
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Scandium-catalysed intermolecular hydroaminoalkylation of olefins with aliphatic tertiary amines†
Adi E.
Nako
 ,
Juzo
Oyamada
,
Masayoshi
Nishiura
and
Zhaomin
Hou
*
,
Juzo
Oyamada
,
Masayoshi
Nishiura
and
Zhaomin
Hou
*
Organometallic Chemistry Laboratory and RIKEN Center for Sustainable Resource Science, RIKEN, 2-1 Hirosawa, Wako, Saitama, 351-0198, Japan. E-mail: houz@riken.jp
First published on 4th July 2016
Abstract
A homoleptic scandium trialkyl complex in combination with a borate compound served as an excellent catalyst for the C–H addition of aliphatic tertiary amines to olefins. This highly regiospecific, 100% atom efficient C–H bond alkylation reaction was applicable to a wide variety of tertiary amines and olefins, including functionalised styrenes and unactivated α-olefins. This work represents the first example of rare-earth catalysed olefin hydroaminoalkylation and also the first example of catalytic C–H addition of aliphatic tertiary amines to olefins with any catalyst.
Introduction
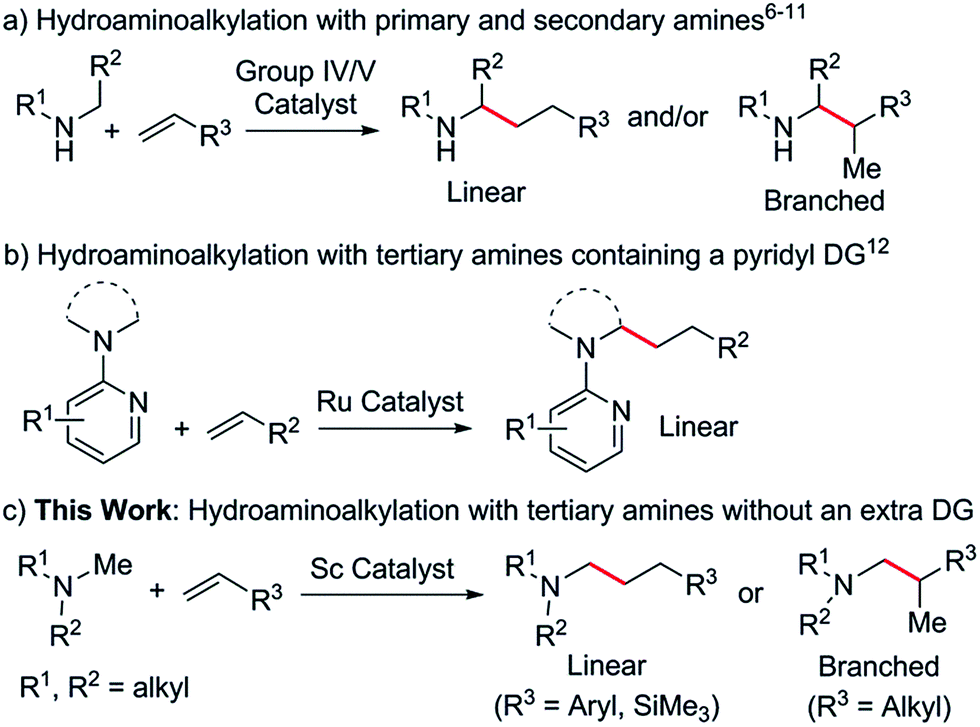
Aliphatic tertiary amines are ubiquitous across the chemical sciences. Therefore, the synthesis of tertiary amines bearing diverse substituents in a selective and atom efficient manner is of great interest and significance. In this regard, the catalytic hydroamination,1 hydroaminomethylation2 and reductive amination3 of unsaturated bonds can serve as useful synthetic routes to these molecules. However, all of these transformations require cleavage of an N–H bond to form a C–N bond, thus limiting potential substrates to primary and secondary amines.4Since the discovery that under certain conditions, group IV and V metal catalysts gave C–C bond formation products instead of the expected C–N coupled products,5 intermolecular hydroaminoalkylation has been rapidly gaining interest as a 100% atom efficient synthetic route to substituted amines.6 It has been shown that N,N-7 and N,O-chelated early transition metal complexes based on 2-pyridonate,8 amidate,9 phosphoramidate10 and related ligands are active catalysts for the hydroaminoalkylation of olefins with a variety of primary and secondary amines (Scheme 1a). The reactions are widely considered to occur via the initial formation of metal amido intermediates through deprotonation of an N–H bond,11 therefore limiting the starting materials to primary and secondary amines. As the N–H bond is restored at the end of the reaction, the corresponding α-C-alkylated primary or secondary amines are obtained as the final products.
 | ||
| Scheme 1 Hydroaminoalkylation of olefins using different amines and different catalysts. | ||
Ruthenium catalysts have been reported to mediate intermolecular hydroaminoalkylation with tertiary amines, but in all cases a pyridyl directing group (DG) was essential for reactivity (Scheme 1b).12 Catalytic hydroaminoalkylation of olefins with tertiary amines that do not contain an extra directing group has not been reported previously. This is most likely due to a lack of suitable catalysts which can not only effectively interact with an unactivated tertiary amine moiety but also promote C–H activation and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond insertion. The search for new catalysts for the hydroaminoalkylation of olefins with aliphatic tertiary amines is therefore of considerable importance.
C double bond insertion. The search for new catalysts for the hydroaminoalkylation of olefins with aliphatic tertiary amines is therefore of considerable importance.
