
Superparamagnetic iron-doped nanocrystalline apatite as a delivery system for doxorubicin†
Michele
Iafisco
*a,
Christophe
Drouet
*b,
Alessio
Adamiano
a,
Patricia
Pascaud‡
b,
Monica
Montesi
a,
Silvia
Panseri
a,
Stephanie
Sarda
b and
Anna
Tampieri
a
aInstitute of Science and Technology for Ceramics (ISTEC), National Research Council (CNR), Via Granarolo 64, 48018 Faenza (RA), Italy. E-mail: michele.iafisco@istec.cnr.it; Tel: +39 0546699730
bCIRIMAT Carnot Institute, University of Toulouse, UMR CNRS/INPT/UPS 5085, Ensiacet, 4 allée Emile Monso, 31030 Toulouse Cedex 4, France. E-mail: christophe.drouet@ensiacet.fr; Tel: +33 0534323411
First published on 18th November 2015
Abstract
The development of non-toxic and biodegradable magnetic nanoparticles (NPs) that can be easily functionalized with drugs or biomolecules and employed, under magnetic fields, as targeted nano-carriers or components of scaffolds with on-demand functionalities, is a big challenge in the biomaterials research. In the present work, the feasibility of previously synthesized iron-doped superparamagnetic apatite (FeHA) NPs to bind and then release the anticancer drug doxorubicin (DOX) under an applied low-frequency pulsed electromagnetic field (PEMF) was investigated. The behavior of FeHA towards DOX has been compared to that of synthetic biomimetic apatite (HA) NPs prepared ad hoc with characteristics close to those of bone mineral. The DOX adsorption kinetics and isotherms on FeHA and HA were explored and fitted according to different mathematical models (Elovich, Sips and Freundlich) revealing enhanced uptake of DOX on FeHA than HA, due to the better interaction of the drug with the surface iron cations and formation of multi-molecular DOX assemblies. In the absence of the PEMF, the quantity of DOX released from HA was higher than that released from FeHA, in agreement with the lower affinity of DOX for HA than FeHA. Interestingly, in the presence of the PEMF, the extent of DOX released from FeHA after 3 and 6 days increased significantly. The higher DOX release from FeHA under PEMF can be explained by the mechanical shacking of superparamagnetic FeHA NPs breaking the bonding with the drug and allowing detachment of DOX assemblies from the NP surface. In vitro assays demonstrated that DOX loaded on HA and FeHA displayed cytotoxicity against the human osteosarcoma cell line (SAOS-2) at the same level as free DOX, for all the concentrations and time points tested. Confocal microscopy analyses showed that drug-loaded NPs were rapidly internalized within cells and released DOX, which accumulated in the nuclei where it exerted the desired cytotoxic activity.
1. Introduction
The use of nanosystems/nanoparticles (that will be described here by the generic abbreviation NPs) as drug carriers has been widely explored during the last 20 years mainly to precisely administer therapeutic molecules in specific parts of the body with the final aim of improving their efficacy (enhancing their concentration at the site of interest and preventing early degradation and clearance) and reducing side effects.1,2 Different types of NPs (metallic, inorganic, polymeric, micelles, liposomes, carbon nanotubes, etc.) have been evaluated as drug delivery systems particularly in cancer therapy, where serious drug side effects compromise the health of patients.3,4More recently, the use of magnetic NPs as drug carriers has strongly emerged since they could be manipulated upon application of magnetic fields.5,6 In particular, the drug attached to magnetic NPs could be efficiently remotely controlled and targeted in desired areas of the body using external magnetic fields.7 Moreover, magnetic NPs can also be employed for several other biomedical applications, such as cell labelling and separation,8 immunoassay,9 MRI contrast agents,10 and hyperthermia.11 Recently, they were also used to prepare scaffolds for bone tissue engineering.12,13 In fact, under magnetic fields, these scaffolds can provide on-demand release of drugs or biomolecules inducing osteogenesis and angiogenesis and at the same time magneto-mechanical stimulation on bone cells favoring proliferation, differentiation and bone healing.14,15
For biomedical applications, a required property of magnetic NPs is the superparamagnetism, which is the ability to be strongly magnetized (i.e., the alignment of all magnetic moments of atoms in parallel along the direction of magnetic fields) when they are exposed to low intensity magnetic fields, without having a remnant or residual magnetization when the magnetic fields are removed.16 This feature is necessary to avoid coagulation and agglomeration of NPs causing possible embolization of capillary vessels when they are used in vivo.17
So far, magnetic NPs employed for biomedical applications are mainly based on super-paramagnetic iron oxide nanoparticles (SPIONs), consisting in mixtures of magnetite, maghemite and hematite, which have been demonstrated successfully both in vitro and in vivo.18 SPIONs can be synthesized with different morphologies, chemical compositions and magnetic properties but some limitations still exist including a low magnetic moment and low cargo capacity.19 In addition, although SPIONs are approved for human in vivo use by the American Food and Drug Administration (FDA),20 several concerns about their cytotoxicity have emerged.21,22 In fact, their chronic administration can lead to the accumulation of a high quantity of iron in soft tissues and organs such as liver and kidney, causing an imbalance in their homeostasis and even cytotoxic and inflammatory effects.23–25
To overcome the limitations of SPIONs, new forms of biocompatible superparamagnetic NPs with a good magnetic moment, high drug loading capability and low iron content are strongly required.
Hydroxyapatite (HA), whose stoichiometric composition corresponds to Ca10(PO4)6(OH)2, is the most extensively used calcium phosphate phase in several biomedical fields because of its chemical similarity with the mineral phase of bone (composed of nanocrystalline non-stoichiometric carbonated apatites) and thanks to its the well-known properties like biocompatibility, biodegradability and pH-dependent dissolution.26 HA is mainly employed in bone tissue engineering in the form of scaffolds, injectable cements, coatings for metallic implants and in nanomedicine as NPs in suspension.26 Whether stoichiometric or not, HA is able to accommodate several substituting ions, while still maintaining its intrinsic structure.27,28 In particular, Ca2+ ions can be substituted to some extent by monovalent to tetravalent cations (e.g. from Na+, to Zn2+, Fe3+ and Ti4+), while PO43− can be replaced by divalent (CO32−, HPO42−), trivalent (AsO43−, VO43−) and tetravalent (SiO44−) anions.27,28 Recently Tampieri et al.29 have developed biocompatible and biodegradable ionic substituted-HA NPs endowed with superparamagnetic and hyperthermia features via doping HA with Fe2+/Fe3+ ions (denoted “FeHA”). Interestingly, even if the iron amount of FeHA NPs is significantly lower than that of maghemite and magnetite NPs, they are featured by comparable superparamagnetic properties and hyperthermia effect.29In vitro studies have also demonstrated that FeHA NPs (alone or combined with poly(L-lactic) acid or poly(ε-caprolactone)) positively influence osteoblast-like cells and mesenchymal stem cells viability.30–32
The aim of the present work was, in essence, to evaluate the ability of superparamagnetic FeHA NPs to interact with the drug doxorubicin (DOX) (known also as adriamycin) monitoring the drug uptake and then the release over the time both in the presence and absence of an applied low-frequency electromagnetic field. DOX is commonly used in chemotherapies due to its therapeutic potential against solid tumors.33 The chemotherapy based on DOX, however, is limited by dose-dependent cardiotoxic side effects that can potentially lead to heart failure.33,34 To avoid this problem, targeted drug delivery could provide therapeutically effective DOX release directly at the tumor site improving the treatment of cancers. Moreover, DOX has strong visible absorption and fluorescence emission, and therefore is one of the best molecules for pursuing the objective of this study, since its uptake and release from NPs can be easily monitored.35
The behavior of FeHA towards DOX has been compared to that of iron-free biomimetic apatite nanocrystals prepared “ad hoc” with characteristics very close to those of bone mineral (denoted HA). The latter material, which has already demonstrated high ability to bind and release several types of drugs such as tetracycline and bisphosphonates,36,37 can be viewed as a biomimetic “golden synthetic standard” for bone mineral. The adsorption of bioactive molecules on the surface of biomimetic apatite (as opposed to simple impregnation) allows the formation of stable bonding with the nano-carrier. These properties were attributed to the existence of a hydrated layer at the surface of biomimetic apatite nanocrystals containing mobile ions with a higher reactivity than those present at the surface of well-crystallized apatite. It thus allows a better control of the dose of the drug released for localized tailored therapeutic applications.38 Finally, the cellular uptake and cytotoxic activity of DOX-loaded FeHA and HA NPs were analyzed, as a proof of concept, in vitro in the human osteosarcoma (SAOS-2) cell line.
