
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrocatalytic amplification of DNA-modified nanoparticle collisions via enzymatic digestion†
Alma D.
Castañeda
,
Donald A.
Robinson
,
Keith J.
Stevenson‡
and
Richard M.
Crooks
*
Department of Chemistry, Center for Electrochemistry, and the Center for Nano- and Molecular Science and Technology, The University of Texas at Austin, 105 E. 24th St., Stop A5300, Austin, TX 78712-1224, USA. E-mail: crooks@cm.utexas.edu; Tel: +1-512-475-8674
First published on 1st July 2016
Abstract
We report a new and general approach that will be useful for adapting the method of electrocatalytic amplification (ECA) to biosensing applications. In ECA, individual collisions of catalytic nanoparticles with a noncatalytic electrode surface lead to bursts of current. In the work described here, the current arises from catalytic electrooxidation of N2H4 at the surface of platinum nanoparticles (PtNPs). The problem with using ECA for biosensing applications heretofore, is that it is necessary to immobilize a receptor, such as DNA (as in the case here) or an antibody on the PtNP surface. This inactivates the colliding NP, however, and leads to very small collision signatures. In the present article, we show that single-stranded DNA (ssDNA) present on the PtNP surface can be detected by selectively removing a fraction of the ssDNA using the enzyme Exonuclease I (Exo I). About half of the current associated with collisions of naked PtNPs can be recovered from fully passivated PtNPs after exposure to Exo I. Experiments carried out using both Au and Hg ultramicroelectrodes reveal some mechanistic aspects of the collision process before and after treatment of the ssDNA-modified PtNPs with Exo I.
Introduction
In this article we report a new and general approach that will be useful for adapting the method of electrocatalytic amplification (ECA) to biosensing applications.1 As shown in Scheme 1a, the original version of ECA occurs when a catalytically active nanoparticle (NP) strikes an appropriately poised inert electrode in the presence of a suitable redox molecule. In this case, significant current flow, arising from a redox indicator reaction, is only observed when the particle is in contact with the electrode. Therefore, it is possible to distinguish individual collisions between nanoparticles and the electrode surface.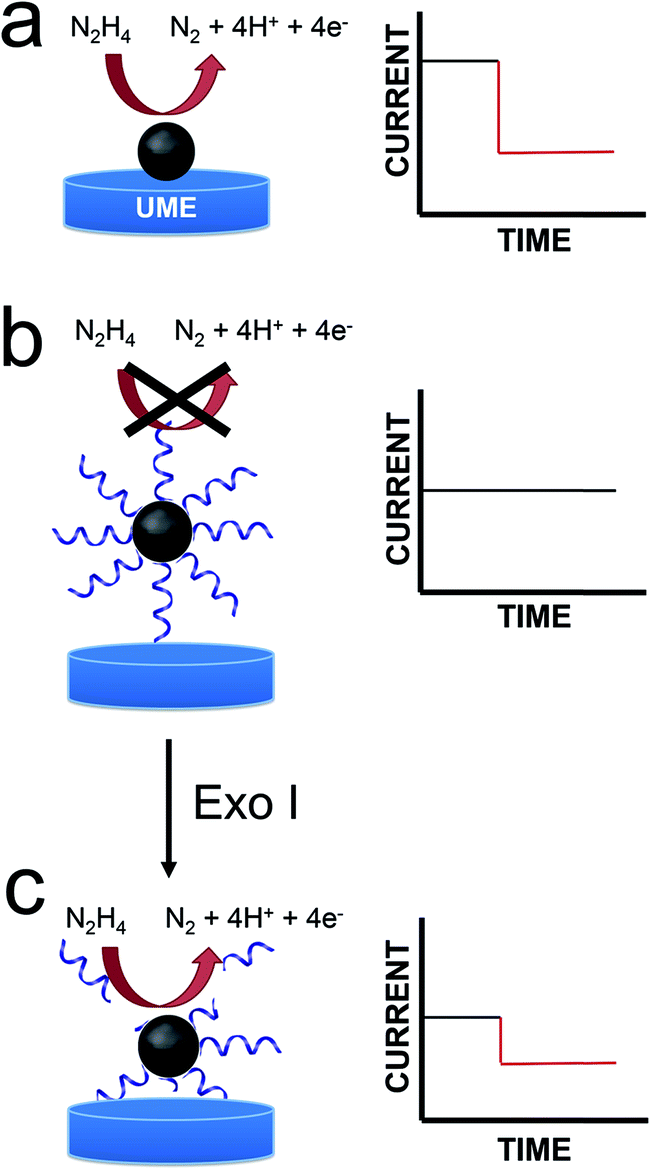 | ||
| Scheme 1 | ||
We are interested in using ECA to detect small numbers of biological molecules such as DNA. Previous work from our lab has shown, however, that when DNA is present on colliding NPs, very little current results.2 This is, of course, a consequence of DNA-induced blocking of catalytically active sites on the NP surface. This finding presents a clear problem for integration of ECA into biosensing schemes: specifically, if a biological recognition element is immobilized on the NPs, then little or no current will be observed upon impact. Here we present a strategy that at least partially addresses this difficulty. Specifically, we start with catalytically inactive DNA-modified NPs, and then detect the presence of DNA using an enzyme (an exonuclease) that degrades DNA sufficiently to reactivate the catalytic properties of the NPs. Importantly, the goal of the present work is to introduce a general methodology that could be useful for a range of future biosensing applications. At this early stage in our research, we do not intend to suggest that the metrics presented here are competitive with the many other DNA sensing methods that have been reported.
There are a number of different experiments that fall into the general category of detection of collisions between particles and an electrode. The first of these3–5 was reported by Scholz and coworkers who began a study of collisions between liposomes and Hg electrodes in 2002. Two years later Lemay and coworkers observed collisions between individual micron-scale, carboxylated latex spheres and an electrode surface arising from partial masking of the electroactive area of the electrode by the insulating spheres.6 Compton and coworkers used a different approach, in which current pulses resulted from oxidation of AgNPs when they struck an electrode surface.7,8 As mentioned earlier, ECA occurs when a colliding NP initiates a catalytic reaction. Two forms of this collision method have been described. In the first reports of ECA, the NPs were tethered to an electrode surface, but this did not result in observation of individual collisions.9,10 In 2007, however, Xiao and Bard showed that when the catalytic NPs were free in solution, individual ECA collisions could be discerned.11 As discussed next, the experiment reported here is most similar to the latter approach. Finally, we note that the research groups of Unwin,12,13 Zhang,14,15 Koper,12,16 Andreescu,17 Alpuche-Aviles,18 Crooks,2,19–21 Stevenson,22–24 and MacPherson25 have all made important contributions to the general field of single-particle collision electrochemistry that have influenced the findings reported here.