We have recently shown that half sandwich mono-cationic rare-earth alkyl species act as efficient catalysts for various transformations, including olefin polymerization,13 the alkylation of sp2 and sp3 C–H bonds of aromatic compounds such as anisoles, pyridines and N,N-dimethylanilines,14 and the C–H polyaddition of 1,4-dimethoxybenzene to unconjugated dienes.15 These C–H functionalisation reactions were all achieved by the initial interaction between a rare-earth metal ion and a substrate heteroatom (such as O and N) followed by deprotonation of an ortho C–H bond with a metal alkyl species and the subsequent insertion of a CC double bond into the resulting M–C bond. This unique catalytic transformation could be ascribed to the strong heteroatom affinity of the rare-earth metal ions as well as to the high activity of the cationic rare-earth metal alkyl species towards both C–H activation and CC double bond insertion. These results encouraged us to examine whether rare-earth catalysts could promote the hydroaminoalkylation of olefins with tertiary amines. Herein we report the first intermolecular hydroaminoalkylation of various olefins with aliphatic tertiary amines using a cationic scandium alkyl catalyst (Scheme 1c).
Results and discussion
Catalyst screening
At first we examined the half-sandwich scandium dialkyl complex [(C5Me5)Sc(CH2C6H4NMe2-o)2]16a together with one equivalent of [Ph3C][B(C6F5)4] for the catalytic C–H addition of N,N-dimethylbutylamine (1a) to norbornene (2a) (Table 1, entry 1). Whilst this reaction was found to give the corresponding hydroaminoalkylation product 3a quantitatively after 24 h at 70 °C, initial attempts to expand the substrate scope to include styrene and 1-hexene were unsuccessful (Table 1, entries 3 and 5). In contrast, when the Cp-free trialkyl compound [Sc(CH2C6H4NMe2-o)3]16b was used instead, significant product formation was observed for both styrene and 1-hexene without a noticeable drop in catalytic activity for norbornene (Table 1, entries 2, 4 and 6).17 These reactions gave the corresponding methyl-functionalised products 3a, 4a′ and 5f′ with no other functionalised products or regioisomers observed by 1H NMR. A significant metal effect was observed for the hydroaminoalkylation of 2a to give 3a. When [Ln(CH2C6H4NMe2-o)3] (Ln = Y, Lu, Gd, Sm) was used as a pre-catalyst with one equivalent of [Ph3C][B(C6F5)4], no reaction was observed, highlighting the importance of a small, highly electropositive metal centre in the activation of tertiary amine methyl groups (Table S1,† entries 3–6). No reaction was seen when either [Sc(CH2C6H4NMe2-o)3] or [Ph3C][B(C6F5)4] alone was used as a catalyst, further highlighting the importance of a cationic scandium alkyl species in this reaction (Table S1,† entries 7 and 8).18 When two equivalents of borate were used in combination with [Sc(CH2C6H4NMe2-o)3], only trace reactivity was observed (Table S1,† entry 9).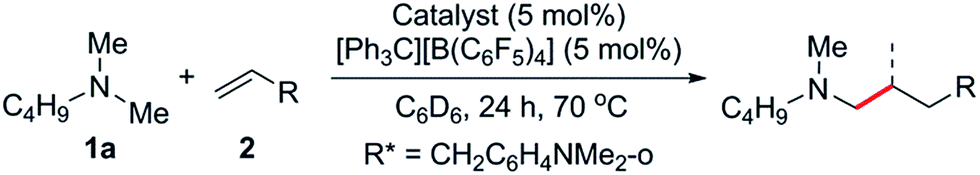
