
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric [4 + 2] annulation of 5H-thiazol-4-ones with a chiral dipeptide-based Brønsted base catalyst†
Bo
Zhu‡
a,
Shuai
Qiu‡
a,
Jiangtao
Li
a,
Michelle L.
Coote
b,
Richmond
Lee
 *b and
Zhiyong
Jiang
*a
*b and
Zhiyong
Jiang
*a
aKey Laboratory of Natural Medicine and Immuno-Engineering of Henan Province, Henan University, Kaifeng, Henan, People's Republic of China. E-mail: chmjzy@henu.edu.cn
bARC Centre of Excellence for Electromaterials Science, Research School of Chemistry, Australian National University, Canberra ACT 2601, Australia. E-mail: richmond.lee@anu.edu.au
First published on 9th June 2016
Abstract
Versatile synthetic strategies that access diverse chemical substrates in a highly chemo- and stereo-selective manner are crucial but demanding. Construction of chiral molecules with multiple (hetero)-quaternary carbon stereocenters in a single fashion is a particularly significant challenge, with important applications in the synthesis of a range of bioactive compounds containing the 1,4-sulfur bridged piperidinone structural motif. The asymmetric synthesis of these entities is complicated due to the need to build at least two hetero-quaternary stereocenters concurrently. In order to achieve this, we have developed a new family of dipeptide-based multifunctional Brønsted base organocatalysts that are highly capable of the first asymmetric [4 + 2] annulation reaction of 5H-thiazol-4-ones with electron-deficient alkenes. This protocol could be applied to a series of alkenes such as nitroalkenes, 4-oxo-4-arylbutenones, 4-oxo-4-arylbutenoates and methyleneindolinones, providing an efficient approach to valuable chiral 1,4-sulfur bridged piperidinones and their derivatives with multiple hetereo-quaternary stereogenic centers in high yields and enantioselectivities. Density functional theory studies involving 5H-thiazol-4-one and nitroolefin catalysis propose stereochemical insights into the origin of enantio- and chemo-selectivity.
Introduction
1,4-Sulfur bridged piperidinones and their related derivatives are important sulfur-containing structural motifs (Fig. 1a).1 They are key intermediates in the synthesis of multifarious biologically important heterocyclic compounds, such as quinazolines,1a–c imidazolidines,1d thiophenes,1e,fetc.1g–j Several of these molecules have been studied as potential fungicides1k and Trypanosoma cruzi dihydrofolate reductase inhibitors.1l Since the use of mesoionic 1,3-thiazolium-4-olates as 1,3-dipolar reagents was reported by Potts and co-workers in 1974,1g,2 cycloaddition reactions of 1,3-thiazolium-4-olates with electron-deficient alkenes under harsh reaction conditions have become the most common strategy for the construction of 1,4-sulfur bridged piperidinones (Fig. 1b).1g–j,3 As an alternative, in 1975, Foucaud and co-workers described an example using 4-hydroxy-1,3-thiazoles as dienes to undergo [4 + 2] annulation with dimethylmaleate and dimethylfumarate.4 Unfortunately, to date, no asymmetric method has yet been developed to access these entities in a non-racemic form.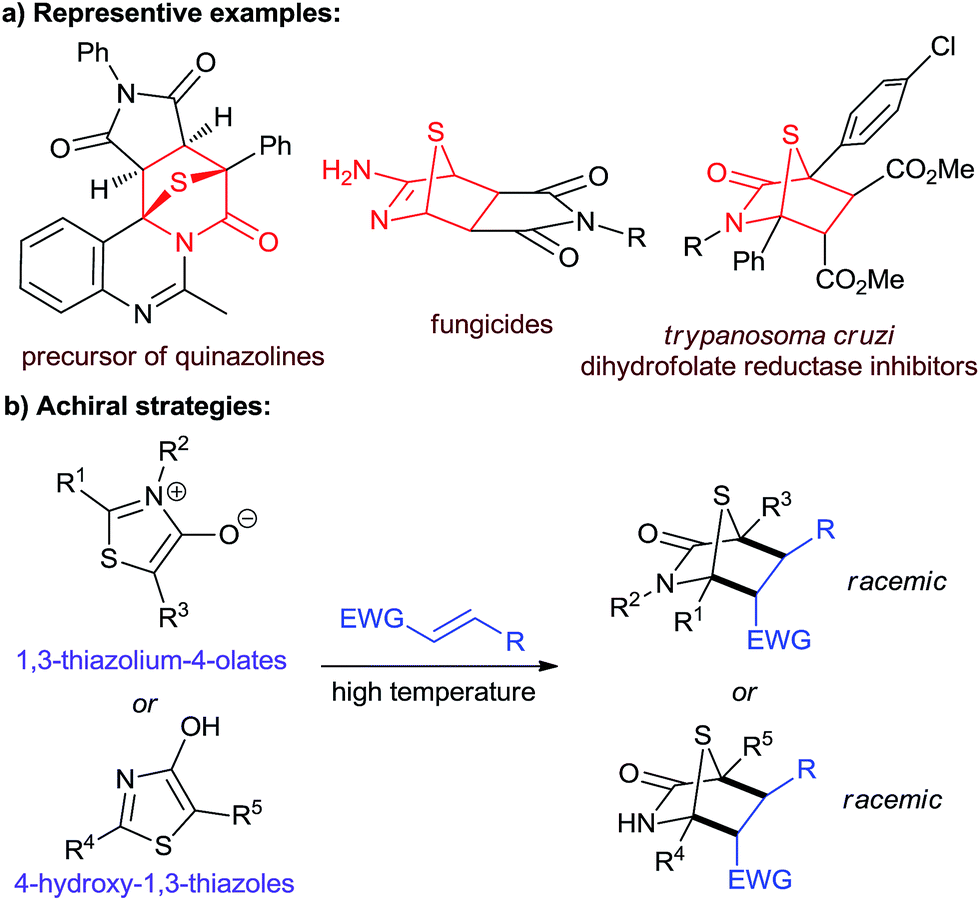 | ||
| Fig. 1 (a) Biologically significant molecules and synthetic targets. (b) Non asymmetric strategies by Foucaud et al. to obtain [4 + 2] products. | ||
The tautomers of 4-hydroxy-1,3-thiazoles, 5H-thiazol-4-ones,4a were featured in the asymmetric catalysis of a recent work by the Palomo group, who, through developing bifunctional ureidopeptide-based Brønsted base catalysts, established highly enantio- and diastereoselective conjugate addition to nitroalkenes.5 Their study demonstrated the efficacy of 5H-thiazol-4-ones as a new class of sulfur-containing pronucleophiles achieving α,α-disubstituted α-mercapto carboxylic acids. Several elegant asymmetric variants have thus been disclosed subsequently, including iridium-catalyzed allylation,6 conjugate addition to enones via hydrogen bonding (H-bonding) catalysis,7 and chiral phosphine-catalyzed γ-addition with allenoates.8 However asymmetric [4 + 2] annulation of 5H-thiazol-4-ones, behaving as 2-azadienes,9 to effectively and simultaneously build two heteroquaternary stereogenic centers still remains elusive.
Intuitively, the addition reaction between 5H-thiazol-4-ones and electron-deficient alkenes first forms an anionic Michael adduct that could undergo two plausible pathways: protonation or a formal Mannich reaction (Fig. 2). Previous examples employing a bifunctional catalytic system5,7 showed that conjugate addition-protonation is favored. So in order for [4 + 2] annulation to proceed preferentially, we believe that the activation of the 5H-thiazol-4-one imine group is crucial in steering the reaction towards Mannich.