For the present study, we chose N2H4 oxidation (eqn (1)) as the redox indicator reaction, because this inner-sphere electron-transfer reaction is catalyzed by PtNPs but not by Au or Hg ultramicroelectrodes (UMEs).
| N2H4 = N2 + 4H+ +4e− | (1) |
Accordingly, when an appropriate interfacial potential is selected for the UME, no current due to eqn (1) is observed until a PtNP strikes the electrode surface. The current transients that result from these collisions are step-shaped for Au UMEs26 and spike-shaped for Hg UMEs.22–24 The rapid current decrease observed for Hg UMEs is due to deactivation of the PtNPs resulting from Hg poisoning.
Enzymes have heretofore not been integrated into ECA sensing schemes, but we were inspired by the surface-enzyme chemistry reported by Corn and coworkers27,28 over the past several years, and we thought coupling the two methods could be quite powerful. This first report focuses on the use of nucleases, which are a family of enzymes capable of hydrolyzing the phosphodiester bonds in DNA chains.29 The difference between endo- and exonucleases lies in the way each class initiates hydrolysis. Endonucleases cleave phosphodiester bonds at a specific site within the middle section of the oligonucleotide, while exonucleases initiate cleavage at a free –OH group on either the 5′ or 3′ end.30 The 5′ to 3′ exonucleases are commonly used in biology to remove RNA primers,31,32 whereas 3′ to 5′ exonucleases are used to help repair DNA mismatches.33 A number of analytical assays incorporate the use of nucleases for operating on nanoparticles conjugated with DNA. These include schemes involving detection via colorimetry,34,35 fluorescence,36 and electrochemistry.37 For this study, we used Exonuclease I (Exo I), a nuclease extracted from E. coli, which is selective for denatured or single-strand DNA (ssDNA). Exo I initiates cleavage at a free 3′-hydroxyl end of ssDNA and cuts nucleotides in a stepwise fashion.38
In the present work, we modified 22 nm-diameter PtNPs with 25-mer ssDNA. Consistent with an earlier report from our group,2 these PtNP@ssDNA conjugates yield few collision signals (Scheme 1b), because DNA restricts access of N2H4 to the electrocatalytic PtNP surface. After incubation of PtNP@ssDNA with 30 U of Exo I, however, about 50% of the collision activity (compared to naked PtNPs) returns (Scheme 1c). This is because Exo I removes much of the ssDNA originally present on the PtNPs. The key result is that these findings point to a general approach for using ECA to detect small molecules, proteins, and DNA.
Experimental section
Chemicals and materials
MgCl2, (NH4)2SO4, 2-mercaptoethanol, Tris–HCl, ZnCl2, glycerol, L-ascorbic acid, citric acid, NaBH4, and N2H4·H2O were purchased from Sigma-Aldrich (St. Louis, MO). Sodium citrate and sodium phosphate monohydrate were purchased from EM Science (Billerica, MA). H2PtCl6·6H2O (99.9%) was purchased from Strem Chemicals (Newburyport, MA). Tween 20 and NaCl were obtained from Fisher Scientific (Pittsburgh, PA). All reagents were used as received. The DNA conjugates, ssDNA (5′–(CH2)3–SH CAC GAC GTT GTA AAA CGA CGG CCA G-3′) and Cy3–ssDNA (5′–(CH2)3–SH CAC GAC GTT GTA AAA CGA CGG CCA G-Cy3-3′), were from Integrated DNA Technologies (Coralville, IA) and received as lyophilized pellets in microcentrifuge tubes. The pellets were centrifuged to ensure no residue on the walls or cap remained, and then suspended in H2O. Deionized (DI) water having a resistivity of 18.2 MΩ cm was used for all experiments (Milli-Q gradient water purification system, Millipore, Bedford, MA). Experiments were conducted at room temperature (23 ± 2 °C). Unless otherwise stated, the phosphate buffer was adjusted to pH 7.Synthesis and characterization of PtNPs
PtNPs were prepared following a previously reported seed-mediated synthesis.39 Briefly, 7.76 mL of a 0.2% (w/v) solution of H2PtCl6 were added to 100 mL of gently boiling H2O. After 1.0 min, 2.37 mL of a solution containing sodium citrate (1%, w/v) and citric acid (0.05%, w/v) were added and the solution was allowed to boil for an additional 30 s. Next, 1.18 mL of a solution containing NaBH4 (0.08%, w/v), sodium citrate (1%, w/v), and citric acid (0.05%, w/v) were added, and boiling was continued for 10 min. After cooling to room temperature, 3–4 nm Pt seed NPs were obtained.A 1.0 mL aliquot of the PtNP seed solution was added to 29.0 mL of H2O at room temperature. With stirring, 0.023 mL of a 0.40 M H2PtCl6 solution and 0.50 mL of a solution containing 1% sodium citrate and 1.25% L-ascorbic acid was added. The solution was then heated to boiling at the rate of 10 °C min−1. The total reaction time was 30 min. After cooling to room temperature, the solution was transferred to a 35 mm dialysis sack (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da MWCO, Sigma-Aldrich) and submerged in 4 L of DI H2O for 24 h to remove excess salts. The PtNPs were characterized by transmission electron microscopy (TEM, FEI Tecnai TEM), and found to have an average diameter of 22 ± 4 nm. A representative TEM image and a histogram showing the NP size distribution are provided in the ESI (Fig. S1†).
000 Da MWCO, Sigma-Aldrich) and submerged in 4 L of DI H2O for 24 h to remove excess salts. The PtNPs were characterized by transmission electron microscopy (TEM, FEI Tecnai TEM), and found to have an average diameter of 22 ± 4 nm. A representative TEM image and a histogram showing the NP size distribution are provided in the ESI (Fig. S1†).
Preparation of PtNP@ssDNA conjugates
PtNP@ssDNA conjugates were prepared using a modified version of a pH-assisted conjugation method previously reported for AuNPs and AgNPs.40,41 Briefly, 400 μL of the 570 pM PtNP solution were mixed with 5.0 μL of 10.0 μM ssDNA (this yields a PtNP:ssDNA ratio of ∼1:220). After 5 min, 25.0 μL of 100 mM citrate–HCl buffer (pH 3) were injected and mixed into the solution. After 5 min, an additional 25.0 μL of citrate–HCl buffer were added. After 25 min at room temperature, 400 μL of 100 mM HEPES buffer (pH 7.4) were added, and the solution was vortexed for ∼5 s. The conjugates were then centrifuged at 16.6 G for 20 min, washed three times with DI water, and resuspended in Taq buffer (75 mM Tris–HCl (pH 8.8), 2 mM MgCl2, 0.01% Tween 20, and 20 mM (NH4)2SO4). For electrochemical measurements, a few tens of microliters of this resuspended solution was diluted in 5–10 mL of PB (pH 7) solution, so the final electrolyte solution contained a small percentage of Taq buffer.