Hydroaminoalkylation of norbornene with various aliphatic amines
Based on these catalyst screening results, the [Sc(CH2C6H4NMe2-o)3]/[Ph3C][B(C6F5)4] combination was then chosen as a catalyst to investigate the C–H addition of various tertiary amines to norbornene. Some representative results are summarised in Table 2. Simple dimethylamine substrates such as N,N-dimethylbutylamine (1a), N,N-dimethyloctylamine (1b) and N,N-dimethylcyclohexylamine (1c) are efficient for this reaction, giving the corresponding N-methyl-alkylation products 3a–c in 87, 91 and 93% isolated yields, respectively (Table 2, entries 1–3). Acyclic monomethylamines such as N-methyl-N-ethylbutylamine (1d) and N-methyldibutylamine (1e) can also be used to give the corresponding products 3d and 3e in 88 and 86% yields, respectively (Table 2, entries 4 and 5).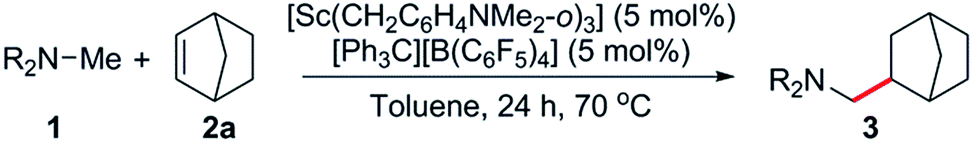
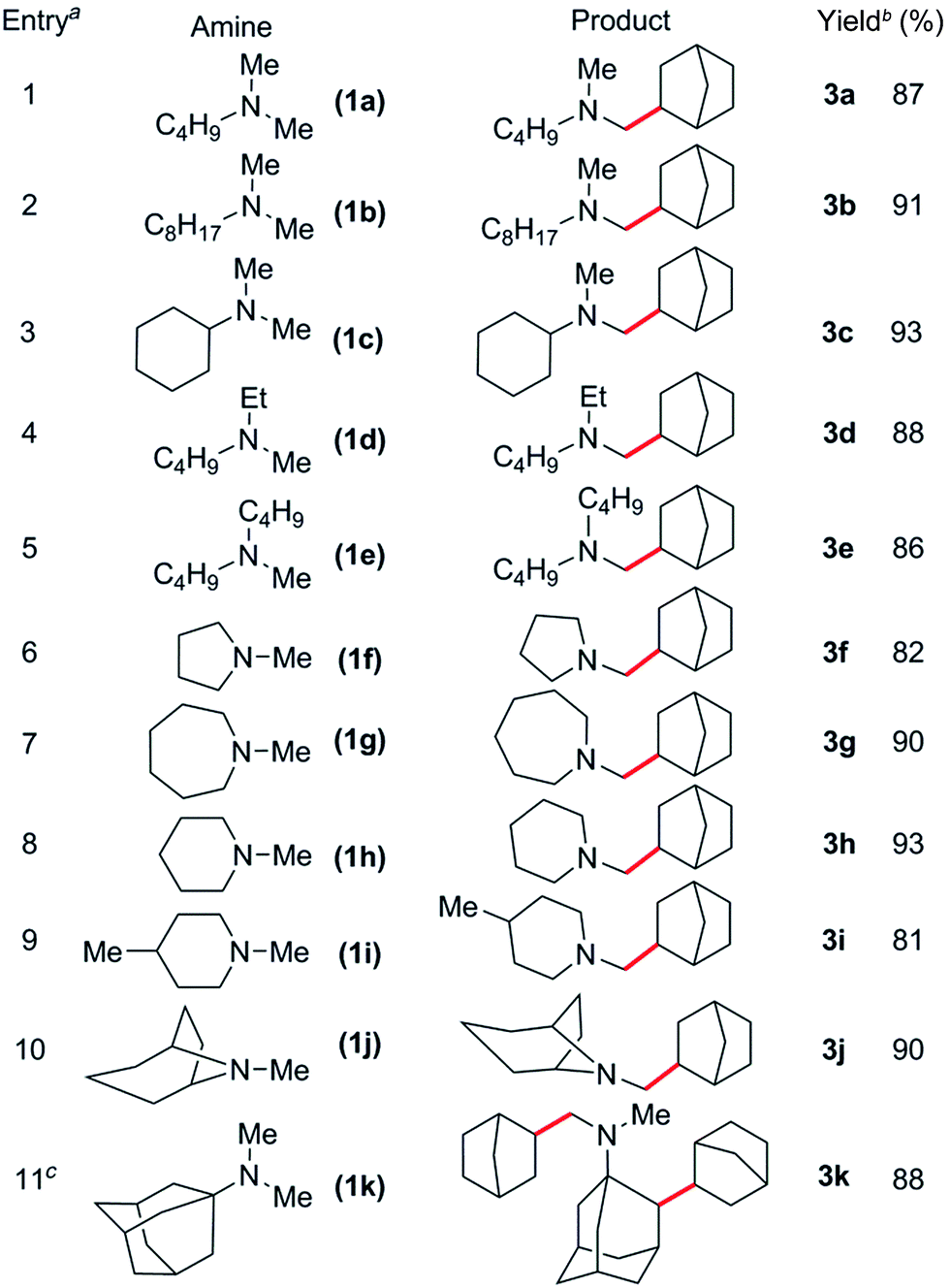
The reaction scope was also expanded to include cyclic methylamines, including the pharmacologically important tropane (1j), to give the products 3f–j in 81 to 93% yields (Table 2, entries 6–10). When N,N-dimethyladamantylamine (1k) was used as a substrate, clean conversion to the dialkylated species 3k was observed in the presence of excess olefin (4 equiv., Table 2, entry 11). Remarkably, subsequent alkylation was found to occur at the secondary β-C–H bond of the adamantyl moiety.19 This could be a result of intramolecular C–H activation of the β-C–H bond following initial alkylation at one amine methyl group (Scheme S1†). Attempts to expand the scope to include sterically congested amines such as N,N-dicyclohexylamine failed, presumably due to steric hindrance, whereas N,N-diethylmethylamine also gave no reaction, most likely due to strong coordination resulting from a lack of steric bulk.14c
Hydroaminoalkylation of various olefins with N-methylpiperidine
Based on these results, N-methylpiperidine (1h) was chosen to explore the olefin scope of this reaction and some representative results are summarised in Tables 3 and 4. The catalyst system [Sc(CH2C6H4NMe2-o)3]/[Ph3C][B(C6F5)4] was found to be very effective for the C–H addition of 1h to styrene (2b), giving exclusively the linear product 4a in 85% yield (Table 3, entry 1). Whilst this regioselectivity is common for related rare-earth catalysed C–H additions to styrenes,14 early transition metal catalysts regularly give branched,5g,7c,7e,11a linear7b or often mixtures of both isomers.6–11 Alkyl-substituted styrene derivatives such as 2c and 2d were also tolerated (Table 3, entries 2 and 3). In the case of the vinylaniline 2e, at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.1 ratio of 1h to 2e the polymerization of 2e was found to be competitive with hydroaminoalkylation.20 However, when