. Nyberg, A. Karlen and A. Hallberg, J. Med. Chem., 2002, 45, 1767 CrossRef CAS PubMed; (b) L. Llauger, H. Z. He, J. Kim, J. Aguirre, N. Rosen, U. Peters, P. Davies and G. Chiosis, J. Med. Chem., 2005, 48, 2892 CrossRef CAS PubMed; (c) A. Gangjee, Y. B. Zheng, T. Talreja, J. J. Mc Guire, R. L. Kisliuk and S. F. Queener, J. Med. Chem., 2007, 50, 2046 Search PubMed.
- For recent examples of C–P bond formation, see: (a) J. Xuan, T. Zeng, J. Chen, L. Lu and W. Xiao, Chem.–Eur. J., 2015, 21, 4962 CrossRef CAS PubMed; (b) G. Hu, W. Chen, T. Fu, Z. Peng, H. Qiao, Y. Gao and Y. Zhao, Org. Lett., 2013, 15, 5362 CrossRef CAS PubMed; (c) O. Berger, C. Petit, E. L. Deal and J. L. Montchamp, Adv. Synth. Catal., 2013, 355, 1361 CrossRef CAS; (d) K. Xu, H. Hu, F. Yang and Y. Wu, Eur. J. Org. Chem., 2013, 2013, 319 CrossRef CAS; (e) A. J. Bloomfield and S. B. Herzon, Org. Lett., 2012, 14, 4370 CrossRef CAS PubMed; (f) S. M. Rummelt, M. Ranocchiari and J. A. van Bokhoven, Org. Lett., 2012, 14, 2188 CrossRef CAS PubMed; (g) C. R. Shen, G. Q. Yang and W. B. Zhang, Org. Biomol. Chem., 2012, 10, 3500 RSC; (h) E. L. Deal, C. Petit and J. L. Montchamp, Org. Lett., 2011, 13, 3270 CrossRef CAS PubMed.
- Y. He, H. Wu and F. D. Toste, Chem. Sci., 2015, 6, 1194 RSC.
- Reaction of L-AuCl with pyridine did not give any [LAu(pyridine)]+ based on NMR, which confirmed that [LAuNu]+ was unlikely to be the actual catalyst for this reaction.
- In the palladium catalyzed C–P coupling with diazonium salts in the presence of CuI, 9a was also isolated as the major by-product. See: R. Berrino, S. Cacchi, G. Fabrizi, A. Goggiamani and P. Stabile, Org. Biomol. Chem., 2010, 8, 4518 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc01742h |
| This journal is © The Royal Society of Chemistry 2016 |