These materials were used for all experiments reported herein, except for those relating to Fig. 2 where lower ratios of PtNP:ssDNA were used to gauge the effect of ssDNA surface concentration on ECA measurements. In those experiments the PtNP@ssDNA were resuspended in 50 mM PB (pH 7) rather than Taq buffer.
Enzymatic digestion of PtNP@ssDNA
30 U of Exo I were combined with 400 μL of the PtNP@ssDNA solution in Taq buffer and immediately vortexed for 5 s. Subsequently, the solution was incubated at 37 °C for 1 h with gentle mixing at 200 rpm before immediate use.UME preparation
Au UMEs having a diameter of 12.5 μm and Pt UMEs having a diameter of 10 μm were purchased from CH Instruments (Austin, TX). Au UMEs were polished by wet sanding for 1 min. The Au UMEs were then submerged in piranha solution (1:3 30% H2O2/H2SO4) Caution!Piranha solution can react violently with organic compounds and should be handled with care. Next, they were electrochemically cleaned by immersing them in 0.1 M H2SO4 and cycling the applied potential between −0.35 and 1.35 V vs. Ag/AgCl (3.4 M KCl) for 25 cycles at 0.30 V s−1 (potentiostat model CH 700D, CH Instruments, Austin, TX). Before use, they were rinsed with H2O and dried under a gentle stream of N2.
Hg UMEs were prepared by electrodepositing Hg onto a Pt UME according to a previously reported method.42 Briefly, the Pt UMEs were polished via wet sanding for 1 min. Hg was then electrodeposited (−0.10 V vs. Ag/AgCl, 3.4 M KCl) for 300 s from a solution containing 5.7 mM Hg2(NO3)2, 0.5% conc. HNO3, and 1 M KNO3. Finally, the Hg UMEs were rinsed with H2O immediately before use.
Electrochemistry
Chronoamperometric (i–t) curves were obtained in a two-electrode cell using a Chem-Clamp voltammeter-amperometer (Dagan Corp., Minneapolis, MN) potentiostat coupled to a PAR 175 Universal Function Generator (Princeton Applied Research, Oak Ridge, TN) to apply the voltage. The potentiostat and function generator were interfaced to a Dell Optiplex 380 computer through a PCI-6251 data acquisition board (National Instruments, Austin, TX) using a BNC-2090A analog breakout accessory (National Instruments, Austin, TX). The electrochemistry data were obtained using a custom LabView program (National Instruments). The sampling time was 0.015 s. All electrochemistry experiments were shielded from environmental noise using a custom Faraday cage constructed from copper plate and mesh. For i–t experiments, the potential was held at −50 mV and 5 mV vs. Ag/AgCl for the Au and Hg UME experiments, respectively. Unless stated otherwise, all potentials are reported vs. a Ag/AgCl reference electrode (3.4 M KCl, model CH111, CH Instruments, Austin, TX) separated from the working electrode by a glass frit to minimize contamination.Nanoparticle tracking analysis
The colloidal stability of PtNPs, PtNP@ssDNA, and PtNP@ssDNA post-Exo I was analyzed using nanoparticle tracking analysis (NTA, Malvern Instruments, Malvern, United Kingdom). NTA is a technique that utilizes light scattering and particle tracking to measure the size distribution and concentration of particles in a solution. Due to the narrow working concentration range of the instrument (<1 pM), NP solutions were diluted to a final concentration of 0.57 pM for PtNPs, 0.89 pM for PtNP@ssDNA, and 0.46 pM for PtNP@ssDNA post-Exo I.Results and discussion
Synthesis and characterization of PtNP@ssDNA
The PtNP@ssDNA conjugates were prepared using the fast, pH-assisted conjugation method discussed in the Experimental section. This procedure results in colloidally stable conjugates having a 22 nm-diameter Pt core and a ssDNA shell in under 1 h.40To estimate the average number of ssDNAs per PtNP, a previously reported technique, which is based on fluorescence quenching by the PtNPs, was used.40,43 For this purpose, the thiolated ssDNA was modified at the 3′ end with the fluorescent dye Cy3 (ssDNA–Cy3), which has a maximum absorption wavelength at 550 nm and a maximum emission wavelength at 570 nm. Adsorption of ssDNA–Cy3 to the PtNP brings the dye sufficiently close to the NP surface to quench its fluorescence. Therefore, after modification of the PtNPs with Cy3-tagged ssDNA, residual fluorescence in solution will arise primarily from unbound ssDNA.
To carry out this analysis we first recorded a calibration curve for ssDNA–Cy3 in Taq buffer (Fig. S2, ESI†), and then measured the fluorescence of solutions containing PtNP@ssDNA–Cy3. By comparison of the residual fluorescence in the latter solution to the calibration curve, the concentration of bound DNA can be determined. Dividing this value by the concentration of PtNPs (determined by NTA) yields a rough estimate of the number of ssDNA–Cy3 per PtNP, which we find to be ∼35. The conditions used to modify PtNPs with unlabeled DNA (e.g., PtNP@ssDNA) were the same as for PtNP@ssDNA–Cy3 to ensure similar coverages.
The poor colloidal stability of nanoparticles in ECA experiments is a very serious problem,16,44,45 and therefore we evaluated this parameter for the PtNP conjugates used in this study prior to carrying out collision experiments. These NTA results (Fig. 1a) indicate a size distribution of 46 ± 27 nm for nominally naked PtNPs (black trace) in 50 mM PB. This value can be compared to the size measured in water (no buffer): 28 ± 20 nm, indicating that the electrostatic shield effect of the buffer leads to some aggregation.46,47 After modification with ssDNA (PtNP@ssDNA), the size of the resulting conjugates in 50 mM PB is a little larger than the nominally naked PtNPs: 57 ± 31 nm (red trace) and 53 ± 29 nm (blue trace) before and after treatment with Exo I, respectively. Note that the aforementioned size distributions are the averages of three independent measurements.
