
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular control over the structural organization of a second-order NLO-active organogelator†
Fátima
Aparicio
,
Lara
Faour
,
Denis
Gindre
 ,
David
Canevet
* and
Marc
Sallé
*
,
David
Canevet
* and
Marc
Sallé
*
Laboratoire MOLTECH-Anjou, Université d'Angers, UMR CNRS 6200, 2 bd Lavoisier, 49045 Angers Cedex, France. E-mail: david.canevet@univ-angers.fr; marc.salle@univ-angers.fr
First published on 27th September 2016
Abstract
A study of the structural parameters which govern the supramolecular organization of an organogelator built from the Disperse Red moiety is proposed. In particular, the key balance between intermolecular H-bonding and/or π–π interactions is addressed by comparing the effect of a secondary amide vs. an ester linker within the molecular structure. Solution 1H-NMR studies show the superiority of the former interaction in promoting the nanostructuring process, allowing it to reach a gel state in toluene. The nanostructures obtained from both the amide and the ester derivatives were also studied in the solid state. In particular, the use of second-harmonic generation microscopy demonstrates that an anisotropic organization of the material can even be observed in the case of the ester derivative, which demonstrates the efficiency of the tris(alkoxy)benzene unit in directing the self-assembly process, independently of additional H-bond interactions.
Nonlinear optically (NLO) active materials have been developed over the last few decades given their interest in photonics1 or optoelectronics.2–4 In the case of second harmonic generation (SHG), a myriad of dipolar π-conjugated push–pull NLO-phores have been described so far.5,6 The non-centrosymmetric organization of the latter within the corresponding materials, which constitutes a requisite for observing a SHG response, can be satisfied through various approaches ranging from the use of chiral NLO-phores to the application of dipole alignment strategies (i.e., Langmuir–Blodgett films,7 self-assembled layers,8 specific matrices9 or corona poling).10 In this context, we recently reported a strategy based on the self-assembly of a urea-based organogelator.11 The latter allows for a spontaneous non-centrosymmetric arrangement of the chromophores, based on a favorable balance of supramolecular forces, without the need of any additional external mediation. However, dipolar chromophores exhibit a strong propensity to self-assemble in a centrosymmetric fashion because of antiparallel donor–acceptor intermolecular interactions. Therefore, maintaining an appropriate balance between supramolecular forces when designing a chromophore to be self-assembled remains a delicate challenge. In this context, studying new organogelators incorporating NLO-phores appears necessary in order to increase knowledge of the forces driving the self-assembly process, as well as for extending the scope of non-linear optical materials obtained by supramolecular polymerization. Herein, we address this issue through a comparative self-assembly study of two new NLO-active dipolar molecules 1 and 2 designed to promote gelation in organic solvents.
Our study began with a benchmark dipolar structure based on the Disperse Red 1 (DR1) unit, a well-known push–pull azobenzene reference compound for SHG. Note that azobenzene-based organogels have already been depicted and allow for a gel–sol transition upon irradiation.12 A tris(alkoxy)benzamide (compound 1) or a tris(alkoxy)benzoester (compound 2) unit was attached to the azobenzene moiety in order to promote supramolecular polymerization assisted by π–π interactions and/or hydrogen bonds or non-covalent interactions.
Compounds 1 and 2 (Scheme 1) were respectively synthesized from the previously reported tris(dodecyloxy)benzoic acid 3,13 which was reacted with amine 4,11 dicyclohexylcarbodiimide (DCC) and 4-(dimethylamino)pyridine (DMAP) (amide 1, 38% yield), or from the commercially available DR1, O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) and diisopropylethylamine (DIPEA) (ester 2, 43% yield). The chemical structures of all unreported compounds were satisfactorily established (see the ESI†).