an excess amount of 1h (5 equiv.) was used, the hydroaminoalkylation product 4d was obtained exclusively (89% yield, Table 3, entry 4).21 No alkylation was observed at the aniline methyl groups,22 and there was no evidence for ortho-alkylation of the aromatic ring.14g Styrene derivatives with para-electron-withdrawing groups such as fluoro, chloro and phenyl were also suitable for this reaction, though an increased catalyst loading was needed to obtain high yields (Table 3, entries 5–7). Oxygen containing substrates such as 4-methoxystyrene and 4-tert-butoxystyrene were incompatible with this catalyst system, likely due to the substantial oxophilicity of the sterically flexible cationic scandium centre. In addition to styrenes, vinyltrimethylsilane (2i) also gave the linear product 4h in 83% yield (Table 3, entry 8).23
:1.1 ratio of 1h to 2e the polymerization of 2e was found to be competitive with hydroaminoalkylation.20 However, when an excess amount of 1h (5 equiv.) was used, the hydroaminoalkylation product 4d was obtained exclusively (89% yield, Table 3, entry 4).21 No alkylation was observed at the aniline methyl groups,22 and there was no evidence for ortho-alkylation of the aromatic ring.14g Styrene derivatives with para-electron-withdrawing groups such as fluoro, chloro and phenyl were also suitable for this reaction, though an increased catalyst loading was needed to obtain high yields (Table 3, entries 5–7). Oxygen containing substrates such as 4-methoxystyrene and 4-tert-butoxystyrene were incompatible with this catalyst system, likely due to the substantial oxophilicity of the sterically flexible cationic scandium centre. In addition to styrenes, vinyltrimethylsilane (2i) also gave the linear product 4h in 83% yield (Table 3, entry 8).23
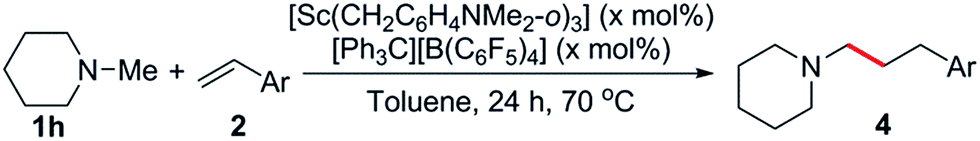
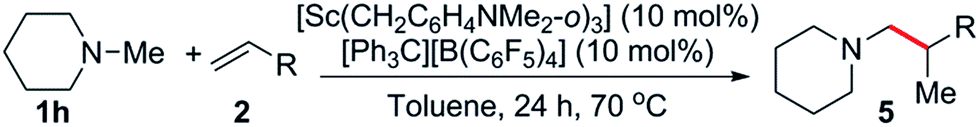
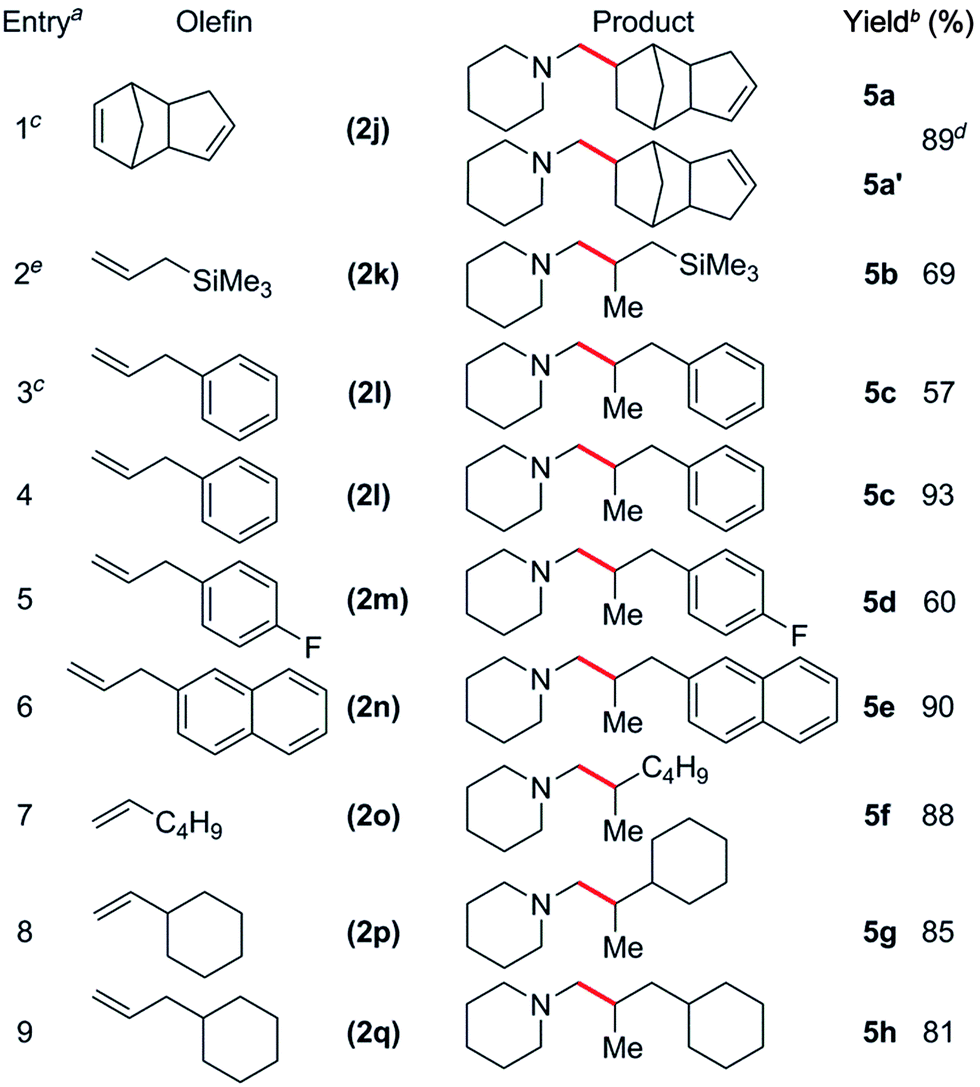
The reaction scope was also expanded to include alkyl substituted olefins, which gave exclusively branched products (Table 4). This is in sharp contrast to late transition metal systems which give linear alkylation products for both alkyl and aryl substituted olefins.12 When dicyclopentadiene (DCPD) was used as a substrate, the reaction occurred exclusively at the norbornene fragment, and a mixture of regioisomers was isolated in a ca. 1:3 5a:5a′ ratio in 89% yield (Table 4, entry 1); similar results have previously been observed in the polymerisation24 and hydropyridinylation14c of DCPD. A variety of allylic substrates were also tolerated, with allyltrimethylsilane giving 5b in 69% yield (Table 4, entry 2). When a 5 mol% catalyst loading was used for the reaction of 1h with allylbenzene (2l), the corresponding branched product (5c) was obtained in 57% yield and this could be improved to 93% yield by increasing the catalyst loading (Table 4, entries 3 and 4). The