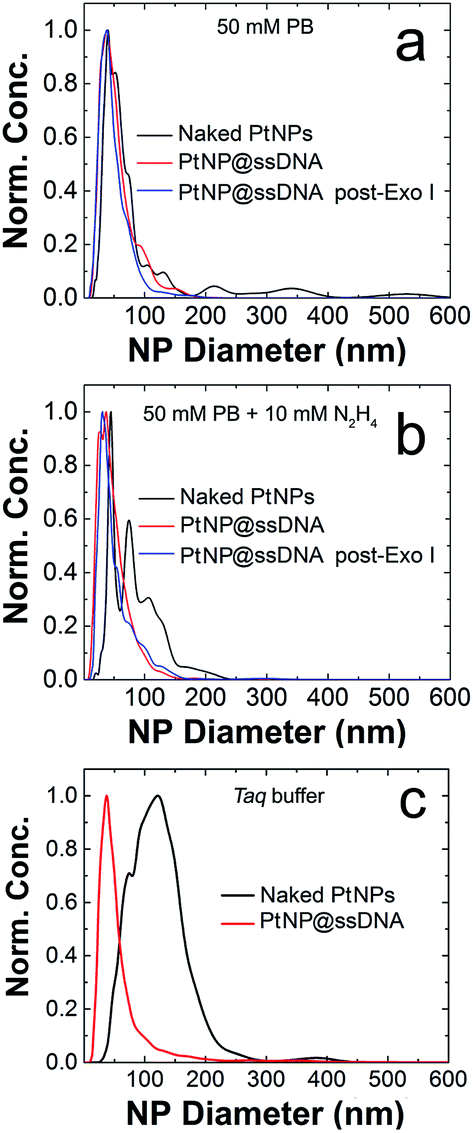 | ||
| Fig. 1 Representative size-distribution histograms, derived from nanoparticle tracking analysis, for the PtNPs used in this study. The types of PtNPs corresponding to each histogram are shown in the legend. The concentrations of PtNPs were: naked PtNPs, 0.57 pM; PtNP@ssDNA, 0.89 pM; and PtNP@ssDNA post-Exo I, 0.46 pM. Measurements were obtained in (a) 50 mM phosphate buffer (PB, pH 7), (b) 50 mM PB (pH 7) + 10 mM N2H4, and (c) Taq buffer. To account for the differences in NP concentration, the histograms are normalized to the highest concentration observed for each species. | ||
It has previously been shown that the presence of N2H4 can increase the degree of aggregation of PtNPs,16 and therefore we repeated the measurements shown in Fig. 1a except included 10.0 mM N2H4 in the solutions (Fig. 1b). The size distribution for naked PtNPs (black trace) in the presence of N2H4 confirms the earlier report. In this case the average PtNP diameter is 100 ± 45 nm. After addition of the ssDNA shell, however, the conjugates are stabilized, and the degree of N2H4-induced aggregation is reduced both before (PtNP@ssDNA, 52 ± 31 nm, red trace) and after (PtNP@ssDNA, 55 ± 35 nm, blue trace) exposure to Exo I. In other words, N2H4 has no significant effect on the colloidal stability of PtNP@ssDNA.
ECA of PtNP@ssDNA at Au UMEs prior to Exo I digestion
We have previously reported that the presence of ssDNA on the surface of 4.6 nm PtNPs reduces both the magnitude and frequency of ECA collision signals arising from N2H4 oxidation on both Au and Hg UMEs.2 To verify that this observation holds true even for the larger NPs used in the present study, we modified PtNPs using solutions containing the following ratios of PtNP:ssDNA—1:10, 1:25, 1:50, and 1:100, and then obtained ECA data using Au UMEs.
Fig. 2a shows representative current–time (i–t) traces for N2H4 + 50 mM PB solutions in the absence of PtNPs (black trace), and in the presence of naked PtNPs (red trace), PtNP@ssDNA prepared using a PtNP:ssDNA ratio of 1:10 (blue trace), and PtNP@ssDNA prepared using a PtNP:ssDNA ratio of 1:100 (green trace). Obviously, no collisions are observed in the absence of PtNPs. Naked PtNPs produced an average ECA current transient frequency of 0.048 ± 0.006 Hz and current magnitude of 51 ± 41 pA (see Fig. S3† for additional i–t curves for collisions of nominally naked PtNPs with Au UMEs). This current is lower than that predicted by eqn (2) (237 pA), which has previously been shown to yield reasonable agreement with experimental measurements involving smaller NPs (∼3–4 nm).1
| i = 4π(ln2)nFDCr | (2) |
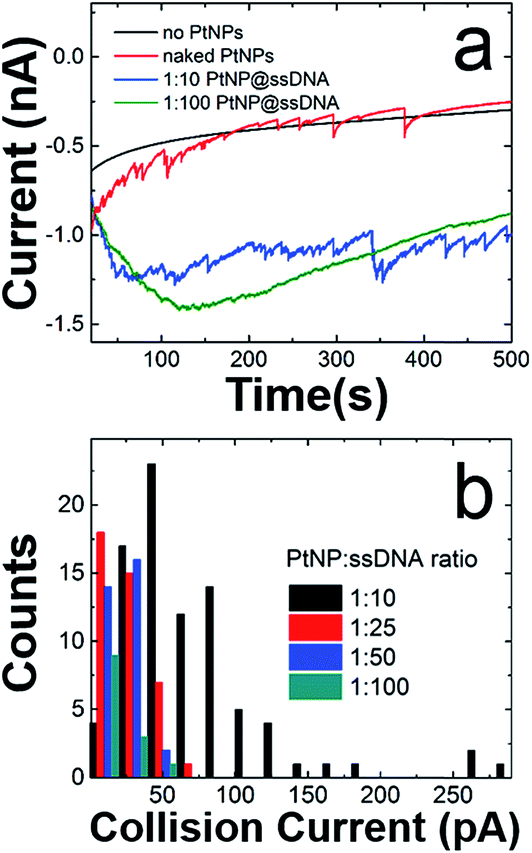 | ||
| Fig. 2 ECA results obtained using a Au UME and a solution containing 50 mM PB (pH 7) + 10.0 mM N2H4. (a) i–t curves obtained at E = −50.0 mV. The types of PtNPs used for each trace are shown in the legend. (b) Histogram showing the distribution of collision currents for the indicated ratios of PtNP:ssDNA. In all cases the indicated ratios refer to the PtNP:ssDNA ratio of the solution used to modify the PtNPs (not the actual surface concentrations of ssDNA on the PtNPs). | ||
The average ECA current and transient frequency resulting from collisions of PtNP@ssDNA (1:10), 51 ± 48 pA and 0.043 ± 0.006 Hz, respectively, were almost identical to those of the naked PtNPs. However, PtNPs with the highest ssDNA modification ratio revealed a much larger decrease in signal. Specifically, the average collision current for PtNP@ssDNA (1:100) was 17 ± 11 pA, a ∼66% decrease relative to naked PtNPs. Additionally, the frequency decreased from 0.048 ± 0.006 Hz to 0.0072 ± 0.004 Hz.