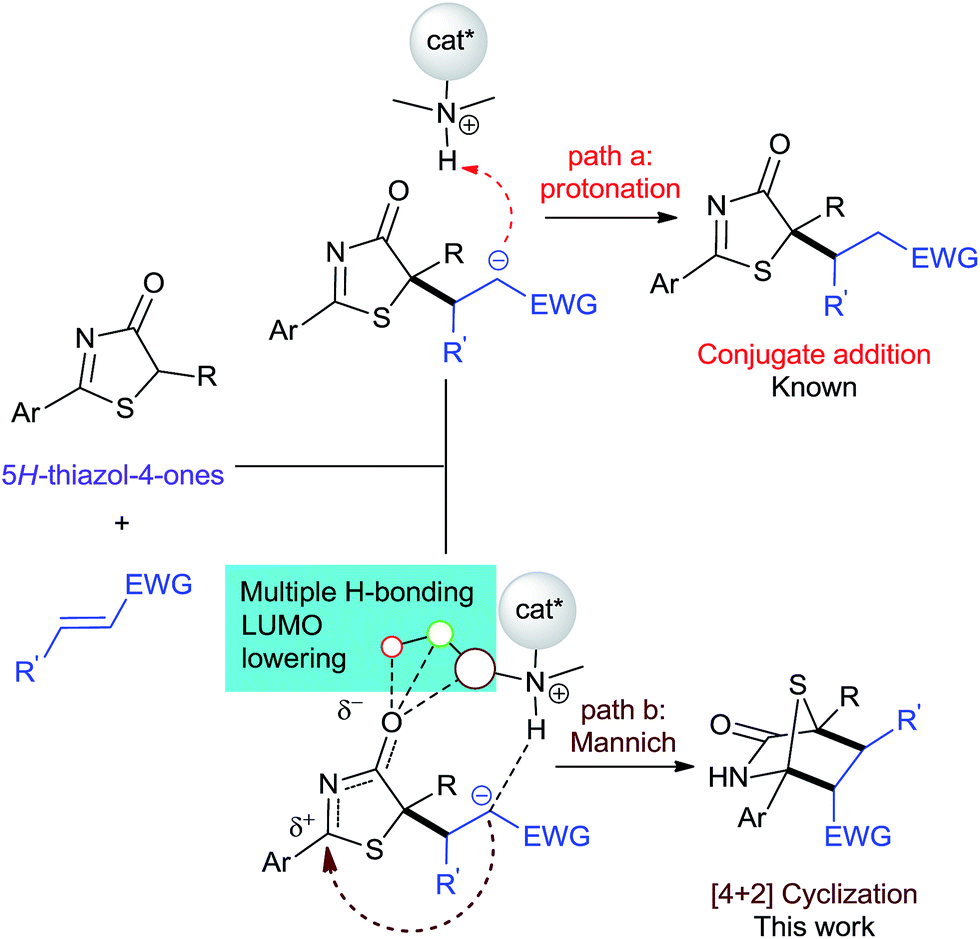 | ||
| Fig. 2 Two plausible paths in the asymmetric reaction between 5H-thiazol-4-ones and activated alkenes. | ||
In this regard, we envisioned that a versatile multifunctional catalyst10 could help realize this through judicious H-bonding interactions. In recent years, we have employed amino acid-based (thio)urea–tertiary amines and their derivatives as bifunctional Brønsted base catalysts for enantioselective reactions.11 In continuation of our pursuits in this burgeoning synthetic field, we herein report a novel family of dipeptide-based10d–g,12,13 (thio)urea–amide–tertiary amine multifunctional Brønsted base catalysts, enabling the first catalytic asymmetric [4 + 2] cyclization reaction between 5H-thiazol-4-ones and electron-deficient alkenes with an extremely broad substrate scope: nitroalkenes, trans-4-oxo-4-arylbutenones, trans-4-oxo-4-arylbutenoates and methyleneindolinones. This new methodology allows highly chemo- and enantio-selective construction of diverse chiral 1,4-sulfur bridged piperidinone architectures, featuring more than two hetereo-quaternary stereogenic centers.
Results and discussion
Optimization of reaction conditions
Our investigations began by examining a model reaction between 5H-thiazol-4-one 1a and nitroalkene 2a (Table 1). Inferring from Palomo's work, vide supra, it was demonstrated that the reaction between 5H-thiazol-4-ones and nitroolefins utilizing a ureidopeptide-based catalyst favors conjugate addition-protonation.5 In the preliminary study, a reaction using achiral Et3N catalyst in CH2Cl2 at 25 °C afforded the conjugate adduct 4 in 36% yield and no trace of the [4 + 2] annulation product (entry 1). When L-tert-leucine-based tertiary amines I,II, were enlisted as catalysts, we observed a slight improvement in the yield of the [4 + 2] annulation product albeit in the region of low enantioselectivity (entries 2 and 3).|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | T (°C) | t (h) |
3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 :4 |
3a | 4 | ||||
| Yieldb (%) | eec (%) | drd | Yieldb (%) | eec (%) | drd | ||||||
| a The reaction was performed using 1a (0.05 mmol) and 2a (0.075 mmol) in 1.0 mL of solvent. b Isolated yield based on 1a after column chromatography. c Enantiomeric ratio of product 3a was determined via chiral phase HPLC analysis. d Determined using crude 1H NMR analysis. e The reaction was performed using 1a (0.2 mmol) and 2a (0.4 mmol) in 4.0 mL of solvent. | |||||||||||
| 1 | Et3N | CH2Cl2 | 25 | 18 | Trace | Trace | n.a. | — | 36 | n.a. | n.a. |
| 2 | I | CH2Cl2 | 25 | 12 | 1:1 |
42 | 0 | 15:1 |
45 | 64 | 1:1 |
| 3 | II | CH2Cl2 | 25 | 12 | 3:1 |
62 | 7 | 9:1 |
20 | 3 | 19:1 |
| 4 | III | CH2Cl2 | 25 | 12 | 4:1 |
73 | 86 | 19:1 |
13 | 88 | 12:1 |
| 5 | IV | CH2Cl2 | 25 | 12 | 4:1 |
68 | 87 | 8:1 |
18 | 87 | 9:1 |
| 6 | III | THF | 25 | 16 | Trace | Trace | n.a. | n.a. | Trace | n.a. | n.a. |
| 7 | III | Toluene | 25 | 16 | 5:1 |
84 | 94 | >19:11 |
8 | 88 | 12:1 |
| 8 | III | Et2O | 25 | 16 | 4:1 |
70 | 92 | >19:1 |
11 | 96 | >19:1 |
| 9 | III | CHCl3 | 25 | 16 | 4.5:1 |
72 | 83 | >19:1 |
9 | 85 | >19:1 |
| 10e | III | Toluene | 10 | 36 | 4:1 |
56 | 95 | >19:1 |
10 | 31 | 3:1 |
| 11 | III | Toluene | 30 | 18 | 4.5:1 |
78 | 92 | 19:1 |
12 | 75 | 19:1 |
| 12e | III | Toluene | 25 | 18 | 5:1 |
83 | 95 | >19:1 |
11 | 88 | 12:1 |
| 13e | V | Toluene | 25 | 18 | 2.5:1 |
68 | −75 | >19:1 |
17 | 13 | >19:1 |
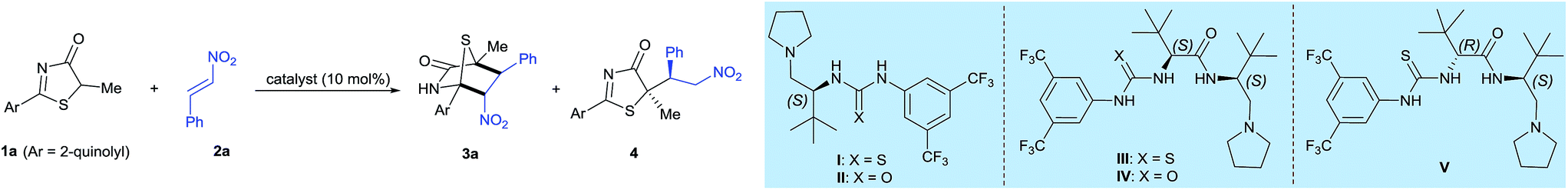
Given the moderate enantioselectivity with L-tert-leucine-based tertiary amine catalysts I,II, we further prototyped a new chiral dipeptide-based thiourea–amide–tertiary amine (DP–TAA) catalyst III constructed with L-tert-leucine units, of which the two molecules of L-tert-leucine are linked by an amido bond (see ESI†). Catalyst III performed extremely well and led to an unprecedented boost in reactivity and stereoselectivity: 3a in 73% yield with 86% enantiomeric excess (ee) was attained (Table 1, entry 4). By modifying the thiourea unit of III to urea, we synthesized a new dipeptide-based urea–amide–tertiary amine (DP–UAA) catalyst IV. Catalyst IV performed less ideally yield-wise but gave a good ee (entry 5). Thus DP–TAA III was selected as the catalyst and tested against various solvents (entries 6–9) but toluene was eventually selected as the optimal solvent. By tuning the reaction temperature (entries 10 and 11), we observed that the yield of 3a was reduced but that ee remained high at 10 °C. At a slightly higher temperature of 30 °C, no large detrimental changes to yield and ee were seen. Using the optimal conditions, the reactions were performed again with higher substrate loadings in toluene at 25 °C, affording the [4 + 2] adduct 3a in 83% yield and 95% ee (entry 12). By modifying the catalyst structure and introducing D-tert-leucine as the second amino acid unit (DP–TAA V), the enantiomer of 3a, ent-3a, was obtained in 68% yield and with moderate enantioselectivity (entry 13).