2. Materials and methods
2.1 Materials
Ammonium phosphate dibasic ((NH4)2HPO4, ACS reagent), calcium hydroxide (Ca(OH)2, ACS reagent), calcium nitrate tetrahydrate (Ca(NO3)·4H2O, BioXtra), dimethyl sulfoxide (DMSO, (CH3)2SO, for molecular biology), doxorubicin hydrochloride (C27H29NO11·HCl, DOX, European Pharmacopoeia (EP) reference standard), HEPES sodium salt (C8H17N2NaO4S, BioXtra), iron(II) chloride tetrahydrate (FeCl2·4H2O, ReagentPlus®12), iron(III) chloride hexahydrate (FeCl3·6H2O, ACS reagent), isopropyl alcohol ((CH3)2CHOH, ≥99.7%, FCC, FG), phosphoric acid (H3PO4, ≥85 wt% in H2O), nitric acid (HNO3, ACS reagent, 70%), potassium chloride (KCl, BioXtra) and sodium bicarbonate (NaHCO3, ACS reagent), thiazolyl blue tetrazolium bromide (MTT, C18H16BrN5S, BioReagent), trypan blue (C34H24N6O14S4Na4, BioReagent) were purchased from Sigma-Aldrich and used as-received without any further purification. Dulbecco's modified Eagle's medium (DMEM F12), fetal bovine serum (FBS), Dulbecco's phosphate buffered saline (DPBS) without calcium and magnesium, penicillin–streptomycin, trypsin–EDTA, and 4,6-diamidino-2-phenylindole dihydrochloride (DAPI) were purchased from Invitrogen Corporation (Carlsbad, USA) and used as-received without any further purification. The human osteosarcoma cell line SAOS-2 was purchased from ATCC (Manassas, USA). Ultrapure water (0.22 μS, 25 °C, Milli-Q, Millipore) was used in all the experiments.2.2 Synthesis of superparamagnetic iron-substituted nanocrystalline apatite (FeHA)
Superparamagnetic iron-substituted apatite (which will be referred to as “FeHA” in this study) was synthesized according to the method developed by Tampieri et al.29 Briefly, an aqueous solution of H3PO4 (44.4 g in 300 mL) was dropped into an aqueous suspension of Ca(OH)2 (50.0 g in 400 mL) containing FeCl2·4H2O (12.7 g) and FeCl3·6H2O (17.9 g) as sources of Fe2+ and Fe3+ ions, respectively, under constant heating and stirring at 40 °C. Once the neutralization reaction was completed, the solution was kept under the same conditions used during the neutralization reaction for 1 h, and then left to age for 24 h at room temperature without further stirring. The precipitate was separated from the mother liquor by centrifugation, washed three times with water by centrifugation and freeze-dried. The powder was then sieved (<125 μm) and stored in a freezer prior to use.2.3 Preparation of iron-free biomimetic nanocrystalline apatite (HA)
The iron-free nanocrystalline apatite powder (which will be referred to as “HA” in this study) was prepared by a double decomposition technique at ambient temperature and close-to-physiological pH (7.47) by pouring an aqueous solution of Ca(NO3)·4H2O (52.2 g in 750 mL) into a carbonate and phosphate aqueous solution (165.8 g of (NH4)2HPO4 and 9.0 g of NaHCO3 in 1500 mL). The precipitate obtained was left to mature for one day at 50 °C, filtered, washed three times with deionized water and freeze-dried. The powder was then sieved (<125 μm) and stored in a freezer prior to use.2.4 Characterization of HA and FeHA
The X-ray diffraction (XRD) patterns of the samples were recorded on a D8 Advance diffractometer (Bruker, Karlsruhe, Germany) equipped with a Lynx-eye position sensitive detector using Cu Kα radiation (λ = 1.54178 Å) generated at 40 kV and 40 mA. XRD spectra were recorded in the 2θ range from 10 to 60° with a step size (2θ) of 0.02° and a counting time of 0.5 s. The crystallite sizes of HA and FeHA along the c-axis and the perpendicular direction were estimated by applying Scherrer's formula39 using 2θ = 25.8° and 2θ = 39.7° diffraction peaks, corresponding to the (002) and (310) reflections, respectively.Fourier transform infrared (FTIR) spectroscopy analyses were achieved on a Nicolet 5700 spectrometer (Thermo Fisher Scientific Inc., Waltham, USA) with a resolution of 4 cm−1 by accumulation of 64 scans covering the 4000 to 400 cm−1 range, using the KBr pellet method.
Calcium, phosphate and iron contents were determined by inductively coupled plasma optical emission spectrometry (ICP-OES) using a Liberty 200 spectrometer (Varian, Palo Alto, USA). An aliquot of 20 mg of sample was dissolved in 50 mL of a 1 wt% HNO3 solution prior the analysis.
Carbonate titration was carried out by coulometry using a CM 5014 coulometer equipped with a CM 5130 acidification unit (UIC Inc., Joliet, USA). All chemical titrations were associated with a relative error of about 1%. A BELSORP mini II apparatus (Microtrac, Krefeld, Germany) (BET method based on nitrogen adsorption) was used to determine the specific surface area (SSABET) of the samples.
The size and ζ-potential were measured by dynamic light scattering (DLS) using a Zetasizer Nano ZS (Malvern, Worcestershire, UK). NPs were suspended in 0.01 M HEPES buffer at pH 7.4 at a concentration of 0.1 mg mL−1. For size distribution measurements, low-volume quartz cuvettes (Hellma, Müllheim, Germany) were used. Ten runs of 30 s were performed for each measurement and four measurements were carried out for each sample. To evaluate the stability of apatite NPs and the corresponding DOX adducts in suspension, the derived count rate (cps) was recorded continuously for 60 min. The ζ-potential was quantified by laser doppler velocimetry as electrophoretic mobility using disposable electrophoretic cells (Malvern, Worcestershire, UK). Twenty runs of 3 s were collected in each measurement.
Transmission electron microscopy (TEM) observations were performed using a Tecnai F20 (FEI, Hillsboro, USA) equipped with a Schottky emitter and operating at 120 keV. The instrument was also equipped with an energy dispersive X-ray spectrometer (EDX) for X-ray microanalysis on selected areas. The powder samples were dispersed in isopropyl alcohol, treated with ultrasound and then a few droplets of the slurry were deposited on holey-carbon foils supported on a conventional copper microgrid (300 mesh).
2.5 Drug adsorption and release experiments
DOX adsorption experiments on apatitic substrates (HA and FeHA) were carried out using a constant solid-to-solution ratio corresponding to 20 mg of apatite in 5 mL of DOX solution at increasing concentrations (0.1–5.0 mg mL−1). The amount of DOX adsorbed (Qads) for any data-point was determined by comparing the amount of DOX remaining in solution after the adsorption experiment (supernatant) to the initial concentration; DOX being directly titrated by visible spectrophotometry (absorption at λ = 496 nm) using a Cary Bio spectrophotometer (Varian, Palo Alto, USA). KCl 0.01 M was used as a solvent in all experiments to provide a nearly constant ionic strength. A calibration curve (linear correlation factor of 0.9998) was previously drawn for standard solutions of DOX concentrations ranging from 0.001 to 0.025 mg mL−1.Adsorption experiments were carried out at 37 °C, placing vials in a horizontal setting to ensure an optimal interaction between the surface of the apatite substrates and the solution. After the contact time was determined in a preliminary step (see the text), the system was centrifuged (5000 rpm for 5 min) to allow retrieval of the solid and sampling of the supernatant for quantification of DOX.