Fig. 2b is a histogram showing the distribution of ECA currents as a function of the PtNP:ssDNA solution ratio used to prepare the conjugates. These data reveal the quantitative attenuation in collision current as a function of increasing ssDNA coverage. Although not shown here, the collision frequency also decreases with increasing ssDNA coverage. This trend is consistent with data previously reported for 4.6 nm PtNPs.2
Colloidal stability during enzymatic reactions
Enzymatic reactions with Exo I are commonly performed in an optimized reaction buffer consisting of 67 mM glycine–KOH (pH 9.5), 6.7 mM MgCl2, and 10 mM 2-mercaptoethanol. However, both naked PtNPs and PtNP@ssDNA precipitated in this reaction buffer within the required incubation period of 1 h. It is known, however, that Exo I also maintains 100% of its activity in a buffer optimized for the Taq polymerase: 75 mM Tris–HCl (pH 8.8), 2.0 mM MgCl2, 0.01% Tween 20, and 20 mM (NH4)2SO4.49 Accordingly, we tested the colloidal stability of PtNPs in the Taq buffer and each of its components by carrying out NTA experiments.Fig. 1c presents normalized NTA data for both naked PtNPs (black trace) and PtNP@ssDNA (red trace). These data were obtained by resuspending the NPs in Taq buffer and incubating at room temperature for 1 h. An aliquot of the NP solution was then further diluted in Taq buffer to an appropriate concentration for NTA measurements (∼0.5–1.0 pM). The important result is that the average size of naked PtNPs in the Taq buffer is 155 ± 119 nm compared to 44 ± 40 nm for those stabilized with ssDNA. The latter size can be compared to 57 ± 31 found for PtNP@ssDNA in 50 mM PB (Fig. 1a). We conclude that PtNP@ssDNA are sufficiently stable in the Taq buffer to carry out the Exo I cleavage. NTA plots for the individual components of Taq buffer (75 mM Tris–HCl (pH 8.8), 2.0 mM MgCl2, 0.01% Tween 20, and 20 mM (NH4)2SO4) can be found in Fig. S4.†
ECA of PtNP@ssDNA at Au UMEs after Exo I digestion
We are interested in the catalytic properties of PtNP@ssDNA after exposure to Exo I, because (as discussed in the Introduction) reactivation of the NPs provides a means of using ECA for biosensing applications. Accordingly, PtNP@ssDNA was incubated with 30 U of Exo I for 1.0 h at 37 °C. Fig. 3a shows ECA i–t traces for collisions of PtNP@ssDNA with a Au UME before (black trace) and after (red trace) Exo I digestion. Prior to digestion, the PtNP@ssDNA produced very small (∼1–2 pA, Fig. 3a inset) and infrequent collisions. Some experiments, typically those carried out at PtNP@ssDNA concentrations <1 pM, produced no collisions at all. After Exo I digestion, which removes a fraction of the ssDNA from the PtNP surface, the anodic background current (red trace) increases with time and step-shaped current transients, corresponding to NP collisions, are observed. This is apparent in the expanded view (Fig. 3b) of a 10 s section of the i–t trace in Fig. 3a.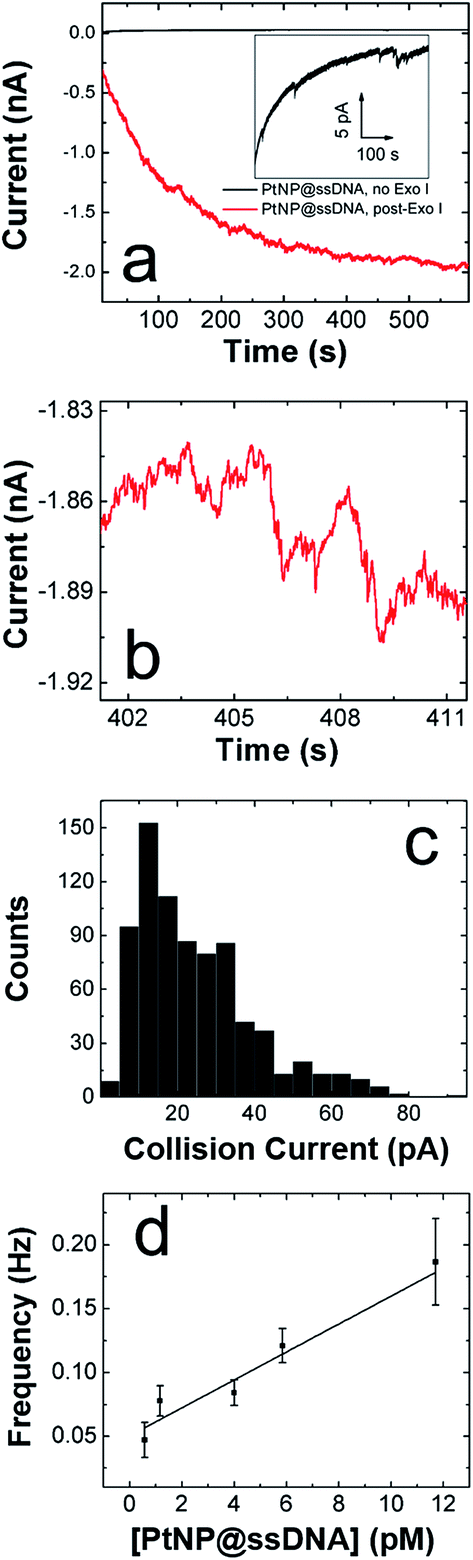 | ||
| Fig. 3 ECA results obtained for PtNP@ssDNA using an Au UME and a solution containing 50 mM PB (pH 7) + 10.0 mM N2H4. (a) i–t curves obtained at E = −50.0 mV for 11.7 pM PtNP@ssDNA before (black) and after (red) ssDNA digestion by Exo I. The inset shows an expanded view of the i–t trace before treatment with Exo I. (b) Expanded view of post-Exo I trace in frame a showing the step-shaped profile characteristic of PtNP collisions on Au. (c) Histogram showing the distribution of currents for collisions of PtNP@ssDNA post-Exo I. The average current is 24 ± 15 pA. (d) Plot of collision frequency as a function of the concentration of PtNP@ssDNA post-Exo I. The error bars represent the standard deviation of current signals obtained from three separate experiments at each concentration. | ||
A histogram showing the frequency of collisions as a function of the ECA current is shown in Fig. 3c. The average collision current for the post-Exo I PtNP–ssDNA is 24 ± 15 pA, which can be compared to the value for naked PtNPs: 51 + 41 pA. Because only about half of the original current is recovered after exposure to the enzyme, we conclude that only about that same fraction of the PtNP surface is available for N2H4 oxidation. The most likely scenario is that a substantial fraction of the ssDNA is reduced in length by Exo I, thereby provide access of N2H4 to the PtNP surface. We wish to emphasize that the ssDNA incorporates a 6-carbon alkylthiol on its 5′ (proximal) end. These alkyl chains will not be removed by Exo I, so it is unlikely that additional catalytic sites are exposed on the Pt surface. Rather, removal of some of bases reduces mass transfer hindrance of N2H4.