Expanding the scope of nitroalkenes
With the optimized reaction conditions, we examined the substrate scope of the DP–TAA III-catalyzed enantioselective [4 + 2] annulation between the 5H-thiazol-4-ones 1 and nitroalkenes 2 (Scheme 1). First with 1a as the model 5H-thiazol-4-one substrate, various aryl bearing nitroalkenes 2, were transformed to the corresponding [4 + 2] cyclo-adducts 3a–j in moderate to excellent yields and superior ee (92–97%). Replacing the R2 substituent on 2 to a 2-furanyl group gave a moderate yield of 76% and high ee of 94%. By reacting 1a with 2l, a nitroolefin cyclohexyl derivative, lower yield and poorer reactivity was observed but the corresponding adduct 3l was achieved with a good 82% ee and moderate chemoselectivity.14 Changing the R1 group on 5H-thiazol-4-one for an ethyl substituent also saw the yield of 3m drop to 55%, possibly due to steric effects during the conjugate addition process. Altering the aryl group of 1 gave low to moderate yields of 3n (45%) and 3o (76%). The absolute configurations of the cycloaddition products were assigned on the basis of the X-ray crystal structural analysis of 3a.14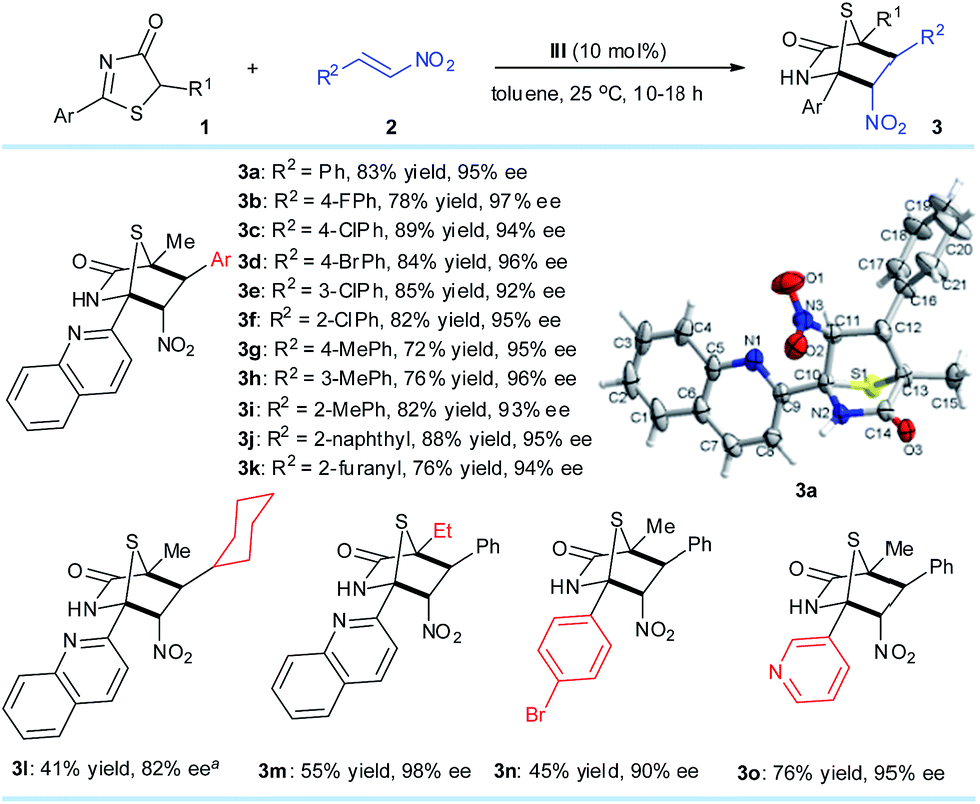 | ||
| Scheme 1 Reaction conditions: 1 (0.2 mmol), 2 (0.4 mmol), III (0.02 mmol), toluene (4.0 mL) at 25 °C. All drs are >20:1. ees were determined via chiral HPLC analysis. a 72 hours. | ||
Mechanistic proposal
To gain mechanistic and stereochemical insights into this highly enantio- and chemo-selective reaction, we first modelled the reaction between 5H-thiazol-4-one 1, nitroalkene 2, and catalyst III with density functional theory (DFT) at a carefully chosen level of theory (for further theoretical details see the ESI†). Preliminary stereochemical-outcomes in generating the conjugate addition adducts were first considered based on the binding of either 1 (mode B) or 2 (mode A) to the main dipeptidyl scaffold (Fig. 3). Only the eclipsed conformation of the reacting substrates was modelled as our previous work has shown that π–π* orbital interactions directly stabilize the transition state by approximately 2.7 kcal mol−1.11e A total of four pathways leading to adducts with S,S- and R,R-configurations and two different binding modes are possible. Transition state (TS) structures were optimized and free energy barriers calculated for each of to the four pathways (Fig. 3). TS1A-S,S (ΔG‡ = 18.5 kcal mol−1) was revealed as the most stable TS followed by TS1A-R,R (20.2). Both TS1B-S,S (23.0) and TS1B-R,R (22.1) have overall higher activation barriers than TS1A-S,S by about 3–5 kcal mol−1. This suggests that the binding of nitroalkene 2 to the urea–thiourea skeleton is preferred due to the stronger oxyanion hole interaction between the positively charged ammonium group and the negative charge of 1.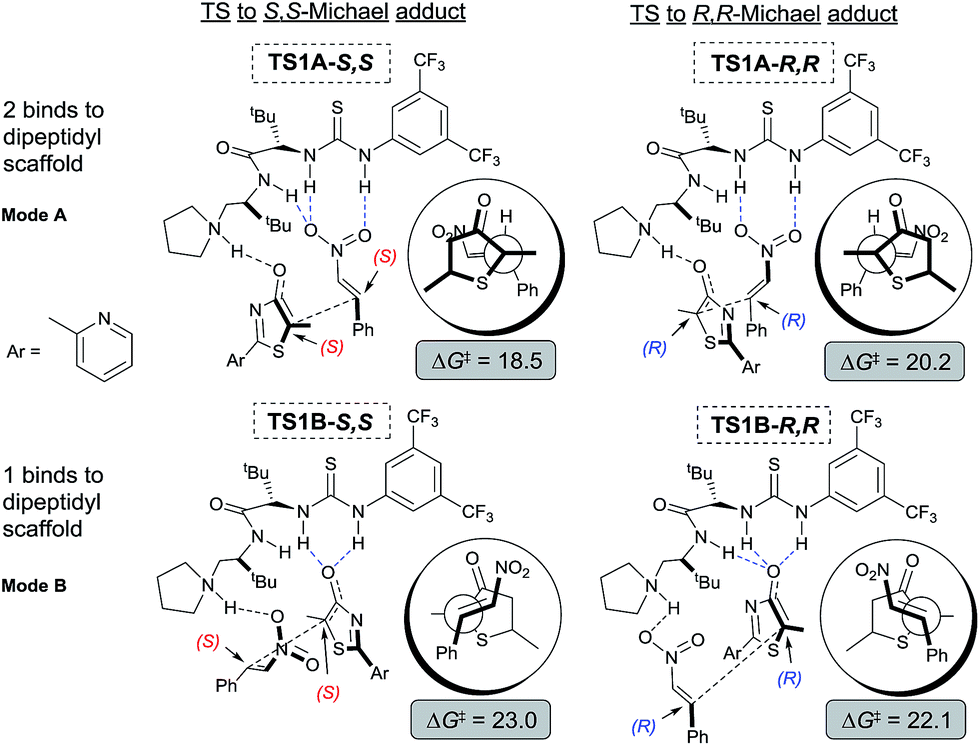 | ||
| Fig. 3 Computational modelling of the possible stereo-outcomes for the first Michael C–C bond formation process. Newman projection diagrams in circles for added perspective. All free energy barriers ΔG‡ and reaction free energies are kcal mol−1 relative to free starting materials 1, 2 and III. | ||
The free energy difference between TS1A-S,S and TS1A-R,R is ΔΔG‡ = 1.7 kcal mol−1, which would correspond to a ∼6:1 S,S to R,R-configuration distribution ratio in the product. However, the first Michael addition TS1 is not the rate determining step. Following the chemical transformation from the two lowest pathways TS1A-S,S and TS1A-R,R through the S,S- and R,R-routes respectively, further bifurcation from the Michael adducts int1A would lead to either the formal Mannich or protonation reaction pathways, processes with higher or equivalent activation free energies (Scheme 2). The Mannich reaction through the S,S pathway is the most kinetically dominant route, ΔG‡ = 18.5 kcal mol−1, and hence yields the majority of the [4 + 2] product 3 from int2A-S,S, consistent with our experiments. The rest of the reaction channels through protonation or Mannich connect to the intermediates int2′A-S,S, int2′A-R,R and int2A-R,R, which have very close free energy barriers: ΔG‡ = 20.3, 20.5 and 20.8 kcal mol−1 respectively. The theoretical ee value is 96%, computed based on the activation free energy difference between TS2A-S,S and TS2A-R,R (ΔΔG‡ = 2.3 kcal mol−1). This value is consistent with the experimental ee value of 95%. For product 4, we rationalize that under conditions of thermodynamic control int2′A-R,R will most likely be favored as having the lowest Gibbs free energy of reaction (Scheme 2). Indeed as experimentally observed when the reaction temperature was lowered (see Table 1 entry 10) more ent-4 (i.e., int2′A-S,S) is formed. This suggests that, when the thermodynamic effect is mitigated, enantioselectivity for the Michael-protonation product will be adversely affected.
