
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoredox-mediated Minisci C–H alkylation of N-heteroarenes using boronic acids and hypervalent iodine†
Guo-Xing
Li
a,
Christian A.
Morales-Rivera
b,
Yaxin
Wang
a,
Fang
Gao
a,
Gang
He
a,
Peng
Liu
*b and
Gong
Chen
*ac
aState Key Laboratory and Institute of Elemento-Organic Chemistry, Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Nankai University, Tianjin 300071, China. E-mail: gongchen@nankai.edu.cn
bDepartment of Chemistry, University of Pittsburgh, Pittsburgh, PA 15260, USA. E-mail: pengliu@pitt.edu
cDepartment of Chemistry, The Pennsylvania State University, 104 Chemistry Building, University Park, PA 16802, USA. E-mail: guc11@psu.edu
First published on 12th July 2016
Abstract
A photoredox-mediated Minisci C–H alkylation reaction of N-heteroarenes with alkyl boronic acids is reported. A broad range of primary and secondary alkyl groups can be efficiently incorporated into various N-heteroarenes using [Ru(bpy)3]Cl2 as photocatalyst and acetoxybenziodoxole as oxidant under mild conditions. The reaction exhibits excellent substrate scope and functional group tolerance, and offers a broadly applicable method for late-stage functionalization of complex substrates. Mechanistic experiments and computational studies suggest that an intramolecularly stabilized ortho-iodobenzoyloxy radical intermediate might play a key role in this reaction system.
Introduction
N-Heteroarenes are common structural motifs in natural products, drug molecules, organic materials and ligands for metal catalysts.1 Synthetic methods which enable the selective functionalization of the C–H bonds of N-heteroarenes could greatly facilitate their applications in these areas.2 Among the different types of C–H functionalizations, C–H alkylations could provide more stereochemically diverse modifications.3 Over the past few years, the C–H functionalization of electron-deficient heteroarenes via addition of carbon-centered radicals under oxidative conditions, known as the Minisci reaction, has undergone a remarkable renaissance, offering increasingly powerful methods for synthesizing alkyl-substituted heteroarenes (Scheme 1).4 While the classical Minisci alkylation reaction involves alkyl carboxylic acids and halides, Baran recently demonstrated that aryl boronic acids are also viable reagents in Minisci-type C–H arylation reactions using Ag(I)/S2O82− oxidant.5 Molander demonstrated that alkyl trifluoroborates, particularly secondary alkyl trifluoroborates, can effect efficient Minisci alkylation using Mn(OAc)3 oxidant.6 In addition, Minisci C–H alkylation transformations have been achieved using a variety of other alkylating reagents, including sulfinates, aldehydes, and even simple alkanes, using different radical initiators and oxidants.7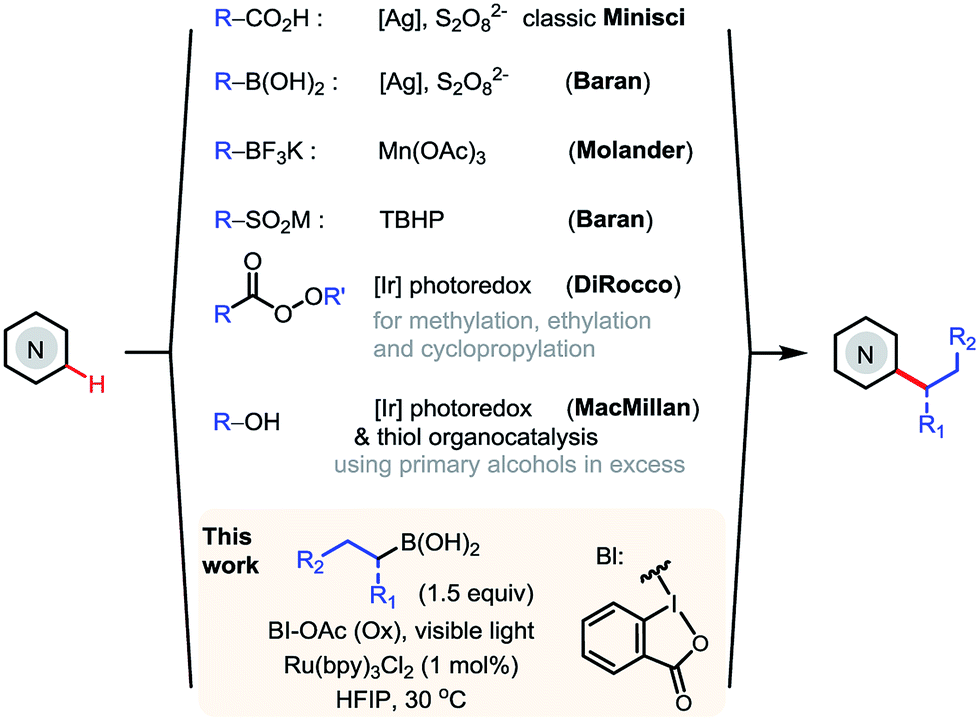 | ||
| Scheme 1 Minisci C–H alkylation of N-heteroarenes. | ||
More recently, DiRocco reported the first photoredox-mediated Minisci alkylation reaction of N-heteroarenes using peroxides as the alkylating reagent.8 MacMillan demonstrated a Minisci alkylation reaction of N-heteroarenes using primary alcohols as the alkylation reagent, via photoredox- and organo- catalysis.9a However, despite these significant advances, practical and broadly applicable methods for Minisci C–H alkylation of N-heteroarenes capable of coupling complex alkyl groups are still lacking. Herein, we report a photoredox-mediated Minisci C–H alkylation reaction of N-heteroarenes with a variety of easily accessible primary and secondary alkyl boronic acids. Its high efficiency, broad substrate scope, excellent functional group tolerance, and mild operation conditions make it particularly suitable for late-stage functionalization of complex substrates such as drug molecules.