Exo I will only initiate cleavage in ssDNA having a free 3′-hydroxyl end. To confirm that the action of Exo I is responsible for the observation of collisions, we immobilized ssDNA on the PtNPs such that only the 5′-hydroxyl end was accessible. For this control experiment, only the orientation of the DNA on the PtNPs was reversed: the ssDNA sequence was unchanged. In this case, no collisions were observed after treatment with Exo I (Fig. S5†). This important finding confirms that the specific enzymatic activity of Exo I for cleavage of 3′-hydroxyl DNA is responsible for the collision currents shown in Fig. 3.
In addition to the collision current, we also studied the collision frequency for PtNP@ssDNA post-Exo I and found that it correlates linearly with concentration in the range of 0.58–11.7 pM (Fig. 3d). This observation is consistent with previous findings for collisions of naked PtNPs at Au UMEs.20 Here, we also found that the collision frequency is higher for PtNP@ssDNA post-Exo I than for naked PtNPs. For example, at a concentration of 11.7 pM, the frequency for PtNP@ssDNA post-Exo I was 0.190 ± 0.033 Hz, compared to 0.048 ± 0.006 Hz for naked PtNPs. We have observed this same trend previously for ssDNA-modified PtNPs colliding with a Au microband electrode.2
The factor-of-three difference in collision frequency may be caused by the difference in the rates of mass transport between the slower diffusing aggregates of naked PtNPs and the smaller, more colloidally stable PtNP@ssDNA. Another observation worth considering is the high noise level observed for collisions arising from PtNP@ssDNA post-Exo I (Fig. 3b). This might suggest that the ssDNA-modified NPs reside on or near the UME surface longer than naked PtNPs, and hence each particle may collide with the electrode multiple times.
ECA of PtNP@ssDNA at Hg UMEs
In an effort to better understand the difference in collision frequency between naked PtNPs and PtNP@ssDNA, we carried out additional ECA experiments using an Hg UME in place of the Au UME. We chose this comparison, because Hg has previously been shown to quickly deactivate, or “poison”, PtNPs upon impact.22 Therefore, each PtNP@ssDNA should only produce one collision signal. Collisions on Hg also provide the advantage of lower background noise, and each signal decays back to the original current baseline rather than producing a step-shaped response.22Fig. 4a shows i–t results for collisions of PtNP@ssDNA with an Hg UME before (black trace) and after (red trace) Exo I digestion of PtNP@ssDNA. The inset in this frame is an expanded view of the black trace over a limited time window. These data show that in the absence of Exo I exposure, PtNP collisions yield small and infrequent collisions having a magnitude of ∼5 pC. Note that due to the spike-shaped i–t transients that occur at Hg UMEs, it is conventional to report their magnitude in terms of charge rather than current.22 After enzymatic digestion, more frequent, spike-shaped collisions are observed as shown in the expanded view of the i–t trace in Fig. 4b. Comparison of the red traces in Fig. 3a (Au UME) and 4a (Hg UME) also shows that the baseline current is much more stable for the latter after digestion.
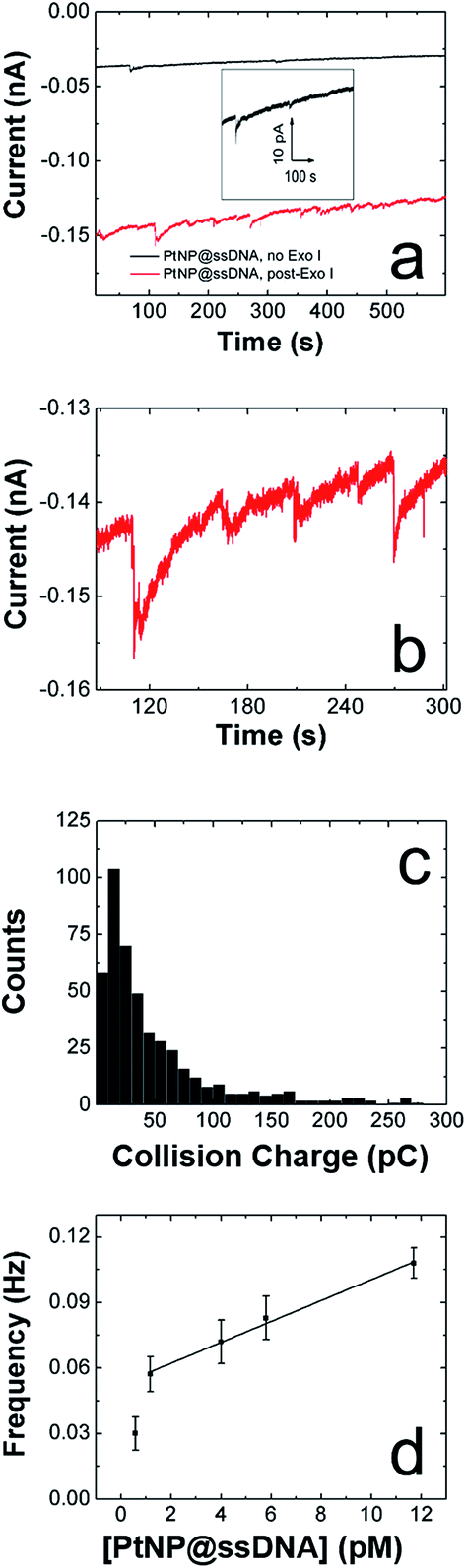 | ||
| Fig. 4 ECA results obtained for PtNP@ssDNA using an Hg UME and a solution containing 50 mM PB (pH 7) + 10.0 mM N2H4. (a) i–t curves obtained at E = 5.0 mV for 1.17 pM PtNP@ssDNA before (black) and after (red) digestion by Exo I. The inset shows an expanded view of the i–t trace before treatment with Exo I. (b) Expanded view of post-Exo I trace in frame a showing the spike-shaped profile characteristic of PtNP collisions on Hg. (c) Histogram showing the distribution of charges for collisions of PtNP@ssDNA post-Exo I. The average charge is 63 ± 98 pC. (d) Plot of collision frequency as a function of the concentration of PtNP@ssDNA post-Exo I. The error bars represent the standard deviation of charge signals obtained from three separate experiments at each concentration. | ||
Fig. 4c is a histogram showing the frequency of collisions as a function of their magnitude for post-Exo I PtNP@ssDNA. The average collision charge for these experiments is 63 ± 98 pC, which is ∼50% less than for naked PtNPs: 118 ± 128 pC. This trend of recovering roughly half the original collision signal is consistent with that observed for Au UMEs. Moreover, the broader distribution of collision charges (determined from the integrated current transients) on Hg is consistent with results reported by Stevenson and coworkers.22
The collision frequency of PtNP@ssDNA post-Exo I as a function of concentration at an Hg UME is shown in Fig. 4d. As for the Au UME data (Fig. 3d), this plot is linear over most of the concentration range studied. The interesting result, however, is that the collision frequencies are significantly lower at every concentration than for the Au UME results (Table 1). This finding is in contrast to previously reported observations in which the larger surface area of the hemispherical Hg UME led to higher collision frequencies.21 It has been found previously that deposition of Hg on Pt yields a hemispherical Hg drop.24 If we make that assumption here too, then the geometric surface area of the Hg electrode is 157 μm2, compared to 123 μm2 for the Au disk. In other words, the relative surface areas of the two electrodes does not account for the observation of lower collision frequencies on the Hg UME. As suggested earlier, however, this difference might be due to specific interactions between the ssDNA-coated NPs and the Au UME that lead to multiple collisions per PtNP at the Au UME. In contrast, Hg deactivates every PtNP upon impact,22 and thus each PtNP@ssDNA post-Exo I will only produce one current transient per collision event.