HA and FeHA functionalized with the maximum loading amount of DOX were analyzed by Raman micro-spectrometry, cross polarization magic angle spinning (CP/MAS) solid-state nuclear magnetic resonance (NMR), FTIR and DLS.
Raman analyses were carried out on a confocal Labram HR800 micro-spectrometer, typically making observations in the range 400–4000 cm−1. The samples were exposed in backscattering mode to a 633 nm He/Ne laser. The measurements were carried out with a spectral resolution of 3 cm−1. An optical objective ×100 was used, conferring to the system a lateral resolution of 0.7 μm and an axial resolution of 2.7 μm.
Solid-state cross-polarization magic angle spinning carbon-13 nuclear magnetic resonance experiments (CP/MAS -NMR) were performed on a Bruker Avance III 400 WB spectrometer operating at 9.4 T, 400 MHz, with Vr = 10 kHz (probe 3.2 mm), and an accumulation performed overnight. 13C chemical shifts were externally referenced to tetramethylsilane (TMS).
FTIR and DLS analyses on the DOX loaded NPs were carried out as reported above for the un-functionalized NPs.
DOX release experiments from HA and FeHA (previously functionalized with the maximum loading amount of DOX) were carried out at 37 °C in the presence or absence of a pulsed electromagnetic field (PEMF). Investigations in the presence of the PEMF were achieved placing the powder samples in an home-made device similar to that previously developed by Fassina et al.40 (Fig. S1, ESI†). Briefly, it consisted of a windowed polymethylmethacrylate tube carrying a well-plate containing the samples and two solenoids (i.e., Helmoltz coils, whose planes were parallel). The generated magnetic field and the induced electric field were perpendicular and parallel to the samples, respectively. Samples were 5 cm distant from each solenoid plane, and a Biostim SPT pulse generator (Igea, Carpi, Italy) was used to power the solenoids. According to the position of the solenoids and the characteristics of the pulse generator, the electromagnetic stimulus had the following parameters: intensity of the magnetic field equal to 2.0 ± 0.2 mT, amplitude of the induced electric tension equal to 5 ± 1 mV, frequency of 75 ± 2 Hz, and pulse duration of 1.3 ms. The samples were prepared using a constant solid-to-solution ratio corresponding to 5 mg of drug–apatite conjugate in 2.5 mL of HEPES buffer at pH 7.4 (0.01 M HEPES, 0.01 M KCl). At the scheduled times ranging from 1 to 6 days, the temperature of the samples were measured (only those exposed to PEMF), centrifuged (5000 rpm for 5 min) to allow retrieval of the solid and sampling of the supernatant for quantification of DOX. The amount of DOX in the medium was determined by visible spectrophotometry as reported above.
Dissolution of HA and FeHA in the presence and in the absence of a PEMF (using the same apparatus described above) was evaluated adding 10 mg of apatite in 10 mL of HEPES buffer at pH 7.4 (0.01 M HEPES, 0.01 M KCl). At scheduled times (up to 7 days), the supernatant (that was well separated from the solid phase by centrifugation) was removed, centrifuged and filtered at 0.25 μm in view of Ca2+ quantification by ICP-OES.
2.6 In vitro cell culture
The human osteosarcoma cell line SAOS-2 was cultured in DMEM F12, 10% FBS and 100 U mL−1 penicillin/streptomycin and kept at 37 °C in an atmosphere of 5% CO2. The cells were detached from culture flasks by trypsinization, centrifuged and re-suspended. The cell number and viability were assessed by the trypan-blue dye exclusion test. For the experiments, the cells were plated at a density of 5 × 103 cells per well in 96-well plates, 24 hours after seeding the samples were added to the culture and the cells were maintained in culture for 24, 48 and 72 hours. HA and FeHA loaded with similar amounts of DOX (475 and 449 μg of DOX on 1 mg of HA and FeHA, respectively) were added to the culture at 10 and 100 μM concentration of attached DOX (Table S1, ESI†). Moreover, the equivalent amounts of un-functionalized HA and FeHA NPs used for each sample and free DOX were tested (Table S1, ESI†). A group of cells only was used as control group. The cells were incubated under standard conditions (37 °C, 5% CO2). All cell-handling procedures were performed in a sterile laminar flow hood.2.7 Cell viability evaluation
The MTT reagent (5 mg mL−1) was prepared in 0.01 M PBS. The cells were incubated with the MTT reagent 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 for 2 h at 37 °C. The medium was collected and cells incubated with 1 mL of DMSO for 15 min. In this assay, the metabolically active cells react with the tetrazolium salt in the MTT reagent to produce a formazan dye that can be observed at absorbance max of 570 nm, using a Multiskan FC microplate photometer (Thermo Scientific, Waltham, USA). This absorbance is directly proportional to the number of metabolically active cells. The mean value of absorbance was determined. The samples of three different experiments were analyzed in triplicate.
:10 for 2 h at 37 °C. The medium was collected and cells incubated with 1 mL of DMSO for 15 min. In this assay, the metabolically active cells react with the tetrazolium salt in the MTT reagent to produce a formazan dye that can be observed at absorbance max of 570 nm, using a Multiskan FC microplate photometer (Thermo Scientific, Waltham, USA). This absorbance is directly proportional to the number of metabolically active cells. The mean value of absorbance was determined. The samples of three different experiments were analyzed in triplicate.
