DOI:
10.1039/C6SC01795A
(Edge Article)
Chem. Sci., 2016,
7, 6176-6181
Benzo[4,5]cyclohepta[1,2-b]fluorene: an isomeric motif for pentacene containing linearly fused five-, six- and seven-membered rings†
Received
25th April 2016
, Accepted 7th June 2016
First published on 7th June 2016
Abstract
Benzo[4,5]cyclohepta[1,2-b]fluorene (5a), a new π-conjugated polycyclic hydrocarbon containing linearly fused six-, five-, six-, seven- and six-membered rings (C6–C5–C6–C7–C6), was designed and its stable derivatives 5b and 5c were synthesized. With 22 π electrons, 5a is an isomer of pentacene with quinoidal, dipolar ionic and diradical resonance forms. Molecules 5b and 5c were experimentally investigated with cyclic voltammetry, electronic absorption spectroscopy and X-ray crystallographic analysis, and theoretically studied by calculating the NICS value, diradical character and dipole moment. A comparison of 5a–c with pentacene and other pentacene analogues containing linearly fused five- or seven-membered rings was also conducted and discussed. It was found that 5b behaved as a p-type organic semiconductor in solution-processed thin film transistors with a field effect mobility of up to 0.025 cm2 V−1 s−1.
Introduction
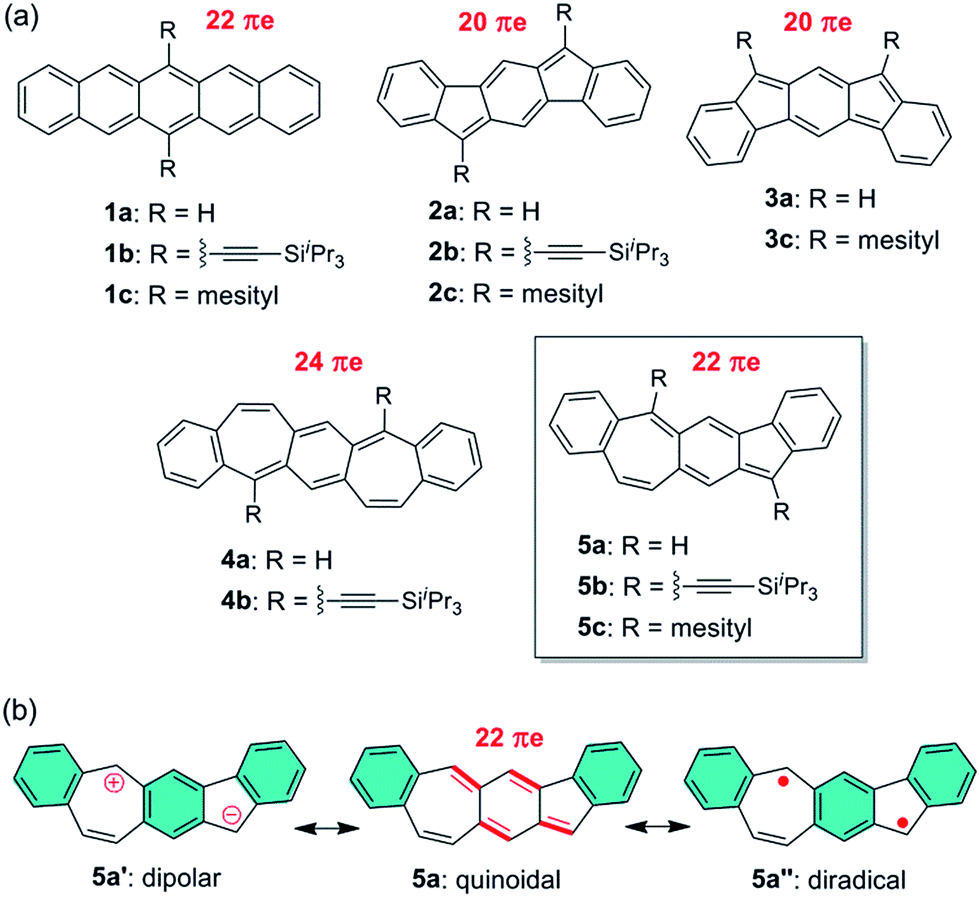
Pentacene (1a in Fig. 1a) is a leading p-type organic semiconductor for applications in light-weight, flexible and low-cost organic electronic devices,1 and has been used as a benchmark in comparison with new materials for applications in organic thin film transistors (OTFTs).2 Pentacene has been molecularly engineered with three strategies in order to modify electronic structure, tune molecular packing in the solid state, improve solubility and stability, and better understand its structure–property relationship. As extensively studied, the first strategy is to substitute H atoms in pentacene with a variety of functional groups.3 The most successful example of this strategy is 6,13-bis((triisopropylsilyl)ethynyl)-pentacene (1b in Fig. 1a),4 which is a solution-processed high-mobility p-type semiconductor5,6 with brickwork arrangement of π-planes. The second strategy is to replace C atoms in pentacene with hetero atoms, such as B,7 N,8 and S.9,10 Among the resultant heteropentacenes, N-heteropentacenes were most extensively studied, and have recently arisen as a class of organic semiconductors with high performance in OTFTs.11 The third strategy is to replace six-membered rings in pentacene with five- or seven-membered rings, leading to recently reported pentacene analogues containing C6–C5–C6–C5–C612–14 and C6–C7–C6–C7–C615 polycyclic frameworks, such as 2–4 in Fig. 1a. With 20 π electrons, 2a and 3a both have two π electrons less than pentacene, while 4a has two more π electrons. Therefore, their electronic structure and physical properties are distinctively different from those of pentacene. In this study, we explore a novel linearly fused pentacene analogue, benzo[4,5]cyclohepta[1,2-b]fluorene (5a in Fig. 1a), which contains an unprecedented C6–C5–C6–C7–C6 polycyclic framework. Unlike other pentacene analogues, 5a is a constitutional isomer of pentacene having both five- and seven-membered rings in the linear π-backbone with 22 π electrons. Besides the quinoidal resonance structure, one dipolar ionic resonance form (5a′) and one open-shell diradical form (5a′′) can be also drawn for 5a (Fig. 1b). The existence of one more aromatic sextet ring (shaded in blue) in 5a′ and 5a′′ suggests that these two resonance forms might make a significant contribution to the ground state structure. Like all other pentacene analogues, bulky triisopropylsilylethynyl (in 5b) or mesityl (in 5c) groups are introduced to the reactive sites so that soluble and stable materials can be obtained. Detailed below are their synthesis, ground-state structures, physical properties and their applications for OTFTs. A comparison with pentacene and other pentacene analogues is also made to better understand the structure–property relationship.
