
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible light mediated metal-free thiol–yne click reaction†
Sergey S.
Zalesskiy
,
Nikita S.
Shlapakov
and
Valentine P.
Ananikov
*
Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Leninsky prospekt, 47, Moscow, 119991, Russia. E-mail: val@ioc.ac.ru
First published on 12th July 2016
Abstract
The carbon–sulfur bond formation reaction is of paramount importance for functionalized materials design, as well as for biochemical applications. The use of expensive metal-based catalysts and the consequent contamination with trace metal impurities are challenging drawbacks of the existing methodologies. Here, we describe the first environmentally friendly metal-free photoredox pathway to the thiol–yne click reaction. Using Eosin Y as a cheap and readily available catalyst, C–S coupling products were obtained in high yields (up to 91%) and excellent selectivity (up to 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). A 3D-printed photoreactor was developed to create arrays of parallel reactions with temperature stabilization to improve the performance of the catalytic system.
:1). A 3D-printed photoreactor was developed to create arrays of parallel reactions with temperature stabilization to improve the performance of the catalytic system.
Introduction
In recent decades, the thiol–yne coupling was established as a powerful and proven tool for carbon–sulfur bond formation.1 Currently, this process is of particular importance for the preparation of polymers,2 dendrimers,3 smart materials1,4 and functionalized surfaces.1b,5 An important prerequisite for most of these applications is flexibility and efficient control with respect to reaction selectivity. Initially, the addition of thiols to alkynes was performed via a free-radical pathway.6 However, free-radical addition suffered from harsh conditions, low selectivity and extensive formation of by-products, which diminished the utility of this method in organic synthesis.6c The application of transitional metal catalysts improved the selectivity in favor of C–S bond formation;1,7 however, leaching of metal species8 and unavoidable contamination with biologically active metal-containing impurities makes the use of metal catalysts unacceptable for biomedical and pharma applications.We propose a practical approach to access valuable S-functionalized products using a cheap and readily available alkyne made by the condensation of acetylene with acetone via the Favorskii reaction (Scheme 1). An atom-economic thiol–yne click reaction yields sulfenylated alkenes, which can be readily converted to dienes by known dehydration protocols in one step (Scheme 1). Sulfenylated dienes are universal building blocks for organic synthesis and polymer science.9 Their high reactivity in Diels–Alder reactions facilitates cycloaddition under mild conditions.10,11 The overall process is totally atom-economic and environmentally benign; the target product is formed from acetylene, acetone and thiol, releasing only one molecule of water. The key step of the overall process is the thiol–yne click reaction, which should be performed with high stereo- and regioselectivity and without a metal catalyst. It is important to exclude transition metal catalysts at this step because their avoidance decreases cost and eliminates the generation of toxic wastes. Thus, carrying out this transformation under metal-free, visible light-mediated conditions would provide an important advance in the synthetic methodology.
 | ||
| Scheme 1 The key light-mediated thiol–yne click reaction for the synthesis of activated dienes from simple precursors. | ||
Herein, we present the first example of a metal-free photoredox thiol–yne click reaction. We have utilized the outstanding advantages of photocatalytic processes to render transformations of functionalized organic molecules.12 A special emphasis was placed on using organic catalysts13 to avoid metal contamination and to increase cost efficiency. To the best of our knowledge, no examples of metal-free photoredox thiol–yne coupling have been reported to date.14
Results and discussion
Initially, we evaluated the performance of various photoredox systems for the thiol–alkyne coupling (Table 1). Carrying out selective transformation is challenging due to a number of parallel processes leading to products 3–6 (Scheme 2). The formation of 4a was eliminated by avoiding an excess of oxygen in the reaction mixture. The addition of pyridine to the reaction mixture allowed us to increase selectivity via suppression of the formation of the double addition products 5a and 6a. The absence of transition metal complexes excluded the formation of another regioisomer 7a.1,7| # | Catalystb | Solvent | LED | 3a, % |