2.8 NPs-DOX uptake and nuclear-localization
After 24 hours, the cells were washed twice with 0.01 M PBS for 5 min, fixed with 4% (w/v) paraformaldehyde for 15 min, washed with 0.01 M PBS for 5 min and incubated with DAPI for 7 min. The images were acquired using an inverted Ti-E fluorescence microscope (Nikon, Chiyoda, Japan). One representative field of each sample and the co-localization of the nuclear marker, DAPI, and DOX were analyzed.2.9 Statistical analysis
The experiments were carried out in triplicate and the results were expressed as mean and standard error of the mean (SEM). Analysis of cell viability was made by two-way analysis of variance (ANOVA), followed by Bonferroni's post hoc test. Statistical analyses were performed using the GraphPad Prism software (version 5.0), with the statistical significance set at p ≤ 0.05.3. Results and discussion
3.1 Chemical-physical and morphological characteristics of FeHA and HA
FeHA NPs exhibiting a saturation magnetization of 4.00 ± 0.01 Am2 Kg−1 and endowed with a hyperthermia feature (temperature increase of about 40 °C in 60 s under an alternating magnetic field of 0.03 N A−1 m−1 at a frequency of 293 kHz) were synthesized according to the procedure reported by Tampieri et al.29 Iron-free biomimetic HA NPs were synthesized ad hoc to possess chemical–physical features (i.e. crystallinity degree, chemical composition, carbonate extent and morphology) similar to those of biogenic bone apatites. An adequate amount of carbonate species (employing NaHCO3) were added in the reaction medium to synthesize HA with a similar carbonate extent to that present in bone apatite (3–6 wt%).26 Carbonate was also included during the preparation of HA as a strategy to favor the formation of small NPs with a low crystallinity degree since carbonate ions are well-known inhibitors of apatite crystal growth and its incorporation leads to a reduction of the crystalline order and dimensions.41,42Both FeHA and HA exhibited similar XRD patterns, with broad and poorly defined peaks (which can be indexed according to the crystallographic features of hydroxyapatite, JCPDS no. 09-432) which are characteristics of bone nanocrystalline apatite (Fig. 1A).43 The presence of the peak at 35.4° (corresponding to (311) plane of Fe3O4; JCPDS no. 00-003-0863) in the XRD pattern of FeHA established the presence of a small amount of magnetite (2.6 ± 0.2 wt%)29 (Fig. 1A). The mean crystallite sizes of FeHA and HA (Table 1), calculated along the (002) and (310) directions applying the Scherrer's formula, evidenced that both samples had the same crystallite dimension along the plane perpendicular to the c-axis, while FeHA was composed of more elongated crystallites along the c-axis with a higher aspect ratio than HA. The lower aspect ratio (i.e. more isometric particles) of HA compared to FeHA was ascribed to the higher presence of carbonate ions inside the crystal lattice as already reported.42,44
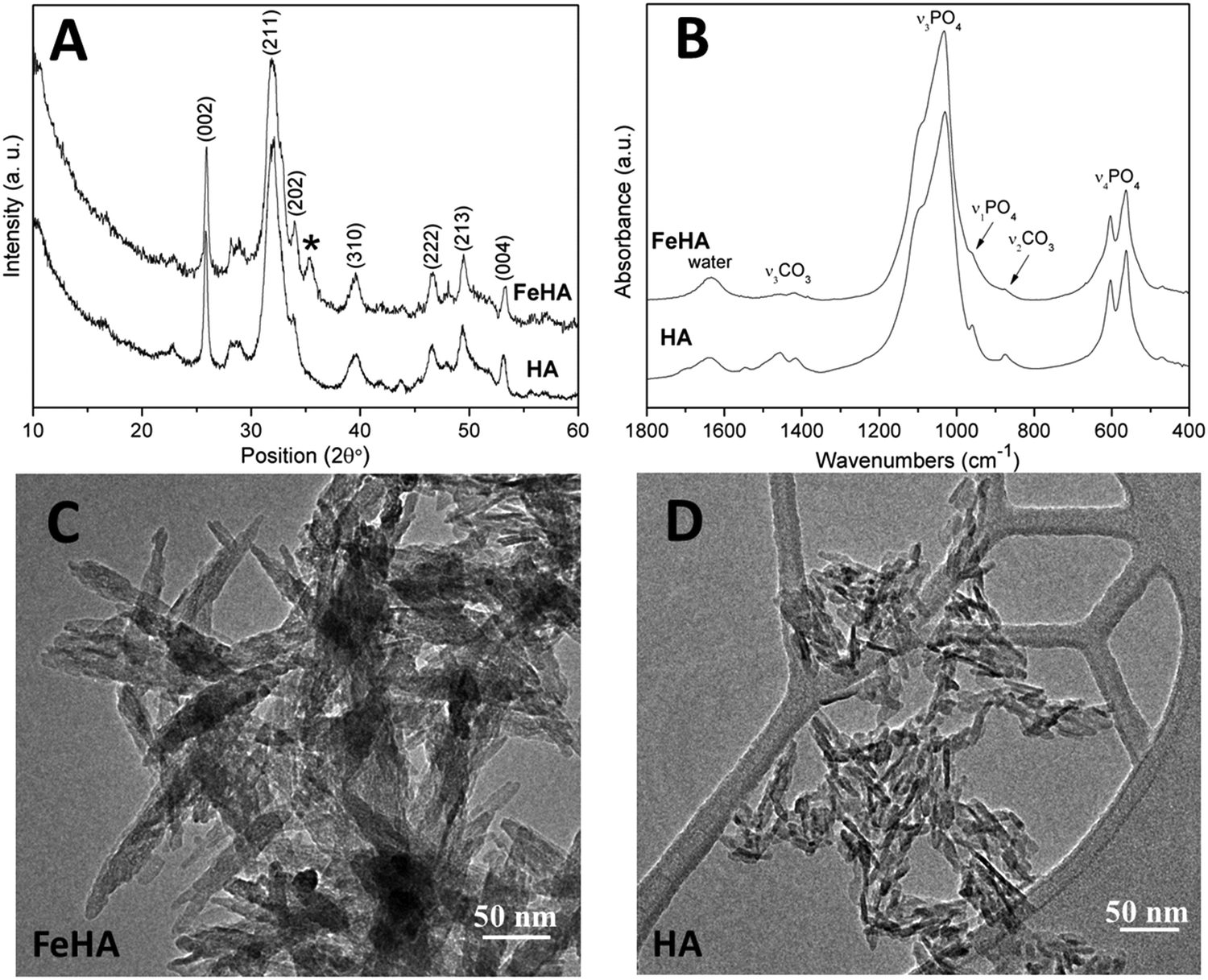 | ||
| Fig. 1 (A) XRD spectra, (B) FTIR spectra and (C and D) TEM images of FeHA and HA NPs. | ||
| Sample | Ca/P ratio | CO3 (wt%) | L 002 (nm) | L 310 (nm) | SSABET (m2 g−1) | ζ-potential (mV) | R H (nm) |
|---|---|---|---|---|---|---|---|
| FeHA | 1.41 ± 0.03 | 1.5 ± 0.1 | 18 ± 3 | 15 ± 3 | 44 ± 1 | −17.6 ± 1.0 | 120 ± 2 |
| HA | 1.34 ± 0.02 | 3.0 ± 0.1 | 31 ± 5 | 18 ± 3 | 102 ± 1 | −18.2 ± 1.3 | 74 ± 1 |
The FTIR spectra of HA and FeHA revealed the typical bands of poorly crystalline biomimetic apatite (i.e. PO43− vibration bands at ca. 560–603 (ν4), 962 (ν1), and 1000–1104 cm−1 (ν3))45 (Fig. 1B). The bands at 870 (ν2), 1420 and 1470 cm−1 (ν3) assignable to CO32− vibrations characteristic of mostly B-type carbonate-apatite (i.e., CO32− replacing PO43−)45 were also present in both samples. The bands assigned to CO32− were more intense in the case of HA as a more limited amount of carbonate, derived from the atmospheric CO2, was adsorbed on the surface and/or entrapped in the lattice of FeHA during the synthesis. The carbonate extent in the FeHA and HA was quantified to be 3.0 ± 0.1 and 1.5 ± 0.1 wt%, respectively (Table 1). The Ca/P ratio of FeHA was remarkably lower (1.41 ± 0.03) compared to that of stoichiometric hydroxyapatite (1.67) because of a partial substitution of Fe2+ and Fe3+ for Ca2+ ions. The Ca/P ratio of HA was also noticeably lower (1.34), pointing to the non-stoichiometry of this compound and its similarity with bone apatite.26
The TEM images of FeHA showed needle-like crystals, rather heterogeneous in size, having dimensions of 10–30 nm in width and 70–100 nm in length mainly composed of smaller aggregated and isometric NPs of about 5–10 nm in width and 10–20 nm in length (Fig. 1C). A very low concentration of dark spots (5–10 nm in size), corresponding to inclusions of Fe-rich phases as demonstrated by EDX analysis was also visible. EDX elementary analysis carried out in selected area of the FeHA sample containing and not containing the dark spots, revealed that the amount of iron in the area rich with black spots is much higher than that in the part without dark spots (Fig. S3, ESI†). The TEM images of HA displayed elongated NPs having dimensions of 5–10 nm in width and 20–30 nm in length (Fig. 1D). HA NPs were less aggregated than FeHA presenting a higher specific surface area (SSABET) (Table 1). The higher tendency of FeHA to form aggregates than HA was ascribed to the greater amount of foreign ions (i.e. Fe2+, Fe3+, CO32−) causing crystal lattice distortion, reduction of the long-range periodic regularity, and possible modifications of surface energy.46 The higher structural disorder can induce the formation of quasi-amorphous parts, especially at the surface, causing junction and adhesion of crystals by inter-crystalline interactions during the synthesis or post synthesis under drying. The similar surface negative charge of FeHA and HA at pH 7.4 (Table 1) was attributed to the existence of a hydrated surface layer (which is a peculiar feature of bone apatite and its biomimetic counterpart) composed of disordered Ca2+, PO43− and HPO42− ionic groups in non-apatitic structural environments, having an impact on the surface charge of the nanocrystals.47 However, it is worthwhile to mention that ζ-potential measurements only estimate the net surface charge of NPs, thus even if a negative value is recorded, this does not imply that only negatively charged sites are present on the NP surface. The mean hydrodynamic radius (RH), calculated by DLS, of FeHA was larger than HA (Table 1) in agreement with the higher tendency of FeHA to form aggregates.