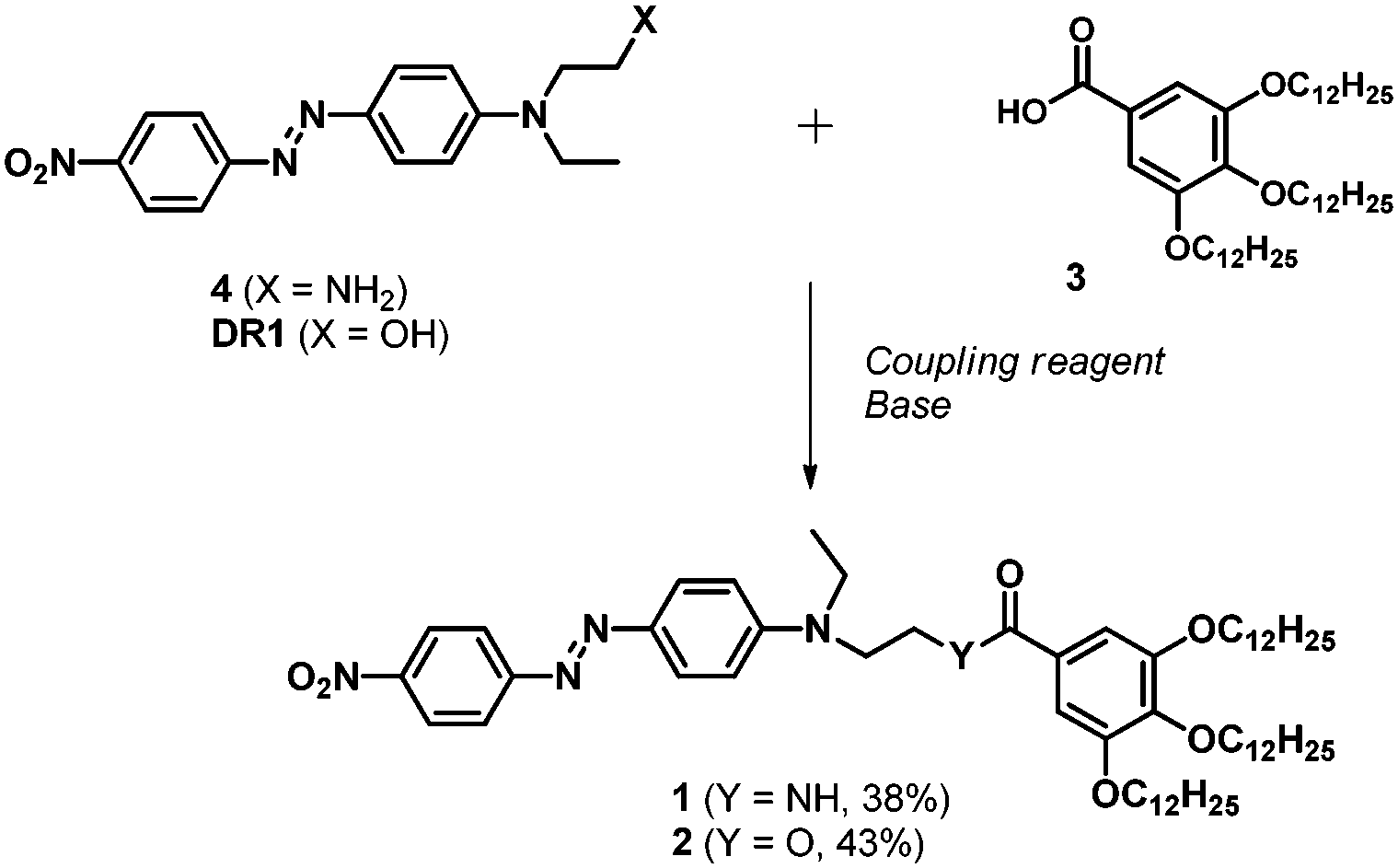 | ||
| Scheme 1 Synthesis of compounds 1 (DCC, DMAP) and 2 (HBTU, DIPEA). | ||
The first indication of the supramolecular polymerization of compound 1 comes from its gelation ability in toluene, with a critical gelification concentration (CGC) of 45 mg mL−1 (46 mmol L−1) (Fig. 1). In contrast, no gelation was observed in this solvent up to a 60 mg mL−1 concentration (62 mM) of compound 2, for which the amide function is replaced by an ester linkage.14 The CGC measured in alternative solvents confirm the general superiority of compound 1 in forming gels (Table S1, ESI†).