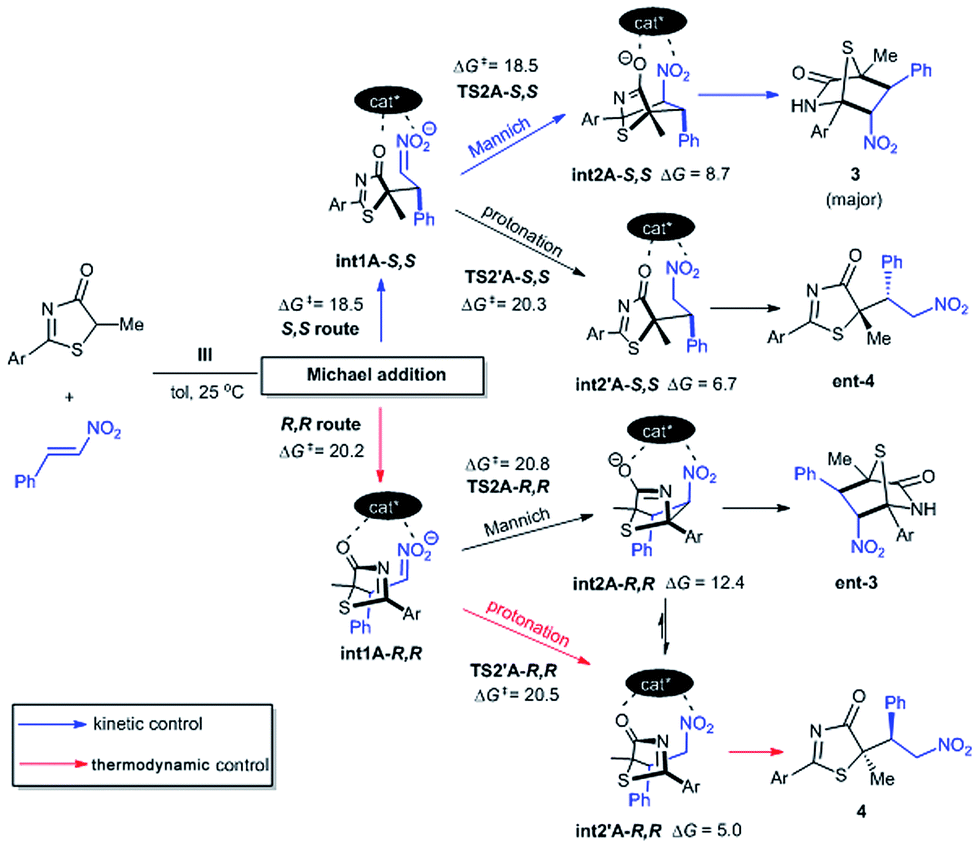 | ||
| Scheme 2 The S,S route affords the kinetically favorable [4 + 2] product 3 as the major product. Competition between the minor Michael products ent-4 and 4 is thermodynamically controlled. All free energy barriers ΔG‡ and reaction free energies are kcal mol−1 relative to free starting materials 1, 2 and III. | ||
Chemically intuitive insights into the stereochemical preference for the rate determining S,S pathway formal Mannich process (TS2A-S,S) were inferred from an examination of the non-covalent interactions (NCI) between the bound catalyst III and the substrates 1 and 2 (see Fig. 4). Thus, Mannich and protonation transition state structures, diverging from the S,S and R,R pathways were analyzed with the reduced density gradient based NCIplot method (see ESI†) to identify these critical NCI areas. These interactions could either be attractive or repulsive depending on the sign of the second density Hessian eigenvalue (sign(λ2), Fig. 4). Visual inspection of the NCI isosurfaces of TS2A-S,S shows a smaller degree of repulsive forces compared with TS2A-R,R, TS2′A-S,S and TS2′A-R,R. More importantly, critical H-bonding features between TS2A-S,S and the rest of the TSs are different. For instance the TS2A-S,S NCI isosurface reveals non-classical H-bonding between the catalyst-pyrrolidinium's activated Cα-H group and the amide group of 1 (Cα–H⋯N = 2.55 Å). The significance of this type of non-classical H-bonding interaction was highlighted by Canizzaro and Houk as playing a crucial role in stereoselective organocatalysis.15 Also, the involvement of the three amide N–H groups of the catalyst core in a tripodal H-bonding fashion would bind 2 in an optimal position for the subsequent C–C bond addition, facilitating the Mannich process. Thus, the chemical influence exerted by the dipeptide catalyst toward activating the substrates for [4 + 2] cyclization was achieved the way we had originally envisaged (Fig. 2).
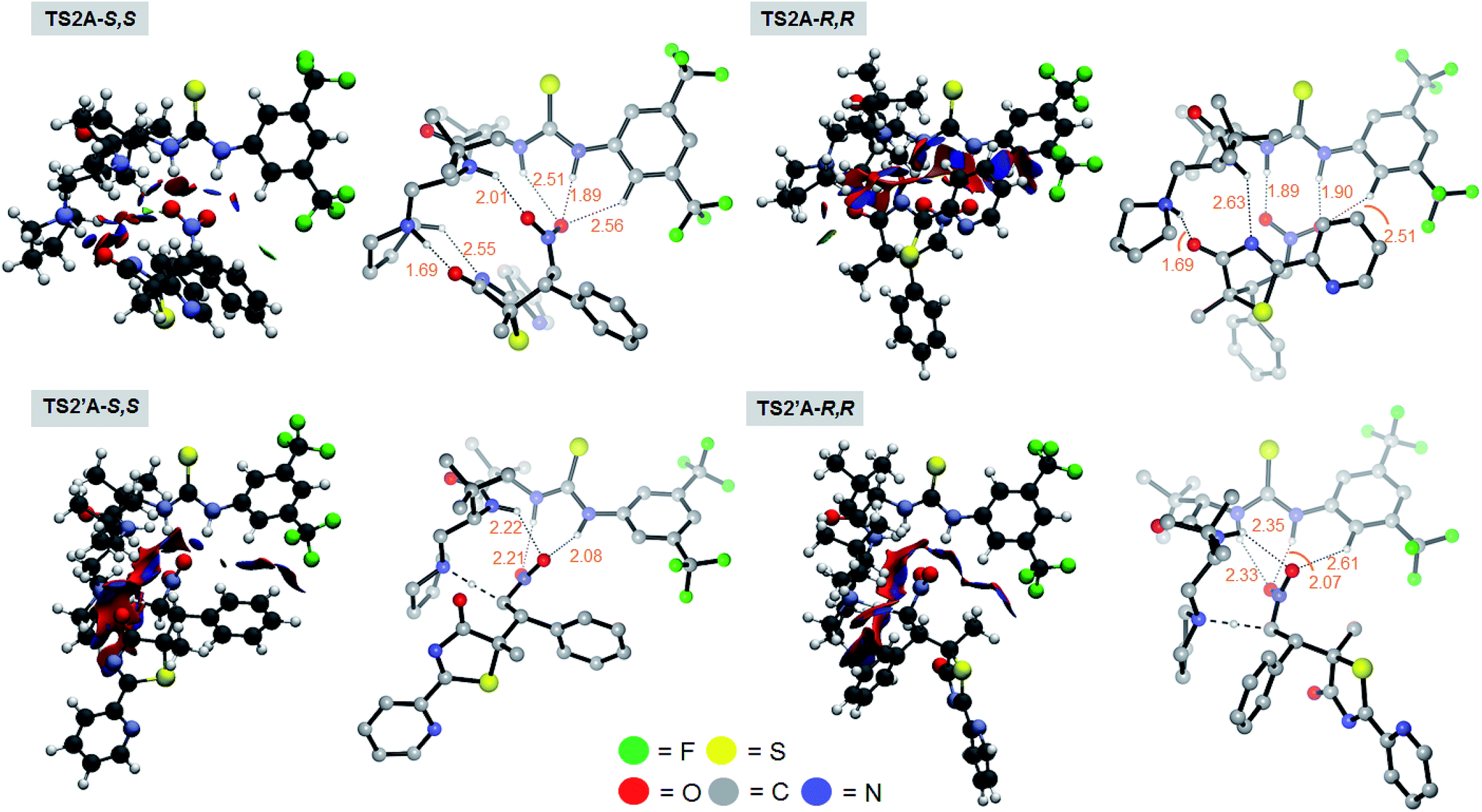 | ||
| Fig. 4 Non-covalent interaction isosurfaces of the optimized TS structures of the Mannich (TS2A-S,S and TS2A-R,R) and the protonation processes (TS2′A-S,S and TS2′A-R,R). Blue-green-red color scale from −0.4 < sign(λ2)ρ(r) > 0.4 au, where blue (positive) is favorable and red (negative) is an unfavorable interaction. Inset: corresponding TS optimized geometries with key H-bonding featured with dotted lines. Non-relevant C–H hydrogens were omitted for visual clarity. Values are bond distances in Å. | ||
Reactivity with trans-4-oxo-4-arylbutenones and trans-4-oxo-4-arylbutenoates
