
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA chiral “Siamese-Twin” calix[4]pyrrole tetramer†
Albano
Galán‡
a,
Gemma
Aragay‡
a and
Pablo
Ballester
*ab
aInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology, Avgda. Països Catalans 16, 43007, Tarragona, Spain. E-mail: pballester@iciq.es
bInstitution for Research and Advanced Studies (ICREA), Passeig Lluís Companys, 23, 08018 Barcelona, Spain
First published on 26th May 2016
Abstract
We describe our results in the attempted template syntheses of oligomacrocycle calix[4]pyrrole dimer 4, using Hay coupling reaction conditions, tetraalkynyl calix[4]pyrrole 5 as starting material and two bipyridyl N-oxides of different length as templates. We found that the short bis-N-oxide 3 was not an efficient template for the macrocyclization reaction producing an insoluble crude reaction mixture containing exclusively oligomerization and polycondensation products. On the other hand, when we used the long bis-N-oxide 6 as template we obtained a soluble crude reaction mixture in which we did not detect the expected calix[4]pyrrole dimer 4. Instead, we isolated, in low yield, an encapsulation complex of the bis-N-oxide 6 in a partially reacted calix[4]pyrrole dimer. The major isolated species was an unprecedented calix[4]pyrrole tetramer encapsulating two molecules of 6. The complex adopted a chiral helical-like conformation in the solid state resembling the previously described so-called “Siamese-Twin porphyrins”.
Introduction
The selective assembly of multiple molecular components into a particular covalent geometry or interlocked topology benefits from the use of organic synthesis with supramolecular assistance.1,2 In this sense, monoatomic and molecular templates have been extensively used to favor the yield of a single product from the mixture typically obtained in the analogous non-templated chemical reaction.3–8 Particularly, in macrocyclization reactions where the formation of several cyclic and linear oligomeric aggregates is possible, the reaction outcome can be controlled by using non-covalent templates that are able to alter the relative thermodynamic stabilities of the initially generated linear products and/or stabilize the transition states leading to the desired cyclic structure.9 Hence, template-assisted chemical synthesis allows the efficient preparation of certain products that otherwise would be obtained in very low yields.10–13In general, the use of a template leads to the formation of a thermodynamically stable substrate–template complex. In most examples the template can be removed after the synthesis yielding the template-free product.14,15 However, in some examples the template might experience tight and constrictive binding and cannot be released from the substrate–template complex, i.e. carceplexes.16,17
Since the earlier work of Cram and co-workers,18 it was shown that the efficient synthesis of carcerands, oligomacrocyclic receptors that hold the guest permanently, was strongly dependent on the use of suitable template molecules i.e. solvents.19–22 The template molecule acted as a mold to shape the reacting substrate in a correct spatial arrangement that favored its intramolecular cyclization and suppressed to a large extent undesired oligomerization and polycondensation reaction pathways.
Recently, we described the synthesis of macrocycle 1 (Fig. 1), which in turn was used for the preparation of molecular assemblies displaying [2]pseudorotaxane topology exhibiting promising anion recognition properties.23,24 Macrocycle 1 was prepared in 60% yield by dimerization of the “two wall” calix[4]pyrrole 2 monomer using Hay reaction conditions (Fig. 1).25,26 One equiv. of 4,4′-bipyridine-N,N′-dioxide 3 was added to the reaction mixture as a putative cyclization template. With the aim to extend the range of polar molecular containers based on aryl-extended calix[4]pyrrole scaffolds, we undertook the synthesis of oligomacrocyclic calix[4]pyrrole dimer container 4 using tetraalkynyl derivative 5 as starting material. We expected that the incorporation of two additional aromatic walls to molecular container 4, compared to 1, would increase the thermodynamic and kinetic stability of its encapsulation complexes providing, simultaneously, a complete isolation of the bound guests from the bulk solution. The closed and polar internal cavity featured in container 4 suggested that it may have potential applications as a reactor vessel.27–29
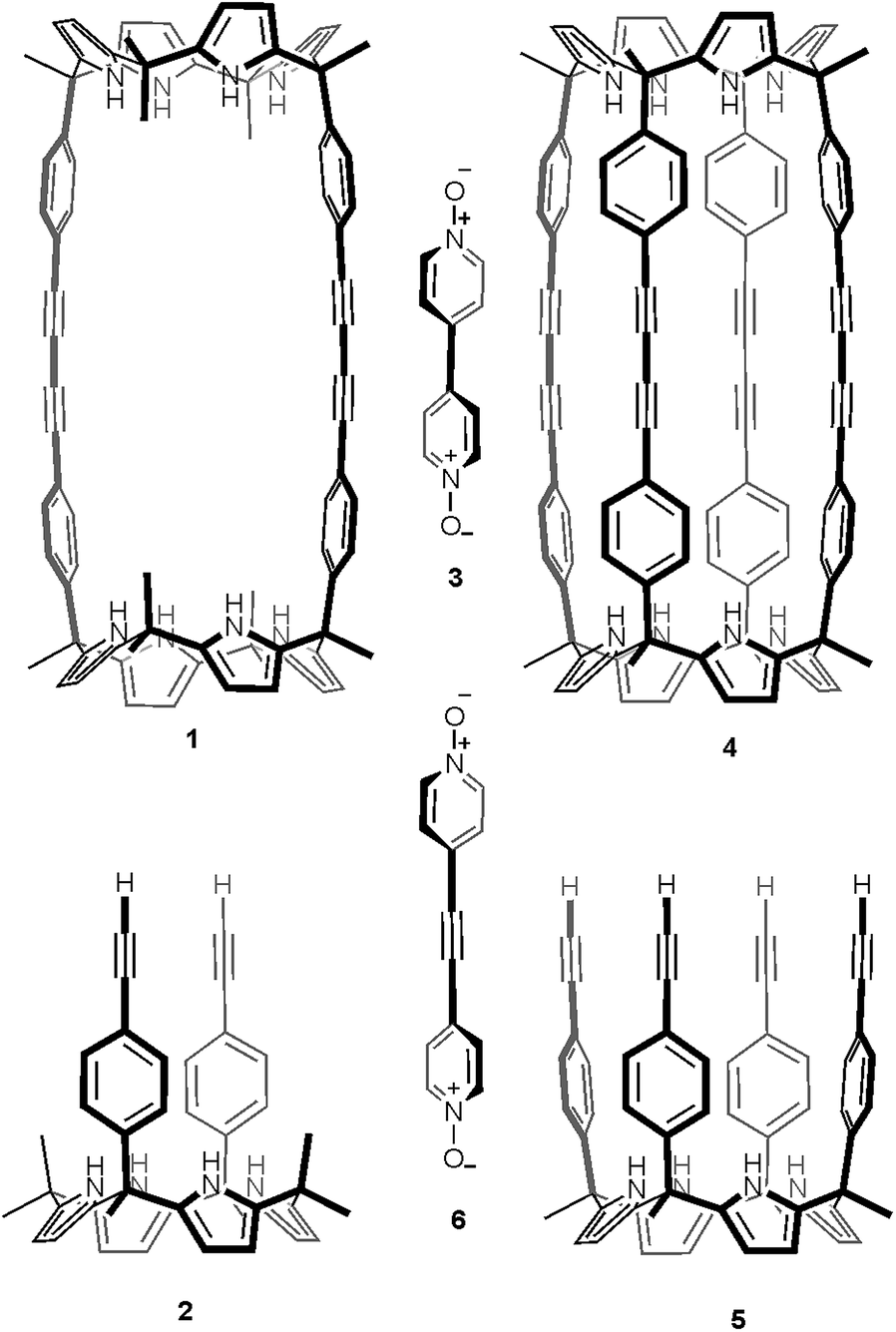 | ||
| Fig. 1 Line-drawing structures of calix[4]pyrrole macrocycles and guests used in the present and past works.23 | ||
Herein, we describe our efforts and results towards the template synthesis of oligomacrocycle 4 using Hay coupling conditions, calix[4]pyrrole 5 and two bipyridine bis-N-oxides, 3 and 6, as templates. On the one hand, we established that bis-N-oxide 3 is not an efficient template for promoting multiple intramolecular coupling reactions between the terminal alkynyl residues at the upper rim of calix[4]pyrrole 5. That is, in the presence of bis-N-oxide 3, calix[4]pyrrole 5 underwent mainly intermolecular oligomerization and polycondensation coupling reactions. On the other hand, bis-N-oxide 6 acted as a positive template favoring three of the four possible alkyne coupling reactions in the dimerization of 5. However, in the presence of 6 the major isolated product was an encapsulation complex of two bis-N-oxide 6 complexes in a tetrameric oligomacrocyclic calix[4]pyrrole. In the solid state, the latter encapsulation complex adopts a chiral conformation.