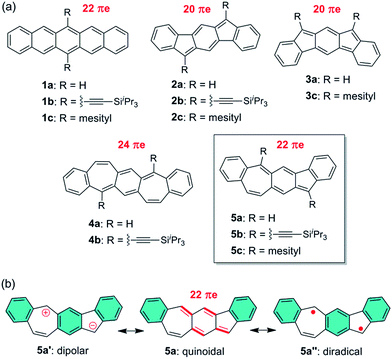 |
| | Fig. 1 (a) Chemical structures of pentacene and its analogues; (b) three typical resonance forms of 5a. | |
Results and discussion
Synthesis and characterization
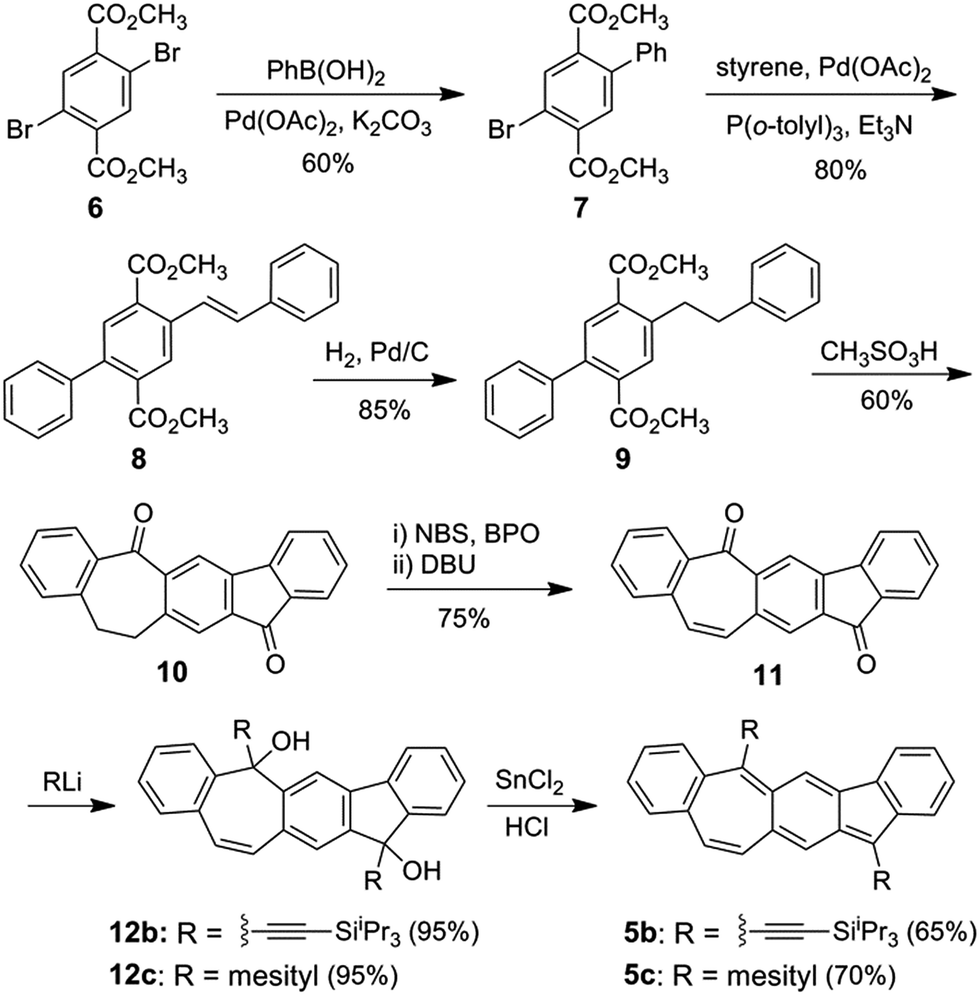
Scheme 1 shows the synthesis of 5b and 5c starting from commercially available dimethyl 2,5-dibromoterephthalate 6, which was coupled with phenyl boronic acid and styrene subsequently in the Suzuki reaction and Heck reaction, respectively, resulting in the diester 8. Pd/C-catalyzed hydrogenation of 8 followed by treatment with methanesulfonic acid at 100 °C led to cyclized product 10. Bromination of 10 and subsequent elimination of HBr yielded the dehydrogenated dione 11. X-Ray crystallographic analysis of the single crystals of 11 revealed a non-planar geometry (Fig. S6 in ESI†), which can explain its moderate solubility in common organic solvents. Nucleophilic addition of (triisopropylsilyl)ethynyl and mesityl lithium to 11 resulted in the diols 12b and 12c, respectively, which both were obtained as a mixture of cis and trans-isomers. Reduction of intermediate diols 12b and 12c in THF with a solution of concentrated HCl that was saturated with SnCl2 led to 5b and 5c, respectively, both as deep green solids in moderate yield. Dione 10 was also synthesized from 2,5-dibromo-p-xylene in a similar approach in higher overall yield but more steps (Scheme S1 in ESI†). The 1H NMR spectra of 5b and 5c (ESI†) both show sharp splitting and narrow line widths indicating that they behave more like closed-shell compounds in the ground state.16
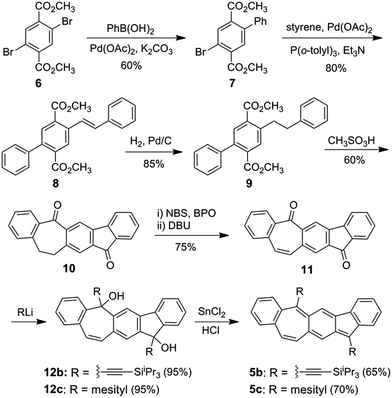 |
| | Scheme 1 Synthesis of 5b/5c. | |
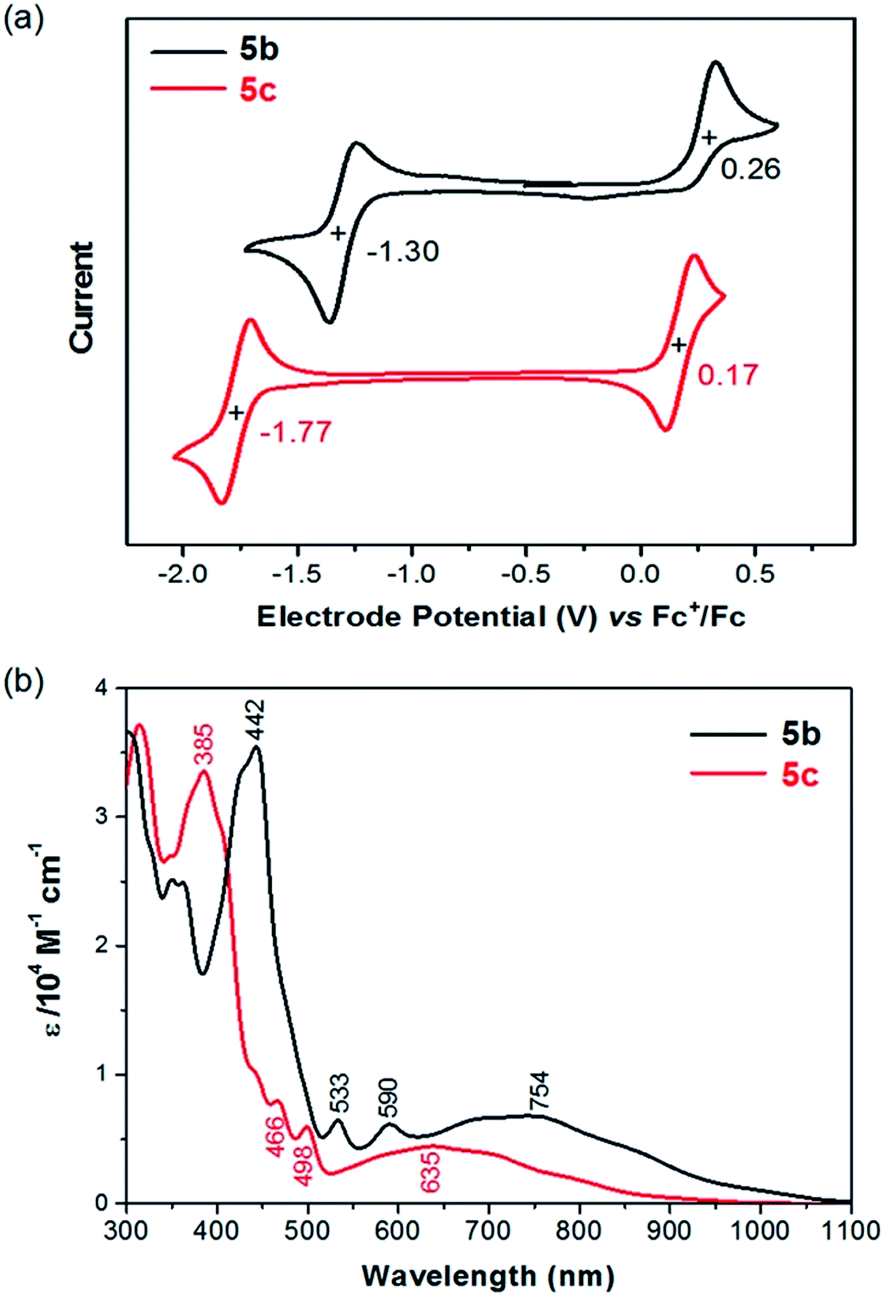