E:Z |
|---|---|---|---|---|---|
| a Conditions: 0.3 mol% catalyst loading, stirring under LED light for 4 h; see ESI for details. b Control experiments in the absence of photocatalyst were also performed; see ESI for details. | |||||
| 1 | [Ru(bpz)3][PF6]2 | MeCN | 465 nm | 34 | 20:1 |
| 2 | [Ru(bpy)3][PF6]2 | MeCN | 465 nm | 40 | 30:1 |
| 3 | Ir(ppy)3 | MeCN | 530 nm | 63 | 40:1 |
| 4 | Fluorescein | MeOH | 465 nm | 60 | 27:1 |
| 5 | Bengal rose | DMF | 530 nm | 53 | 25:1 |
| 6 | Eosin Y | MeOH | 530 nm | 53 | 25:1 |
| 7 | Eosin Y | DMF | 530 nm | 15 | 14:1 |
| 8 | Eosin Y | MeCN | 530 nm | 56 | 27:1 |
| 9 | Eosin Y | Et2O | 530 nm | 20 | 20:1 |
| 10 | Eosin Y | DMSO | 530 nm | 63 | 30:1 |
 | ||
| Scheme 2 Plausible products in the thiol–yne coupling process. | ||
Known ruthenium (entries 1 and 2, Table 1) and iridium-based (entry 3) photoredox systems gave good yields and E:Z selectivities of the desired product. Three different organic dyes demonstrated similar yields but afforded lower selectivity (entries 4–6, Table 1) relative to the Ir catalyst (entry 3, Table 1). Varying the solvent (entries 6–10, Table 1) allowed us to achieve 63% yield of 3a and 30:1 selectivity using Eosin Y as a photosensitizer (entry 10, Table 1). Although initial screening did not favor organic dyes as viable catalysts due to lower selectivities (cf. entries 3 and 10, Table 1), we developed this approach further to meet the requirements of cost-efficiency and avoiding metal contamination.
Variation of the amount of photocatalyst revealed a non-uniform trend (Table 2). Decreasing the amount of Eosin Y initially resulted in an increase in both the yield and selectivity (entries 1, 2 and 4, Table 2) but further lowering the amount of the dye led to a decrease in both parameters (entries 4 and 5, Table 2). It is very likely that the combination of two factors, namely excitation of only the surface layer of the solution due to increased optical density (because of the large amount of the dye in solution) and reaction stoppage due to degradation of Eosin Y (especially for very low dye loadings), is responsible for the observed behavior. Indeed, rapid degradation of the photocatalyst was clearly visible at low catalyst loadings, which resulted in discoloration of the solution.
| # | Eosin Y, mol% | Solvent | Yield, % |
E:Z |
|---|---|---|---|---|
| a The reaction conditions were similar to those in Table 1. b The reaction time was 5 h (longer reaction times did not further improve the yield). | ||||
| 1 | 2 | DMSO | 15 | 30:1 |
| 2 | 0.3 | DMSO | 38 | 30:1 |
| 3 | 0.3b | DMSO | 65 | 30:1 |
| 4 | 0.02 | DMSO | 46 | 45:1 |
| 5 | 0.001 | DMSO | 33 | 32:1 |
| 6 | 0.3 | Hexane | 70 | 37:1 |
| 7 | 0.3b | Hexane | 85 | 50:1 |
| 8 | 0.02 | Hexane | 11 | 10:1 |
| 9 | 0.001 | Hexane | 9 | 8:1 |
We found that the dye degradation issues can be overcome by switching the reaction solvent to hexane (Table 2, entries 6 and 7). Eosin Y exhibits very low solubility in hexane, and at the beginning of the reaction, the majority of the Eosin Y remains undissolved. A low concentration of the dissolved catalyst results in a reaction mixture with optimal optical density and ensures uniform excitation of the reaction volume.
The dissolved photocatalyst slowly degrades during the course of the reaction, which makes the solution unsaturated. This naturally leads to the dissolution of subsequent portions of Eosin Y from the solid phase. This “saturation feedback” automatically maintains the concentration of the photocatalyst at the optimal level, thus compensating for the loss of active species. Under such self-regulating conditions, yields and E:Z selectivities as high as 85% and 50:1, respectively, were achieved (entry 7, Table 2).
Thus, a typical photoredox system with Eosin Y suffers from gradual photocatalyst degradation during the course of the reaction when all of the catalyst is dissolved in the reaction mixture (Fig. S1, ESI†). This degradation can halt the progress of the reaction at the middle or even initial stages of the reaction (entries 8 and 9, Table 2). In the developed approach, the low solubility of the photocatalyst increases the efficiency of light utilization by maximizing reactive excitations. The inevitable degradation of the photocatalyst is immediately compensated by the undissolved portion (Fig. S1†). It should be noted that when identical catalyst loadings were used in different solvents, namely DMSO and hexane (i.e., 2.1 mg of Eosin Y for 0.3 mol%), the reaction was governed by the solubility of the photocatalyst. Notably, the reaction in hexane proceeded with higher selectivity and yield than the reaction conducted in DMSO (entries 3 and 7, Table 2).