Encouraged by the promising potential of the DP–TAA type catalysts for asymmetric [4 + 2] annulation, we shifted our focus to an alkene electrophile trans-4-oxo-4-phenylbutenone 5,16 a new substrate for the asymmetric [4 + 2] cyclization (Scheme 3). The reactions were first performed between the 5H-thiazol-4-one substrate 1a and the trans-4-oxo-4-arylbutenones 5 bearing various aryls, (hetero)aryls, and ethyl under slightly modified reaction conditions (in CHCl3 at −10 °C). The corresponding [4 + 2] cyclo-adducts 6a–l were obtained in moderate to excellent yields and superior ee (94–99%). Replacing the 2-quinolyl Ar1 substituent of 1a at the 2-position to other (hetero)aryl and aryl groups also gave products 6m–q with excellent ee values (98–99%). When the methyl R1 group of 6a was replaced by ethyl (6r) or benzyl (6s), the reaction was sluggish possibly due to steric effects, but satisfactory results were obtained when the reaction was carried out at 25 °C. The absolute configurations of the cycloaddition products were assigned on the basis of the X-ray crystal structural analysis of 6b.14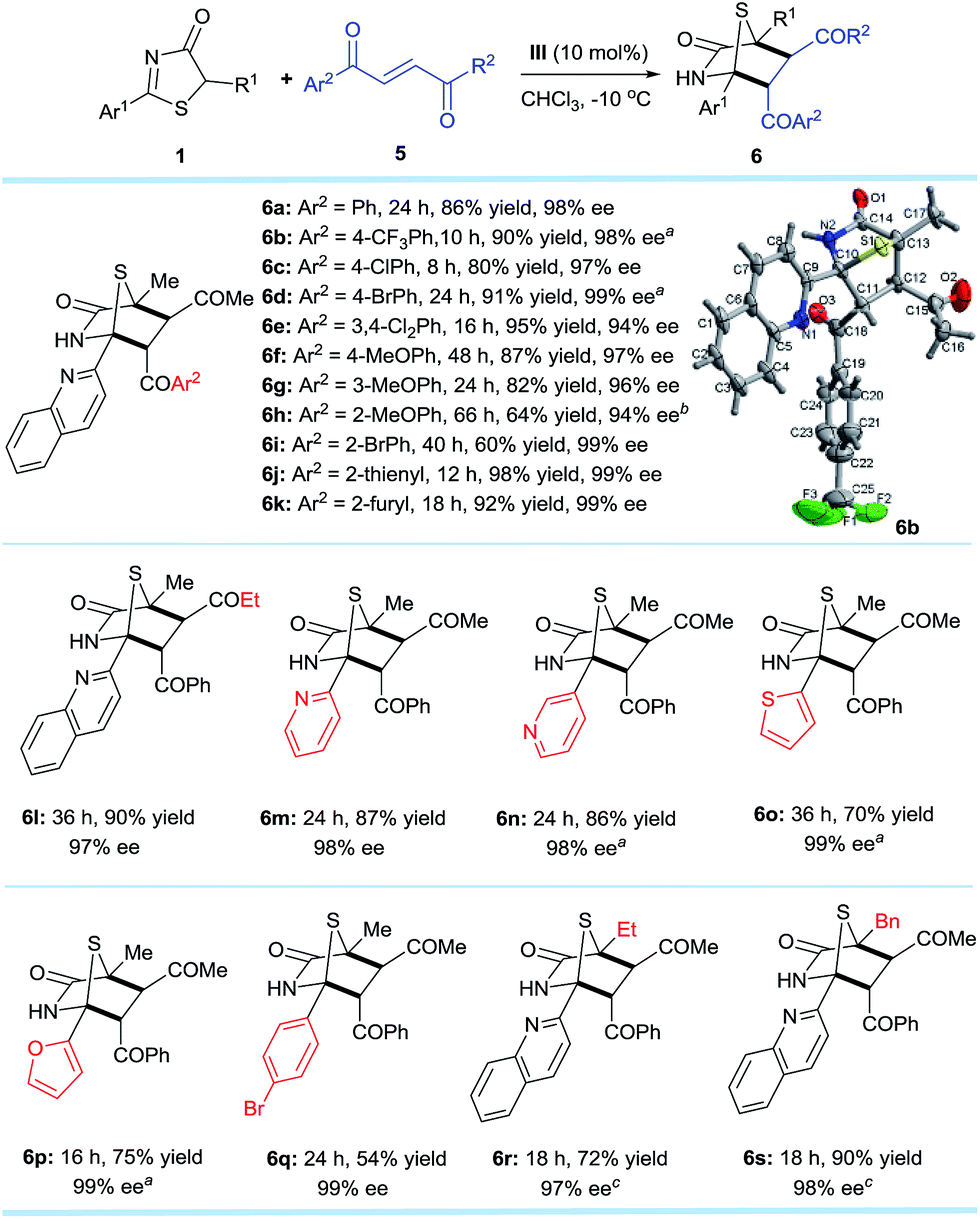 | ||
| Scheme 3 Reaction conditions: 1 (0.2 mmol), 5 (0.3 mmol), III (0.02 mmol), CHCl3 (4.0 mL) at −10 °C. All drs are >20:1. ees were determined via chiral phase high-performance liquid chromatography analysis. aT = −30 °C. bT = 20 °C. cT = 25 °C. | ||
The established methodology was also successfully applied to trans-4-oxo-4-arylbutenoates10h,16,177 that led to [4 + 2] annulation adducts 8a–h in 67–97% yields and excellent enantioselectivities, unaffected by steric and electronic effects on the aromatic ring substituents of the trans-4-oxo-4-arylbutenoates (Scheme 4). It is noteworthy that both annulation adducts 6 and 8 are analogues of Trypanosoma cruzi dihydrofolate reductase inhibitors1l (Fig. 1a).
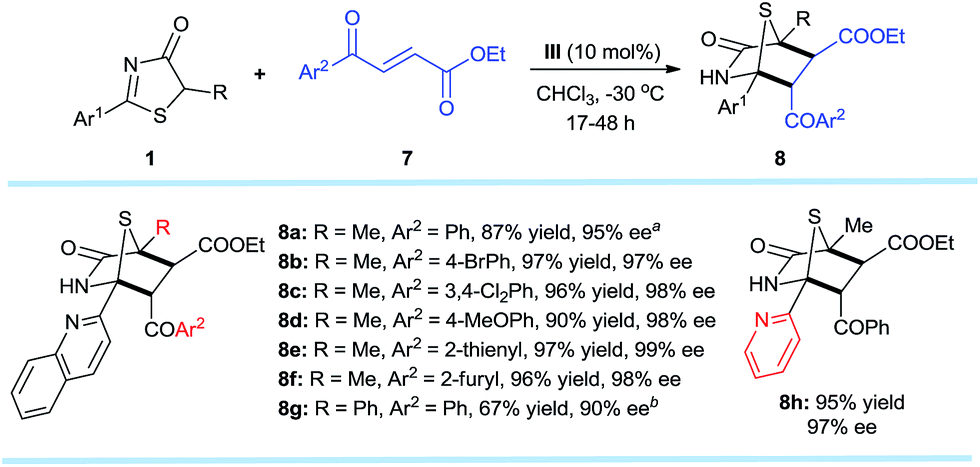 | ||
| Scheme 4
1 (0.2 mmol), 7 (0.3 mmol), III (0.02 mmol), CHCl3 (4.0 mL) at −30 °C. All drs are >20:1. ee values were determined via chiral phase high-performance liquid chromatography analysis. aT = −20 °C. b 0.04 mmol of III, 0.2 mmol of Li3PO4, T = 30 °C. | ||
Reactivity with methyleneindolinones
Methyleneindolinones have been widely used as dipolarophiles in [2 + 3] cyclizations to build spirooxindoles with multiple stereocenters.18 However, few catalytic asymmetric strategies19 are suitable for the [4 + 2] process. Inspired by the success of [4 + 2] annulation from the DP–TAA catalysts, the reaction between 5H-thiazol-4-one substrate 1a and methyleneindolinone 9a was first evaluated under the established reaction conditions (10 mol% of catalyst III in CHCl3 at −30 °C). As expected, the 1,4-sulfur bridged piperidinonespirooxindole 10a, which has three quaternary and one tertiary stereogenic centers, was obtained in 96% yield with 95% ee and >20:1 dr (Scheme 5).