The redox behaviors of 5b/5c in solution were investigated with cyclic voltammetry. In the test window of cyclic voltammetry, 5b exhibits a reversible reduction (5b/5b−) wave and an irreversible oxidation (5b/5b+) wave, while 5c exhibits a reversible reduction (5c/5c−) wave and a reversible oxidation (5c/5c+) wave as shown in Fig. 2a. The half-wave reduction potentials (Ered1/2) of 5b and 5c are −1.30 V and −1.77 V versus the ferrocenium/ferrocene (Fc+/Fc) redox couple, respectively, from which the lowest unoccupied molecular orbital (LUMO) energy levels of 5b and 5c are estimated as −3.80 eV and −3.33 eV, respectively.17 Similarly, the highest occupied molecular orbital (HOMO) energy levels of 5b and 5c are estimated as −5.36 eV and −5.27 eV from the half-wave oxidation potential (Eox1/2 = 0.26 V and 0.17 V vs. Fc+/Fc, respectively).17 The lower LUMO and HOMO energy levels of 5b in comparison with 5c can be attributed to the facts that the ethynyl substituents with sp hybridized carbons in 5b are electron withdrawing and the substituting phenyl groups in 5c are almost orthogonal to the polycyclic backbone with poor conjugation. Table 1 compares 5b/5c with those of the related molecules 1–4 in terms of electrochemical potentials and frontier molecular orbital energy levels. It is found that 5b and 5c have a higher HOMO energy level and a lower LUMO energy level than the corresponding pentacene derivatives 1b and 1c, respectively. Furthermore, the oxidation potential of 5b is almost the same as that of 4b, and the reduction potential of 5b is close to that of 2b. Molecule 5c has a reduction potential close to that of 3c, which has the same mesityl substituents. These findings are in agreement with the assumption that the first reduction of 5b/5c occurs on the five-membered ring leading to an aromatic cyclopentadienide anion and the first oxidation of 5b/5c occurs on the seven-membered ring leading to an aromatic cycloheptatrienium cation.