Results and discussion
Although alkyl boron reagents are readily available and are well-known precursors for alkyl radicals, they have been rarely applied in photoredox-mediated C–C coupling reactions.10–13 In 2012, Akita reported that alkyl trifluoroborates or cyclic triolborates can couple with 2,2,6,6-tetramethylpiperidinyl-1-oxy (TEMPO) or Michael acceptors under Ru or Ir photoredox catalysis.11 In 2015, Chen reported a decarboxylative alkenylation of alkyl trifluoroborates with vinyl carboxylic acids using a hypervalent iodine oxidant, acetoxybenziodoxole (BI-OAc), under Ru photoredox catalysis.12a More recently, Molander achieved coupling of alkyl trifluoroborates with aryl halides by merging photoredox with Ni cross-coupling catalysis.13 During our recent investigation of radical-mediated sp3 C–H azidation reactions, we discovered that azidobenziodoxole (Bl-N3) can be readily activated by visible light in the presence of [Ru(bpy)3]Cl2, initiating a radical chain reaction.14 Intrigued by the unique radical reactivity of benziodoxole reagents with photocatalysts, we questioned whether they can facilitate Minisci C–H alkylation with alkyl boron reagents under photoredox-mediated conditions.15–17We commenced our investigation with C–H butylation of 4-chloroquinoline 1, a common model substrate for Minisci reactions, using butyl boronic acid 2a or trifluoroborate 2b under the visible light (VL) irradiation (Table 1). We were delighted to find that the desired C2-alkylated product 3a can be formed in excellent yield with 2a using [Ru(bpy)3]Cl2 photocatalyst and BI-OAc oxidant under optimized conditions (entry 5). Alkylation with 2b proceeded in lower yield (entry 6). In comparison with Bl-OAc, hydroxylbenziodoxole (BI-OH) gave slightly lower yield, methoxylbenziodoxole (Bl-OMe) was notably less effective, BI-N3 gave low yield, chlorobenziodoxole (Bl-Cl) and PhI(OAc)2 showed little reactivity (entries 7–11). Hexafluoroisopropanol (HFIP) solvent is critical for obtaining high yield (entries 3–5). No 3a was formed in the absence of either Ru catalysis or light irradiation (entries 13–14). Formation of 3a was completely suppressed when 2 equiv of TEMPO was added, forming side product O-butyl TEMPO in 16% yield (entry 15).
|
|
||||
|---|---|---|---|---|
| Entry | Reagents (equiv.) | Solvents | t (°C)/time (h) | Yielda (%) 3a |
| a Yields are based on 1H-NMR analysis on a 0.2 mmol scale. b VL: compact household fluorescent bulb, 20 W. c Isolated yield. | ||||
| 1 | 2a (1.5), AgNO3 (0.2), K2S2O8 (3) | DCM/H2O | 30/24 | 18 |
| 2 | 2b (1.5), Mn(OAc)3 (2.5), TFA (1) | AcOH/H2O | 50/18 | 30 |
| 3 | 2a (1.5), Bl-OAc (2), Ru(bpy)3Cl2 (0.01), VLb | DCM | 30/24 | 33 |
| 4 | 2a (1.5), Bl-OAc (2), Ru(bpy)3Cl2 (0.01), VL | CH3CN | 30/24 | 38 |
| 5 | 2a (1.5), Bl-OAc (2), Ru(bpy)3Cl2 (0.01), VL, Ar | HFIP | 30/24 | 88 (82c) |
| 6 | 2b (1.5), Bl-OAc (2), Ru(bpy)3Cl2 (0.01), VL | HFIP | 30/24 | 59 |
| 7 | 2a (1.5), Bl-OH (2), Ru(bpy)3Cl2 (0.01), VL | HFIP | 30/24 | 82 |
| 8 | 2a (1.5), Bl-OMe (2), Ru(bpy)3Cl2 (0.01), VL | HFIP | 30/24 | 61 |
| 9 | 2a (1.5), Bl-N3 (2), Ru(bpy)3Cl2 (0.01), VL | HFIP | 30/24 | 25 |
| 10 | 2a (1.5), Bl-Cl (2), Ru(bpy)3Cl2 (0.01), VL | HFIP | 30/24 | <2 |
| 11 | 2a (1.5), PhI(OAc)2 (2), Ru(bpy)3Cl2 (0.01), VL | HFIP | 30/24 | <2 |
| 12 | 2a (1.5), Bl-OAc (2), Ir(ppy)3 (0.01), VL | HFIP | 30/24 | 22 |
| 13 | 2a (1.5), Bl-OAc (2), VL | HFIP | 30/24 | <2 |
| 14 | 2a (1.5), Bl-OAc (2), Ru(bpy)3Cl2 (0.01), in darkness | HFIP | 30/24 | <2 |
| 15 | 2a (1.5), Bl-OAc (2), TEMPO (2), Ru(bpy)3Cl2 (0.01), VL | HFIP | 30/24 | <2 |
With the optimized conditions in hand, we next explored the substrate scope (Scheme 2). As seen in 3c–3l, a range of primary alkyl boronic acids reacted with 4-chloroquinoline 3 to give C2-alkylated products in good to excellent yield. Methylation with MeB(OH)2 gave moderate yield (see 3b). Primary alkyl radicals are more challenging reactants in Minisci reactions than secondary alkyl radicals due to their lower stability and nucleophilicity.18 We were pleased to observe that primary alkyl substituents carrying various functional groups, including alkyl bromide, aryl iodide, ester, amide, carbamate, terminal alkyne, and benzyl chloride, can be incorporated in good yield (see 3g–3l). As seen in 3m–3r, the alkylation reactions of secondary alkyl boronic acids are much faster than the primary and typically proceed in good to excellent yield under the standard conditions. In contrast to alkylation, arylation with PhB(OH)2 gave product 3s in low yield (21%).