Results and discussion
Initially, we attempted the cyclic dimerization of calix[4]pyrrole 5 in CH2Cl2 solution using Hay conditions in the absence of bis-N-oxide 3.§ The obtained crude reaction mixture showed very limited solubility in most organic solvents. We hypothesized that the almost exclusive presence of oligomeric and ill-defined aggregates as components of the crude reaction mixture was responsible for its limited solubility. Being aware of the excellent results reported by Sanders and co-workers on the use of templates for the synthesis of porphyrin cyclic oligomers using Hay coupling conditions, we decided to attempt the dimerization of 5 using bis-N-oxide 3 as template (Scheme 1).10,30 Our expectations were that after the formation of the linear dimer 7, connected through a single butadiyne link, bis-N-oxide 3 would act as an efficient intramolecular cyclization template. The ditopic 3⊂7 complex (Scheme 1 and Fig. 2b) should adopt a conformation in which the six remaining reactive acetylene groups are forced to be in close proximity and in a geometry that might accelerate intramolecular couplings with respect to intermolecular ones. Nevertheless, all these predictions assume that the binding of the template (N-oxide) to the linear calix[4]pyrrole dimer matches that observed in pure CH2Cl2 solution for related systems31 (Fig. 2b).¶ It is worth noting that from the multiple sequences of intermolecular connections that are possible for the six terminal alkynes in 3⊂7 only one (a·a′, b·b′ and c·c′, Scheme 1) affords the desired cyclic dimer 4. Other intramolecular connectivities i.e. a·b′ and b·c′ would favor the formation of dangling reacting alkynes, a′ and c′, prone to be involved in intermolecular oligomerization reactions. Disappointingly, when we repeated the cyclization reaction of calix[4]pyrrole 5 (2.6 mM) under Hay conditions in the presence of 0.5 equiv. of bis-N-oxide 3 the isolated reaction crude mixture did not show signs of solubility improvement. We concluded that bis-N-oxide 3 was not acting as a positive template for the formation of the cyclic dimer 4 featuring four butadiynyl linkers. Owing to the synthetic difficulties associated with the preparation of mono-linked linear dimer 7, we decided to investigate the binding properties of bis-N-oxide 3 directly with the tetraethynyl calix[4]pyrrole monomer 5.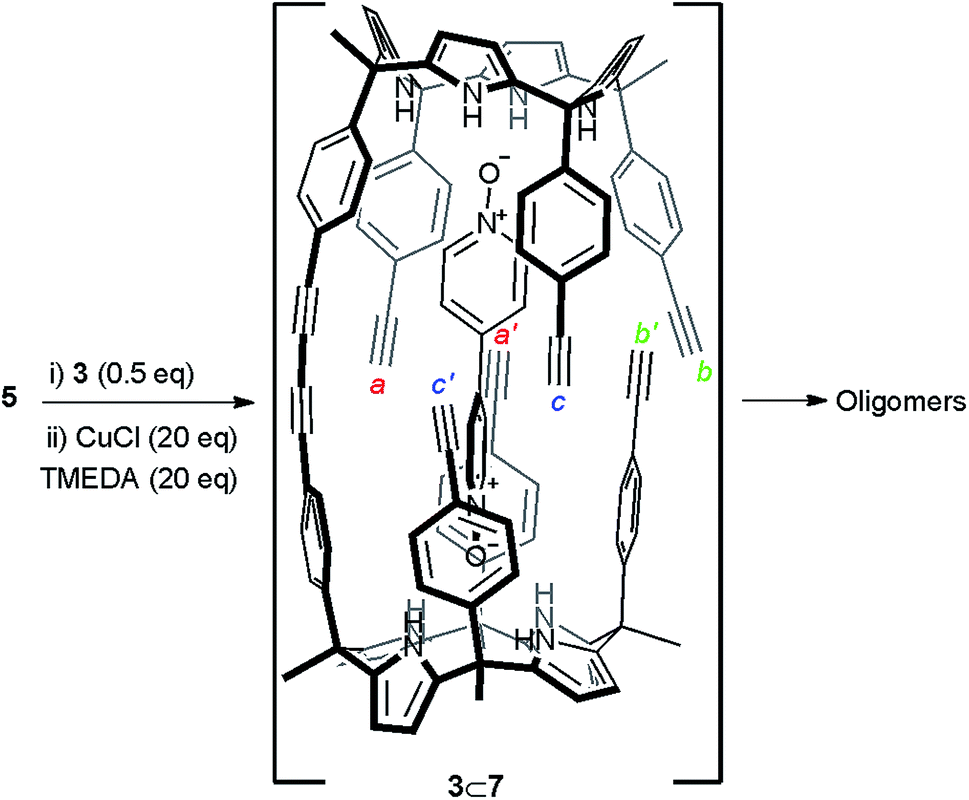 | ||
| Scheme 1 Reaction scheme of the attempted cyclic dimerization of 5 using Hay coupling conditions in the presence of template 3. The structure of the putative 3⊂7 dimer is shown. | ||
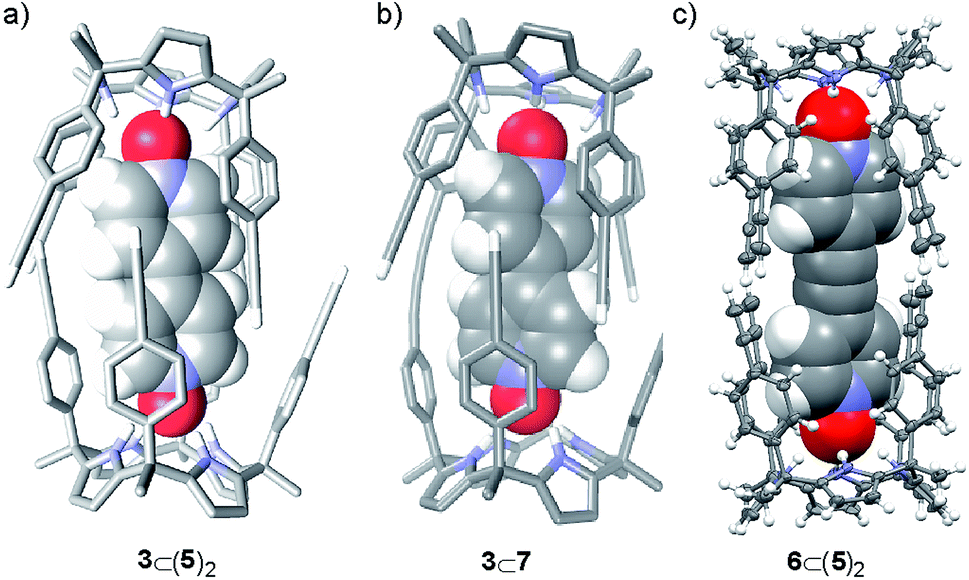 | ||
| Fig. 2 (a) PM6 energy minimized structure of the 3⊂(5)2 homocapsule. (b) PM6 energy minimized structure of the putative complex formed by dimeric species 7 and guest 3. For clarity, some non-polar hydrogen atoms from the host are removed. Guest molecules are shown as CPK models and calix[4]pyrrole 5 is depicted in stick representation. (c) X-ray structure of the 6⊂(5)2 homocapsule shown in ORTEP representation with thermal ellipsoids set at 50% probability and hydrogen atoms as fixed spheres of 0.3 Å radius. | ||