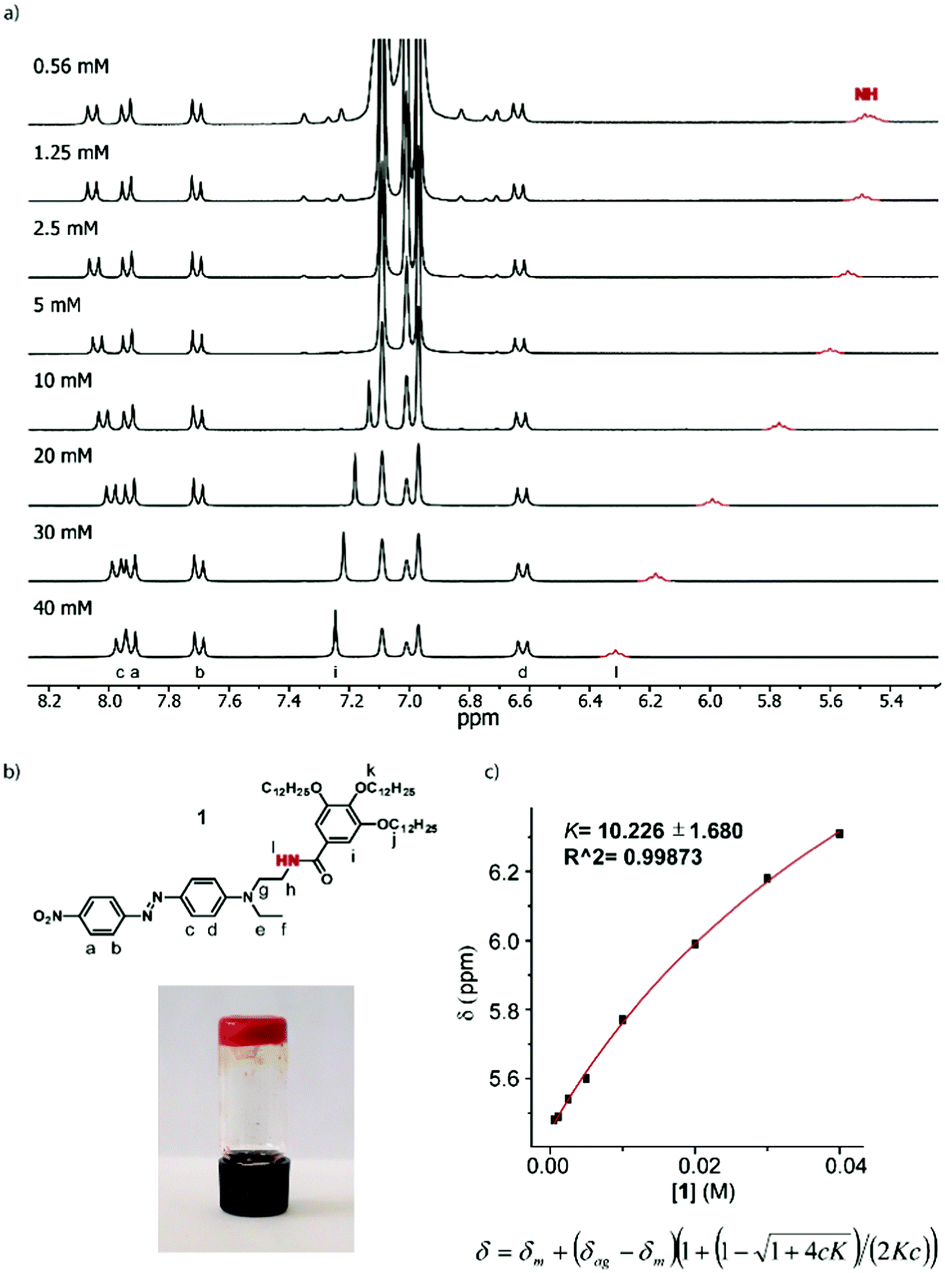 | ||
| Fig. 1 (a) Partial 1H NMR spectra (300 MHz, 298 K) of 1 at different concentrations in toluene-D8; the resonance in red corresponds to the N–H signal; (b) molecular structure and assignment of the corresponding protons (top) and the image of the gel formed by 1 in toluene at 45 mg mL−1 (bottom); (c) fit of the variation of the N–H chemical shift with concentration (top) and the equation for the isodesmic mechanism, where c and K are the concentrations of the amide and binding constant, respectively (bottom). | ||
NMR constitutes a valuable tool for evaluating the non-covalent forces driving supramolecular polymerization. Concentration-dependent 1H NMR experiments in toluene-D8 (Fig. 1 and Fig. S1, ESI†) nicely illustrate the aggregation of amide 1. These studies were carried out at concentrations ranging from 0.56 mM to 40 mM, i.e. just below the critical gelification concentration. Increasing the concentration within this concentration range leads to a clear deshielding of the N–H proton signal (in red, Fig. 1), assignable to the intermolecular H-bonding interactions taking place. Plotting the variation of the chemical shift against the concentration results in a non-linear behaviour (Fig. 1), which can be fitted to an equation accounting for an isodesmic mechanism,15,16 affording a low binding constant (K = 10). The intermolecular character of the N–H based hydrogen bond was confirmed using FTIR analysis, which shows a dilution-dependent shift of the N–H wavenumber value from 3285 cm−1 to 3270 cm−1 (Fig. S11, ESI†). Finally, some signals in the aromatic region are also impacted upon increasing the concentration (Fig. S1, ESI†). In particular, a significant deshielding of the tris(alkoxy)benzene signal in 1 (7.25 ppm, 40 mM) is observed, concomitantly to H-bond formation. This is explained by the modified electron density on the amide functional group engaged in a H-bond (i.e. upon increasing concentration), which