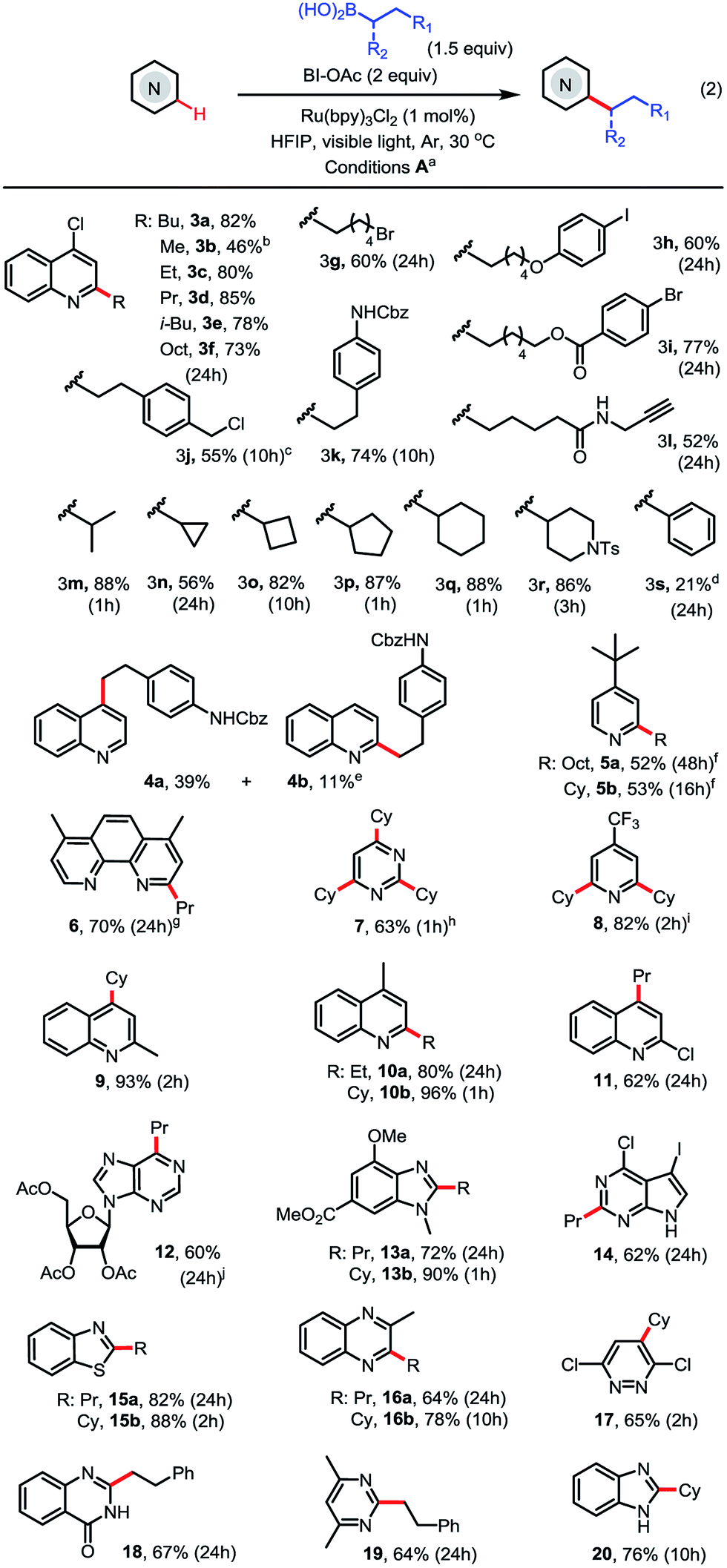 | ||
| Scheme 2 Substrate scope of photoredox-mediated C–H alkylation of N-heteroarenes. (a) Isolated yield on 0.2 mmol scale; (b) 2 equiv. of MeB(OH)2, 48 h; (c) 2 equiv. of RB(OH)2; (d) 1.5 equiv. of PhB(OH)2; (e) 1 equiv. of RB(OH), 2,4-dialkylated product was formed in <5% yield; (f) 2,6-dialkylated product was formed in <3% yield; (g) 2,2′-dialkylated product was formed in <5% yield; (h) 4.5 equiv. of CyB(OH)2; (i) 3 equiv. of CyB(OH)2. (j) other alkylated regioisomer was formed in <5% yield. | ||
As seen in 4–11, alkylation of pyridines and pyridine-based heteroarenes selectively took place at C2 and/or C4 positions. A mixture of C2-substituted 4a and C4-substituted 4b was obtained for unsubstituted quinoline. Alkylation of 2-methylquinoline selectively occurred at C4 to give 9 in excellent yield. In general, electron-deficient N-heteroarenes show higher reactivity toward alkylation. For instance, cyclohexylation of 4-t-butylpyridine predominantly resulted in mono-alkylation (see 5b) while cyclohexylation of 4-CF3-pyridine gave 2,6-dialkylated 8 in excellent yield using 3 equiv. of boronic acid. A variety of other N-heteroarenes can also be alkylated in good yield and regioselectivity (see 12–20). For instance, a purine riboside substrate was selectively alkylated at C6 to give 12 in excellent selectivity;7d and a pyrrole-fused pyrimidine was selectively alkylated on the electron-deficient pyrimidine ring to give 14 in good yield. Without protection of the NH group, benzimidazole can be alkylated at C2 to give 20 in good yield.