After establishing the optimal reaction conditions, efforts were focused toward performing thiol–yne click reactions with different substrates. The developed synthetic approach provides excellent opportunities for incorporating various sulfur substituents (R) by using readily available thiols (Scheme 1). However, investigation of the substrate scope has shown that the process suffered from low reproducibility due to poor temperature control and variations in light intensity from one LED set up to another.
A closer examination revealed that experimental conditions (e.g., temperature and light intensity) can significantly vary depending on the particular setup. Commonly used LED strips wrapped around the photocatalytic system substantially heat the reaction vessel. The inner temperature of such an assembly can reach up to 60 °C, with large temperature variations of ±10 °C (Fig. 1A). Another standard setup involves placement of the reaction vessel on top of a single LED or LED matrix (Fig. 1B), which may increase the temperature up to 45 °C with temperature variations of ±5 °C. The optimal solution would provide irradiation of the reaction mixture from top, with the LED source incorporated into the vessel's cap (Fig. 1C). In such a setup, temperature stabilization and stirring can be easily performed using a standard oil or water bath equipped with a magnetic stirrer.
 | ||
| Fig. 1 Comparison of the possible designs of the photoreactor: (A) side irradiation (LED strip); (B) bottom irradiation (LED matrix); (C) developed design with temperature stabilization. | ||
Because LED sources incorporated into reaction vessel caps are not readily available, we designed a custom cap using 3D computer-aided modeling. The outstanding advantage of computer-aided modeling is the ability to virtually assemble all components of the setup together and to check for possible clearance issues and design pitfalls prior to manufacturing. We have developed a concept optimized for photoredox applications that is designed for compatibility with standard glass reaction vessels (Fig. 2A). The designed photoreactor was fabricated using a commonly available FDM 3D printer (Fig. 2B).
 | ||
| Fig. 2 Computer model of designed photoreactor (A) and ready to use assembled photoreactor (B). | ||
The reactor includes an efficient twist-lock LED mount that allows the LED light source to be changed easily. The bottom part can house a range of common glass vials available in the lab, and the presence of an O-ring ensures a tight fit with the reaction vessel. A detailed description of the reactor and the 3D model are provided in the ESI.† The developed model can be directly printed on a regular 3D printer in approximately 30 minutes and requires only 6 grams of the plastic source material (<$1 cost). The developed photoreactor was totally compatible with common magnetic stirrers and allowed for very good temperature stabilization at 25 ± 0.5 °C (Fig. 1C). The ability to maintain stable experimental conditions (e.g., temperature and light intensity) drastically improves the selectivity and reproducibility of photochemical reactions. An array of the custom-built photoreactors allowed us to execute a study of the substrate scope involving various substituents in a parallel fashion (Scheme 3). Four important advantages deserve particular attention: (i) the catalytic system showed excellent performance for various aryl thiols;9 (ii) excellent stereoselectivity in the range of 60:1–25:1 was achieved; (iii) complete regioselectivity was maintained and the formation of another regioisomer (7a) was not observed; and (iv) the hydroxyl group remained intact during the reaction, which is a necessary prerequisite to access the diene backbone after dehydration (Scheme 1). The developed synthetic approach was tolerant toward electronic and steric effects of the substituents in the thiol moiety (Scheme 3). Even for thiols containing strongly electron-withdrawing substituents, such as CF3 or COOMe, the desired products were formed in very high yield and high selectivity (i.e., 3e, 3g, 3h). We also tested aliphatic thiols in this reaction (R1 = Alk); however, significantly lower selectivity was observed (<10:1). An important factor is the structure of the alkyne, where steric effect played a key role in obtaining high selectivity (see ESI†). Amazingly, high selectivity was observed for the alkynes bearing α-substituted carbon atom, which is required to implement the synthetic methodology proposed in the present study (Scheme 1). For the studied class of alkynes the corresponding products 3a–3m were synthesized in good to high yields and excellent selectivity (Scheme 3) including sterically hindered alkynes, where the products 3n–3p were formed in good yields (89–96%) and high selectivity (35:1–40:1).