| PtNP–ssDNA concentration (pM) | Frequency (Hz) Au UME | Frequency (Hz) Hg UME |
|---|---|---|
| 0.58 | 0.047 ± 0.014 | 0.03 ± 0.08 |
| 1.17 | 0.077 ± 0.012 | 0.057 ± 0.008 |
| 4.0 | 0.084 ± 0.011 | 0.072 ± 0.010 |
| 5.8 | 0.12 ± 0.01 | 0.083 ± 0.010 |
| 11.7 | 0.19 ± 0.03 | 0.11 ± 0.01 |
Summary and conclusions
The method of ECA relies on access of a redox probe molecule, N2H4 in this report, to the surface of a catalytic NP. This fact has serious consequences for adaptation of ECA to biosensing applications, because in general the latter require surface modification with a recognition element such as DNA or antibodies. In ECA, the presence of these recognition elements decreases the collision signal in proportion to their surface concentration.2 To overcome this problem, we have introduced a new approach to ECA in which the colliding PtNP is pre-poisoned with the receptor (ssDNA in this case), but in the presence of an appropriate enzyme the catalytic properties of the PtNP are reactivated. In the present case, one can consider the target of this “turn-on sensor” to be either the ssDNA or the enzyme. As Corn and coworkers have shown,50 there is a vast array of surface enzyme operations, and we believe that many of these could be adapted to the ECA methodology described herein. Hence, this approach could be quite general in resolving the apparent paradox of adapting ECA to biosensing applications. Indeed, at the present time we are working on a method, based on the approach described here, for detection of microRNA.Finally, there are three specific conclusions that can be drawn from the results presented here. First, PtNPs modified with ssDNA are not irreversibly passivated, and at least ∼50% of the original current can be recovered after treatment with Exo I. Second, PtNP@ssDNA remain colloidally stable under the conditions required for enzymatic digestion (e.g., high salt concentration). Third, the lower collision frequency observed at Hg UMEs, compared to Au UMEs, may be indicative of single NPs producing more than one signal per collision event at Au electrodes. This is an important point, because practical applications of ECA require quantitative correlation of the concentration of a target with the collision frequency and/or signal magnitude. These findings set the stage for future biosensing applications of ECA, and we will report the results of those studies in due course.
Acknowledgements
We gratefully acknowledge financial support from the U.S. Defense Threat Reduction Agency (Grant No. HDTRA1-11-1-0005). We also thank the Robert A. Welch Foundation (Grant F-0032) for sustained research support. We gratefully acknowledge Dwight Romanovicz (ICMB, UT Austin) for assistance with TEM imaging and Timothy M. Alligrant for helpful discussions.References
- A. J. Bard, H. Zhou and S. J. Kwon, Isr. J. Chem., 2010, 50, 267–276 CrossRef CAS.
- T. M. Alligrant, R. Dasari, K. J. Stevenson and R. M. Crooks, Langmuir, 2015, 31, 11724–11733 CrossRef CAS PubMed.
- D. Hellberg, F. Scholz, F. Schauer and W. Weitschies, Electrochem. Commun., 2002, 4, 305–309 CrossRef CAS.
- F. Scholz, D. Hellberg, F. Harnisch, A. Hummel and U. Hasse, Electrochem. Commun., 2004, 6, 929–933 CrossRef CAS.
- D. Hellberg, F. Scholz, F. Schubert, M. Lovrić, D. Omanović, V. A. Hernández and R. Thede, J. Phys. Chem. B, 2005, 109, 14715–14726 CrossRef CAS PubMed.
- B. M. Quinn, P. G. Van't Hof and S. G. Lemay, J. Am. Chem. Soc., 2004, 126, 8360–8361 CrossRef CAS PubMed.
- Y.-G. Zhou, N. V. Rees and R. G. Compton, Angew. Chem., Int. Ed., 2011, 50, 4219–4221 CrossRef CAS PubMed.
- I. J. Cutress, N. V. Rees, Y.-G. Zhou and R. G. Compton, Chem. Phys. Lett., 2011, 514, 58–61 CrossRef CAS.
- R. Polsky, R. Gill, L. Kaganovsky and I. Willner, Anal. Chem., 2006, 78, 2268–2271 CrossRef CAS PubMed.
- K.-F. Chow, F. Mavré and R. M. Crooks, J. Am. Chem. Soc., 2008, 130, 7544–7545 CrossRef CAS PubMed.
- X. Xiao and A. J. Bard, J. Am. Chem. Soc., 2007, 129, 9610–9612 CrossRef CAS PubMed.
- S. E. F. Kleijn, S. C. S. Lai, T. S. Miller, A. I. Yanson, M. T. M. Koper and P. R. Unwin, J. Am. Chem. Soc., 2012, 134, 18558–18561 CrossRef CAS PubMed.
- C. Chen, E. R. Ravenhill, D. Momotenko, Y.-R. Kim, S. C. S. Lai and P. R. Unwin, Langmuir, 2015, 31, 11932–11942 CrossRef CAS PubMed.
- J. H. Park, S. N. Thorgaard, B. Zhang and A. J. Bard, J. Am. Chem. Soc., 2013, 135, 5258–5261 CrossRef CAS PubMed.