We performed a 1H NMR titration of calix[4]pyrrole 5 (1 mM) in CD2Cl2 solution by adding incremental amounts of bis-N-oxide 3. The addition of 0.5 equiv. of 3 resulted in broadening of the calix[4]pyrrole proton signals. This observation indicated that the chemical exchange between free and bound calix[4]pyrrole 5 was intermediate on the chemical shift time scale (Fig. S1†). The addition of 1 equiv. of bis-N-oxide 3 induced the observation of sharp proton signals for both the calix[4]pyrrole 5 and the bis-N-oxide 3. The hydrogen atoms of the bis-N-oxide resonated as four separate doublets at δ = 8.27, 7.56, 6.99 and 4.55 ppm. Only two of the four signals of the bis-N-oxide 3 protons experienced a significant upfield shift compared to those from the free guest. Moreover, the signal corresponding to pyrrole NHs of the calix[4]pyrrole 5 resonates downfield shifted compared to that of free host (Δδ = 2.1 ppm) suggesting the establishment of hydrogen bonding interactions (N–H⋯O) between the pyrrole NHs and the oxygen atom of the N-oxide. The addition of more than 1 equiv. of bis-N-oxide 3 did not induce noticeable changes in the signals corresponding to the protons of the calix[4]pyrrole 5 and produced the emergence of a new set of signals corresponding to free 3. Taken together, these observations supported the quantitative formation of an inclusion 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 3⊂5 complex in an equimolar mixture of the two components at 1 mM concentration. They also indicated that the association constant for the 3⊂5 complex must be larger than 104 M−1. The binding geometry of the 1:1 complex positioned one pyridyl N-oxide group of 3 hydrogen bonded and deeply included in the aromatic cavity defined by the cone conformation of the calix[4]pyrrole 5 where it experienced the strong magnetic shielding exerted by the four meso-phenyl substituents. The other pyridyl residue of bound 3 must be fully exposed to the bulk solvent.
:1 3⊂5 complex in an equimolar mixture of the two components at 1 mM concentration. They also indicated that the association constant for the 3⊂5 complex must be larger than 104 M−1. The binding geometry of the 1:1 complex positioned one pyridyl N-oxide group of 3 hydrogen bonded and deeply included in the aromatic cavity defined by the cone conformation of the calix[4]pyrrole 5 where it experienced the strong magnetic shielding exerted by the four meso-phenyl substituents. The other pyridyl residue of bound 3 must be fully exposed to the bulk solvent.
We performed variable temperature 1H NMR experiments using CD2Cl2 solution that contained 3 and 5 in a 2:1 molar ratio. At 213 K, some of the broad proton signals observed for the calix[4]pyrrole 5 at room temperature split in three different sets of sharp signals. Two of the three sets of signals were easily assigned to protons of free 5 and bound 5 in the 1:1 3⊂5 complex, respectively. Based on the chemical shifts and number of proton signals that composed the remaining set of proton signals, we assigned it to a 1:2 complex, 3⊂(5)2, featuring D4h symmetry (Fig. 2a). Integration of selected proton signals of calix[4]pyrrole 5 in the three different sets of signals allowed us to calculate its distribution in three different stoichiometries, [5]:[3⊂5]:[3⊂(5)2], at 213 K as 0.12:0.25:0.63. From these studies we concluded that the formation of a putative 1:1 complex between the mono-linked linear dimer 7 and bis-N-oxide 3 was geometrically feasible because the corresponding sandwich 3⊂(5)2 complex was indeed formed, even suffering from a negative allosteric cooperativity (K1 > 104 M−1 and K1/K2 ∼ 10). Most likely, the fact that the bis-N-oxide 3 was not an efficient (positive) template for the formation of the cyclic dimer 4 was related to the inappropriate placement of the reactive alkyne ends in the 3⊂7 complex, which disfavored the intramolecular reactions (Scheme 1). This hypothesis is supported by the energy minimized geometry obtained for the 3⊂7 complex using simple theoretical calculations (PM6) (Fig. 2b).