The higher sizes determined by DLS in comparison to those quantified by TEM were consistent with the fact that TEM provides information on individual NPs and the sample is dehydrated and immobilized on a solid support, and in contrast DLS gives the average of the hydrodynamic size of an ensemble of dispersed NPs.48
All the chemical–physical characteristics of the apatitic NPs reported above highlighted the biomimetic features of HA thanks to the evident morphological and structural similarities with bone apatite. FeHA NPs were primarily different from HA NPs in terms of chemical structure, surface area and tendency to form aggregates, while they had comparable surface charge and morphology.
3.2 Adsorption of DOX on HA and FeHA
The adsorption study was organized in two steps aiming, firstly, at inspecting the kinetics of adsorption, and secondly, at determining the adsorption isotherms (at 37 °C).In the first stage, the adsorption kinetics of DOX on HA and FeHA substrates were investigated. The kinetic overview was made possible by following the amount of DOX adsorbed (Qads) as a function of contact time between the inorganic substrate and the drug. The evolution of Qads (given in μmol DOX m−2 apatite) versus time is plotted on Fig. 2 in the case of a constant DOX concentration of ca. 5 mmol L−1.
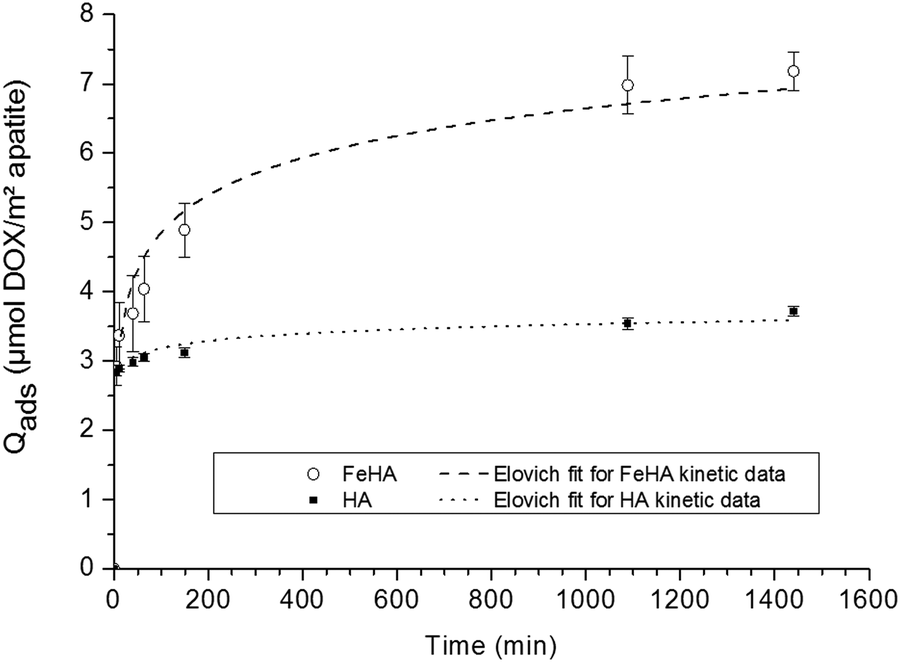 | ||
| Fig. 2 Adsorption kinetics of DOX on (○) FeHA and (■) HA NPs. Separate points are the experimental data; dotted lines indicate Elovich fits of kinetic data. | ||
These data indicated that in both cases the adsorbed amount increased as a function of time, although in different ranges, with a first rapid rise followed by a progressive stabilization: up to around 3.5 μmol DOX m−2 apatite (∼2 mg m−2) in the case of HA compared to a two-fold value of ca. 7 μmol m−2 (∼4 mg m−2) for FeHA. The shape of these kinetic curves points to a rather rapid adsorption of the DOX molecules, although faster for HA (around 200 min) than for FeHA (around 1000 min). The data have been tentatively fitted to the kinetic models often encountered, namely the pseudo-first-order and pseudo-second-order as well as a general kinetic law of order “n”, and finally to the Elovich model, in a similar way as was done recently in the study of tetracycline adsorption on biomimetic apatite.36 Only the latter led to satisfactory correlation coefficients (R2 greater than 0.97) with R2 = 0.9766 for FeHA and 0.9945 for HA. In contrast, kinetic models of order “n” (including with n = 1 and n = 2) led to uncorrelated data showing that the adsorption kinetics followed a more complex process. A relatively good fit (see dotted lines in Fig. 2) to the Elovich model, a logarithmic function of time, is often encountered for the adsorption of polyelectrolytes on heterogeneous surfaces.49,50
The molecular structure of DOX is represented in Fig. S2, ESI.† It is composed of an (aromatic) anthracycline ring linked to an amino-sugar: the pK value of the amino group was reported to be around 8.2,51,52 while other pK values of the molecule (typically for –OH groups) were found to be beyond 9.5. This indicates that, under physiological conditions, the DOX molecule exists mainly in its acidic form, that is with a protonated amino group. The multiple oxygen atoms of the molecule offer on the other hand many polar groups (especially thanks to the conjugation of several double bonds and non-bonding doublets), but the hydrogen atoms linked to the –OH groups of the molecule are expected to remain present at physiological pH (7.4). This leads to a situation where the DOX molecule exposes an –NH3+ group and several oxygen atoms providing multiple potential “interaction points” with apatite nanocrystals, despite the lack of anionic end-groups with well-known high affinity for apatitic surfaces such as phosphates, phosphonates or even carboxylates.53,54 This multiplicity of interaction sites on the DOX molecule may explain, in conjunction with the relative heterogeneity of the substrates, the relatively good fit with the Elovich equation and the absence of any satisfying correlation with other kinetic models.
The modeling of adsorption data over a long period of time however poses the potential issue of progressive modifications of non-stoichiometric apatitic substrates, which can further mature or else undergo dissolution or partial re-precipitation phenomena. In this study, we selected the value of 90 min as the contact time for the adsorption isotherms to limit the significant post-maturation effect in the solids and dissolution.
In the second stage, using this selected contact time, the adsorption isotherms of DOX on both FeHA and HA have been analyzed by performing experiments with increasing DOX concentrations. Fig. 3 reports the obtained isotherms (at 37 °C in KCl 10−2 M) for Qads expressed in μmol DOX m−2 apatite and Ceq (equilibrium concentration) in mmol l−1. Interestingly, these two isotherms were found to exhibit distinct shapes. The different mechanisms of adsorption observed can reasonably be attributed to the presence of iron within the apatite lattice as well as the small amount of a secondary iron oxide phase in the FeHA sample, thus leading to different types of accessible surface sites.