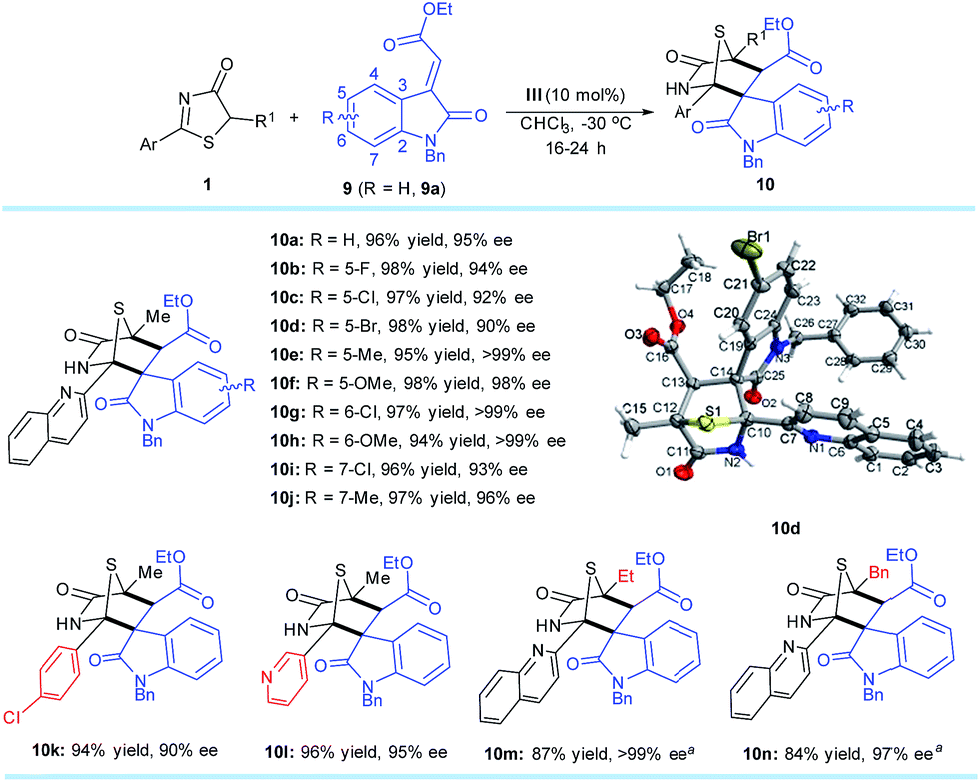 | ||
| Scheme 5
1 (0.2 mmol), 9 (0.3 mmol), III (0.02 mmol), CHCl3 (4.0 mL) at −30 °C. All drs are >20:1. ees were determined via chiral phase high-performance liquid chromatography analysis. aT = −20 °C. | ||
A number of 5H-thiazol-4-ones and methyleneindolinones were next examined to determine the substrate generality and limitations of this strategy. Various adducts 10b–n were attained in excellent yields (84–98%) and enantioselectivities (90–>99% ee). Moreover, only one diastereomer was observed as determined using 1H NMR spectroscopy of the crude reaction mixtures for each product. It is worth noting that these synthesized chiral compounds are known as drug candidates for antiproliferative and anticancer agents.20 The absolute configurations of the cycloaddition products were assigned on the basis of the X-ray crystal structural analysis of 10d.14
The synthetic value of this novel catalyst in enantioselective synthesis was further explored for the targeted synthesis of a hexa-substituted tetrahydrothiophene 11, which features four contiguous chiral centers including two heteroquaternary stereogenic centers (eqn (1)). Such a tetrahydrothiophene is an important structural scaffold existing in several molecules displaying interesting biological activities.21
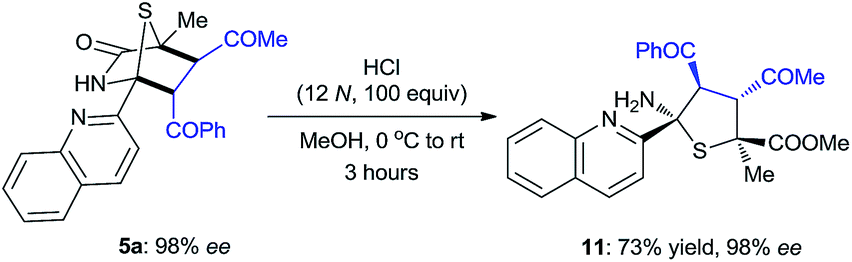 | (1) |
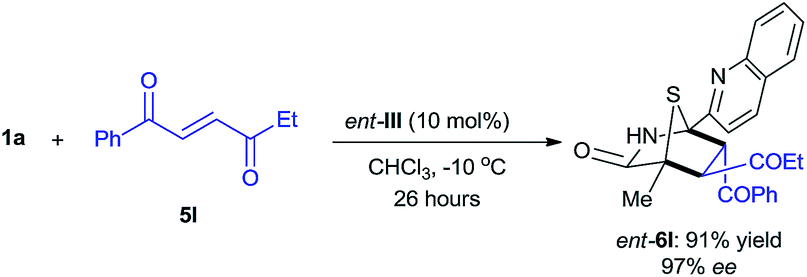 | (2) |
The reaction between 5H-thiazol-4-one 1a and trans-4-oxo-4-phenylbutenone 5l was conducted with 10 mol% of D-tert-leucine-based ent-III catalyst in chloroform at −10 °C (eqn (2)). The enantiomer of 6l (ent-6l) was successfully obtained in 91% yield with 97% ee after 26 hours, thus demonstrating the potential of these dipeptide-based catalysts for enantiodivergent synthesis.