addition of a para-fluoro group had a significant effect on the yield of 5d (60%, Table 4, entry 5), whereas reaction with allylnaphthalene gave 5e in 90% yield. Unactivated, simple α-olefins such as 1-hexene (2o), vinylcyclohexene (2p) and allylcyclohexene (2q) all gave the corresponding products 5f, 5g and 5h in high yields (81–88%) despite the reactions being performed with a 1:1.1 amine:olefin ratio (Table 4, entries 7–9). This is in contrast to the alkylation of pyridines by Cp-supported rare-earth catalysts that often require a significant excess of olefin (10–30 equiv.) to give high yields.14 Attempts to expand the substrate scope to include ethylene and conjugated dienes such as isoprene and 1,3-cyclohexadiene resulted in the polymerization of the olefin monomers,25 with no consumption of N-methylpiperidine observed by 1H NMR spectroscopy. Attempts to favour hydroaminoalkylation by using an excess of amine (5 equiv., vide supra) failed for both the aforementioned dienes.
Possible mechanism of C–H functionalisation including isotopic labelling studies
The reaction of a 1:1:1 mixture of d3-N-methylpyrrolidine, N-methylpyrrolidine and styrene showed a significant kinetic isotope effect (kH/kD = 2.70) (eqn (1), Fig. S1†). A kinetic isotope effect was also observed when the initial rates of the separate C–H (eqn (2)) and C–D (eqn (3)) addition reactions were compared (kH/kD = 1.97), implying that C–H activation could be involved in the rate determining step in this reaction (Fig. S2†).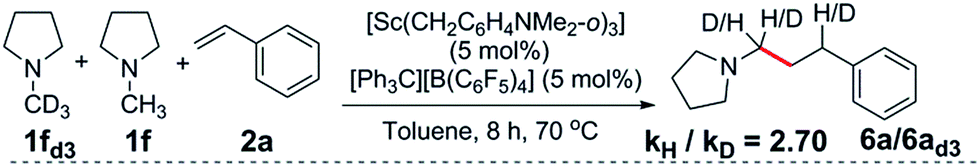 | (1) |
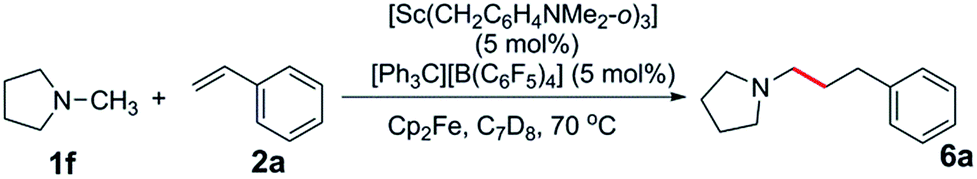 | (2) |
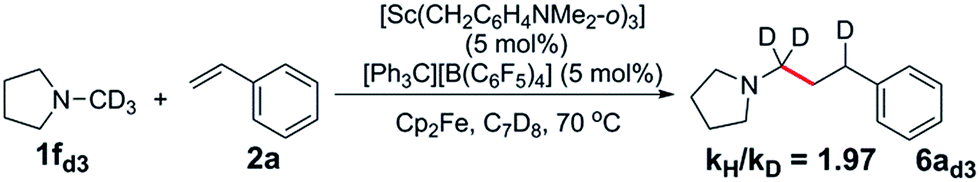 | (3) |
A plausible catalytic mechanism is shown in Scheme 2. The reaction of [Sc(CH2C6H4NMe2-o)3] with an equiv of [Ph3C][B(C6F5)4] would afford a mono-cationic scandium bis(aminobenzyl) species A, which upon coordination of the amine NMe2 group would form an adduct such as B.14g Deprotonation of an amine methyl group by an aminobenzyl ligand (R*) would then give an η2-azametallacyclic intermediate, C, along with release of one equivalent of N,N-dimethyl-o-toluidine. This highly strained species would then undergo 1,2-addition to an α-olefin to give the corresponding branched ring-expanded azametallocyclic intermediate, D. Subsequent C–H activation of another molecule of amine substrate would release the corresponding branched product and complete the catalytic cycle. When styrene derivatives are used as substrates, 2,1-insertion instead occurs, possibly due to stabilization of the Sc centre through interaction with the aromatic ring of E, which undergoes subsequent activation of a further amine equivalent to release the corresponding linear product.14
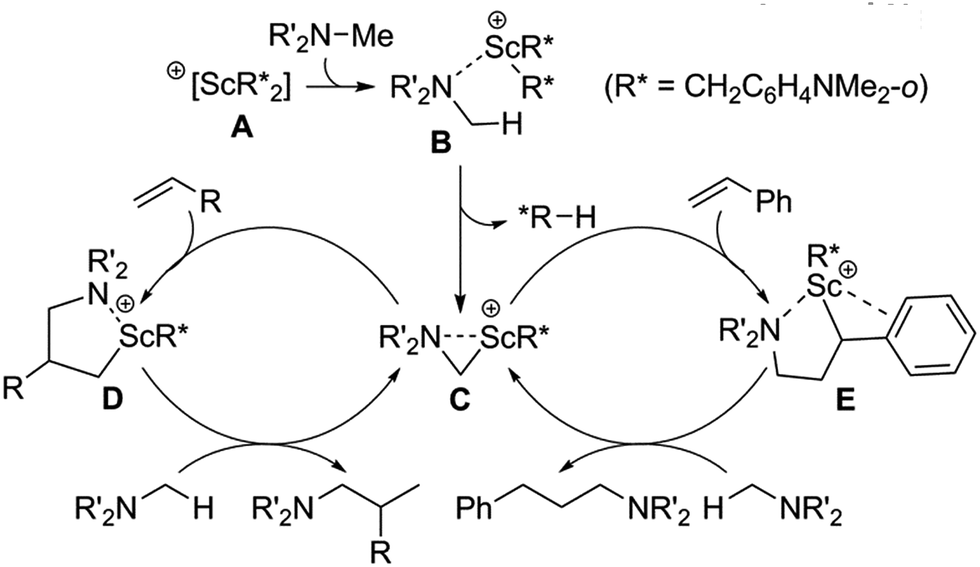 | ||
| Scheme 2 Plausible mechanisms of scandium-catalysed intermolecular hydroaminoalkylation of both α-olefins and styrenes. | ||