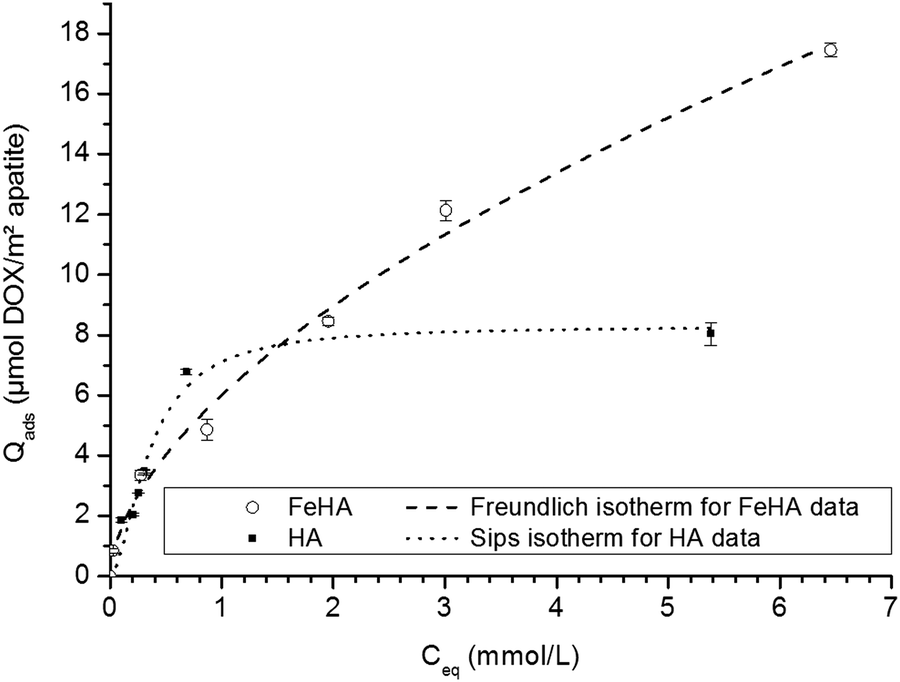 | ||
| Fig. 3 Adsorption isotherms of DOX on FeHA (○) and HA (■). Separate points are the experimental data; dotted lines indicate Freundlich and Sips fits of isotherm data for FeHA and HA, respectively. | ||
In the case of the iron-free HA substrate, the isotherm curve showed a first steep increase of the amount of DOX adsorbed at an equilibrium concentration of up to Ceq ∼1 mmol L−1, followed by stabilization at greater concentrations. This general tendency points to an adsorption behavior with a progressive filling of surface sites and towards a maximal degree of coverage. This type of evolution is noticed in particular for the Langmuir model and for the more general equation of Sips (Langmuir–Freundlich):
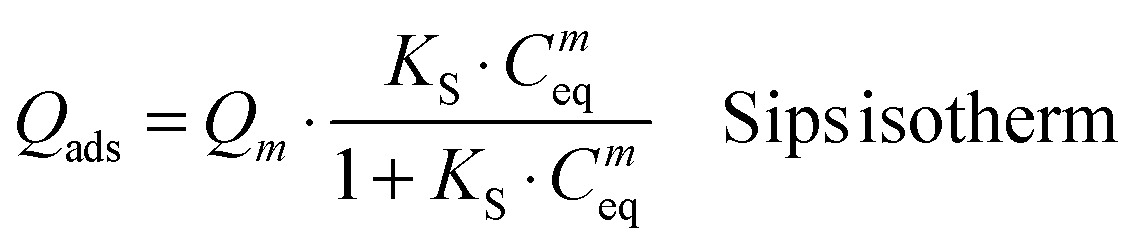
A mathematical analysis indicates that the adsorption data for the system HA–DOX can best be fitted with the Sips model (R2 = 0.9703) with KS = 5.96 ± 3.27 and m = 1.7 ± 0.4. Despite a non-negligible uncertainty, the value of “m” appears to be noticeably distinct from unity, thus suggesting a neat departure from the standard Langmuirian case. A value of “m” greater than 1 can suggest the existence of significant positive interactions (molecular cooperativity) of adsorbed molecules among each other.55,56 This situation was recently also encountered for the adsorption of other biomolecules on biomimetic apatites, such as tetracycline36 and cytidine monophosphate,57 but the maximal adsorbed amounts for these two molecules were significantly lower than the one observed here. As in the case of tetracycline, DOX molecules do not expose high-affinity end-groups for the surface of apatite; the amount of DOX adsorbed on HA is however found to be particularly high, reaching ∼8 μmol DOX m−2 apatite. It suggests a rather high affinity of DOX for apatitic substrates, in agreement with a preliminary work indicating the value 8.2 μmol m−2,58 but in contrast to what could have been expected from such molecules without specific high-affinity end-groups for apatite surfaces. The range of adsorbed quantities can be compared to other adsorption data obtained for rather similar conditions/substrates. For example, the maximal adsorbed amount reached ca. 2.7 μmol m−2 for the adsorption of tiludronate (an anti-osteoporotic bisphosphonate compound) on carbonated nanocrystalline apatite,37,59 or else ∼2.4 μmol m−2 for platinum bisphosphonate complexes.60 The observation of a high affinity of DOX for the apatitic surface is surprising taking into account the absence of known high-affinity anionic end-groups such as phosphates, phosphonates or carboxylates, and also in comparison with the adsorption of tetracycline which led to a maximal coverage of 0.66 μmol m−2 (on a carbonated apatite matured for 1 week at room temperature).36 DOX molecules are expected to interact with apatite surfaces mostly through their multiple polar sites, but these sites are also susceptible to allow interactions among neighboring DOX molecules for a cooperative adsorption, as already suggested by the value of the Sips parameter “m”.
DOX molecules however present an important peculiarity over other molecules tested so far in adsorption experiments involving apatites: these water-soluble molecules are indeed composed of a large hydrophobic anthracycline ring composed of 4 coplanar carbon cycles (see Fig. S2, ESI†), attached to a smaller hydrophilic side chain. In this context, hydrophobic/hydrophilic properties are expected to significantly affect the behavior of DOX molecules in aqueous-based experiments.61 The presence of this large planar aromatic portion of the molecules is at the origin of the therapeutic activity of DOX via intercalation within DNA strands. Several studies have pointed out the possibility for DOX molecules to self-assemble, forming dimers, trimers or even much larger polymolecular units.52,61,62 This type of molecular interaction between DOX molecules is believed to involve π–π stacking processes of their coplanar aromatic domains;61 and both parallel and antiparallel stacking can be considered between interacting DOX molecules.62 Such strong molecular interactions/stacking (dictated by optimal spatial distribution of hydrophobic and hydrophilic parts of the molecules) are also likely to occur in an adsorption context, where the molecules are contacted not only with an aqueous solution but also with a solid surface. This phenomenon could indeed explain the exceptionally high adsorbed amounts noticed for the HA–DOX system compared to the case of other organic molecules: instead of having only individual “monomeric” DOX molecules adsorbed on the apatitic surface, strongly-interacting molecules could organize in a stable manner on the surface of the solid and among each other, leading to what could be finally considered as a “high affinity” system with favored basal and lateral interactions. The fact that the HA–DOX isotherm reaches a stabilized plateau indicates that the adsorption process finally leads to a DOX-saturated surface where no further molecules can be added (at this temperature), and the relatively good fit to the Sips model suggests that, at least macroscopically, the adsorption process can be satisfactorily treated by considering the adsorption of initially monomeric DOX with significant cooperativity between adsorbed DOX molecules (although the exact orientation and positioning of the species cannot be categorized at this point).
The Sips affinity constant KS (recalculated for Ceq expressed in mol L−1) is itself related to the standard Gibbs free energy 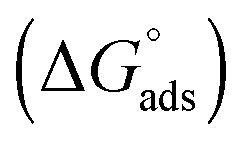 of the global adsorption process by the relationship:
of the global adsorption process by the relationship:
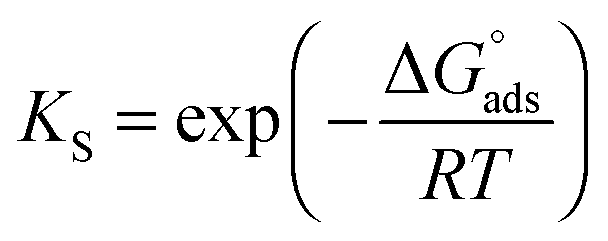
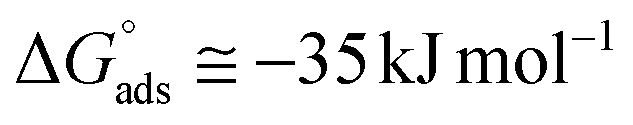 . Although this value relates to standard conditions, it allows comparisons with the same property for other adsorption systems. It is in particular more negative than the
. Although this value relates to standard conditions, it allows comparisons with the same property for other adsorption systems. It is in particular more negative than the 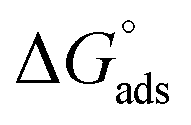 values estimated for the systems tetracycline/apatite36 and cytidine monophosphate/apatite57 (of the order of −20 kJ mol−1), indicating a more energetically favorable process. These findings are in agreement with the above experimental results and discussion, pointing out high adsorbed amounts and strongly suggesting the existence of interactions between DOX molecules.
values estimated for the systems tetracycline/apatite36 and cytidine monophosphate/apatite57 (of the order of −20 kJ mol−1), indicating a more energetically favorable process. These findings are in agreement with the above experimental results and discussion, pointing out high adsorbed amounts and strongly suggesting the existence of interactions between DOX molecules.