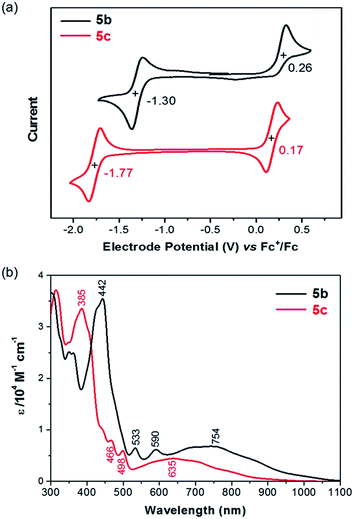 |
| | Fig. 2 (a) Cyclic voltammograms of 5b and 5c recorded in CH2Cl2 with Fc+/Fc as the external standard at a scan rate of 50 mV s−1; (b) UV-vis-NIR absorption of 5b and 5c in CH2Cl2. | |
Table 1 Electrochemical potentials and frontier molecular orbital energy levels of 1–5
| |
E
red1/2
/V |
E
ox1/2
/V |
LUMOb/eV |
HOMOc/eV |
E
ECg
/eV |
|
E
red1/2 and Eox1/2 are the half-wave potential (vs. Fc+/Fc) of the first oxidation and reduction wave, respectively.
Estimated from LUMO = −5.10 − Ered (eV).
Estimated from HOMO = −5.10 − Eox (eV).
E
ECg = LUMO − HOMO.
|
|
1b
18
|
−1.50 |
0.37 |
−3.60 |
−5.47 |
1.87 |
|
1c
10
|
−1.92 |
0.22 |
−3.18 |
−5.32 |
2.14 |
|
2b
12
|
−1.15 |
0.74 |
−3.95 |
−5.84 |
1.89 |
|
2c
13
|
−1.58 |
0.64 |
−3.52 |
−5.74 |
2.22 |
|
3c
14
|
−1.13 |
0.13 |
−3.97 |
−5.23 |
1.26 |
|
4b
15
|
−1.66 |
0.12 |
−3.44 |
−5.32 |
1.78 |
|
5b
|
−1.30 |
0.26 |
−3.80 |
−5.36 |
1.56 |
|
5c
|
−1.77 |
0.17 |
−3.33 |
−5.27 |
1.94 |
As shown in Fig. 2b, 5b and 5c in CH2Cl2 exhibit electronic absorption spectra very different from those of pentacene and other analogues. The broad absorption band in the visible-near infrared (vis-NIR) region could be attributed to the HOMO → LUMO transition based on time-dependent density functional theory (TDDFT) calculations (ESI†). The intense absorption band at the UV-vis region can be mainly attributed to the HOMO−1 → LUMO and HOMO → LUMO+1 transitions. The optical energy gaps (EOptg) of 5b and 5c were estimated to be 1.13 eV and 1.25 eV, respectively, from the lowest energy absorption onset. The optical energy gap of 5b/5c is significantly smaller than the HOMO–LUMO gap (EECg) as estimated from electrochemical potentials. A similar phenomenon was also observed from azulene, which has an optical energy gap of 1.75 eV (about 710 nm)19 and an electrochemical energy gap of 2.35 eV.20 Azulene has a lower transition energy than anticipated from the HOMO–LUMO gap because the excited state of azulene has a smaller repulsive energy between the two electrons occupying HOMO and LUMO due to the nonalternant nature of azulene.21–23 This explanation may also account for the smaller optical energy gap of 5b/5c, whose pentacyclic backbone is also nonalternant.
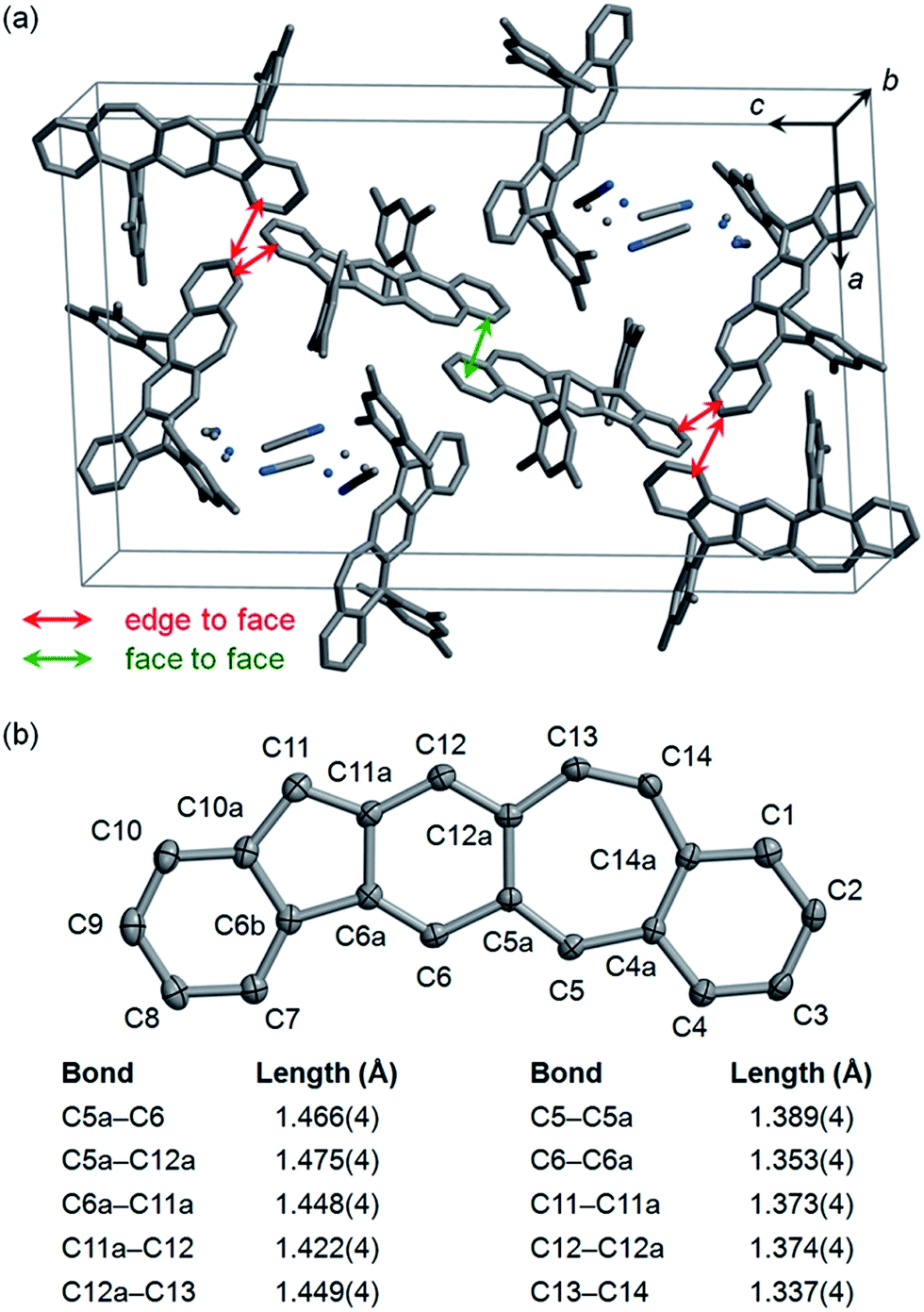