 | ||
| Scheme 3 The scope of the developed photocatalytic thiol–yne click reactiona. (a Isolated yields are given in parentheses, reaction time – 5 h. b Reaction time – 10 h. c Reaction time – 20 h). | ||
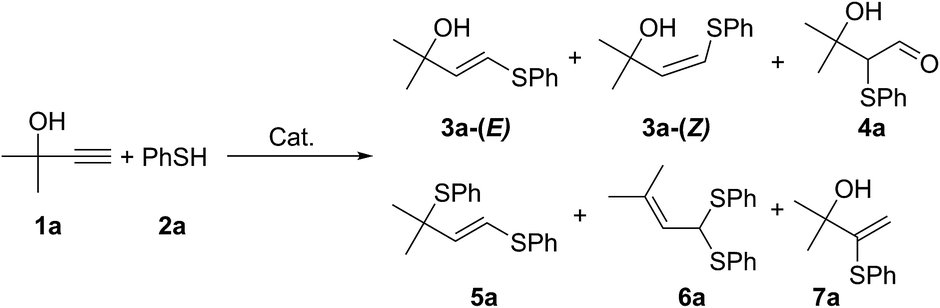
A plausible mechanism for the photoredox thiol–yne click reaction is presented in Scheme 4A.13,15 A photoexcited molecule of Eosin Y (E*) undergoes oxidative quenching, furnishing a radical cation of arylthiol. Pyridine abstracts a proton from this species, yielding thiyl radicals and preventing side reactions of ArSH˙+. In the next step, ArS˙ is involved in the radical addition to the alkyne, giving the desired product 3 and regenerating ArS˙ radicals. Within the mechanism the E-species of Eosin Y are regenerated upon interaction with molecular oxygen from ambient air. According to thermodynamic properties single electron oxidation of benzenethiol by excited state of Eosin Y is a feasible process: the potential EE*/E− = 1.18 V vs. SCE is much higher as compared to EPhSH/PhSH+ ≈ 0.95 V vs. SCE.16
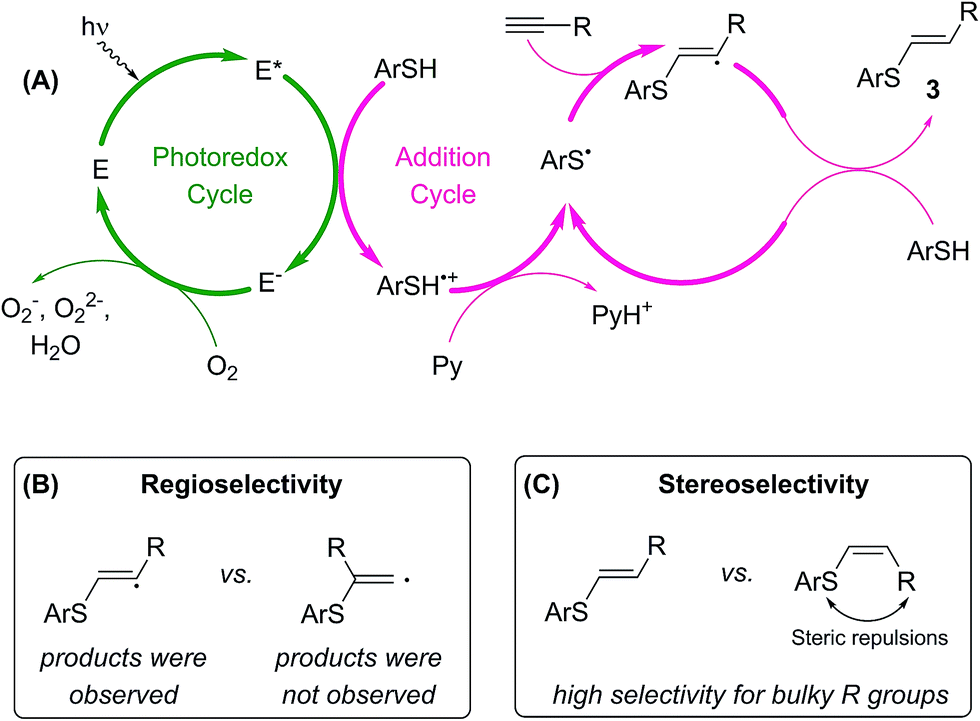 | ||
| Scheme 4 The proposed mechanism (A) for the Eosin Y mediated thiol–yne photoredox coupling (Eosin Y is denoted with E), plausible origins of reaction regioselectivity (B) and stereoselectivity (C). | ||
Rather different stability of the primary and secondary intermediate radical species governs high regioselectivity in the studied reaction (Scheme 4B). According to DFT calculations secondary radical is more stable by >8 kcal mol−1 compared to primary radical (see ESI†). In total agreement with relative stability of the radical species linear (anti-Markovnikov type) products 3 were formed in the reactions, whereas formation of branched (Markovnikov type) products was not observed.