Replacing bis-N-oxide 3 by the longer analogue 6 (Fig. 1) provided an energy minimized structure (PM6) of the mono-linked linear dimer 6⊂7 complex in which the reactive alkyne groups seemed to be better positioned to undergo multiple intramolecular cyclization reactions (Scheme 2 and Fig. S17a†). First, we probed the complexation of bis-N-oxide 6 with monomer 5 using 1H NMR spectroscopy. Contrary to our previous observation using 3, a mM CD2Cl2 solution containing a 1:2 mixture of 6 (0.5 mM) and 5 (1 mM) produced sharp signals for both the protons of the bis-N-oxide and the calix[4]pyrrole.
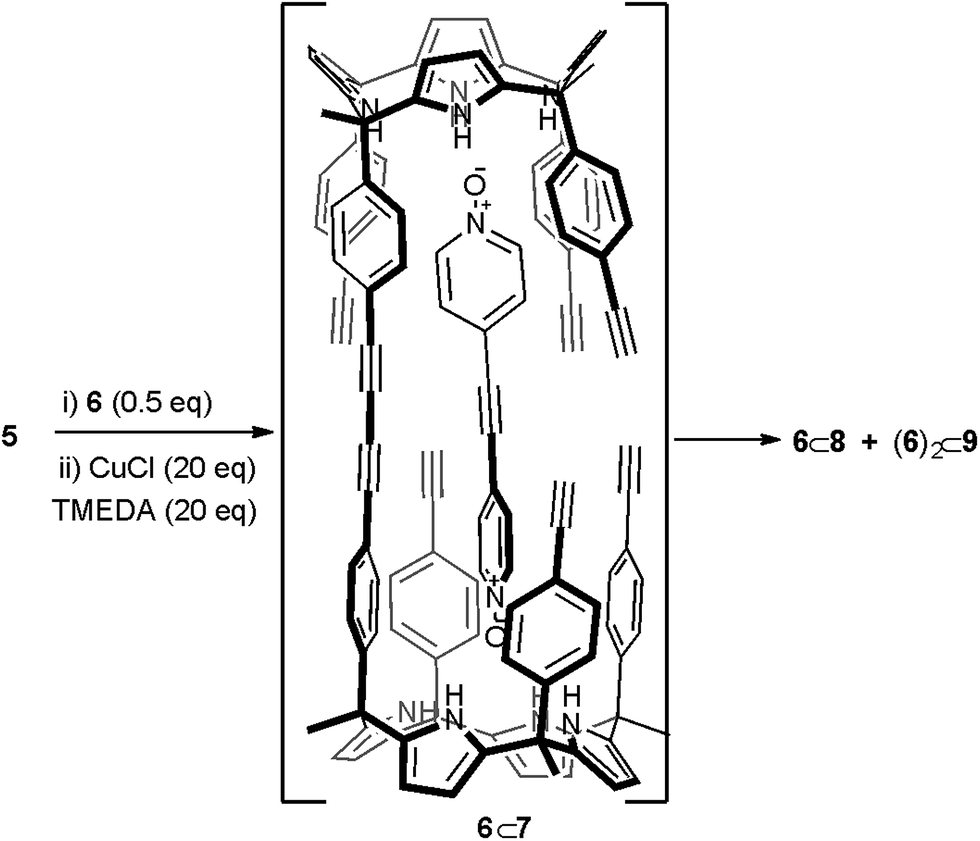 | ||
| Scheme 2 Reaction scheme of the attempted cyclic dimerization of 5 using Hay coupling conditions in the presence of template 6. The structure of the putative 6⊂7 dimer is shown. | ||
The number of proton signals and their chemical shift values were fully consistent with the quantitative assembly in solution of the 1:2 complex, 6⊂(5)2 under these conditions. The exclusive assembly of the 1:1 complex 6⊂5 required the addition to the above solution of more than three equiv. of bis-N-oxide 6. These results indicated that the assembly of the 1:2 sandwich complex, 6⊂(5)2, did not experience negative allosteric cooperativity (K1 ≈ K2 > 104 M−1). The sandwich complex 6⊂(5)2 was further characterized in solution by DOSY spectroscopy and in the solid-state by X-ray diffraction. The solid-state structure of the 6⊂(5)2 complex was in complete agreement with the proposed structure in solution (Fig. 2c). In the solid state, bis-N-oxide 6 is sandwiched between two calix[4]pyrrole units 5. Each N-oxide pyridyl residue in 6 establishes four hydrogen bonds with the calix[4]pyrrole core of two distal molecules of bound 5 in cone conformation. The averaged N–H⋯O distance of the four hydrogen bonds is 2.95 Å.
The obtained results encouraged us to investigate the use of bis-N-oxide 6 as a potential template for the dimerization of calix[4]pyrrole 5 to afford macrocycle 4. We reacted a 2:1 molar mixture of calix[4]pyrrole 5 (2.6 mM) and bis-N-oxide 6 (1.3 mM) in dichloromethane solution for five hours in the presence of CuCl·TMEDA (26 mM) (Scheme 2). The reaction was quenched with water and the organic solution was further washed with water, dried and evaporated. We isolated two fractions from the column chromatography purification of the now soluble reaction crude mixture. Both fractions produced white solids upon solvent evaporation. The obtained solids were characterized using a combination of high resolution 1H NMR spectroscopy, HRMS spectrometry and single-crystal X-ray crystallographic analysis.
The 1H NMR spectrum in CDCl3 solution of the first eluted solid showed two singlets resonating at δ = 10.08 and 9.67 ppm that were attributed to pyrrole NHs (Fig. 3b). The large downfield shift experienced by these protons, compared to free 5 (Fig. 3a), and the observation of two doublets, attributed to the aromatic protons of bis-N-oxide 6, which in turn experienced significant upfield shifts (Δδ = −1.41 and −2.96 ppm), suggested that the two pyridyl N-oxide residues of 6 were hydrogen bonded and deeply included in two separate but symmetry related aryl-extended calix[4]pyrrole units adopting a cone conformation. Because the isolated complex had survived column chromatography purification, we surmised that the two calix[4]pyrrole units had to be covalently connected.