Single crystals of 5c selected for X-ray crystallographic analysis were grown by slow diffusion of acetonitrile into a solution in CH2Cl2.24 It is found that the unit cell of this crystal contains crystallized solvent (CH3CN) molecules with disorder as shown in Fig. 3a. In the crystal structure of 5c·CH3CN, the pentacyclic backbone of 5c (Fig. 3b) is essentially flat and is almost perpendicular to the substituting mesityl groups with dihedral angles of 80.2° and 87.9°. Examination of the bond lengths in the central six-membered ring reveals four C–C single bonds (C5a–C12a, C5a–C6, C6a–C11a, C11a–C12) with bond lengths of 1.42–1.48 Å and two C–C double bonds (C6–C6a, C12–C12a) with bond lengths of 1.35–1.37 Å.25 Moreover, the central six-membered ring is bonded to C5 and C11 with relatively short bond lengths (C5–C5a: 1.39 Å; C11–C11a: 1.37 Å). The above bond lengths are similar to the corresponding bond lengths in the crystallographic structures of 2b,122c,13 and 4b,15 indicating a p-quinodimethane structure with large bond length alternation. In addition to the C5–C5a bond, the seven-membered ring contains another C–C double bond (C13–C14) with a bond length (1.34 Å) typical for alkenes. Neighboring molecules of 5c exhibit poor π–π interactions between the pentacyclic backbones presumably because the bulky mesityl substituting groups block π–π interactions. Only a small face-to-face overlap with a π-to-π distance of 3.40 Å and a small number of edge-to-face contacts are observed as shown in Fig. 3a.
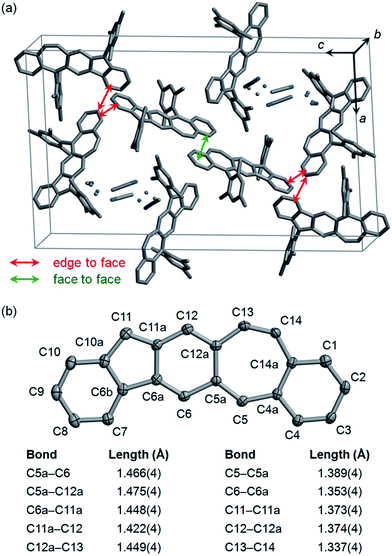 |
| | Fig. 3 Crystallographic structure of 5c·CH3CN with hydrogen atoms removed for clarification: (a) a unit cell with disordered atoms of CH3CN shown as dots; (b) the pentacyclic backbone of 5c with carbon atoms labeled and some bond lengths highlighted (carbon atoms are shown as ellipsoids at the 50% probability level). | |
Computational studies
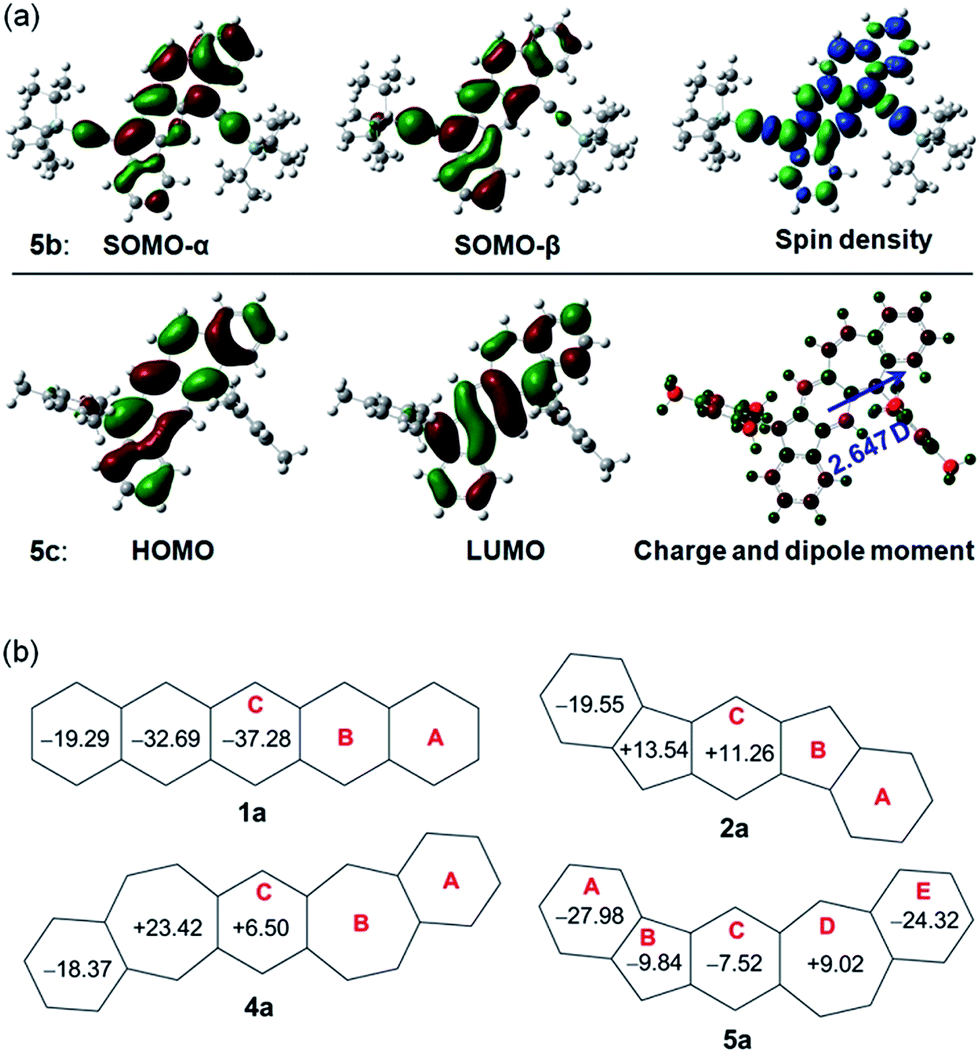
Density functional theory (DFT) calculations at the (U)CAM-B3LYP/6-31G* level of theory were conducted to better understand the ground state structures of 5a–c. It is found that the solution of the open-shell singlet (OS) state has a lower energy than the closed-shell (CS) state for 5b, thus defining an open-shell singlet ground state. The singly occupied molecular orbitals (SOMO) of the α and β spins are partially disjointed (Fig. 4a), in accordance with a calculated small diradical character (y0 = 4.7%). The spins are delocalized throughout the whole π-conjugated framework, including the C–C triple bonds (Fig. 4a). This result indicates that the diradical resonance form 5a′′ indeed contributes to the ground state of 5b to a certain