Conclusions
In summary, we have developed a new family of dipeptide-based multifunctional Brønsted base catalysts that are DP–TAAs and DP–UAAs. By employing DP–TAA III derived from L-tert-leucine as the catalyst, the asymmetric reaction of 5H-thiazol-4-ones with a broad range of electron-deficient alkenes including nitroolefins, trans-4-oxo-4-arylbutenones, trans-4-oxo-4-arylbutenoates and methyleneindolinones, unprecedented [4 + 2] annulation with high chemo- and enantio-selectivities (up to 98% yield and >99% ee) was realized. The current method furnishes an expedient approach to chiral 1,4-sulfur bridged piperidinones and their derivatives, such as hexa-substituted tetrahydrothiophenes, which feature two or even three quaternary carbon stereocenters and are potentially important for medicinal chemistry research. Furthermore, mechanistic insights gained from computational modelling studies of III, 1 and nitroolefin 2 provide valuable information on the stereo- and chemo-selection origins. Finally, given the highly modular approach in synthesizing the catalyst by varying the amino acid chiral backbones and appending various Brønsted bases, we anticipate that such dipeptide catalysts will find prolific application in more challenging organic transformations.Acknowledgements
Grants from NSFC (21072044), NCET-11-0938 and the Program for Innovative Research Team from the University of Henan Province (14IRTSTHN006) are gratefully acknowledged by Z. J. M. L. C. and R. L. also gratefully acknowledge generous allocations of supercomputing time on the National Facility of the National Computational Infrastructure and financial support from the Australian Research Council Centre of Excellence for Electromaterials Science.Notes and references
- For selected examples, see: (a) K. T. Potts, K. G. Bordeaux, W. R. Kuehnling and R. L. Salsbury, J. Org. Chem., 1985, 50, 1666 CrossRef CAS; (b) M. J. Arévalo, M. Ávalos, R. Babiano, P. Cintas, J. L. Jiménez, M. E. Light and J. C. Palacios, Tetrahedron, 2006, 62, 6909 CrossRef; (c) D. R. Bobeck, H. I. Lee, A. C. Flick and A. Padwa, J. Org. Chem., 2009, 74, 7389 CrossRef CAS PubMed; (d) T. Sheradsky and N. Itzhak, J. Chem. Soc., Perkin Trans. 1, 1989, 33 RSC; (e) M. Avalos, R. Babiano, A. Cabanillas, P. Cintas, F. J. Higes, J. L. Jiménez and J. C. Palacios, J. Org. Chem., 1996, 61, 3738 CrossRef CAS PubMed; (f) M. Avalos, R. Babiano, P. Cintas, F. R. Clemente, R. Gordillo, J. L. Jiménez, J. C. Palacios and P. R. Raithby, J. Org. Chem., 2000, 65, 5089 CrossRef CAS PubMed; (g) K. T. Potts, J. Baum and E. Houghton, J. Org. Chem., 1974, 39, 3631 CrossRef CAS; (h) O. Tsuge, H. Shiraishi and M. Noguchi, Chem. Lett., 1981, 213 CrossRef CAS; (i) D. L. Hertzog, W. R. Nadler, Z. J. Zhang and A. Padwa, Tetrahedron Lett., 1992, 33, 5877 CrossRef CAS; (j) A. Padwa, L. S. Beall, T. M. Heidelbaugh, B. Liu and S. M. Sheehan, J. Org. Chem., 2000, 65, 2684 CrossRef CAS PubMed; (k) S. N. Lopez, M. Sortino, A. Escalante, F. de Campos, R. Correa, F. V. Cechinel, R. J. Nunes and S. A. Zacchino, Arzneim. Forsch., 2003, 53, 280 CAS; (l) F. Zuccotto, M. Zvelebil, R. Brun, S. F. Chowdhury, R. D. Lucrezia, I. Leal, L. Maes, L. M. Ruiz-Perez, D. G. Pacanowska and I. H. Gilbert, Eur. J. Med. Chem., 2001, 36, 395 CrossRef CAS PubMed.
- K. T. Potts, E. Houghton and U. P. Singh, J. Org. Chem., 1974, 39, 3627 CrossRef CAS.
- (a) H. Matsukubo and H. Kato, J. Chem. Soc., Chem. Commun., 1975, 840 RSC; (b) M. Baudy, A. Robert and G. Guimon, Tetrahedron, 1982, 38, 1241 CrossRef CAS; (c) T. Takido and K. Itabashi, Synthesis, 1985, 430 CrossRef CAS; (d) A. Padwa, S. R. Harring, D. L. Hertzog and W. R. Nadler, Synthesis, 1994, 993 CrossRef CAS; (e) T. Takido, S. Tamura, K. Sato, H. Kamijo, T. Nakazawa, T. Hata and M. Seno, J. Heterocycl. Chem., 1998, 35, 437 CrossRef CAS.
- (a) A. Robert, M. Ferrey and A. Foucaud, Tetrahedron Lett., 1975, 16, 1377 CrossRef; (b) M. Ferrey, A. Robert and A. Foucaud, Synthesis, 1976, 261 CrossRef CAS; (c) A. Robert, M. Baudy and A. Foucaud, Tetrahedron, 1978, 34, 3525 CrossRef CAS.
- (a) S. Diosdado, J. Etxabe, J. Izquierdo, A. landa, A. Mielgo, I. Olaizola, R. López and C. Palomo, Angew. Chem., Int. Ed., 2013, 52, 11846 CrossRef CAS PubMed; (b) S. Diosdado, R. López and C. Palomo, Chem.–Eur. J., 2014, 20, 6526 CrossRef CAS PubMed.
- W. Chen and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 377 CrossRef CAS PubMed.
- E. Badiola, B. Fiser, E. Gómez-Bengoa, A. Mielgo, I. Olaizola, I. Urruzuno, J. M. García, J. M. Odriozola, J. Razkin, M. Oiarbide and C. Palomo, J. Am. Chem. Soc., 2014, 136, 17869 CrossRef CAS PubMed.
- T. Wang, Z. Yu, D. L. Hoon, K.-W. Huang, Y. Lan and Y. Lu, Chem. Sci., 2015, 6, 4912 RSC.
- D. L. Boger, Tetrahedron, 1983, 39, 2869 CrossRef CAS.
- For selected reviews, see: (a) J. M. Garnier, C. Anstiss and F. Liu, Adv. Synth. Catal., 2009, 351, 331 CrossRef CAS; (b) R. Kenny and F. Liu, Eur. J. Org. Chem., 2015, 5304 CrossRef CAS. And the references therein. For selected examples, see: (c) Q. Zhu and Y. Lu, Angew. Chem., Int. Ed., 2010, 49, 7753 CrossRef CAS PubMed; (d) X. Han, Y. Wang, F. Zhong and Y. Lu, J. Am. Chem. Soc., 2011, 133, 1726 CrossRef CAS PubMed; (e) X. Han, F. Zhong, Y. Wang and Y. Lu, Angew. Chem., Int. Ed., 2012, 51, 767 CrossRef CAS PubMed; (f) T. Wang, W. Yao, F. Zhong, G. H. Pang and Y. Lu, Angew. Chem., Int. Ed., 2014, 53, 2964 CrossRef CAS PubMed; (g) W. Yao, X. Dou and Y. Lu, J. Am. Chem. Soc., 2015, 137, 55 Search PubMed; (h) X. Dong, L. Liang, E. Li and Y. Huang, Angew. Chem., Int. Ed., 2015, 54, 1621 CrossRef CAS PubMed.
- (a) W. Zhang, D. Tan, R. Lee, G. Tong, W. Chen, B. Qi, K.-W. Huang, C.-H. Tan and Z. Jiang, Angew. Chem., Int. Ed., 2012, 51, 10069 CrossRef CAS PubMed; (b) B. Zhu, W. Zhang, R. Lee, Z. Han, W. Yang, D. Tan, K.-W. Huang and Z. Jiang, Angew. Chem., Int. Ed., 2013, 52, 6666 CrossRef CAS PubMed; (c) B. Qiao, Y. An, Q. Liu, W. Yang, H. Liu, J. Shen, L. Yan and Z. Jiang, Org. Lett., 2013, 15, 2358 CrossRef CAS PubMed; (d) S. Duan, S. Li, X. Ye, N.-N. Du, C.-H. Tan and Z. Jiang, J. Org. Chem., 2015, 80, 7770 CrossRef CAS PubMed; (e) X. Zhao, B. Zhu and Z. Jiang, Synlett, 2015, 26, 2216 CrossRef CAS.
- For selected reviews, see: (a) Z. Chai and G. Zhao, Catal. Sci. Technol., 2012, 2, 29 RSC; (b) O. V. Serdyuk, C. M. Heckel and S. B. Tsogoeva, Org. Biomol. Chem., 2013, 11, 7051 RSC.