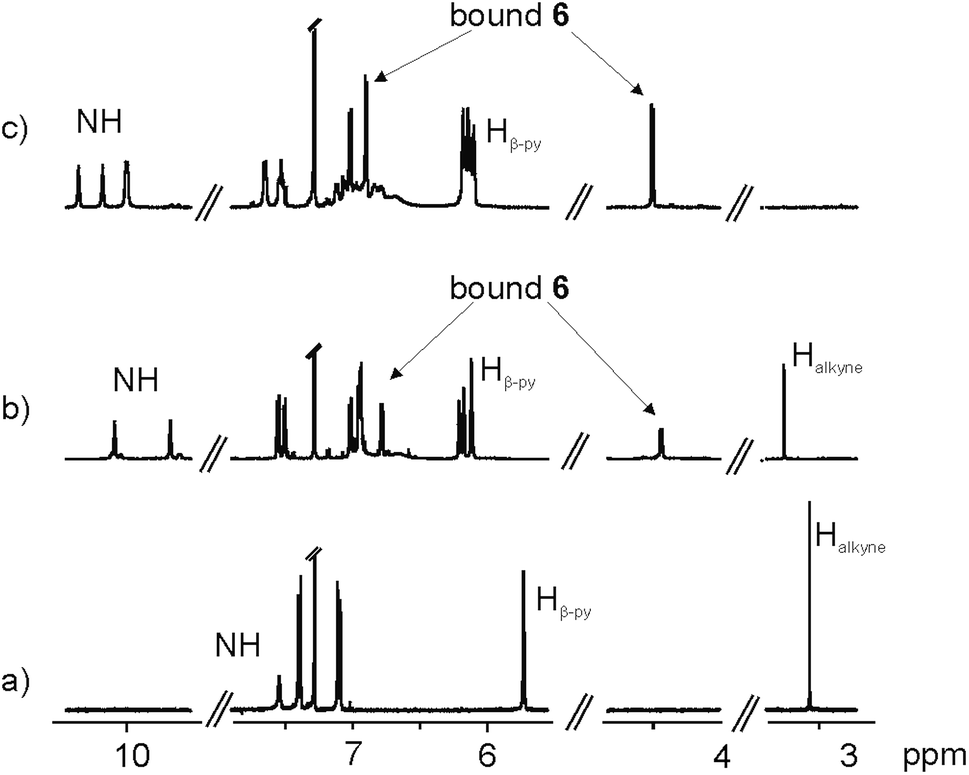 | ||
| Fig. 3 Selected regions of 1H NMR spectra of compounds 5 (a), 6⊂8 (b) and (6)2⊂9 (c). | ||
However, the number of signals displayed in the 1H NMR spectrum of the solid was not in agreement with the D4h symmetry expected for the encapsulation complex 6⊂4. We observed a sharp singlet resonating at δ = 3.26 ppm, which we attributed to non-reacted acetylenic protons.
By integration, we determined that one acetylenic residue in each of the two calix[4]pyrrole units had not reacted. Taking into consideration the NMR and MS data, we assigned to this first eluted solid the structure of the encapsulation complex 6⊂8 (5% yield) (Fig. 4a). In this complex, the bis-N-oxide 6 is ditopically encapsulated in an oligocyclic calix[4]pyrrole dimer 8 covalently connected by only three butadiynyl linkers. In short, bis-N-oxide 6 acted as a positive but moderate cyclization template for the dimerization of 5. It induced two of the three possible intramolecular alkyne couplings in the putative bound linear calix[4]pyrrole dimer forming the 6⊂7 complex. On the other hand, bis-N-oxide 6 seemed to be a very efficient negative template for the complete cyclization of the linear dimer 7 to afford 4.
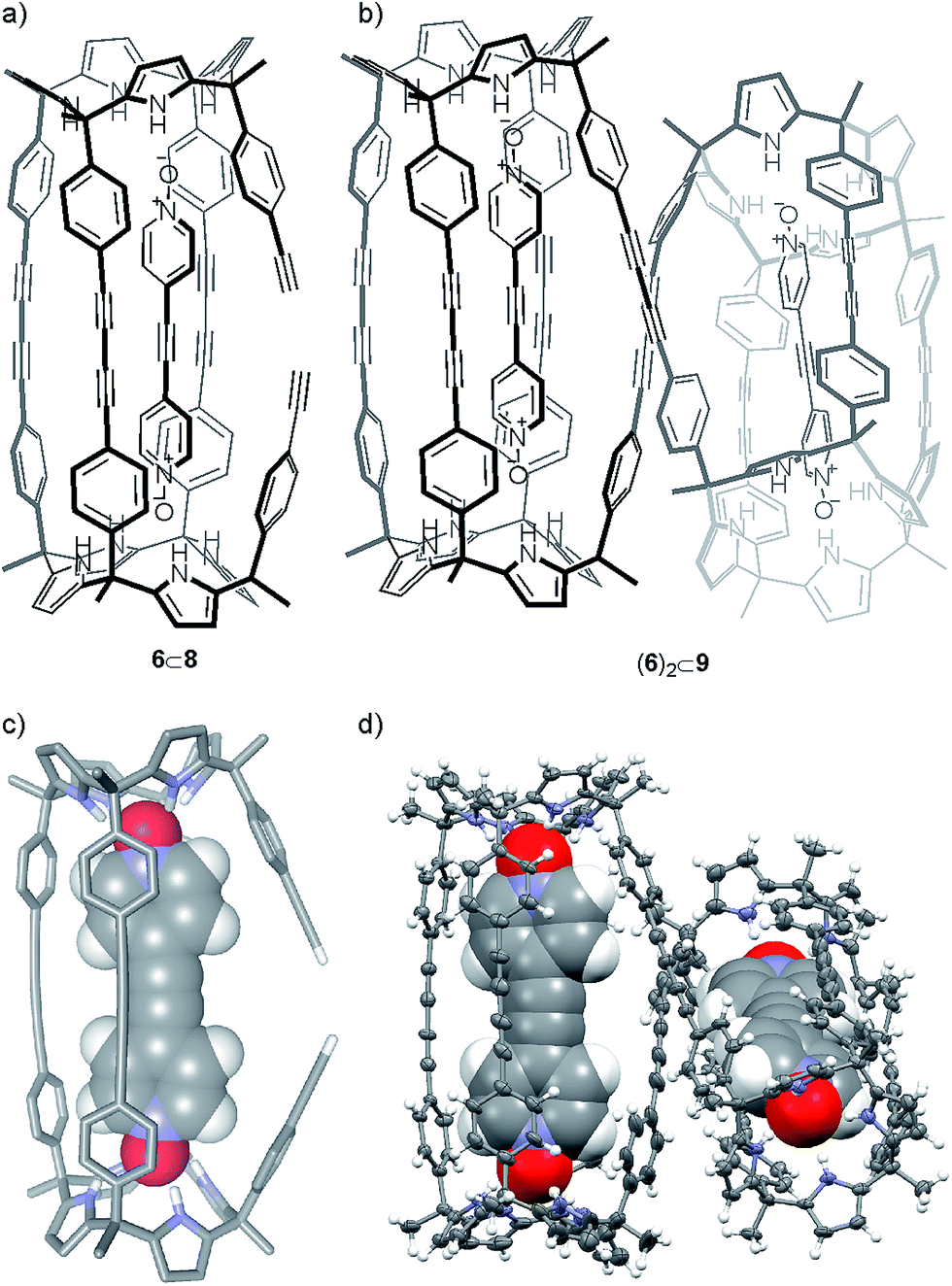 | ||
| Fig. 4 Line-drawing structures of compounds: (a) 6⊂8 and (b) (6)2⊂9; (c) PM6 energy minimized structure of 6⊂8 and (d) X-ray crystal structure of (6)2⊂9 shown in ORTEP representation with thermal ellipsoids set at 50% probability and hydrogen atoms as fixed spheres of 0.3 Å radius. | ||
A CDCl3 solution of the solid eluted in the second fraction displayed a significantly more complex 1H NMR spectrum (Fig. 3c). It showed four singlets resonating at δ = 10.35, 10.17, 10.01 and 9.99 ppm that we assigned to hydrogen bonded pyrrole NHs. We also observed two doublets centered at δ = 6.89 and 4.49 ppm, which based on our previous assignment must correspond to protons of ditopically encapsulated N-oxide 6. In the case at hand, we did not detect any signal that could be assigned to unreacted acetylenic protons. Moreover, some of the signals of the aromatic protons of the aryl extended calix[4]pyrrole unit appeared as broad bands resonating at ∼7 ppm. Most likely, this is due to intermediate rotation on the NMR time scale of the Cmeso–phenyl bond caused by steric clashes. The 1H NMR spectrum acquired at higher temperatures (358 K) showed that some of these broad proton signals sharpened, indicating a change in the rate of rotation of the Cmeso–phenyl bonds at this temperature (from intermediate to fast in the NMR time scale) (Fig. S19†).