As shown in Scheme 3, this Minisci C–H alkylation can be readily applied to functionalize complex natural products and drug molecules.2c,8,9a For instance, quinine with a free OH and vinyl group can be selectively alkylated at C2 position with both ethyl and cyclohexyl groups in excellent yield (see 21). Camptothecin can be selectively alkylated at the C4 position of the pyridine ring (see 23). Caffeine, a challenging substrate for previous Minisci reactions, can be selectively alkylated at C2 (see 22).7b Alkyl chains carrying an alkynyl or alkyl bromide group can be installed at C6 of Famciclovir in good yield (see 24a, 24b). Fasudil carrying a free secondary NH group was selectively alkylated at C1 in good yield (see 26).
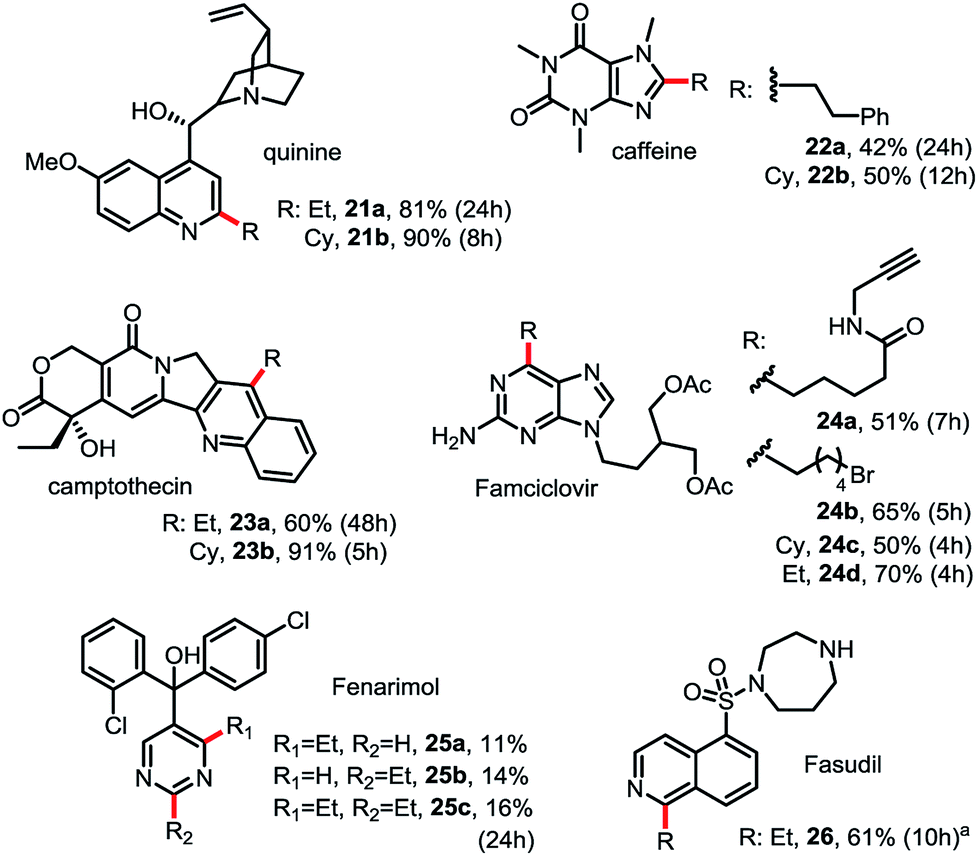 | ||
| Scheme 3 Minisci C–H alkylation for functionalization of natural products and drug molecules. Isolated yield on 0.2 mmol scale under the standard conditions with 1.5 equiv. of R-B(OH)2 (see Scheme 2). (a) Product was isolated in N-Boc protected form. | ||
Control experiments and density functional theory (DFT) calculations have been carried out to probe the mechanism of this photoredox-mediated Minisci alkylation with alkyl boronic acids and Bl-OAc oxidant.19 As shown in Scheme 4A, we were surprised to observe that reaction of 1 with BuB(OH)2 and 1 equiv. of benzoyl peroxide in HFIP under visible light irradiation without photocatalyst also gave the alkylated product 3a in 20% yield along with 11% yield of arylated side product 3s, which is presumably formed from Ph˙ via the decarboxylation of BzO˙. In comparison with BuB(OH)2, BuBF3K showed much lower reactivity. Furthermore, 3a was formed as the only C–H functionalized product in 38% yield when ortho-iodobenzoyl peroxide 27 (highly explosive) was used as the oxidant for reaction of 1 with BuB(OH)2 under the same conditions.20 These experiments suggest that benzoyloxy radicals can react with alkyl boronic acids to generate the requisite alkyl radical for the subsequent C–H alkylation. As shown in Scheme 4B, our DFT calculation showed that oxidant Bl-OAc can be readily reduced by photoexcited Ru(II)* via single electron transfer (SET) to form a radical anion intermediate Bl-1, which then can undergo I–O bond cleavage to form radical Bl-2 and acetate anion via pathway a or form Bl-3 and acetoxy radical AcO˙ via pathway b.21 Formation of Bl-2 is considerably more thermodynamically favorable than formation of AcO˙. Although a pair of interconvertible radical species, I-centered radical Bl-4 and O-centered radical Bl-2, have been invoked in a number of previous studies,22 the postulated cyclic structure of Bl-4 with a typical I–O bond length of ca. 2.1–2.2 Å cannot be located in our DFT calculation (Scheme 4C).23 Instead, the acyclic radical intermediate Bl-2 is stabilized by a secondary I–O bonding interaction (∼2.6 Å) and its spin density is distributed between the O and I atoms.24 Calculation also revealed that Bl-2 is notably more stable than benzoyloxy radical BzO˙ and is much less prone to undergo decarboxylation to form the corresponding aryl radical, which could cause the C–H arylation side reaction.25 Similar to the nucleo-homolytic substitution reaction of more reactive alkylboranes with O-centered radicals, Bl-2 could react with the less Lewis-acidic boronic acids to form an alkyl radical R˙ via a radical “ate” transition state.26–28 The DFT calculation showed that this is a facile process at ambient temperature and highly exothermic (Scheme 4D).