Conclusion
In summary, we have achieved the efficient and highly selective catalytic hydroaminoalkylation of various olefins with tertiary amines by using a homoleptic Sc alkyl pre-catalyst. This represents not only the first example of rare-earth catalysed olefin hydroaminoalkylation but also the first example of 100% atom efficient, intermolecular catalytic C–H alkylation of aliphatic tertiary amines with any catalyst. The reaction occurs between a wide variety of cyclic and acyclic amines and olefins, including functionalised styrenes and unactivated α-olefins, affording a new family of tertiary amines with diversified substituents. The C–H alkylation of a β-C–H bond was also observed, demonstrating that this chemistry may be extended to amine-directed functionalisation of remote C–H bonds. The success of these transformations is obviously due to the unique affinity and reactivity of cationic scandium alkyl species towards tertiary amino groups and C–H and CC bonds. Moreover, the observation that a homoleptic trialkyl scandium complex such as [Sc(CH2C6H4NMe2-o)3] can show superior performance to its Cp-ligated analogue [(C5Me5)Sc(CH2C6H4NMe2-o)2] for C–H functionalisation is also remarkable. These results suggest that cationic homoleptic rare earth alkyls, which have previously received little attention as catalysts for organic synthesis, may activate and functionalise C–H bonds that do not work with other catalysts. Further studies along this direction are currently in progress.
Acknowledgements
A. E. N. is grateful to JSPS London for a short-term postdoctoral fellowship, and RIKEN for a postdoctoral fellowship. This work was supported in part by a Grant-in-Aid for Scientific Research (S) (26220802) from JSPS.Notes and references
- Recent reviews on hydroamination of olefins: (a) L. Huang, M. Arndt, K. Gooßen, H. Heydt and L. Gooßen, Chem. Rev., 2015, 115, 2596 CrossRef CAS PubMed; (b) M. Willa and A. J. V. Wangelin, Angew. Chem., Int. Ed., 2015, 54, 11906 CrossRef PubMed; (c) M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972 RSC; (d) M. T. Pirnot, Y. M. Wang and S. L. Buchwald, Angew. Chem., Int. Ed., 2016, 55, 48 CrossRef CAS PubMed; (e) V. Rodriguez-Ruiz, R. Carlino, S. Bezzenine-Lafollée, R. Gil, D. Prim, E. Schulz and J. Hannedouche, Dalton Trans., 2015, 44, 12029 RSC.
- Recent reviews on hydroaminomethylation (hydroformylation and subsequent reductive amination) of olefins: (a) S. Raoufmoghaddam, Org. Biomol. Chem., 2014, 12, 7179 RSC; (b) X. F. Wu, X. Fang, L. Wu, R. Jackstell, H. Neumann and M. Beller, Acc. Chem. Res., 2014, 47, 1041 CrossRef CAS PubMed; (c) A. Behr and A. J. Vorholt, Top. Organomet. Chem., 2012, 39, 103 CrossRef CAS; (d) D. Crozet, M. Urrutigoity and P. Kalck, ChemCatChem, 2011, 3, 1102 CrossRef CAS; (e) J. L. Klinkenberg and J. F. Hartwig, Angew. Chem., Int. Ed., 2011, 50, 86 CrossRef CAS PubMed.