In the case of the FeHA, even greater adsorbed amounts were observed compared to HA, and the shapes of both isotherms were significantly different (Fig. 3). When expressed in μmol m−2, the adsorbed amounts achieved for FeHA appear to be somewhat lower than the values obtained for HA only up to an equilibrium concentration of ca. 1.5 mmol L−1; beyond this concentration limit, the adsorbed amount continues however to increase without stabilizing in the rather large concentration window 1.5–7 mmol L−1. The values of Nads achieved here (up to 18 μmol m−2 when expressed per unit surface area) are particularly high for apatitic surfaces and especially in comparison to the adsorption data obtained previously on the HA substrate. In both cases (FeHA and HA), the formation of multi-molecular DOX assemblies adsorbed has to be considered to explain these observations, as was developed above for the HA–DOX system, but the significant further increase observed for FeHA can only be assigned to the presence of iron doping (as iron-substituted apatite but also as small percentage of iron oxide secondary phases, see Fig. 1), thus providing additional adsorption sites. In solution, the presence of divalent and trivalent ions was shown to favor the interaction among DOX molecules towards the formation of multimolecular self-assemblies, and the case of Fe3+ was particularly noticed:61 quelamycin (triferric doxorubicin) is a derivative of DOX exploiting this specificity of iron–DOX interaction.63–65 The good affinity of DOX for iron cations is thus likely to play a large part in the greater adsorbed amount noticed for FeHA–DOX in comparison to HA–DOX.
The mathematical modeling of the FeHA–DOX adsorption isotherm is delicate because various types of adsorption sites coexist, in the form of separate crystalline phases. The overall adsorption process is thus composed of superimposition of various adsorption phenomena involving the various types of surface sites accessible to DOX molecules. In this context, the global isotherm corresponds to a sum of events and should only be seen as a “macroscopic” result. However, being able to describe it mathematically could prove to be useful, from a practical viewpoint, for example for being able to estimate the amount of DOX molecules susceptible to adsorb on such a substrate in a given concentration scenario. The application of the Sips model did not allow here a good description of the FeHA–DOX data without very large uncertainties on Sips parameters. This can be related to the coexistence on this sample of different crystalline phases with distinct adsorptive behaviors. The mathematical modeling of the global FeHA–DOX isotherm using Freundlich's equation (Qads = KF.Ceq1/n) led, on the contrary, to a satisfactory overall fit, with a value of “n” of 1.7 ± 0.1 and a correlation coefficient of R2 = 0.9919. Freundlich's model has initially been established for explaining adsorption data on non-equivalent adsorption sites corresponding to a range of adsorption energies (rather than a constant value as is the case in Langmuir's theory). The goodness of fit noticed here with the Freundlich model probably arises from the existence, in the FeHA substrate, of a series of adsorption sites with a non-negligible span in energy. Even if it is not possible at this stage to give more detail on the type of interaction existing between the multi-phased FeHA sample and DOX nor between adjacent DOX molecules, this mathematical modelling at least allows one to satisfactorily describe the overall evolution of Qadsversus Ceq.
3.3 Characterization of HA and FeHA functionalized with DOX
In order to get more information on the interaction mechanism of DOX with HA and FeHA and to evaluate how the functionalization can affect the surface properties and the size of the NPs, the ζ-potential and mean RH of the HA and FeHA functionalized with the maximum amount of DOX were measured. The coupling of HA and FeHA with DOX altered their surface charge, in particular this functionalization caused a shift of the ζ-potential of HA and FeHA (−5.6 ± 2.0 and −8.1 ± 3.5 mV, respectively) toward less negative values. This finding is in agreement with a possible interaction of the positively charged –NH3+ groups of the DOX dimers with the negatively charged surface groups of apatites (i.e. phosphates, phosphonates or even carboxylates) as reported above. The surface uptake of DOX provided the NPs with a less negative surface charge, which in turn also decreased the inter-particles repulsion. Consequently, HA–DOX and FeHA–DOX were more aggregated, forming less stable suspensions than the bare ones with increased mean RH of 281 ± 24 and 354 ± 52 nm, respectively. The stability in an aqueous suspension of bare and functionalized NPs (0.1 mg mL−1) as a function of time has been evaluated by DLS, measuring continuously for 60 min the derived count rate (Fig. S4, ESI†). In the case of HA and FeHA, the recorded values remained nearly constant, corroborating that neither significant aggregation nor fast sedimentation occurred. On the contrary, in the case of functionalized NPs the fast decrease of the derived count rate revealed their lower stability in suspension.Comparison of the FTIR spectra of FeHA–DOX, HA–DOX and free DOX (Fig. S5, ESI†) was not useful to shed some light on the interaction mechanisms of DOX molecules with the inorganic phase. Nevertheless, the appearance of the DOX main signal at 1284 cm−1 (C–O–C asymmetric stretching66) in the spectra of FeHA–DOX and HA–DOX confirmed the effective uptake of the drug on the NPs.
Raman microanalyses were run on the solids after DOX adsorption, but no useable data on the position of bands and eventual shifts were obtained due to strong fluorescence artifacts from DOX with the employed laser wavelength at our disposal (Fig. S6, ESI†). Moreover, 13C CP/MAS solid-state NMR analyses were tentatively run to observe the interaction of DOX molecules with the surface of apatite nanocrystals. However, although the recorded signals can be well correlated to the presence of DOX (Fig. S7, ESI†), they only showed a low-intensity noisy signal not exploitable for drawing conclusive statements on the chemical interaction of the drug with the apatitic substrate, which can be linked to the limited amount of DOX on the samples and low isotopic abundance of 13C.
3.4 Release of DOX from HA and FeHA
HA and FeHA were functionalized with similar amounts of DOX (i.e. 475 and 449 μg of DOX on 1 mg of HA and FeHA, respectively) corresponding to the maximum drug loading capacity of HA. The DOX release efficacy (DR) from drug-loaded HA and FeHA, defined as the ratio (wt%) between the amount of drug released at different times (Q(t)) and the initial drug loading capacity Qi, was calculated using the following formula:DR of DOX from HA and FeHA as a function of time under near physiological conditions (pH 7.4) and in the absence or presence of a PEMF is shown in Fig. 4. In the absence of the PEMF, the percentage of DOX released from HA was higher than that released from FeHA. DOX was gradually released from HA as a function of time reaching after 6 days the value of about 15 wt% of the initial DOX loaded. On the contrary, the maximum amount of DOX released from FeHA in the absence of the PEMF (about 3 wt% of the initial DOX loaded) was achieved after 2 days since no significant difference was found with the drug released after 3 and 6 days. The lower amount of drug released from FeHA with respect to HA is in good agreement with the higher affinity of DOX for FeHA, as already mentioned in the discussion on the adsorption isotherms.