- For selected reviews, see: (a) S. J. Miller, Acc. Chem. Res., 2004, 37, 601 CrossRef CAS PubMed; (b) S. B. Tsogoeva, Lett. Org. Chem., 2005, 2, 208 CrossRef CAS; (c) E. A. C. Davie, S. M. Mennen, Y. Xu and S. J. Miller, Chem. Rev., 2007, 107, 5759 CrossRef PubMed; (d) J. D. Revell and H. Wennemers, Curr. Opin. Chem. Biol., 2007, 11, 269 CrossRef CAS PubMed; (e) H. Wennemers, Chem. Commun., 2011, 47, 12036 RSC; For selected examples involving peptide-derived catalysis, see: (f) S. Gilbertson, S. Collibee and A. Agarkov, J. Am. Chem. Soc., 2000, 122, 6522 CrossRef CAS; (g) H. J. Martin and B. List, Synlett, 2003, 1901 CAS; (h) S. B. Tsogoeva, S. B. Jagtap, Z. A. Ardemasova and V. N. Kalikhevich, Eur. J. Org. Chem., 2004, 4014 CrossRef CAS; (i) S. B. Tsogoeva and S.-W. Wei, Tetrahedron: Asymmetry, 2005, 16, 1947 CrossRef CAS; (j) Y. Xu, W. Zou, H. Sundén, I. Ibrahem and A. Córdova, Adv. Synth. Catal., 2006, 348, 418 CrossRef CAS; (k) M. Wiesner, J. D. Revell and H. Wennemers, Angew. Chem., Int. Ed., 2008, 47, 1871 CrossRef CAS PubMed; (l) M. Wiesner, J. D. Revell, S. Tonazzi and H. Wennemers, J. Am. Chem. Soc., 2008, 130, 5610 CrossRef CAS PubMed; (m) K. W. Fiori, A. L. A. Puchlopek and S. J. Miller, Nat. Chem., 2009, 1, 630 CrossRef CAS PubMed; (n) B. J. Cowen, L. B. Saunders and S. J. Miller, J. Am. Chem. Soc., 2009, 131, 6105 CrossRef CAS PubMed; (o) M. Freund, S. Schenker and S. B. Tsogoeva, Org. Biomol. Chem., 2009, 7, 4279 RSC; (p) J. Gustafson, D. Lim and S. J. Miller, Science, 2010, 328, 1251 CrossRef CAS PubMed; (q) B. S. Fowler, P. J. Mikochik and S. J. Miller, J. Am. Chem. Soc., 2010, 132, 2870 CrossRef CAS PubMed; (r) M. Wiesner, G. Upert, G. Angelici and H. Wennemers, J. Am. Chem. Soc., 2010, 132, 6 CrossRef CAS PubMed; (s) E. Garand, M. Z. Kamrath, P. A. Jordan, A. B. Wolk, A. B. McCoy, S. J. Miller and M. A. Johnson, Science, 2012, 335, 694 CrossRef CAS PubMed; (t) K. T. Barrett and S. J. Miller, J. Am. Chem. Soc., 2013, 135, 2963 CrossRef CAS PubMed; (u) K. T. Barrett, A. J. Metrano, P. R. Rablen and S. J. Miller, Nature, 2014, 509, 71 CrossRef CAS PubMed; (v) T. Wang, W. Yao, F. Zhong, G. H. Pang and Y. Lu, Angew. Chem., Int. Ed., 2014, 53, 2964 CrossRef CAS PubMed; (w) D. Cao, J. Zhang, H. Wang and G. Zhao, Chem.–Eur. J., 2015, 21, 9998 CrossRef CAS PubMed; (x) M. E. Diener, A. J. Metrano, S. Kusano and S. J. Miller, J. Am. Chem. Soc., 2015, 137, 12369 CrossRef CAS PubMed.
- See the ESI† for details.
- C. E. Cannizzaro and K. N. Houk, J. Am. Chem. Soc., 2002, 124, 7163 CrossRef CAS PubMed.
- For examples using trans-4-oxo-4-arylbutenones as electrophiles, see: (a) Z. Jiang, Y. Yang, Y. Pan, Y. Zhao, H. Liu and C.-H. Tan, Chem.–Eur. J., 2009, 15, 4925 CrossRef CAS PubMed; (b) G. Blay, I. Fernández, A. Monleón, M. C. Muñoz, J. R. Pedro and C. Vila, Adv. Synth. Catal., 2009, 351, 2433 CrossRef CAS; (c) F. Zhao, W. Zhang, Y. Yang, Y. Pan, W. Chen, H. Liu, L. Yan, C.-H. Tan and Z. Jiang, Adv. Synth. Catal., 2011, 353, 2624 CAS; (d) Y.-R. Chen, U. Das, M.-H. Liu and W. Lin, J. Org. Chem., 2015, 80, 1985 CrossRef CAS PubMed.
- For examples using trans-4-oxo-4-arylbutenoates as electrophiles, see: (a) J. Shen, T. T. Nguyen, Y.-P. Goh, W. Ye, X. Fu, J. Xu and C.-H. Tan, J. Am. Chem. Soc., 2006, 128, 13692 CrossRef CAS PubMed; (b) W. Ye, Z. Jiang, Y. Zhao, S. L. M. Goh, D. Leow, Y.-T. Soh and C.-H. Tan, Adv. Synth. Catal., 2007, 349, 2454 CrossRef CAS; (c) T. R. Wu and J. M. Choon, J. Am. Chem. Soc., 2007, 129, 4908 CrossRef CAS PubMed; (d) H.-H. Lu, X.-F. Wang, C.-J. Yao, J.-M. Zhang, H. Wu and W.-J. Xiao, Chem. Commun., 2009, 4251 RSC; (e) M. Sugiura, M. Tokudomi and M. Nakajima, Chem. Commun., 2010, 46, 7799 RSC; (f) J. Jiang, Y. Cai, W. Chen, L. Lin, X. Liu and X. Feng, Chem. Commun., 2011, 47, 4016 RSC; (g) Z. Wang, Z. Yang, D. Chen, X. Liu, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2011, 50, 4928 CrossRef CAS PubMed; (h) K. M. Steward, M. T. Corbett, C. G. Goodman and J. S. Johnson, J. Am. Chem. Soc., 2012, 134, 20197 CrossRef CAS PubMed; (i) S. Bai, X. Liu, Z. Wang, W. Cao, L. Lin and X. Feng, Adv. Synth. Catal., 2012, 354, 2096 CrossRef CAS; (j) Y.-H. Liao, X.-L. Liu, Z.-J. Wu, X.-L. Du, X.-M. Zhang and W.-C. Yuan, Chem.–Eur. J., 2012, 18, 6679 CrossRef CAS PubMed; (k) U. Das, Y.-R. Chen, Y.-L. Tsai and W. Lin, Chem.–Eur. J., 2013, 19, 7713 CrossRef CAS PubMed; (l) Z. Jin, J. Xu, S. Yang, B.-A. Song and Y. R. Chi, Angew. Chem., Int. Ed., 2013, 52, 12354 CrossRef CAS PubMed; (m) J. Ji, L. Lin, L. Zhou, Y. Zhang, Y. Liu, X. Liu and X. Feng, Adv. Synth. Catal., 2013, 355, 2764 CrossRef CAS; (n) R. Kowalczyk, A. J. Wierzba, P. J. Boratyński and J. Bąkowicz, Tetrahedron, 2014, 70, 5834 CrossRef CAS; (o) Z. Wang, Q. Yao, T. Kang, J. Feng, X. Liu, L. Lin and X. Feng, Chem. Commun., 2014, 50, 4918 RSC; (p) X. Dong, L. Liang, E. Li and Y. Huang, Angew. Chem., Int. Ed., 2015, 54, 1621 CrossRef CAS PubMed.
- (a) B. Tan, N. R. Candeias III and C. F. Barbas, Nat. Chem., 2011, 3, 473 CAS; (b) Y. Cao, X. Jiang, L. Liu, F. Shen, F. Zhang and R. Wang, Angew. Chem., Int. Ed., 2011, 50, 9124 CrossRef CAS PubMed; (c) W. Sun, G. Zhu, C. Wu, G. Li, L. Hong and R. Wang, Angew. Chem., Int. Ed., 2013, 52, 8633 CrossRef CAS PubMed; (d) Q.-S. Sun, H. Zhu, Y.-J. Chen, X.-D. Yang, X.-W. Sun and G.-Q. Lin, Angew. Chem., Int. Ed., 2015, 54, 13253 CrossRef CAS PubMed.
- (a) G. Bencivenni, L.-Y. Wu, A. Mazzanti, B. Giannichi, F. Pesciaioli, M.-P. Song, G. Bartoli and P. Melchiorre, Angew. Chem., Int. Ed., 2009, 48, 7200 CrossRef CAS PubMed; (b) Z.-J. Jia, H. Jiang, J.-L. Li, B. Gschwend, Q.-Z. Li, X. Yin, J. Grouleff, Y.-C. Chen and K. A. Jørgensen, J. Am. Chem. Soc., 2011, 133, 5053 CrossRef CAS PubMed; (c) B. Tan, G. Hernández-Torres III and C. F. Barbas, J. Am. Chem. Soc., 2011, 133, 12354 CrossRef CAS PubMed.
- (a) L. Chen, Q. Ding, J.-J. Liu, S. Yang, Z. Zhang, US Pat., 20070213341 A1 20070913, 2007; (b) L. Chen, Q. Ding, J.-J. Liu, S. Yang, Z. Zhang, US Pat., 20080009486 A1 20080110, 2008.
- (a) A. Hamza, X.-Y. Zhao, M. Tong, H.-H. Tai and C.-G. Zhan, Bioorg. Med. Chem., 2011, 19, 6077 CrossRef CAS PubMed; (b) R. D. Cone, J. A. Sebag, J. Pantel, S. Y. Williams, C. D. Weaver, G. A. Sulikowski, C. W. Lindsley, P. S. Portonova, C. Zhang, PCT Int. Appl., WO 2013134376 A1 20130912, 2013.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1419574, 1419583, and 1463891. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc02039a |
| ‡ The authors made equal contributions. |
| This journal is © The Royal Society of Chemistry 2016 |