extent. On the other hand, 5a and 5c are calculated to have a closed-shell ground state with zero diradical character. The HOMO and LUMO of 5a and 5c are delocalized through the whole backbone with slight segregation as shown in Fig. S1 (ESI)† and 4a, respectively. The above results suggest that the ethynyl substituents can help to stabilize the diradical resonance form. The dipole moments of 5b and 5c were calculated to be 3.179 and 2.647 debye, respectively, at the CAM-B3LYP/6-31G* level of DFT, which are larger than that of azulene (1.268 debye) as calculated with the same method. This reflects the contribution of the dipolar ionic form 5a′ to the ground state of both 5b and 5c.
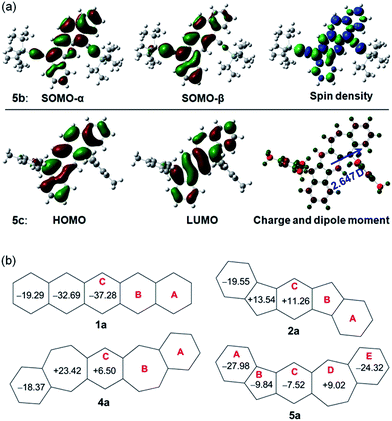 |
| | Fig. 4 (a) Calculated frontier MO profiles of 5b and 5c, spin density map of singlet diradical of 5b, and Mulliken charge distribution (−0.528 (red) to 0.528 (green)) and dipole moment of 5c; (b) calculated NICS(1)zz values for pentacene and its analogues. | |
To provide further insight into the aromaticity of each individual ring of these π-conjugated polycyclic hydrocarbons, nucleus independent chemical shift (NICS) of 1a, 2a, 4a and 5a were also calculated. Fig. 4b compares the calculated NICS(1)zz values of these molecules. Large negative values are found for all rings in 1a, in agreement with its known aromatic character. In 2a, a large negative value is calculated for ring A while both ring B and ring C show positive values, indicating that it can be regarded as a dibenzo-fused anti-aromatic s-indacene structure. In 4a, the central ring C is less positive compared with that in 2a, indicating its less anti-aromatic character. The seven-membered ring B however has a large positive value. In 5a, the central ring C and the five-membered ring B both become negative, and the seven-membered ring D is much less positive than that in 4a, indicating that a balance of three resonance forms leads to a weak aromatic character of the central C5–C6–C7 framework. The outmost benzenoid rings (A and E) are aromatic with large negative values. In agreement with the negative NICS value for the central ring C in 5a, the protons on the same ring in 5b exhibit a downfield singlet peak at 8.95 ppm as well as a singlet peak 7.10 ppm in the 1H NMR spectrum. In comparison to this, the corresponding protons on the central ring C in 2b12 and 4b15 exhibit singlet peaks at 7.26 and 7.16 ppm, respectively, in the 1H NMR spectra taken from the same solution (CDCl3).