Steric effect of the substituent R provides the necessary control for stereoselectivity in the studied transformation (Scheme 4C). Decreasing steric bulk resulted in lower reaction selectivity or non-selective transformation (see ESI†). The presence of OH group and two other substituents at the α-carbon atom in the developed concept (Scheme 1) perfectly matches the structure of the alkyne towards getting a high stereoselectivity.
Radical nature of the studied process was confirmed by performing a control experiment using a radical trap. Indeed, addition of γ-terpinene suppressed the reaction between the thiol and alkyne. Several other control experiments were also carried out to confirm the proposed mechanism and to demonstrate regeneration of the dye in the presence of oxygen (see ESI†).
Finally, as a proof of concept, we have prepared the target diene 8a using the industrially available starting materials acetylene, acetone and benzenethiol (Scheme 5). In the first step, alkyne 1a was prepared according to the published procedure.17 Alkyne 1a was converted to vinyl sulfide 3a using the developed photochemical protocol. Dehydration of 3a under mild conditions furnished diene 8a in 94% isolated yield. Thus, a demanding S-functionalized 1,3-diene was prepared starting from simple precursors through a sequence of atom-economic addition reactions with the only release of water (Scheme 5).
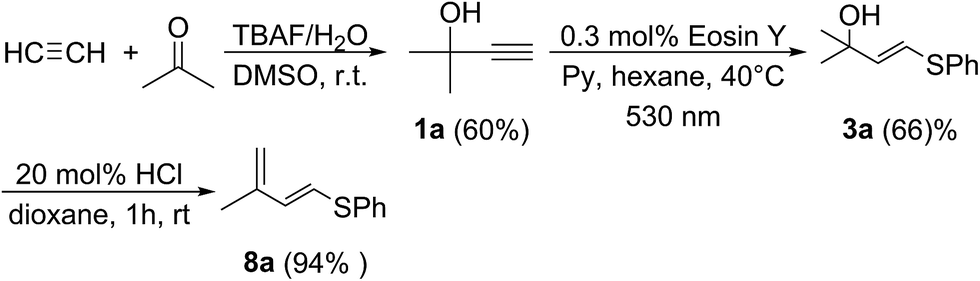 | ||
| Scheme 5 The synthesis of diene 8a using the developed approach. | ||
Conclusions
To summarize, the use of cheap and commercially available Eosin Y as a photocatalyst provided an attractive and environmentally friendly protocol for thiol–yne coupling, furnishing valuable sulfur-functionalized products in good yields. The developed catalytic system demonstrated unprecedented selectivities (up to 60:1) considering the broad scope of substituted aryl thiols that could be used. The use of a custom-designed 3D-printed chemical photoreactor facilitated the rapid assessment of optimal reaction conditions to improve the performance of the catalytic system.
An efficient photocatalytic system was created using a combination of Eosin Y and hexane as solvent. Utilization of the “saturation feedback” approach described here to maintain the optimal concentration of the catalyst may substantially facilitate development of a variety of other photocatalytic transformations.
Experimental details
General procedures
Photochemical reactions were carried in 2 mL glass vials covered with aluminium tape. Magnetic stirrer bars were cleaned with boiling solution of alkali followed by boiling solution of aqua regia and further rinsing with distilled water to ensure that all traces of absorbed catalyst were removed. All reagents and catalysts were purchased from commercial sources and used without further purification. Solvents were purificated by standard procedures. Flash chromatography was performed using Merck 60 μm silica. LC-MS-grade solvents (MeCN and DCM) for ESI-MS experiments were ordered from Merck and used as received. All samples (solutions in MeCN or DCM) for the ESI-MS experiments were prepared in 1.5 mL Eppendorf tubes. All plastic disposables (Eppendorf tubes and tips) used for sample preparation were washed with MeCN or DCM prior to use. 1H and 13C NMR data are referenced to TMS and residual solvent signal, respectively.Formation of double addition products was observed in a few studied cases at a trace level. Purification was easily performed using a regular flash chromatography as described below.