We performed DOSY NMR experiments on CDCl3 solutions of the two isolated solids. The calculated diffusion constants were very different, 6.62 ± 0.10 × 10−10 m2 s−1 for the 6⊂8 complex and 4.79 ± 0.16 × 10−10 m2 s−1 for the solid with a more complex 1H NMR spectrum. The diffusion constant value determined for the 6⊂8 complex was very similar to the one obtained for the supramolecular sandwich 6⊂(5)2. This result is in total agreement with the size (hydrodynamic radii) similarity of both species. The small diffusion constant measured for the second eluted solid implied the formation of a covalent assembly larger than a dimer. The analysis of the solid using ESI-HRMS+ spectrum showed a high intensity peak at a m/z ratio corresponding to the [2M + 2Na]2+ ion, M being the expected mass for macrocyclic encapsulation complex 6⊂4 (Fig. S16†). For us, the elucidation of the structure of this solid was not evident based on the available spectroscopic data.
Gratifyingly, we were able to grow single crystals for the two solids isolated in the column chromatography purification, which were analyzed by X-ray diffraction methods. For the first eluted solid we could only obtain an incomplete crystal structure model. The incomplete model clearly showed that the oligocyclic calix[4]pyrrole dimer 8 was covalently connected through only three adjacent butadiynyl linkers. Based on these preliminary X-ray results, we modeled the structure of 6⊂8 (Fig. 4c). The energy minimized structure of the 6⊂8 complex showed, in agreement with the X-ray structure, that the encapsulated bis-N-oxide 6 must bend to adapt its length to the dimensions provided by the dimer 8.
The encapsulation complex 6⊂8 adopts a conformation in which the two unreacted alkynyl ends are held apart. The energy minimized structure of 6⊂8 complex and its preliminary X-ray solution nicely explain the role of 6 as negative template for the dimerization reaction of 5 through the four terminal acetylenes.
The X-ray crystal structure of the second isolated solid (Fig. 4d) was crucial for the assignment of its structure. The solution of the diffraction data revealed a calix[4]pyrrole tetramer displaying a helical-like conformation and encapsulating two bis-N-oxides, (6)2⊂9 (Fig. 4b). Probably, this aggregate resulted from the dimerization of two 6⊂8 complexes by coupling their dangling reactive alkyne groups, which were unable to couple intramolecularly. To us, the structure of the calix[4]pyrrole tetramer complex (6)2⊂9 resembled the previously described porphyrin structures with a persistent helical conformation, the so-called “Siamese-Twin porphyrins”.32,33 The tetrameric structure (6)2⊂9 fully explained the molecular ion observed in the HRMS spectrum of the second isolated solid (39% yield), as well as the small diffusion constant derived from DOSY NMR experiments.
Next, we attempted the conversion of 5 to macrocycle 4 using 6 as template under more diluted Hay conditions. However, we did not observe any evidence of the formation of macrocycle 4.||
The conformation adopted by the (6)2⊂9 complex in the solid state is dissymmetric (D2-like symmetry), which explains the observation of its pyrrole NHs as three separate signals in the 1H NMR spectrum. In the complex, the calix[4]pyrrole tetramer 9, owing to the asymmetry provided by the persistent helical twist, can exist as two conformational enantiomers. Related examples of conformational chirality have been previously described for other compounds.34,35 Not surprisingly, we observed the two enantiomeric conformers of (6)2⊂9 (M and P, Fig. 5) in the crystal packing.**
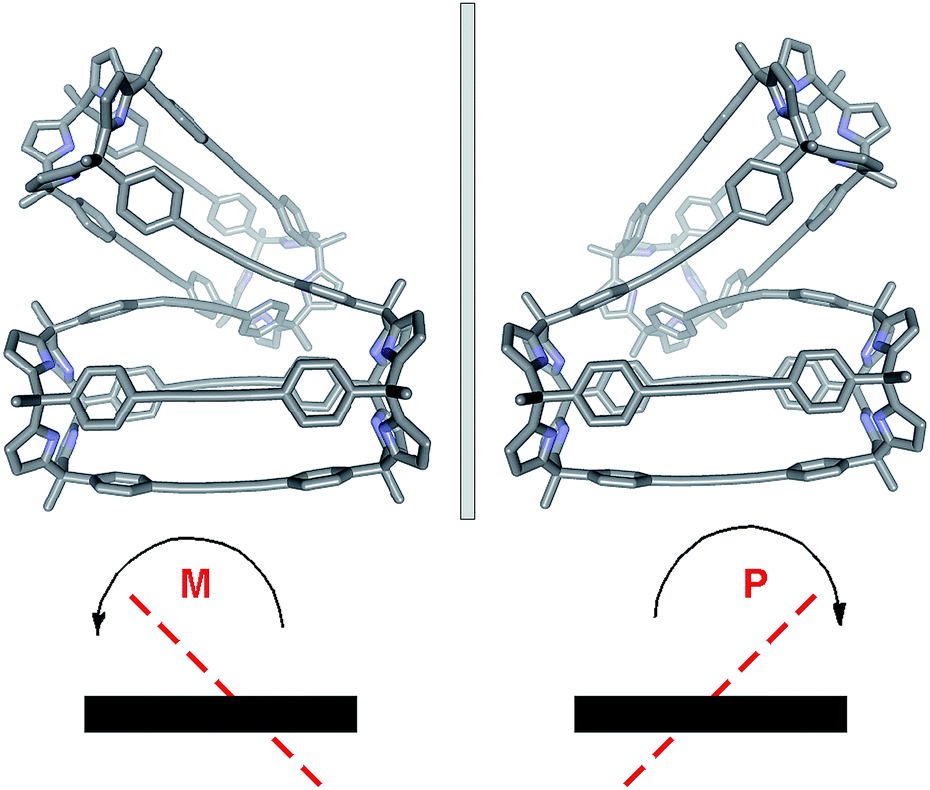 | ||
| Fig. 5 X-ray crystal structures of the enantiomeric conformers of the (6)2⊂9 tetramer present in the crystal lattice. The M and P nomenclature was used to assign the absolute configuration of the two helimers. Hydrogen atoms and the encapsulated guest molecules have been omitted for clarity. | ||
We performed variable temperature 1H NMR experiments using a Cl2CDCDCl2 solution of complex (6)2⊂9 in order to determine the value of the racemization barrier for the conformational chirality (M/P enantiomers). Although we were unable to reach coalescence for any of the proton signals, we observed significant broadening for the most downfield shifted diastereotopic pyrrole NHs at the highest temperature we could reach (408 K, krac > 102 s−1) (Fig. 6b). Based on this result, we estimated that the energy barrier for the racemization process of complex (6)2⊂9 must be larger than 20 kcal mol−1.