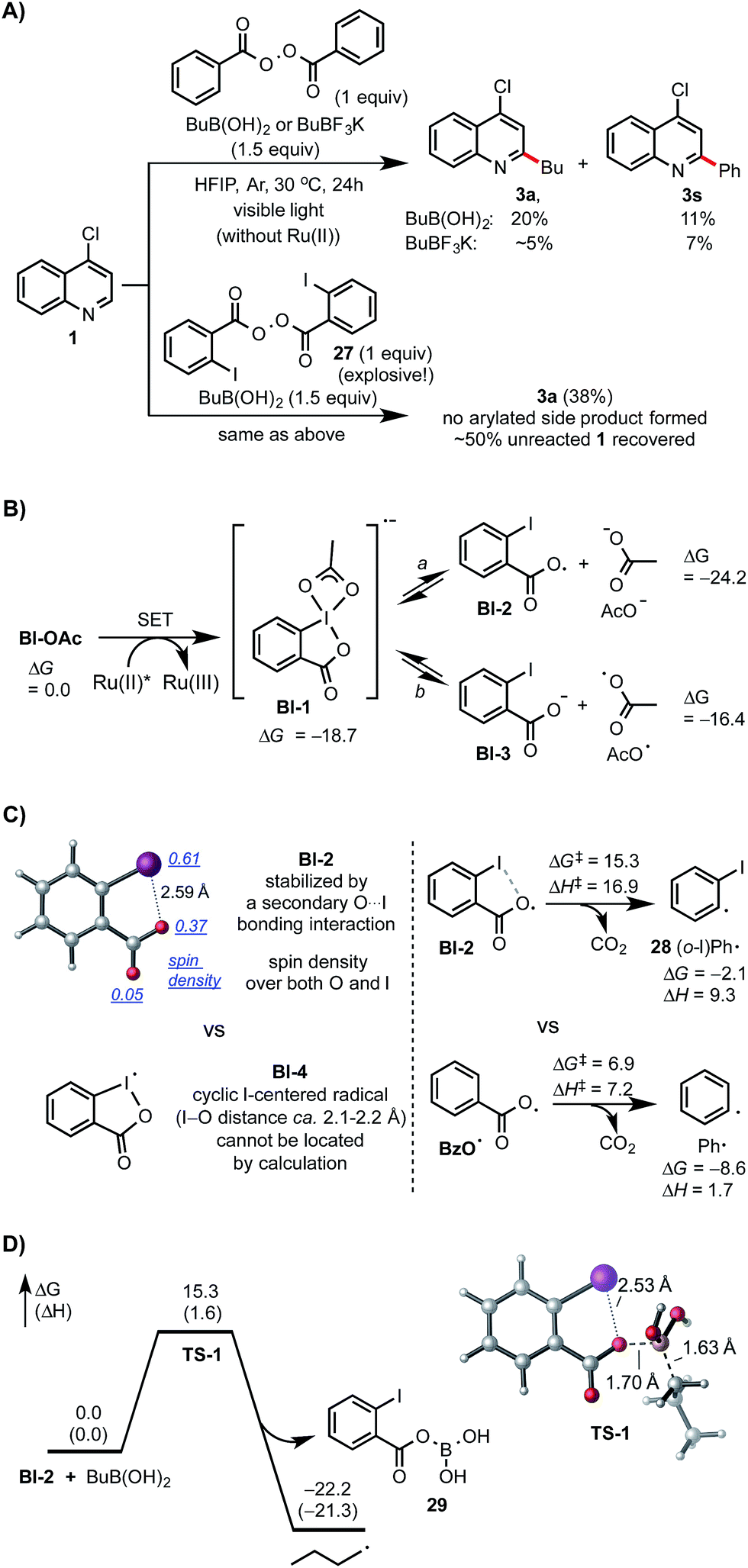 | ||
| Scheme 4 Mechanistic studies. DFT calculations were performed at the M06-2X/6-311++G(d,p)-SDD/SMD(HFIP)//M06-2X/6-31+G(d)-SDD level of theory. All energies are in kcal mol−1. | ||
Based on the above studies, we propose that the reaction with boronic acid substrates is initiated with the SET from photoexcited Ru(II)* to Bl-OAc (Scheme 5). The resulting Bl-2 reacts with boronic acid to form a R˙, which then undergoes nucleophilic addition reaction with protonated N-heteroarenes to form a σ-complex. Single-electron oxidation of this intermediate by Ru(III) and deprotonation gives the final C–H alkylated product and closes the photoredox cycle.29
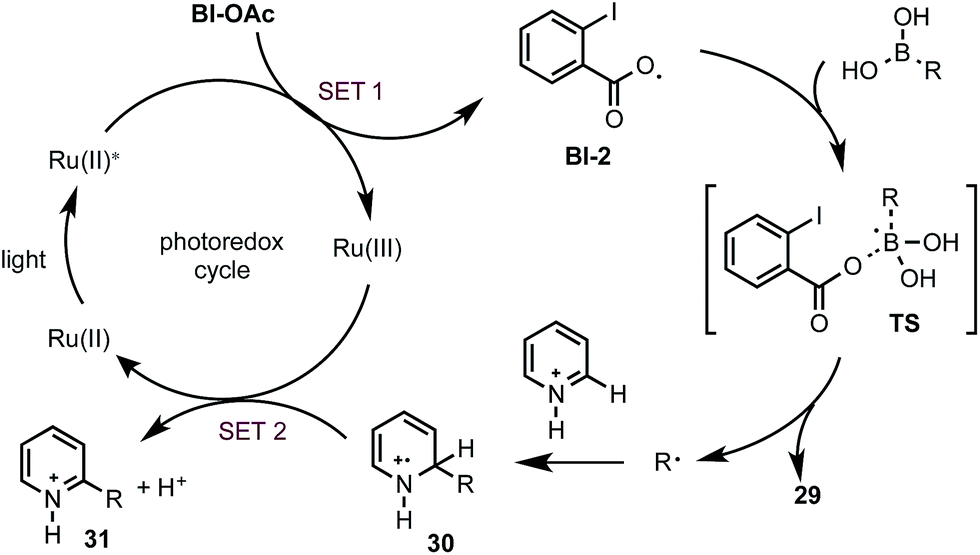 | ||
| Scheme 5 Proposed mechanism. | ||
Conclusions
In summary, we have developed a photoredox-mediated Minisci C–H alkylation reaction of N-heteroarenes with easily accessible alkyl boronic acids. A broad range of alkyl groups, including challenging primary alkyl groups, can be readily incorporated into various N-heteroarenes with high efficiency under mild conditions. These reactions exhibit excellent substrate scope and functional group tolerance, and offer a broadly applicable method for the late-stage functionalization of complex substrates. Mechanistic studies have revealed that acetoxybenziodoxole serves as a facile precursor for an ortho-iodobenzoyloxy radical intermediate under photoredox catalysis. The unique property of this intramolecularly stabilized benzoyloxy radical might be critical for the efficient transformation of usually less reactive alkyl boronic acids to form alkyl radicals. Further mechanistic studies and application of benziodoxole reagents in other photoredox-mediated reaction systems are currently underway.Acknowledgements
We gratefully thank the State Key Laboratory of Elemento-Organic Chemistry at Nankai University and the University of Pittsburgh for financial support of this work.Notes and references