- S. J. Kwon, H. Zhou, F.-R. F. Fan, V. Vorobyev, B. Zhang and A. J. Bard, Phys. Chem. Chem. Phys., 2011, 13, 5394–5402 RSC.
- S. E. F. Kleijn, B. Serrano-Bou, A. I. Yanson and M. T. M. Koper, Langmuir, 2013, 29, 2054–2064 CrossRef CAS PubMed.
- N. P. Sardesai, D. Andreescu and S. Andreescu, J. Am. Chem. Soc., 2013, 135, 16770–16773 CrossRef CAS PubMed.
- A. Fernando, S. Parajuli and M. A. Alpuche-Aviles, J. Am. Chem. Soc., 2013, 135, 10894–10897 CrossRef CAS PubMed.
- T. M. Alligrant, E. G. Nettleton and R. M. Crooks, Lab Chip, 2013, 13, 349–354 RSC.
- A. D. Castañeda, T. M. Alligrant, J. A. Loussaert and R. M. Crooks, Langmuir, 2015, 31, 876–885 CrossRef PubMed.
- T. M. Alligrant, M. J. Anderson, R. Dasari, K. J. Stevenson and R. M. Crooks, Langmuir, 2014, 30, 13462–13469 CrossRef CAS PubMed.
- R. Dasari, D. A. Robinson and K. J. Stevenson, J. Am. Chem. Soc., 2013, 135, 570–573 CrossRef CAS PubMed.
- R. Dasari, B. Walther, D. A. Robinson and K. J. Stevenson, Langmuir, 2013, 29, 15100–15106 CrossRef CAS PubMed.
- R. Dasari, K. Tai, D. A. Robinson and K. J. Stevenson, ACS Nano, 2014, 8, 4539–4546 CrossRef CAS PubMed.
- D. Wakerley, A. G. Güell, L. A. Hutton, T. S. Miller, A. J. Bard and J. V. Macpherson, Chem. Commun., 2013, 49, 5657–5659 RSC.
- X. Xiao, F.-R. F. Fan, J. Zhou and A. J. Bard, J. Am. Chem. Soc., 2008, 130, 16669–16677 CrossRef CAS PubMed.
- J. B. Fasoli and R. M. Corn, Langmuir, 2015, 31, 9527–9536 CrossRef CAS PubMed.
- H. J. Lee, A. W. Wark, T. T. Goodrich, S. Fang and R. M. Corn, Langmuir, 2005, 21, 4050–4057 CrossRef CAS PubMed.
- W. Yang, Q. Rev. Biophys., 2011, 44, 1–93 CrossRef CAS PubMed.
- N. C. Mishra, Nucleases, John Wiley & Sons, Inc., Hoboken, NJ, 2002 Search PubMed.
- H.-I. Kao and R. A. Bambara, Crit. Rev. Biochem. Mol. Biol., 2008, 38, 433–452 CrossRef PubMed.
- B. Shen, P. Singh, R. Liu, J. Qiu, L. Zheng, L. D. Finger and S. Alas, BioEssays, 2005, 27, 717–729 CrossRef CAS PubMed.
- L. J. Reha-Krantz, Biochim. Biophys. Acta, 2010, 1804, 1049–1063 CrossRef CAS PubMed.
- X. Xu, M. S. Han and C. A. Mirkin, Angew. Chem., Int. Ed., 2007, 46, 3468–3470 CrossRef CAS PubMed.
- M. Liu, M. Yuan, X. Lou, H. Mao, D. Zheng, R. Zou, N. Zou, X. Tang and J. Zhao, Biosens. Bioelectron., 2011, 26, 4294–4300 CrossRef CAS PubMed.
- A. E. Prigodich, A. H. Alhasan and C. A. Mirkin, J. Am. Chem. Soc., 2011, 133, 2120–2123 CrossRef CAS PubMed.
- X.-M. Li, L.-L. Wang, J. Luo and Q.-L. Wei, Biosens. Bioelectron., 2014, 65, 245–250 CrossRef PubMed.
- I. R. Lehman and A. L. Nussbaum, J. Biol. Chem., 1964, 239, 2628–2636 CAS.
- N. C. Bigall, T. Härtling, M. Klose, P. Simon, L. M. Eng and A. Eychmüller, Nano Lett., 2008, 8, 4588–4592 CrossRef CAS PubMed.
- X. Zhang, M. R. Servos and J. Liu, J. Am. Chem. Soc., 2012, 134, 7266–7269 CrossRef CAS PubMed.
- X. Zhang, M. R. Servos and J. Liu, Chem. Commun., 2012, 48, 10114–10116 RSC.
- K. R. Wehmeyer and R. M. Wightman, Anal. Chem., 1985, 57, 1989–1993 CrossRef CAS.
- H. Li and L. J. Rothberg, Anal. Chem., 2004, 76, 5414–5417 CrossRef CAS PubMed.
- D. A. Robinson, J. J. Yoo, A. D. Castañeda, B. Gu, R. Dasari, R. M. Crooks and K. J. Stevenson, ACS Nano, 2015, 9, 7583–7595 CrossRef CAS PubMed.
- D. A. Robinson, A. M. Kondajji, A. D. Castañeda, R. Dasari, R. M. Crooks and K. J. Stevenson, J. Phys. Chem. Lett., 2016, 7, 2512–2517 CrossRef CAS PubMed.
- E. J. W. Verwey, J. T. G. Overbeek and J. T. G. Overbeek, Theory of the Stability of Lyophobic Colloids, Dover Publications, 1999 Search PubMed.
- W. B. Russel, D. A. Saville and W. R. Schowalter, Colloidal Dispersions, Cambridge University Press, 1992 Search PubMed.
- L. Aldous and R. G. Compton, Phys. Chem. Chem. Phys., 2011, 13, 5279–5287 RSC.
- ThermoFisher Scientific, modifying enzyme buffers. https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/thermo-scientific-restriction-modifying-enzymes/modifying-enzymes-thermo-scientific/modifying-enzyme-buffers.html, accessed Feb 21, 2016.
- H. J. Lee, A. W. Wark and R. M. Corn, Langmuir, 2006, 22, 5241–5450 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supporting data including size distribution analysis of the PtNPs by TEM, a fluorescence calibration curve for estimating the coverage of ssDNA on PtNPs, ECA of PtNP@ssDNA modified with thiol on the 3′-end, and i–t curves for collisions of naked PtNPs at Au UMEs. See DOI: 10.1039/c6sc02165d |
| ‡ Current address: Skolkovo Institute of Science and Technology, 3 Nobel Street, Moscow, Russia 143026. |
| This journal is © The Royal Society of Chemistry 2016 |