results in a decreased electron density on the tris(alkoxy) benzene ring. Interestingly, a progressive change is also observed on the DR-based unit, but only for the hydrogen atoms located on the meta positions of the tertiary amine function. In this case, a high-field shift is observed from 8.05 ppm (0.56 mM) to 7.96 ppm (40 mM), which can be assigned to a π–π interaction involving the DR1 unit and which is promoted by the proximal H-bond interaction center. It is noteworthy that a similar study using the ester analogue 2 (Fig. S2, ESI†) shows only a very weak chemical shift variation, for the protons of the aminophenyl (DR) unit at 8.01 ppm (60 mM) or for the tris(alkoxy)benzene moiety at 7.49 ppm. Such an observation is consistent with the self-assembly process of compound 1, which is highly dominated by H-bond interactions, combined with π–π interactions. On the other hand, compound 2 only displays the latter type of interactions but at a much lower extent than 1.
Diffusion NMR spectroscopy is a valuable tool for evaluating the size of supramolecular aggregates.17–19 The diffusion coefficients (D) for both compounds 1 and 2 have been measured at different concentrations in toluene-D8 as the solvent (Fig. 2, Fig. S3–S5 and Table S2, ESI†). In both cases, upon increasing the concentration, the diffusion coefficient decreases (Fig. 2), which is consistent with a size increase of the corresponding aggregates. Both compounds exhibit similar D values at low concentrations (∼5.1 × 10−10 m2 s−1), as expected from their very similar structures and molecular sizes. On the other hand, in both cases, the curves converge to a plateau (∼4.3 × 10−10 m2 s−1 for 1 and ∼4.7 × 10−10 m2 s−1 for compound 2) at high concentrations, values which can therefore be considered as corresponding to the largest assemblies attainable under these conditions. Comparing those D values demonstrates that supramolecular structures from amide 1 are larger than those formed from the ester derivative 2.