- (a) Bioactive heterocycles V, Topics in Heterocyclic Chemistry, ed. R. R. Gupta, Springer Verlag, New York, 2008, vol. 11 Search PubMed; (b) M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347 CrossRef CAS PubMed.
- (a) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173 RSC; (b) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS PubMed; (c) T. Bruckl, R. D. Baxter, R. Y. Ishihara and P. S. Baran, Acc. Chem. Res., 2012, 45, 826 CrossRef CAS PubMed; (d) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed.
- For selected reviews and examples on C–H alkylation of N-heteroarenes: (a) L. Ackermann, Chem. Commun., 2010, 46, 4866 RSC; (b) J. C. Lewis, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2007, 129, 5332 CrossRef CAS PubMed; (c) D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224 CrossRef CAS PubMed; (d) H. Schonherr and T. Cernak, Angew. Chem., Int. Ed., 2013, 52, 12256 CrossRef PubMed; (e) B. Xiao, Z.-J. Liu, L. Liu and Y. Fu, J. Am. Chem. Soc., 2013, 135, 616 CrossRef CAS PubMed; (f) Y. Schramm, M. Takeuchi, K. Semba, Y. Nakao and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 12215 CrossRef CAS PubMed; (g) L. Bering and A. P. Antonchick, Org. Lett., 2015, 17, 3134 CrossRef CAS PubMed.
- (a) F. Minisci, E. Vismara and F. Fontana, Heterocycles, 1989, 28, 489 CrossRef CAS; (b) M. A. J. Duncton, Med. Chem. Commun., 2011, 2, 1135 RSC.
- (a) I. B. Seiple, S. Su, R. A. Rodriguez, R. Gianatassio, Y. Fujiwara, A. L. Sobel and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 13194 CrossRef CAS PubMed; Baran also demonstrated alkylation of 1,4-quinone with alkyl boronic acids under Ag(I)/S2O82− conditions, see: (b) Y. Fujiwara, V. Domingo, I. B. Seiple, R. Gianatassio, M. D. Bel and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 3292 CrossRef CAS PubMed.
- (a) G. A. Molander, V. Colombel and V. A. Braz, Org. Lett., 2011, 13, 1852 CrossRef CAS PubMed; (b) M. Presset, N. Fleury-Bregeot, D. Oehlrich, F. Rombouts and G. A. Molander, J. Org. Chem., 2013, 78, 4615 CrossRef CAS PubMed.