- Recent reviews on reductive amination of aldehydes: (a) M. O. Sydnes, Curr. Green Chem., 2016, 3, 101 CrossRef CAS; (b) H. Alinezhad, H. Yavari and F. Salehian, Curr. Org. Chem., 2015, 19, 1021 CrossRef CAS; (c) C. Wang and J. Xiao, Top. Curr. Chem., 2014, 343, 261 CrossRef CAS PubMed; (d) R. P. Tripathi, S. S. Verma, J. Pandey and V. K. Tiwari, Curr. Org. Chem., 2008, 12, 1093 CrossRef CAS.
- (a) K. R. Campos, Chem. Soc. Rev., 2007, 36, 1069 RSC; (b) E. A. Mitchell, A. Peschiulli, N. Lefevre, L. Meerpoel and B. U. W. Maes, Chem.–Eur. J., 2012, 18, 10092 CrossRef CAS PubMed.
- (a) M. G. Clerici and F. Maspero, Synthesis, 1980, 4, 305 CrossRef; (b) W. A. Nugent, D. W. Ovenall and S. J. Holmes, Organometallics, 1983, 2, 162 CrossRef; (c) S. B. Herzon and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 6690 CrossRef CAS PubMed; (d) S. B. Herzon and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 14940 CrossRef CAS PubMed; (e) R. Kubiak, I. Prochnow and S. Doye, Angew. Chem., Int. Ed., 2009, 48, 1153 CrossRef CAS PubMed; (f) T. Preuß, W. Saak and S. Doye, Chem.–Eur. J., 2013, 19, 3833 CrossRef PubMed; (g) Z. Zhang, J. Hamel and L. L. Schafer, Chem.–Eur. J., 2013, 19, 8751 CrossRef CAS PubMed; (h) J. Dörfler and S. Doye, Eur. J. Org. Chem., 2014, 2790 CrossRef.
- Recent reviews on hydroaminoalkylation of olefins: (a) P. W. Roesky, Angew. Chem., Int. Ed., 2009, 48, 4892 CrossRef CAS PubMed; (b) E. Chong, P. Garcia and L. L. Schafer, Synthesis, 2014, 46, 2884 CrossRef.
- (a) J. Dörfler and S. Doye, Angew. Chem., Int. Ed., 2013, 52, 1806 CrossRef PubMed; (b) J. Dörfler, T. Preuß, A. Schischko, M. Schmidtmann and S. Doye, Angew. Chem., Int. Ed., 2014, 53, 7918 CrossRef PubMed; (c) J. Dörfler, T. Preuß, C. Brahms, D. Scheuer and S. Doye, Dalton Trans., 2015, 44, 12149 RSC; (d) J. Dörfler, B. Bytyqi, S. Hüller, N. M. Mann, C. Brahms, M. Schmidtmann and S. Doye, Adv. Synth. Catal., 2015, 357, 2265 CrossRef; (e) J. Dörfler and S. Doye, Eur. J. Org. Chem., 2014, 2790 CrossRef.
- (a) E. Chong, J. W. Brandt and L. L. Schafer, J. Am. Chem. Soc., 2014, 136, 10898 CrossRef CAS PubMed; (b) E. Chong and L. L. Schaffer, Org. Lett., 2013, 15, 6002 CrossRef CAS PubMed.
- (a) P. Eisenberger, R. O. Ayinla, J. M. P. Lauzon and L. L. Schafer, Angew. Chem., Int. Ed., 2009, 48, 8361 CrossRef CAS PubMed; (b) P. R. Payne, P. Garcia, P. Eisenberger, J. C. H. Yim and L. L. Schafer, Org. Lett., 2013, 15, 2182 CrossRef CAS PubMed; (c) P. Eisenberger, R. O. Ayinla, J. M. P. Lauzon and L. L. Schafer, Angew. Chem., Int. Ed., 2009, 48, 8361 CrossRef CAS PubMed.
- (a) P. Garcia, Y. Y. Lau, M. R. Perry and L. L. Schafer, Angew. Chem., Int. Ed., 2013, 52, 9144 CrossRef CAS PubMed; (b) A. L. Reznichenko and K. C. Hultzsch, J. Am. Chem. Soc., 2012, 134, 3300 CrossRef CAS PubMed; (c) J. M. P. Lauzon and L. L. Schafer, Dalton Trans., 2012, 41, 11539 RSC; (d) I. Prochnow, P. Zark, T. Müller and S. Doye, Angew. Chem., Int. Ed., 2011, 50, 6401 CrossRef CAS PubMed.
- (a) A. L. Reznichenko and K. C. Hultzsch, J. Am. Chem. Soc., 2012, 134, 3300 CrossRef CAS PubMed; (b) J. M. P. Lauzon and L. L. Schafer, Dalton Trans., 2012, 41, 11539 RSC; (c) I. Prochnow, P. Zark, T. Müller and S. Doye, Angew. Chem., Int. Ed., 2011, 50, 6401 CrossRef CAS PubMed.