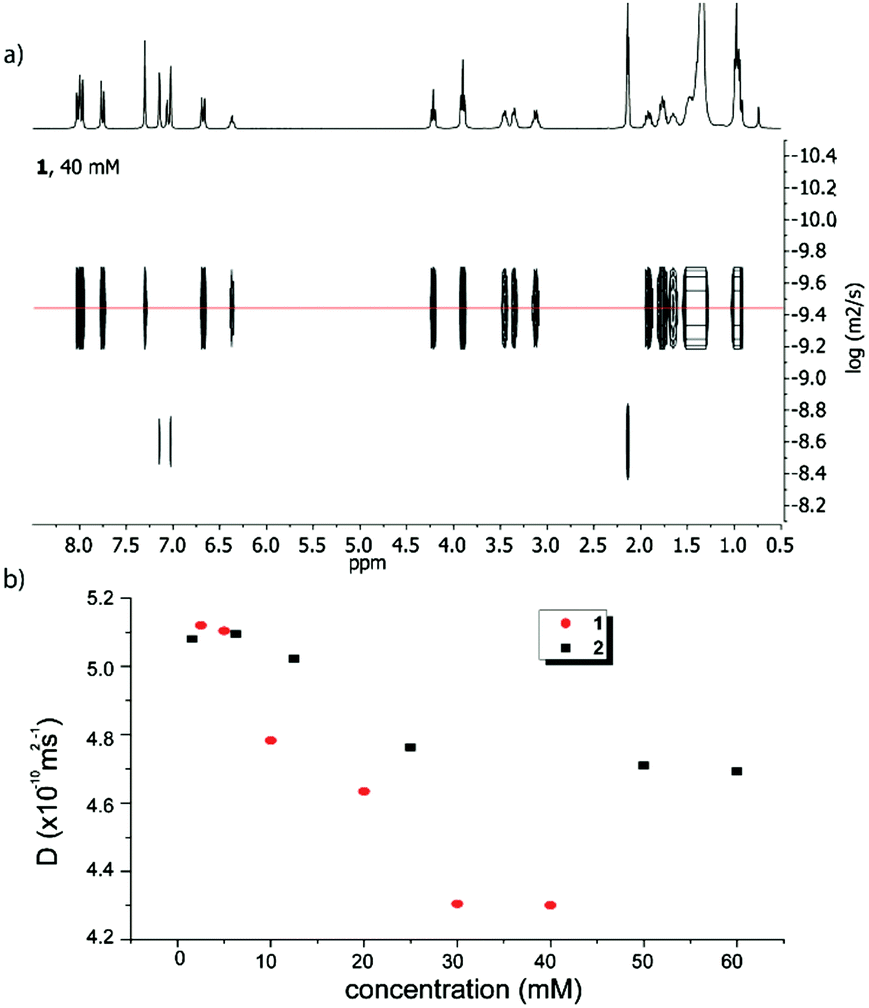 | ||
| Fig. 2 Top: DOSY NMR spectrum of 1 (toluene-D8, 40 mM); bottom: evolution of the diffusion coefficients of amide 1 (red circles) and ester 2 (black squares) as a function of the concentration. | ||
In order to gain a deeper insight into the structural parameters governing the molecular organization of aggregates 1 and 2 in solution, selective NOE experiments were carried out.20–22 To this end, highly concentrated toluene solutions (in which the biggest aggregates are obtained) of 1 and 2 as well as diluted solutions (in which only isolated molecules are present) were studied. Both structures 1 and 2 consist of three substituted benzene rings (namely, the “nitro-ring”, “amino-ring” and “alkoxy-ring” respectively), whose relative interactions have been evaluated. Two distinguishable behaviors are observed for compounds 1 and 2. Compound 2 recorded at a high concentration (61 mM) exhibits reciprocal NOE effects upon selective irradiation of protons “b” and “c” of the DR unit (Fig. 3b and Fig. S9 in the ESI†). It is noteworthy that when similar experiments are carried out under diluted conditions (1 mM) (Fig. S6 and S10, ESI†), these NOE effects are cancelled out, which unambiguously demonstrates the intermolecular nature of the interactions between an electron-rich “amino-ring” and an electron-poor “nitro-ring”, belonging to two distinctive DR units for concentrated solutions. On the other hand, the NOE contact found between protons “d” and “i” of 2 does not disappear upon dilution (Fig. S9 and S10, ESI†), which indicates an intramolecular contact between the “amino-ring” and the “alkoxy-ring” whatever the concentration. This series of NOE experiments suggests the aggregation of ester 2 through intermolecular interactions between the DR units combined with a folding of the molecule around the ester linkage with π–π interactions occurring between the “amino-ring” and the “alkoxy-ring”.