Semiconductor properties
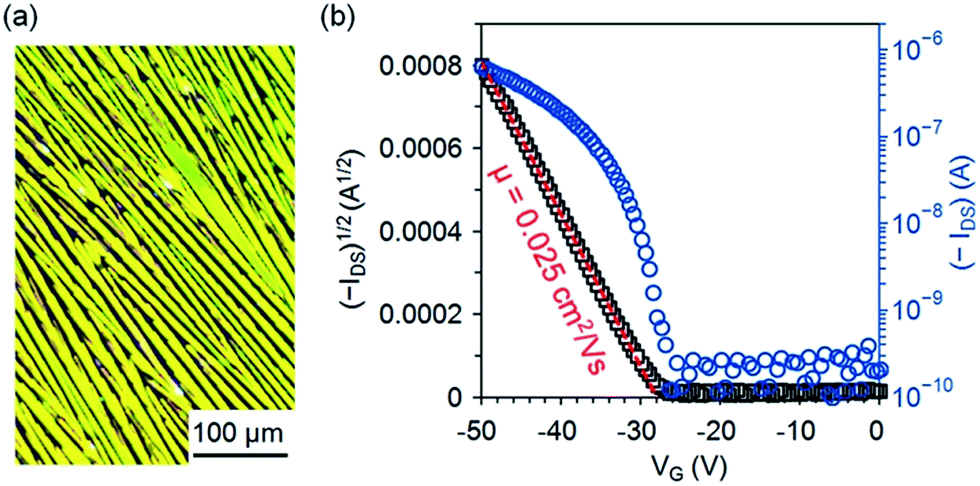
One interesting aspect of 5b is its semiconducting properties since it is a constitutional isomer of pentacene 1b, a well-known solution-processed p-type organic semiconductor. To test the semiconducting properties of 5b, top-contact transistors were fabricated on dip-coated films of 5b, which were formed by immersing a SiO2/Si substrate in a solution of 5b (2.5 mg mL−1) in n-hexane and then pulling it up with a constant speed of 5.3 μm s−1. As shown in the polarized-light micrograph in Fig. 5a, the dip-coated films of 5b on SiO2 are composed of crystalline fibers roughly aligned in the pulling direction; X-ray diffraction patterns from the films of 5b (Fig. S4 in ESI†) exhibit an intense peak at d-spacing of 18.88 Å (2θ = 4.68°) accompanied with three higher-order peaks at 9.44 Å (2θ = 9.37°), 6.29 Å (2θ = 14.07°), and 4.72 Å (2θ = 18.80°), indicating a crystalline film with a layered structure. As measured in air from these devices, 5b functions as a p-type semiconductor with a field-effect mobility of up to 0.025 cm2 V−1 s−1 (average 0.018 ± 0.003 cm2 V−1 s−1). Fig. 5b shows the transfer I–V curve in the saturation region for one of the best-performing OTFTs of 5b measured in air. From this transfer I–V curve, the field mobility is extracted using the equation: IDS = (μWCi/2L)(VG − VT)2, where IDS is the drain current, μ is field-effect mobility, Ci is the capacitance per unit area (11 nF cm−2) for the 300 nm-thick dielectric layer of SiO2, W is the channel width, L is the channel length, and VG and VT are the gate and threshold voltage, respectively. The mobility of 5b is lower than those of 1b26 and 4b15 in solution-processed OTFTs on bare SiO2 by one order of magnitude likely because of the unsymmetrical arrangement of silylethynyl substituting groups, which presumably leads to unfavorable molecular packing with poor π–π interactions.
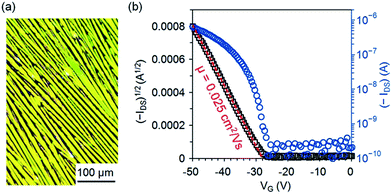 |
| | Fig. 5 (a) Reflection polarized-light micrograph for a dip-coated film of 5b on SiO2; (b) drain current (IDS) versus gate voltage (VG) with drain voltage (VDS) at −50 V for an OTFT of 5b with an active channel of W = 1 mm and L = 100 μm as measured in air. | |
Conclusions
In summary, the above study puts forth a new class of conjugated polycyclic molecules that contain a C6–C5–C6–C7–C6 framework isomeric to pentacene. The benzo[4,5]cyclohepta[1,2-b]fluorene derivatives 5b/5c display different optical and electrochemical properties in comparison with pentacene and its analogues 2–4. As found from the crystal structure, 5b has a nearly flat pentacyclic π-backbone with a quinoidal core. The computational studies indicate that the dipolar ionic resonance form contributes to the ground states of both 5b and 5c, while the diradical characters in the ground state depends on the substituting groups. 5b has a calculated diradical character (y0) in the ground state as small as 4.7%, which is not spectroscopically detectable, while 5c has a closed-shell ground state with zero diradical character. As a constitutional isomer of pentacene 1b, 5b functions as a p-type organic semiconductor in solution-processed OTFTs with field effect mobility of up to 0.025 cm2 V−1 s−1. As an extension from this study, synthesis of novel polycyclic arenes containing both five- and seven membered rings is in progress in our laboratories. These molecules may exhibit interesting physical properties that are not available for their benzenoid analogues as suggested by a recent theoretical study.27
Acknowledgements
We are grateful to Prof. Michael M. Haley (University of Oregon) and Prof. Yoshito Tobe (Osaka University) for their kind support and helpful discussion. X. Yang and Q. Miao thank financial support from the Research Grants Council of Hong Kong (project number: GRF402412, CRF C4030-14G). X. Shi and C. Chi thank financial support from MOE Tier 1 grant (R-143-000-623-112), Tier 2 grant (MOE2014-T2-1-080) and Tier 3 programme (MOE2014-T3-1-004). N. Aratani and H. Yamada thank support from the Grants-in-Aid for Scientific Research from JSPS (No. 16H02286, 26105004 and 26288038) and CREST, JST. T. P. Gonçalves and K.-W. Huang acknowledge the financial support from KAUST including NOOR 2 and Shaheen 2 HPC Facilities.
Notes and references
- T. Sekitani, U. Zschieschang, H. Klauk and T. Someya, Nat. Mater., 2010, 9, 1015–1022 CrossRef CAS PubMed.
- A. R. Murphy and J. M. J. Fréchet, Chem. Rev., 2007, 107, 1066–1096 CrossRef CAS PubMed.