- (a) N. Chatani, T. Asaumi, S. Yorimitsu, T. Ikeda, F. Kakiuchi and S. Murai, J. Am. Chem. Soc., 2001, 123, 10935 CrossRef CAS PubMed; (b) S. D. Bergman, T. E. Storr, H. Prokopcová, K. Aelvoet, G. Diels, L. Meerpoel and B. U. W. Maes, Chem.–Eur. J., 2012, 18, 10393 CrossRef CAS PubMed; (c) M. Schinkel, L. Wang, K. Bielefeld and L. Ackermann, Org. Lett., 2014, 16, 1876 CrossRef CAS PubMed.
- (a) M. Nishiura and Z. Hou, Nat. Chem., 2010, 2, 257 CrossRef CAS PubMed; (b) M. Nishiura, F. Guo and Z. Hou, Acc. Chem. Res., 2015, 48, 2209 CrossRef CAS PubMed.
- (a) J. Oyamada and Z. Hou, Angew. Chem., Int. Ed., 2012, 51, 12828 CrossRef CAS PubMed; (b) B. T. Guan, B. Wang, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2013, 52, 4418 CrossRef CAS PubMed; (c) B. T. Guan and Z. Hou, J. Am. Chem. Soc., 2011, 133, 18086 CrossRef CAS PubMed; (d) G. Luo, Y. Luo, J. Qu and Z. Hou, Organometallics, 2012, 31, 3930 CrossRef CAS; (e) G. Song, W. N. O and Z. Hou, J. Am. Chem. Soc., 2014, 136, 12209 CrossRef CAS PubMed; (f) G. Song, B. Wang, M. Nishiura and Z. Hou, Chem.–Eur. J., 2015, 21, 8394 CrossRef CAS PubMed; (g) G. Song, G. Luo, J. Oyamada, Y. Luo and Z. Hou, Chem. Sci., 2016 10.1039/C6SC00833J; (h) A. Yamamoto, M. Nishiura, J. Oyamada, H. Koshino and Z. Hou, Macromolecules, 2016, 49, 2458 CrossRef CAS.
- X. Shi, M. Nishiura and Z. Hou, J. Am. Chem. Soc., 2016, 138, 6147 CrossRef CAS PubMed.
- (a) X. Li, M. Nishiura, K. Mori, T. Mashiko and Z. Hou, Chem. Commun., 2007, 4137 RSC; (b) L. E. Manzer, J. Am. Chem. Soc., 1978, 100, 8068 CrossRef CAS.
- This difference in reactivity could be attributed to a less hindered scandium coordination sphere due to the presence of flexible chelating ligands (compared to a rigid Cp* ligand).
- For examples of C–H activation using neutral rare earth alkyl complexes, see: (a) A. D. Sadow and T. D. Tilley, J. Am. Chem. Soc., 2003, 125, 7971 CrossRef CAS PubMed; (b) F. G. Fontaine and T. D. Tilley, Organometallics, 2005, 24, 4340 CrossRef CAS; (c) J. E. Bercaw, M. E. Thompson, S. M. Baxter, R. A. Bulls, B. J. Burger, M. C. Nolan, B. D. Santarsiero and W. P. Schaefer, J. Am. Chem. Soc., 1987, 109, 203 CrossRef; (d) H. Tsurugi, K. Yamamoto, H. Nagae, H. Kaneko and K. Mashima, Dalton Trans., 2014, 43, 2331 RSC.
- The attempted activation of secondary C–H bonds in substrates containing ethyl groups such as N-ethylpiperidine gave no reaction. This is likely due to steric congestion preventing the formation of the proposed highly strained 3 membered ring intermediate (Scheme 2, C).
- For the polymerization of 2e by a scandium catalyst, see: Z. Shi, F. Guo, Y. Li and Z. Hou, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 5 CrossRef CAS.
- The catalytic intermolecular hydroaminoalkylation of amine containing olefins has not been previously reported.
- Other aniline derived substrates such as N,N-dimethylaniline and N,N-dimethyl-o-toluidine do not add to norbornene under the same conditions.
- The preferred linear selectivity in C–H addition to vinyltrimethylsilane was also observed in previously related systems and has been proposed to be electronic in nature. See ref. 14a and S. Bi, Z. Lin and R. F. Jordan, Organometallics, 2004, 23, 4882 CrossRef CAS.
- X. Li and Z. Hou, Macromolecules, 2005, 38, 6767 CrossRef CAS.
- For examples of polymerization of ethylene, isoprene and 1,3-cyclohexadiene catalysed by cationic rare-earth alkyls, see: (a) S. Arndt, T. P. Spaniol and J. Okuda, Angew. Chem., Int. Ed., 2003, 42, 5075 CrossRef CAS PubMed; (b) B. D. Ward, S. Bellemin-Laponnaz and L. H. Gade, Angew. Chem., Int. Ed., 2005, 44, 1668 CrossRef CAS PubMed; (c) C. S. Tredget, F. Bonnet, A. R. Cowley and P. Mountford, Chem. Commun., 2005, 3301 RSC; (d) L. Zhang, T. Suzuki, Y. Luo, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2007, 46, 1909 CrossRef CAS PubMed; (e) X. Li, M. Nishiura, L. Hu, K. Mori and Z. Hou, J. Am. Chem. Soc., 2009, 131, 13870 CrossRef CAS PubMed; (f) D. Robert, E. Abinet, T. P. Spaniol and J. Okuda, Chem.–Eur. J., 2009, 15, 11937 CrossRef CAS PubMed; (g) X. Li and Z. Hou, Macromolecules, 2010, 43, 8904 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details for catalytic procedures and characterization data. See DOI: 10.1039/c6sc02129h |
| This journal is © The Royal Society of Chemistry 2016 |