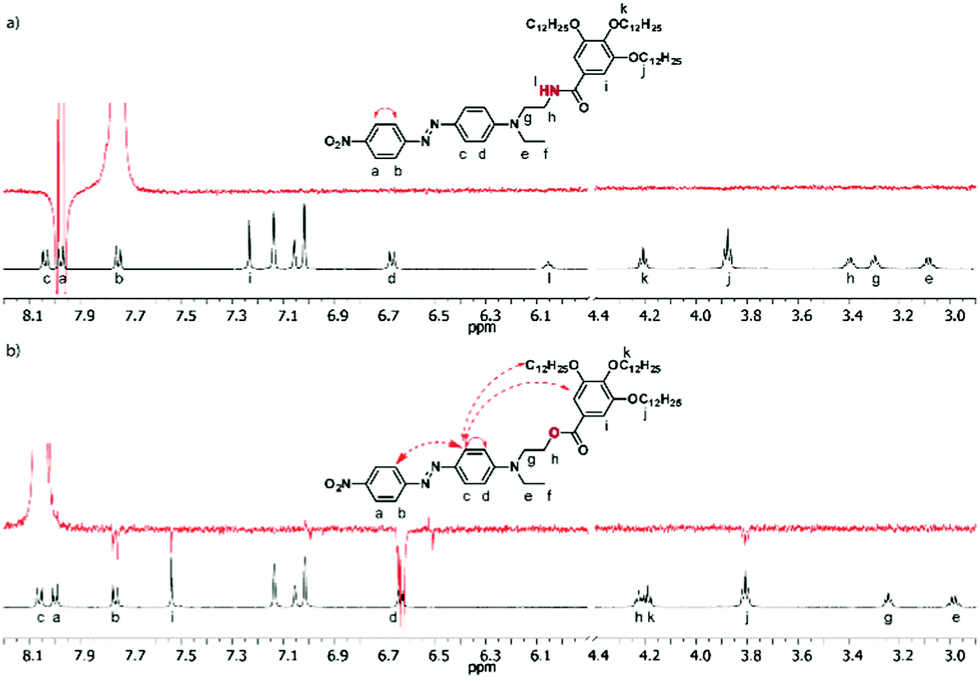 | ||
| Fig. 3 Partial 1H NMR spectra (300 MHz, 298 K, toluene-D8) (black) and NOE experiments (red) of 1 (28 mM) (a) and 2 (61 mM) (b). NOE irradiations were performed at 7.75 ppm (protons “b”, in the case of 1) and 8.06 ppm (protons “c”, in the case of 2). The solid arrows represent the NOE contacts for the intramolecular effects, and the dashed arrows represent the intermolecular effects. | ||
A significantly different behavior was observed from the NOE experiments with derivative 1. Although the irradiating proton “c” was not possible in a selective way in this case, the irradiation of proton “b” only shows a NOE effect with proton “a”, be that under concentrated or diluted conditions (Fig. 3a and Fig. S6a, ESI†). Unlike compound 2, no interaction was observed between the “amino-ring” and the “nitro-ring” protons. This fact indicates a different mode of aggregation for the amide derivative 1, which is indeed predominantly governed by H-bonds rather than by π–π interactions as in the case of 2.
The morphology of the supramolecular objects formed from both compounds was investigated using optical and scanning electron microscopy (SEM). Each derivative was deposited by drop casting onto a glass substrate at a concentration allowing it to reach the largest aggregates. The SEM and optical images show the presence of fibers in both cases (Fig. 4 and Fig. S12 and S13, ESI†), but which are significantly different in their size and shape. The fibers appear larger for compound 1, which is consistent with the above-mentioned diffusion NMR experiments. In addition, they correspond to a network of long coaxially organized fibers as seen clearly from the SEM images (Fig. 4 and Fig. S12, ESI†). On the other hand, the supramolecular aggregates issued from ester 2 are organized in microdomains of more densely packed materials (Fig. S12 and S13, ESI†). Such structural organization is less favorable to allow entrapping of solvent molecules and accounts for the differences in the gelation ability of both compounds. In addition, one can note the occurrence of a birefringence state for both materials in polarized light optical microscopy (Fig. S13, ESI†), an observation which demonstrates the anisotropic organization of both materials.