High-resolution mass spectra were recorded on a Bruker maXis Q-TOF instrument (Bruker Daltonik GmbH, Bremen, Germany) equipped with electrospray ionization (ESI) ion source. The measurements were performed in a positive (+MS; +MS/MS) ion mode (HV capillary: 4500 V; HV end plate offset: −500 V) with a scan range m/z: 50–3000. External calibration of the mass spectrometer was performed using an electrospray calibrant solution (Fluka). A direct syringe injection was used for all of the analyzed solutions in MeCN or DCM (flow rate: 3 μL min−1). Nitrogen was used as the nebulizer gas (0.4 bar), dry gas (4.0 L min−1) and collision gas for all of the MS/MS analyses and experiments; the dry temperature was set at 180 °C. All of the recorded spectra were processed using the Bruker DataAnalysis 4.0 software package.
Synthesis of 3a–p
Alkyne (1 mmol), thiol (1.1 mmol), Eosin Y (0.003 mmol), pyridine (0.25 mmol) and 250 μL of hexane were placed into a 2 mL glass vial (covered with aluminium tape) equipped with a stirrer bar. The mixture was suspended with ultrasound. The vial was placed on a magnetic stirrer and fitted with the developed photoreactor reactor equipped with green LED (λmax = 533 nm). After completing the reaction, the solvent was removed under reduced pressure. The remaining viscous bright pink oil was purified with flash chromatography (petroleum ether/ethyl acetate/triethylamine = 4:1:0.075 elution) to obtain a pure product.
Synthesis of 8a
Vinylsulfide 3a (10.6 mg, 5.45 × 10−2 mmol) was dissolved in 2 mL of 1,4-dioxane, followed by addition of HCl (1 μL of 38% aqueos solution). The reaction mixture was stirred at room temperature for 4 hours. The reaction mixture was diluted with 10 mL of dioxane and the solvent was removed on a rotary evaporator. The water bath temperature should not exceed 30 °C during evaporation to avoid product decomposition. Product 8a was isolated as a slightly yellow oil and identified according to the published NMR data.18 Product yield – 9.3 mg (94%).Acknowledgements
This work was supported by the Russian Science Foundation (RSF grant 14-13-01030). The authors thank Dr Evgeny Gordeev for helpful discussion.Notes and references
- (a) R. Hoogenboom, Angew. Chem., Int. Ed., 2010, 49, 3415 CrossRef CAS PubMed; (b) A. Ogawa, T. Ikeda, K. Kimura and T. Hirao, J. Am. Chem. Soc., 1999, 121, 5108 CrossRef CAS; (c) A. Massi and D. Nanni, Org. Biomol. Chem., 2012, 10, 3791 RSC; (d) A. B. Lowe, C. E. Hoyle and C. N. Bowman, J. Mater. Chem., 2010, 20, 4745 RSC; (e) P. Bichler and J. A. Love, Top. Organomet. Chem., 2010, 31, 39 CrossRef CAS; (f) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596 CrossRef CAS PubMed; (g) A. Ogawa, Top. Organomet. Chem., 2011, 43, 325 CrossRef; (h) A. Ishii and N. Nakata, Top. Organomet. Chem., 2011, 43, 21 CrossRef; (i) R. Castarlenas, A. Di Giuseppe, J. J. Perez-Torrente and L. A. Oro, Angew. Chem., Int. Ed., 2013, 52, 211 CrossRef CAS PubMed.
- (a) D. Konkolewicz, A. Gray-Weale and S. Perrier, J. Am. Chem. Soc., 2009, 131, 18075 CrossRef CAS PubMed; (b) B. Yao, J. Mei, J. Li, J. Wang, H. Wu, J. Z. Sun, A. Qin and B. Z. Tang, Macromolecules, 2014, 47, 1325 CrossRef CAS; (c) B. C. Yao, J. Z. Sun, A. J. Qin and B. Z. Tang, Chin. Sci. Bull., 2013, 58, 2711 CrossRef CAS; (d) B. D. Fairbanks, T. F. Scott, C. J. Kloxin, K. S. Anseth and C. N. Bowman, Macromolecules, 2009, 42, 211 CrossRef CAS PubMed.