- For selected recent examples of Minisci alkylation: (a) C. A. Correia, L. Yang and C.-J. Li, Org. Lett., 2011, 13, 4581 CrossRef CAS PubMed; (b) Y. Fujiwara, J. A. Dixon, F. O'Hara, E. D. Funder, D. D. Dixon, R. A. Rodriguez, R. D. Baxter, B. Herle, N. Sach, M. R. Collins, Y. Ishihara and P. S. Baran, Nature, 2012, 492, 95 CrossRef CAS PubMed; (c) A. P. Antonchick and L. Burgmann, Angew. Chem., Int. Ed., 2013, 52, 3267 CrossRef CAS PubMed; (d) R. Xia, M.-S. Xie, H.-Y. Niu, G.-R. Qu and H.-M. Guo, Org. Lett., 2014, 16, 447 Search PubMed; (e) W.-M. Zhao, X.-L. Chen, J.-W. Yuan, L.-B. Qu, L.-K. Duan and Y.-F. Zhao, Chem. Commun., 2014, 50, 2018 RSC; (f) R.-J. Tang, L. Kang and L. Yang, Adv. Synth. Catal., 2015, 357, 2055 CrossRef CAS; (g) T. McCallum and L. Barriault, Chem. Sci., 2016, 7, 4754 RSC.
- D. A. DiRocco, K. Dykstra, S. Krska, P. Vachal, D. V. Conway and M. Tudge, Angew. Chem., Int. Ed., 2014, 53, 4802 CrossRef CAS PubMed.
- (a) J. Jin and D. W. C. MacMillan, Nature, 2015, 525, 87 CrossRef CAS PubMed; (b) J. Jin and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 1565 CrossRef CAS PubMed.
- (a) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58 CrossRef CAS PubMed; (b) T. Koike and M. Akita, Org. Biomol. Chem., 2016 10.1039/C6OB00996D.
- Y. Yasu, T. Koike and M. Akita, Adv. Synth. Catal., 2012, 354, 3414 CrossRef CAS.
- (a) H. Huang, K. Jia and Y. Chen, Angew. Chem., Int. Ed., 2015, 54, 1881 CrossRef CAS PubMed; (b) H. Huang, G. Zhang, L. Gong, S. Zhang and Y. Chen, J. Am. Chem. Soc., 2014, 136, 2280 CrossRef CAS PubMed.
- (a) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433 CrossRef CAS PubMed; (b) M. El Khatib, R. A. M. Serafim and G. A. Molander, Angew. Chem., Int. Ed., 2016, 55, 254 CrossRef CAS PubMed.
- Y. Wang, G.-X. Li, G. Yang, G. He and G. Chen, Chem. Sci., 2016, 7, 2679 RSC.
- For other examples of using benziodoxole reagents in visible light-mediated reactions: (a) S. A. Moteki, A. Usui, S. Selvakumar, T. Zhang and K. Maruoka, Angew. Chem., Int. Ed., 2014, 53, 11060 CrossRef CAS PubMed; (b) H. Tan, H. Li, W. Ji and L. Wang, Angew. Chem., Int. Ed., 2015, 54, 8374 CrossRef CAS PubMed; (c) Q.-Q. Zhou, W. Guo, W. Ding, X. Wu, X. Chen, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 11196 CrossRef CAS PubMed.
- For select reviews on hypervalent iodine chemistry: (a) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328 CrossRef CAS PubMed; (b) H. Tohma and Y. Kita, Adv. Synth. Catal., 2004, 346, 111 CrossRef CAS; (c) R. Narayan, S. Manna and A. P. Antonchick, Synlett, 2015, 26, 1785 CrossRef CAS; For selected reviews on benziodoxole chemistry: (d) V. V. Zhdankin, Curr. Org. Synth., 2005, 2, 121 CrossRef CAS; (e) Y. Li, D. P. Hari, M. V. Vita and J. Waser, Angew. Chem., Int. Ed., 2016, 55, 4436 CrossRef CAS PubMed.
- For selected reviews on visible light-mediated organic reactions: (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (b) T. P. Yoon, M. A. Ischay and J. N. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed; (c) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (d) J. Xie, H. Jin, P. Xu and C. Zhu, Tetrahedron Lett., 2014, 55, 36 CrossRef CAS.
- H. Togo, Advanced Free Radical Reactions for Organic Synthesis, Elsevier, 2004 Search PubMed.
- (a) M. J. Frisch, et al., DFT calculations were performed using Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed. Geometries were optimized at the M06-2X/6-31+G(d)-SDD level of theory in the gas phase. Single point energies were calculated at the M06-2X/6-311++G(d,p)-SDD level with the SMD solvation model in HFIP. See ESI† for computational details. For recent computational studies on photoredox mediated C–C bond formation reactions, see: (b) O. Gutierrez, J. C. Tellis, D. N. Primer, G. A. Molander and M. C. Kozlowski, J. Am. Chem. Soc., 2015, 137, 4896 CrossRef CAS PubMed; (c) T. B. Demissie, K. Ruud and J. H. Hansen, Organometallics, 2015, 34, 4218 CrossRef CAS; (d) X. Pan, C. Fang, M. Fantin, N. Malhotra, W. Y. So, L. A. Peteanu, A. A. Isse, A. Gennaro, P. Liu and K. Matyjaszewski, J. Am. Chem. Soc., 2016, 138, 2411 CrossRef CAS PubMed.