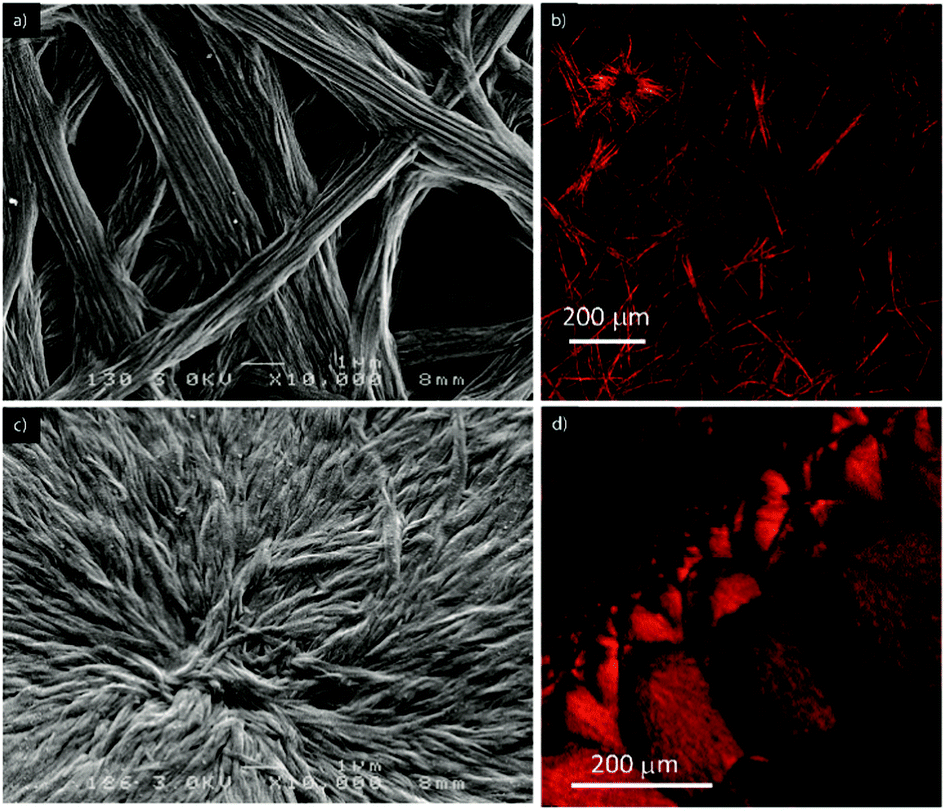 | ||
| Fig. 4 SEM (left) and SHG (right) micrographs of the fibers formed by the self-assembly of compound 1 (28 mM) (a and b) and 2 (61 mM) (c and d) in toluene. | ||
The same samples used for optical and SEM microscopy were subsequently studied via second harmonic generation microscopy. This technique provides information regarding the SHG activity at the microscale level. The experimental setup for recording the SHG images was used as described previously.11 Both samples prepared from compounds 1 and 2 showed SHG responses in accordance with a local non-centrosymmetric organization and whose distribution is reminiscent of the anisotropic organization of the respective corresponding materials (Fig. 4b, d and 5; Fig. S14 and S15 and Movies S1 and S2, ESI†). Note that no change of the morphology of the fibrillar xerogel was detected when comparing the optical microscopy images from before and after irradiation. This is assigned to the more robust character of the xerogel state compared to the azobenzene-based organogels, which possibly undergo Z/E isomerization effects.12 It is worth noting from Fig. 5 that SHG is polarization dependent and consequently all objects cannot be SHG active simultaneously with the same intensity. This result appears particularly significant when one considers that no SHG signal was detected upon analyzing a film of the reference N,N-diethyl-4-(4-nitrophenylazo)aniline, i.e. devoid of auxiliary organizing substituents, drop-casted from a toluene solution.11
 | ||
| Fig. 5 Evolution of the SHG response of compound 1 in toluene as a function of the linear polarization angle of the excitation laser beam. The polarisation angle is indicated at the top left corner of each picture. The evolution of the SHG response with the rotation of the polarization plane is shown in Movie S1 (ESI†). | ||
Even though no gelation could be observed with compound 2, this observation illustrates the prominent role of the tris(alkoxy)benzamide and tris(alkoxy)benzoester units in directing the self-assembly process.
Conclusion
In summary, two compounds incorporating a secondary amide and an ester linker respectively and derived from the benchmark SHG-active Disperse Red unit were synthesized. A gel could only be observed in the case of the amide derivative 1, indicating the prominent role of intermolecular H-bond interactions along with the supramolecular polymerization process. Importantly, an anisotropic organization of the material could be characterized using second harmonic generation microscopy in both cases, showing that even compound 2, which does not display intermolecular H-bond interactions, can support a local non-centrosymmetric organization thanks to the contribution of tris(alkoxy)benzene units.Acknowledgements
The authors sincerely acknowledge Julia Buendía and Prof. Luis Sánchez, and also the members of CAI RMN of Facultad de Ciencias Químicas from Universidad Complutense de Madrid for their help with the NOE experiments. The Region Pays-de-la-Loire is also acknowledged for a LUMOMAT postdoctoral fellowship, and the authors are grateful to the PIAM and the SCIAM platforms for their help.Notes and references
- G. Zerbi, Organic Materials for Photonics: Science and Technology, Elsevier, 1993 Search PubMed.