- (a) C. Wendeln, S. Rinnen, C. Schulz, H. F. Arlinghaus and B. J. Ravoo, Langmuir, 2010, 26, 15966 CrossRef CAS PubMed; (b) G. Chen, J. Kumar, A. Gregory and M. H. Stenzel, Chem. Commun., 2009, 6291 RSC.
- (a) J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Am. Chem. Soc., 2009, 131, 5751 CrossRef CAS PubMed; (b) C. Y. Zhou, H. Wu and N. K. Devaraj, Chem. Sci., 2015, 6, 4365 RSC; (c) A. S. Quick, A. de los Santos Pereira, M. Bruns, T. Buckmann, C. Rodriguez-Emmenegger, M. Wegener and C. Barner-Kowollik, Adv. Funct. Mater., 2015, 25, 3735 CrossRef CAS; (d) V. T. Huynh, G. Chen, P. D. Souza and M. H. Stenzel, Biomacromolecules, 2011, 12, 1738 CrossRef CAS PubMed; (e) S. S. Naik, J. W. Chan, C. Comer, C. E. Hoyle and D. A. Savin, Polym. Chem., 2011, 2, 303 RSC.
- M. Guerrouache, S. Mahouche-Chergui, M. M. Chehimi and B. Carbonnier, Chem. Commun., 2012, 48, 7486 RSC.
- (a) L. Benati, L. Capella, P. C. Montevecchi and P. Spagnolo, J. Org. Chem., 1995, 60, 7941 CrossRef CAS; (b) D. Melandri, P. C. Montevecchi and M. L. Navacchia, Tetrahedron, 1999, 55, 12227 CrossRef CAS; (c) K. Griesbaum, Angew. Chem., Int. Ed., 1970, 9, 273 CrossRef CAS.
- (a) H. Kuniyasu, A. Ogawa, K. Sato, I. Ryu, N. Kambe and N. Sonoda, J. Am. Chem. Soc., 1992, 114, 5902 CrossRef CAS; (b) V. P. Ananikov, N. V. Orlov, I. P. Beletskaya, V. N. Khrustalev, M. Y. Antipin and T. V. Timofeeva, J. Am. Chem. Soc., 2007, 129, 7252 CrossRef CAS PubMed; (c) V. P. Ananikov, N. V. Orlov, S. S. Zalesskiy, I. P. Beletskaya, V. N. Khrustalev, K. Morokuma and D. G. Musaev, J. Am. Chem. Soc., 2012, 134, 6637 CrossRef CAS PubMed; (d) C. J. Weiss and T. J. Marks, J. Am. Chem. Soc., 2010, 132, 10533 CrossRef CAS PubMed; (e) C. J. Weiss, S. D. Wobser and T. J. Marks, Organometallics, 2010, 29, 6308 CrossRef CAS; (f) C. J. Weiss, S. D. Wobser and T. J. Marks, J. Am. Chem. Soc., 2009, 131, 2062 CrossRef CAS PubMed.
- (a) C. Deraedt and D. Astruc, Acc. Chem. Res., 2014, 47, 494 CrossRef CAS PubMed; (b) N. T. S. Phan, M. Van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609 CrossRef CAS; (c) I. Pryjomska-Ray, A. Gniewek, A. M. Trzeciak, J. J. Ziółkowski and W. Tylus, Top. Catal., 2006, 40, 173 CrossRef CAS; (d) R. H. Crabtree, Chem. Rev., 2012, 112, 1536 CrossRef CAS PubMed; (e) S. Hübner, J. G. de Vries and V. Farina, Adv. Synth. Catal., 2016, 358, 3 CrossRef; (f) A. S. Kashin and V. P. Ananikov, J. Org. Chem., 2013, 78, 11117 CrossRef CAS PubMed.
- Aryl derivatives (R = Ar) of sulfenylated dienes are particularly valuable due to their stronger S–Csp2 bond and have more reliable applications in the synthesis of sulfur-functionalized compounds. Alkyl derivatives (R = Alk) are less stable due to a relatively weaker S–Csp3 bond. For the difference in bond energy see: V. P. Ananikov, K. A. Gayduk, I. P. Beletskaya, V. N. Khrustalev and M. Y. Antipin, Chem.–Eur. J., 2008, 14, 2420 CrossRef CAS PubMed .