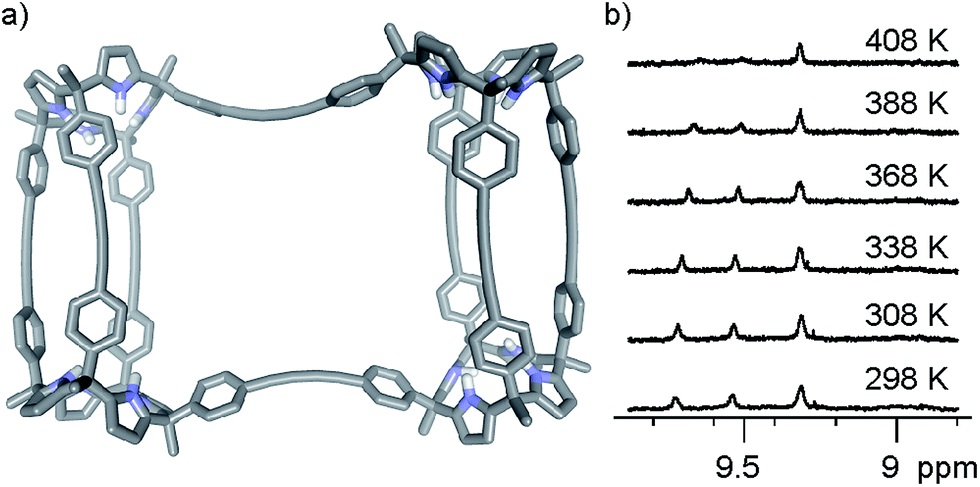 | ||
| Fig. 6 (a) MM3 energy minimized structure of a putative transition state structure for the racemization process of (M,P)-(6)2⊂9. Guest molecules and hydrogens atoms have been omitted for clarity. (b) Selected 1H NMR regions (NH pyrrole protons) of the variable temperature experiments performed using a solution of (6)2⊂9 in Cl2CDCDCl2. | ||
We also performed an EXSY experiment at room temperature using the Cl2CDCDCl2 solution of complex (6)2⊂9. We did not observe chemical exchange cross peaks between the diastereotopic NH signals. This result indicated that at room temperature the racemization process is slow on the EXSY time scale (krac < 0.3 s−1 at r.t.) setting a minimum value of 18.4 kcal mol−1 for the conformational chirality racemization barrier, which is fully consistent with the estimate described above.
In agreement with these experimental results, simple molecular modeling studies (MM3) indicated that the energy of the enantiomeric conformers was 22 kcal mol−1 lower than that of a conformer for a putative transition state. The proposed racemization mechanism assumes that the two covalently connected calix[4]pyrrole dimers adopted a parallel arrangement in the transition state (Fig. 6a). Based on the relatively low energy barrier estimated for the conformational chirality racemization process, we speculate that at room temperature this process should take place at a rate that is fast on the human time scale. In fact, all our efforts to separate the two enantiomeric conformers of (6)2⊂9 using chiral HPLC were unfruitful.
Unfortunately, we were not able to remove bis-N-oxide 6 from any of its templated encapsulation complexes, 6⊂8 and (6)2⊂9, either by purification using silica column chromatography or by the addition of protic and polar solvents to their CDCl3 solutions (i.e. MeOH).
Conclusions
In summary, we attempted the synthesis of oligomacrocycle 4 by template-synthesis and using two different bipyridyl bis-N-oxides, 3 and 6, as templates. The obtained results evidenced that 3 was not an efficient template to favor intramolecular cyclization reactions of 5 over intermolecular oligomerization processes. On the other hand, bis-N-oxide 6 acted as a positive cyclization template able to induce two of the three possible intramolecular alkyne dimerization in a putative encapsulation complex of the linear calix[4]pyrrole dimer 7. However, the fourth intramolecular reaction yielding oligomacrocycle 4 was never observed. Most likely, the length of bis-N-oxide 6 is slightly too large to hold the two last alkyne groups close together. This allowed the isolation, in low yield, of the 6⊂8 complex featuring a partially reacted calix[4]pyrrole oligocyclic dimer. Remarkably, we isolated as the major reaction product a non-expected encapsulation complex (6)2⊂9. Possibly, the isolated product derived from the dimerization of initially formed 6⊂8 complexes. The oligomacrocyclic container 9 is an unprecedented calix[4]pyrrole tetramer, in which the solid state adopts a chiral helical-like conformation resembling “Siamese-Twin porphyrins”. We estimated that the energy barrier for the racemization process of the enantiomeric conformers (P/M) of the (6)2⊂9 complex, detected in the crystalline packing, was higher than 20 kcal mol−1. At room temperature the racemization process is slow on the EXSY time scale but most likely it is fast on the human time scale.The chiral conformation adopted by 9 displays two well-defined polar cavities that are covalently connected. The induction of a preferred chiral conformer of 9 through reversible encapsulation of chiral N-oxides represents a challenging endeavor. Nevertheless, it will have to wait for the finding of successful conditions to exchange/remove the trapped template bis-N-oxide 6. This topic is currently under investigation in our laboratory.
Acknowledgements
We dedicate this work to Prof. Miquel A. Pericàs, Institute of Chemical Research of Catalonia (ICIQ), on the occasion of his 65th anniversary. The authors thank Gobierno de España MINECO for the project CTQ2014-56295-R, Severo Ochoa Excellence Accreditation (2014-2018 SEV-2013-0319), FEDER funds (project CTQ2014-56295-R) and the ICIQ Foundation for funding. A. G. thanks MINECO for a FPU fellowship. We thank Eduardo C. Escudero-Adán for help with the analysis of the X-ray crystallographic data.Notes and references
-
S. Anderson and H. L. Anderson, in Templated Organic Synthesis, Wiley-VCH Verlag GmbH, 2007, pp. 1–38 Search PubMed
.