- (a) Extreme caution should be used for preparation and handling of 27: J. E. Leffler, R. D. Faulkner and C. C. Petropoulos, J. Am. Chem. Soc., 1958, 80, 5435 CrossRef CAS; (b) In comparison with 27, Bl-OAc is very stable and can be readily prepared in two steps from ortho-iodobenzoic acid: P. Eisenberger, S. Gischig and A. Togni, Chem.–Eur. J., 2006, 12, 2579 CrossRef CAS PubMed.
-
(a) The experimental redox potential of
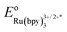 (−0.81 V vs. SCE in MeCN) was used in the computation of the SET reaction energy with Ru(II)*. See: C. R. Bock, T. J. Meyer and D. G. Whitten, J. Am. Chem. Soc., 1975, 97, 2909 CrossRef CAS;
(b) Dissociative single electron transfer (DSET) processes to directly form Bl-2 and AcO−Bl-3 and AcO˙ are also possible. For DFT calculations of redox potentials, see:
(c) P. Winget, C. J. Cramer and D. G. Truhlar, Theor. Chem. Acc., 2004, 112, 217 CrossRef CAS;
(d) A. A. Isse, C. Y. Lin, M. L. Coote and A. Gennaro, J. Phys. Chem. B, 2011, 115, 678 CrossRef CAS PubMed;
(e) H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714 CAS.
(−0.81 V vs. SCE in MeCN) was used in the computation of the SET reaction energy with Ru(II)*. See: C. R. Bock, T. J. Meyer and D. G. Whitten, J. Am. Chem. Soc., 1975, 97, 2909 CrossRef CAS;
(b) Dissociative single electron transfer (DSET) processes to directly form Bl-2 and AcO−Bl-3 and AcO˙ are also possible. For DFT calculations of redox potentials, see:
(c) P. Winget, C. J. Cramer and D. G. Truhlar, Theor. Chem. Acc., 2004, 112, 217 CrossRef CAS;
(d) A. A. Isse, C. Y. Lin, M. L. Coote and A. Gennaro, J. Phys. Chem. B, 2011, 115, 678 CrossRef CAS PubMed;
(e) H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714 CAS. - (a) V. V. Zhdankin, A. P. Krasutsky, C. J. Kuehl, A. J. Simonsen, J. K. Woodward, B. Mismash and J. T. Bolz, J. Am. Chem. Soc., 1996, 118, 5192 CrossRef CAS; (b) M. Ochiai, T. Ito, H. Takahashi, A. Nakanishi, M. Toyonari, T. Sueda, S. Goto and M. Shiro, J. Am. Chem. Soc., 1996, 118, 7716 CrossRef CAS; (c) F. Le Vaillant, T. Courant and J. Waser, Angew. Chem., Int. Ed., 2015, 54, 11200 CrossRef CAS PubMed; (d) K. Jia, F. Zhang, H. Huang and Y. Chen, J. Am. Chem. Soc., 2016, 138, 1514 CrossRef CAS PubMed; (e) Ref. 15c and 20a .
- The length of I–O bond in known carboxyl benziodoxoles typically lie in the range of 2.1 to 2.2 Å. see: (a) J. Z. Gougoutas and L. Lessinger, J. Solid State Chem., 1974, 9, 155 CrossRef CAS; (b) Ref. 12a and 16a ; (c) An unusually long I–O bond of 2.48 Å was observed in an arylbenziodoxle compound; the I–O interaction was described as a weak primary bond or strong intramolecular secondary bond: R. J. Batchelor, T. Birchall and J. F. Sawyer, Inorg. Chem., 1986, 25, 1415 CrossRef CAS.
- Bl-2 is probably best described as a resonance structure between the O-centered and I-centered radicals.
- No detectable C–H ortho-iodoarylated side products were observed in our alkylation reaction system.
- Although the intramolecular I−O interaction stabilizes Bl-2, there is still sufficient radical character on the O atom that manifests similar reactivity as that of typical O-centered radicals.
- (a) C. Ollivier and P. Renaud, Chem. Rev., 2001, 101, 3415 CrossRef CAS PubMed; (b) A.-P. Schaffner and P. Renaud, Eur. J. Org. Chem., 2004, 2291 CrossRef CAS; (c) C. Carra and J. C. Scaiano, Eur. J. Org. Chem., 2008, 4454 CrossRef CAS; (d) D. Liu, Y. Li, X. Qi, C. Liu, Y. Lan and A. Lei, Org. Lett., 2015, 17, 998 CrossRef CAS PubMed.
- For a leading review on homolytic substitution reaction: A. G. Davies and B. P. Roberts, Acc. Chem. Res., 1972, 5, 387 CrossRef CAS.
- The control experiment in Scheme 4A suggest that the reaction of alkyl boronic acids and trifluobororates might proceed with different mechanisms under our reaction conditions. It is plausible that Ru(III) formed via the oxidation by Bl-OAc might react with the easily oxidizable alkyl trifluoroborates to give the alkyl radical and Ru(II). Such process has been proposed in the previous studies of Bl-mediated photoredox-catalyzed reaction system using alkyl trifluoroborates (ref. 12). Formation of alkyl radical via SET oxidation of alkyl trifluoroborates by Ru(III) or Ir(x) have been proposed in other photoredox-mediated system, see ref. 11 and 13. As seen in the control experiment with benzoyl peroxide, benzoyloxy radical might also be able to react with σ complex 30via H-abstraction to form the final alkylated product and close the photoredox cycle.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc02653b |
| This journal is © The Royal Society of Chemistry 2016 |