- (a) S. Danishefsky and T. Kitahara, J. Am. Chem. Soc., 1974, 96, 7807 CrossRef CAS; (b) J. Choi, H. Park, H. J. Yoo, S. Kim, E. J. Sorensen and C. Lee, J. Am. Chem. Soc., 2014, 136, 9918 CrossRef CAS PubMed; (c) S. Zhou, E. Sánchez-Larios and M. Gravel, J. Org. Chem., 2012, 77, 3576 CrossRef CAS PubMed; (d) J. M. Holmes, A. L. Albert and M. Gravel, J. Org. Chem., 2009, 74, 6406 CrossRef CAS PubMed; (e) S. A. Kozmin and V. H. Rawal, J. Org. Chem., 1997, 62, 5252 CrossRef CAS; (f) J. Barluenga, F. Aznar and M. Fernández, Tetrahedron Lett., 1995, 36, 6551 CrossRef CAS; (g) R. Gompper and U. Heinemann, Angew. Chem., Int. Ed., 1980, 19, 217 CrossRef.
- Representative reviews: (a) K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668 CrossRef CAS; (b) T. G. Back, K. N. Clary and D. Gao, Chem. Rev., 2010, 110, 4498 CrossRef CAS PubMed; (c) M. A. Tasdelen, Polym. Chem., 2011, 2, 2133 RSC; (d) M. M. Heravi and V. F. Vavsari, RSC Adv., 2015, 5, 50890 RSC; (e) A. C. Aragonès, N. L. Haworth, N. Darwish, S. Ciampi, N. J. Bloomfield, G. G. Wallace, I. Diez-Perez and M. L. Coote, Nature, 2016, 531, 88 CrossRef PubMed.
- Selected representative examples: (a) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437 CrossRef CAS PubMed; (b) M. N. Hopkinson, B. Sahoo, J. L. Li and F. Glorius, Chem.–Eur. J., 2014, 3874 CrossRef CAS PubMed; (c) J. Li, H. Wang, L. Liu and J. Sun, RSC Adv., 2014, 4, 49974 RSC; (d) M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951 CrossRef CAS PubMed; (e) S. P. Pitre, C. D. McTiernan, H. Ismaili and J. C. Scaiano, ACS Catal., 2014, 4, 2530 CrossRef CAS; (f) M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593 CrossRef CAS PubMed; (g) I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725 CrossRef CAS PubMed; (h) M. Cherevatskaya and B. König, Russ. Chem. Rev., 2014, 83, 183 CrossRef; (i) G. Palmisano, V. Augugliaro, M. Pagliaro and L. Palmisano, Chem. Commun., 2007, 3425 RSC; (j) R. Ciriminna, R. Delisi, Y.-J. Xu and M. Pagliaro, Org. Process Res. Dev., 2016, 20, 403 CrossRef CAS.
- (a) D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2014, 4, 355 CrossRef CAS; (b) D. Ravelli and M. Fagnoni, ChemCatChem, 2012, 4, 169 CrossRef CAS; (c) D. P. Hari and B. König, Org. Lett., 2011, 13, 3852 CrossRef CAS PubMed; (d) D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688 RSC.
- (a) E. L. Tyson, M. S. Ament and T. P. Yoon, J. Org. Chem., 2013, 78, 2046 CrossRef CAS PubMed; (b) W.-J. Yoo and S. Kobayashi, Green Chem., 2013, 15, 1844 RSC.
- (a) J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617 CrossRef CAS PubMed; (b) H. Görner, Photochem. Photobiol. Sci., 2008, 7, 371 RSC.
- (a) D. Ravelli and M. Fagnoni, ChemCatChem, 2012, 4, 169 CrossRef CAS; (b) F. G. Bordwell, X.-M. Zhang, A. V. Satish and J.-P. Cheng, J. Am. Chem. Soc., 1994, 116, 6605 CrossRef CAS.
- A. Hosseini, D. Seidel, A. Miska and P. R. Schreiner, Org. Lett., 2015, 17, 2808 CrossRef CAS PubMed.
- P. G. Jr Guerrero, M. J. Dabdoub, F. A. Marques, C. L. Wosch, A. C. M. Baroni and A. G. Ferreira, Synth. Commun., 2008, 38(24), 4379 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc02132h |
| This journal is © The Royal Society of Chemistry 2016 |