- C. A. Schalley, F. Voegtle and K. H. Doetz, Templates in Chemistry II, Top. Curr. Chem., 2005, 249, 2005 Search PubMed
- D. V. Kondratuk, L. M. A. Perdigão, A. M. S. Esmail, J. N. O'Shea, P. H. Beton and H. L. Anderson, Nat. Chem., 2015, 7, 317–322 CrossRef CAS PubMed
- A.-J. Avestro, M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 5881–5895 RSC
- J. D. Crowley, S. M. Goldup, A.-L. Lee, D. A. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530–1541 RSC
- M. J. Langton, J. D. Matichak, A. L. Thompson and H. L. Anderson, Chem. Sci., 2011, 2, 1897–1901 RSC
- J. F. Ayme, J. E. Beves, C. J. Campbell and D. A. Leigh, Chem. Soc. Rev., 2013, 42, 1700–1712 RSC
- M. R. Sambrook, P. D. Beer, J. A. Wisner, R. L. Paul, A. R. Cowley, F. Szemes and M. G. B. Drew, J. Am. Chem. Soc., 2005, 127, 2292–2302 CrossRef CAS PubMed
- R. L. E. Furlan, S. Otto and J. K. M. Sanders, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4801–4804 CrossRef CAS PubMed
- H. L. Anderson, S. Anderson and J. K. M. Sanders, J. Chem. Soc., Perkin Trans. 1, 1995, 2231–2245 RSC
- S. Anderson, H. L. Anderson and J. K. M. Sanders, J. Chem. Soc., Perkin Trans. 1, 1995, 2255–2267 RSC
- S. Anderson, H. L. Anderson and J. K. M. Sanders, J. Chem. Soc., Perkin Trans. 1, 1995, 2247–2254 RSC
- D. Lucas, T. Minami, G. Iannuzzi, L. Cao, J. B. Wittenberg, P. Anzenbacher and L. Isaacs, J. Am. Chem. Soc., 2011, 133, 17966–17976 CrossRef CAS PubMed
- G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, Angew. Chem., Int. Ed., 2015, 54, 6110–6150 CrossRef PubMed
- M. Hoffmann, J. Karnbratt, M. H. Chang, L. M. Herz, B. Albinsson and H. L. Anderson, Angew. Chem., Int. Ed., 2008, 47, 4993–4996 CrossRef CAS PubMed
-
R. Warmuth, in Supramolecular Chemistry, John Wiley & Sons, Ltd, 2012 Search PubMed
- J. Sherman, Chem. Commun., 2003, 1617–1623 RSC
- D. J. Cram, S. Karbach, Y. H. Kim, L. Baczynskyj and G. W. Kalleymeyn, J. Am. Chem. Soc., 1985, 107, 2575–2576 CrossRef CAS
-
D. M. Rudkevich, in Functional Synthetic Receptors, Wiley-VCH Verlag GmbH & Co. KGaA, 2005, pp. 257–298 Search PubMed
- R. G. Chapman and J. C. Sherman, J. Org. Chem., 1998, 63, 4103–4110 CrossRef CAS
- R. G. Chapman, N. Chopra, E. D. Cochien and J. C. Sherman, J. Am. Chem. Soc., 1994, 116, 369–370 CrossRef CAS
- J. C. Sherman, C. B. Knobler and D. J. Cram, J. Am. Chem. Soc., 1991, 113, 2194–2204 CrossRef CAS
- V. Valderrey, E. C. Escudero-Adan and P. Ballester, J. Am. Chem. Soc., 2012, 134, 10733–10736 CrossRef CAS PubMed
- V. Valderrey, E. C. Escudero-Adan and P. Ballester, Angew. Chem., Int. Ed., 2013, 52, 6898–6902 CrossRef CAS PubMed
- A. S. Hay, J. Org. Chem., 1962, 27, 3320–3321 CrossRef CAS
- P. Siemsen, R. C. Livingston and F. Diederich, Angew. Chem., Int. Ed., 2000, 39, 2633–2657 CrossRef
- B. Breiner, J. K. Clegg and J. R. Nitschke, Chem. Sci., 2011, 2, 51–56 RSC
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed
- B. W. Purse, P. Ballester and J. Rebek, J. Am. Chem. Soc., 2003, 125, 14682–14683 CrossRef CAS PubMed
- S. Anderson, H. L. Anderson and J. K. M. Sanders, Acc. Chem. Res., 1993, 26, 469–475 CrossRef CAS
- A. Galan, E. C. Escudero-Adan, A. Frontera and P. Ballester, J. Org. Chem., 2014, 79, 5545–5557 CrossRef CAS PubMed
- L. K. Frensch, K. Pröpper, M. John, S. Demeshko, C. Brückner and F. Meyer, Angew. Chem., Int. Ed., 2011, 50, 1420–1424 CrossRef CAS PubMed
- L. K. Blusch, Y. Hemberger, K. Propper, B. Dittrich, F. Witterauf, M. John, G. Bringmann, C. Bruckner and F. Meyer, Chem.–Eur. J., 2013, 19, 5868–5880 CrossRef CAS PubMed
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed
- O. Pattawong, M. Q. Salih, N. T. Rosson, C. M. Beaudry and P. H.-Y. Cheong, Org. Biomol. Chem., 2014, 12, 3303–3309 CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, 1H NMR titrations, DOSY and 2D NMR experiments, HRMS experiments and modelling experiments. CCDC 1476421 and 1476422. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc01843b |
| ‡ These authors contributed equally to this work. |
| § Recently, we learnt that the use of bis-N-oxide 3 was completely irrelevant with respect to the yield of cyclic dimer 1. |
| ¶ Experimentally, the addition of 20 equiv. of CuCl·TMEDA to an equimolar mixture of a tetraiodo calix[4]pyrrole 10 (see ESI†) and 4-methylpyridine N-oxide in CD2Cl2 at mM concentration did not disrupt the 1:1 inclusion complex formed between the calix[4]pyrrole and the N-oxide. |
| || Higher dilution conditions (0.32 mM) required longer reaction times (up to one week) and yielded the formation of a larger number of species whose structures were not possible to assign. |
| ** With one of the two dimers of the tetramer placed horizontally and close to the viewer and the second dimer placed behind it, we assigned the absolute configuration (P and M) of the conformer considering the sense of the shortest twist (clockwise or counter-clockwise, respectively) that we need to apply to superimpose the dimer in the back with the one in the front. |
| This journal is © The Royal Society of Chemistry 2016 |