- P. N. Prasad and D. R. Ulrich, Nonlinear optical and electro-active polymers, Plenum Press, New York, 1988 Search PubMed.
- Y. Zhang, J. Ortega, U. Baumeister, C. L. Folcia, G. Sanz-Enguita, C. Walker, S. Rodriguez-Conde, J. Etxebarria, M. J. O'Callaghan and K. More, J. Am. Chem. Soc., 2012, 134, 6298–16306 Search PubMed.
- K. Iliopoulos, O. Krupka, D. Gindre and M. Sallé, J. Am. Chem. Soc., 2010, 132, 14343–14345 CrossRef CAS PubMed.
- F. Bures, RSC Adv., 2014, 4, 58826–58851 RSC.
- A. V. Kulinich and A. A. Ischchenko, Russ. Chem. Rev., 2009, 78, 141 CrossRef CAS.
- G. J. Ashwell, R. C. Hargreaves, C. E. Baldwin, G. S. Bahra and C. R. Brown, Nature, 1992, 357, 393–395 CrossRef CAS.
- W. Lin, W. Lin, G. K. Wong and T. J. Marks, J. Am. Chem. Soc., 1996, 118, 8034–8042 CrossRef CAS.
- J. Yu, Y. Cui, C. Wu, Y. Yang, Z. Wang, M. O'Keeffe, B. Chen and G. Qian, Angew. Chem., Int. Ed., 2012, 51, 10542–10545 CrossRef CAS PubMed.
- L. R. Dalton, P. A. Sullivan and D. H. Bale, Chem. Rev., 2010, 110, 25–55 CrossRef CAS PubMed.
- A. B. Marco, F. Aparicio, L. Faour, K. Iliopoulos, Y. Morille, M. Allain, S. Franco, R. Andreu, B. Sahraoui, D. Gindre, D. Canevet and M. Sallé, J. Am. Chem. Soc., 2016, 138, 9025–9028 CrossRef CAS PubMed.
- C. Wang, Q. Chen, F. Sun, D. Zhang, G. Zhang, Y. Huang, R. Zhao and D. Zhu, J. Am. Chem. Soc., 2010, 132, 3092–3096 CrossRef CAS PubMed.
- J. P. A. Custers, M. C. Hersmis, J. Meuldijk, J. A. J. M. Vekemans and L. A. Hulshof, Org. Process Res. Dev., 2002, 6, 645–651 CrossRef CAS.
- D. Canevet, V. Repussard and M. Sallé, Asian J. Org. Chem., 2014, 3, 216–224 CrossRef CAS.
- R. B. Martin, Chem. Rev., 1996, 96, 3043–3064 CrossRef CAS PubMed.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- Y. Cohen, L. Avram and L. Frish, Angew. Chem., Int. Ed., 2005, 44, 520–554 CrossRef CAS PubMed.
- J. S. Valera, J. Calbo, R. Gomez, E. Orti and L. Sanchez, Chem. Commun., 2015, 51, 10142–10145 RSC.
- M. L. Pellizzaro, J. Fisher and A. J. Wilson, RSC Adv., 2013, 3, 3103–3108 RSC.
- T. Rehm, V. Stepanenko, X. Zhang, F. Wurthner, F. Grohn, K. Klein and C. Schmuck, Org. Lett., 2008, 10, 1469–1472 CrossRef CAS PubMed.
- R. Gavara, J. Llorca, J. C. Lima and L. Rodriguez, Chem. Commun., 2013, 49, 72–74 RSC.
- F. Aparicio and L. Sánchez, Chem. – Eur. J., 2013, 19, 10482–10486 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, 1H and 13C NMR, infrared absorption information, additional DOSY and NOE experiments, optical, electron and SHG micrographs and movies. See DOI: 10.1039/c6sm01836j |
| This journal is © The Royal Society of Chemistry 2016 |