-
(a) M. Bendikov, F. Wudl and D. F. Perepichka, Chem. Rev., 2004, 104, 4891–4946 CrossRef CAS PubMed;
(b) J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452–483 CrossRef CAS PubMed.
- J. E. Anthony, J. S. Brooks, D. L. Eaton and S. R. Parkin, J. Am. Chem. Soc., 2001, 123, 9482–9483 CrossRef CAS PubMed.
- Y. Diao, B. C.-K. Tee, G. Giri, J. Xu, D. H. Kim, H. A. Becerril, R. M. Stoltenberg, T. H. Lee, G. Xue, S. C. B. Mannsfeld and Z. Bao, Nat. Mater., 2013, 12, 665–671 CrossRef CAS PubMed.
- D. Liu, Z. He, Y. Su, Y. Diao, S. C. B. Mannsfeld, Z. Bao, J. Xu and Q. Miao, Adv. Mater., 2014, 26, 7190–7196 CrossRef CAS PubMed.
- A. Caruso Jr., M. A. Siegler and J. D. Tovar, Angew. Chem., Int. Ed., 2010, 49, 4213–4217 CrossRef PubMed.
- U. H. F. Bunz, J. U. Engelhart, B. D. Lindner and M. Schaffroth, Angew. Chem., Int. Ed., 2013, 52, 3810–3821 CrossRef CAS PubMed.
- Q. Ye, J. Chang, X. Shi, G. Dai, W. Zhang, K.-W. Huang and C. Chi, Org. Lett., 2014, 16, 3966–3969 CrossRef CAS PubMed.
- X. Shi, W. Kueh, B. Zheng, K.-W. Huang and C. Chi, Angew. Chem., Int. Ed., 2015, 54, 14412–14415 CrossRef CAS PubMed.
-
(a) Q. Miao, Adv. Mater., 2014, 26, 5541–5549 CrossRef CAS PubMed;
(b) Q. Miao, Synlett, 2012, 23, 326–336 CrossRef CAS.
- D. T. Chase, A. G. Fix, B. D. Rose, C. D. Weber, S. Nobusue, C. E. Stockwell, L. N. Zakharov, M. C. Lonergan and M. M. Haley, Angew. Chem., Int. Ed., 2011, 50, 11103–11106 CrossRef CAS PubMed.
- D. T. Chase, A. G. Fix, S. J. Kang, B. D. Rose, C. D. Weber, Y. Zhong, L. N. Zakharov, M. C. Lonergan, C. Nuckolls and M. M. Haley, J. Am. Chem. Soc., 2012, 134, 10349–10352 CrossRef CAS PubMed.
- A. Shimizu, R. Kishi, M. Nakano, D. Shiomi, K. Sato, T. Takui, I. Hisaki, M. Miyata and Y. Tobe, Angew. Chem., Int. Ed., 2013, 52, 6076–6079 CrossRef CAS PubMed.
- X. Yang, D. Liu and Q. Miao, Angew. Chem., Int. Ed., 2014, 53, 6786–6790 CrossRef CAS PubMed.
-
S. Das and J. Wu, in Polycyclic Arenes and Heteroarenes: Synthesis, Properties and Applications, ed. Q. Miao, Wiley VCH, Weinheim, 2016, ch. 1, pp. 3–36 Search PubMed.
- The commonly used formal potential of the redox couple of ferrocenium/ferrocene (Fc+/Fc) in the Fermi scale is −5.1 eV, which is calculated on the basis of an approximation neglecting solvent effects using a work function of 4.46 eV for the normal hydrogen electrode (NHE) and an electrochemical potential of 0.64 V for (Fc+/Fc) versus NHE. C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed.
- Z. Liang, Q. Tang, J. Xu and Q. Miao, Adv. Mater., 2011, 23, 1535–1539 CrossRef CAS PubMed.
- R. S. H. Liu, J. Chem. Educ., 2002, 79, 183–185 CrossRef CAS.
- S. Förster, T. Hahn, C. Loose, C. Röder, S. Liebing, W. Seichter, F. Eißmann, J. Kortus and E. Weber, J. Phys. Org. Chem., 2012, 25, 856–863 CrossRef.
- D. M. Lemal and G. D. Goldman, J. Chem. Educ., 1988, 65, 923–925 CrossRef CAS.
- J. Michl and E. W. Thulstrup., Tetrahedron, 1976, 32, 205–209 CrossRef CAS.
-
R. Gleiter, G. Haberhauer, Aromaticity and Other Conjugation Effects, Wiley VCH, Weinheim, 2012, pp. 70–72 Search PubMed.
- Crystallographic data for 5c·CH3CN: C42H37N. Mw: 555.72; monoclinic; space group P21/n; a = 20.925(2) Å, b = 8.4456(10) Å, c = 34.677(4) Å, β = 91.177(3)°, V = 6126.9(12) Å3; Z = 8; ρcalcd = 1.205 Mg m−3; R1 = 0.0652, wR2 = 0.1536 (I > 2σ(I)); R1 = 0.1248, wR2 = 0.1851 (all data). CCDC No. 1468927.
- The bond length is 1.45–1.46 Å for a typical single bond between two sp2 carbon atoms, 1.38–1.40 Å for a typical C–C 1.5 bond in arenes, and 1.31–1.34 Å for a typical C–C double bond in alkenes. See: E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, Sausalito, 2004, ch. 1, p. 22 Search PubMed.
- M. M. Payne, S. R. Parkin, J. E. Anthony, C.-C. Kuo and T. N. Jackson, J. Am. Chem. Soc., 2005, 127, 4986–4987 CrossRef CAS PubMed.
- M. Nakano, K. Fukuda and B. Champagne, J. Phys. Chem. C, 2016, 120, 1193–1207 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterization data for all new compounds; general experimental method; additional spectroscopic data; DFT calculation details; crystallographic data; and OTFT characterizations. CCDC 1468926 and 1468927. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc01795a |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a