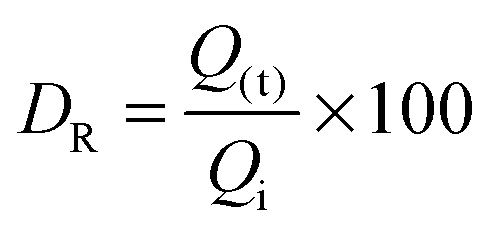
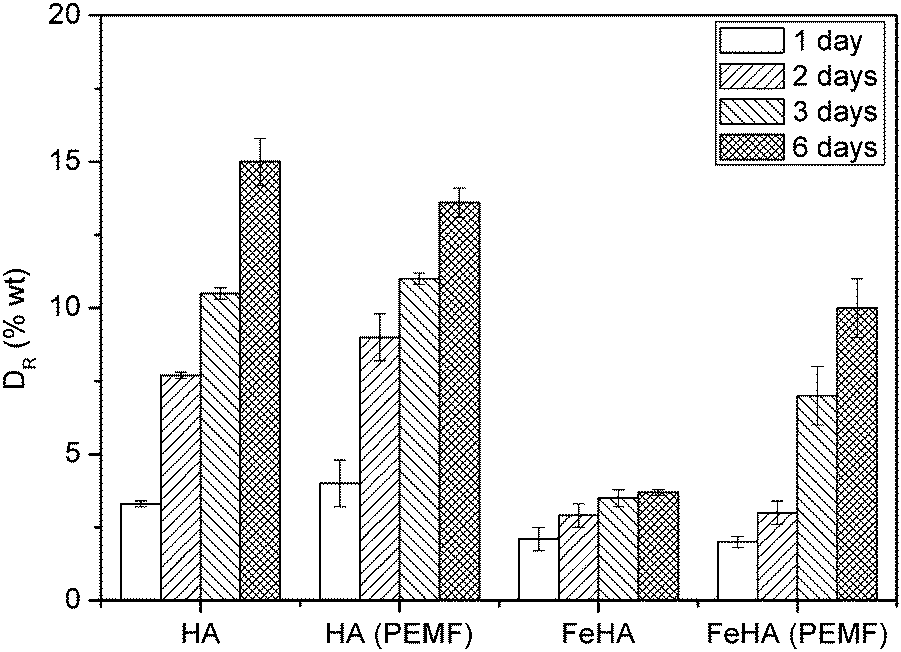 | ||
| Fig. 4 Kinetics of DOX release from HA and FeHA in the absence or presence of a pulsed electromagnetic field (PEMF). | ||
Generally speaking, the relatively low amounts of DOX released from HA and FeHA at pH 7.4 are in agreement with the data previously reported in other studies58,67 and confirm the strength of the bonding between the apatite substrate and the drug molecules. The possibility to move FeHA NPs (and even entire cells after their internalization) upon the application of a static magnetic field of 320 mT (Fig. S8, ESI†) has been recently reported,68 corroborating their potential ability to be targeted in specific body systems by magnetic fields. The high affinity of FeHA towards DOX and the low amount of the drug released at pH 7.4 points out that even FeHA–DOX could be guided without any concomitant loss of the therapeutic agent.
Magnetic NPs can be directed by static magnetic fields, but also provide remote controlled release of drugs or biomolecules, i.e. in scaffold formulation by magnetic stimulation. Therefore the release of DOX from FeHA, in comparison with that from HA, has been evaluated applying a low frequency PEMF. Interestingly, in the presence of the PEMF, the extent of DOX released from FeHA after 3 and 6 days increased significantly (3.5 ± 0.3 vs. 7.0 ± 1.0 wt% after 3 days and 3.7 ± 0.1 vs. 10.0 ± 1.0 wt% after 6 days, in the absence and presence of the PEMF, respectively). This increase was directly related to the superparamagnetic feature of FeHA, since no statistical difference between DOX released from HA in the presence and in absence of the PEMF was observed.
It was well demonstrated in the last years that upon application of an alternating electromagnetic field, superparamagnetic NPs could generate local internal heating, causing structure disassembling and allowing the cargo (drug) to be released.69,70 In order to assess if a similar effect has been exhibited in this work, dissolution of FeHA in the presence or absence of a PEMF was monitored. The extent of released Ca2+, which can be considered as an index of the FeHA dissolution, was quantified after 3 and 6 days (Fig. S9, ESI†). No significant differences were observed between the amounts of Ca2+ released in the presence or absence of the PEMF, indicating that the increase of DOX released was not triggered by a more rapid degradation of the carriers. Moreover, during the PEMF exposure, FeHA suspension did not show any significant increase in temperature in the bulk solution. The PEMF used in this work was of low frequency comparable to that commercially used to stimulate and activate bone healing71 and according to previous magnetic characterizations,29 it was too weak to induce a hyperthermia effect on FeHA. Therefore, the higher release of DOX from FeHA in the presence of the PEMF can presumably be explained by the mechanical movement (i.e. shaking and flipping) of superparamagnetic NPs breaking the bonding with the drug or allowing detachment of multi-molecular DOX assemblies from the NPs surface, rather than the destabilization of the crystal structure or the increase of temperature. This hypothesis is coherent with the fact that the appreciable differences of drug release in comparison to the experiments carried out without the PEMF were evident only at long time points (i.e. 3 and 6 days) when the strong bonding between the drug and NPs was weakened by the continuous pulsed stimulation.
The DOX release kinetics data obtained for both HA and FeHA (whether in the absence or presence of the PEMF) were analyzed from a mathematical viewpoint by fitting with several release models (zero order, first order, gas-like desorption, Higuchi, Korsmeyer–Peppas (KP) and Hixson–Crowell (HC) models), described in the literature.72–74 A screening of these models was done using their linearized forms and considering the same number of data points to get comparable R2 correlation coefficients based on the same number of degrees of freedom (the origin (0;0) was omitted here due to logarithmic calculations tending to infinity in some of the models). The obtained correlation coefficients are listed in Table S2, ESI.† The results systematically indicate that the diffusion-based models, i.e. Higuchi and KP models, were the most appropriate to fit the experimental release data among the models tested. In contrast, only a poor correlation was found for gas-like desorption (the model supposing a low adsorbent–adsorbate affinity) or the matrix-dissolution model like the HC model. These results suggest that the release kinetics of DOX from HA or FeHA are mainly controlled by DOX diffusion from the substrate (inter)granular surfaces/spaces up to the solution rather than by the progressive dissolution of the substrates (which is indeed expected to be limited at physiological pH). The KP model is described by a power law of the form DR = k·tn where DR is the cumulative release described above, “t” is the time, and “k” and “n” are constants (the Higuchi model being a special case of the KP model with n = 0.5). The exponential factor “n” was found to vary in the range 0.29–0.86 (Table S2, ESI†). Except for the unusually low value of 0.29 (lower than 0.5 and thus probably not representative), all other cases fall in the 0.5 ≤ n ≤ 1.0 domain often considered as “anomalous transport”, corresponding to complex release mechanisms with non-Fickian diffusivity. A similar situation was for example found in the case of vancomycin release from brushite.75 It may be noted that no clear trend allows here distinguishing the release mechanisms of the different conditions studied (HA or FeHA; with or without PEMF application); only the amount of DOX released seems to be affected.
3.5 In vitro cell culture
Assessment of SAOS-2 cell viability grown in contact with 10 and 100 μM of DOX, either free or loaded onto HA and FeHA NPs, as well as with equivalent amounts of un-functionalized NPs, was carried out by quantification of metabolically active cells by the use of the MTT assay.76 The results indicated that the DOX, loaded on HA and FeHA, was able to exert its cytotoxic activity on SAOS-2 cells. In fact, HA–DOX and FeHA–DOX, at all the concentrations tested (Table S1, ESI†), showed reduced cell viability compared to the sample with only cells, with statistic differences after 48 and 72 hours of culture (Fig. 5). Moreover, no significant difference existed between the cytotoxic effect exerted by DOX free or loaded on HA and FeHA NPs at all the concentrations and each time points tested (Fig. 5) indicating that the bonding with the NPs did not affect the DOX antitumor activity.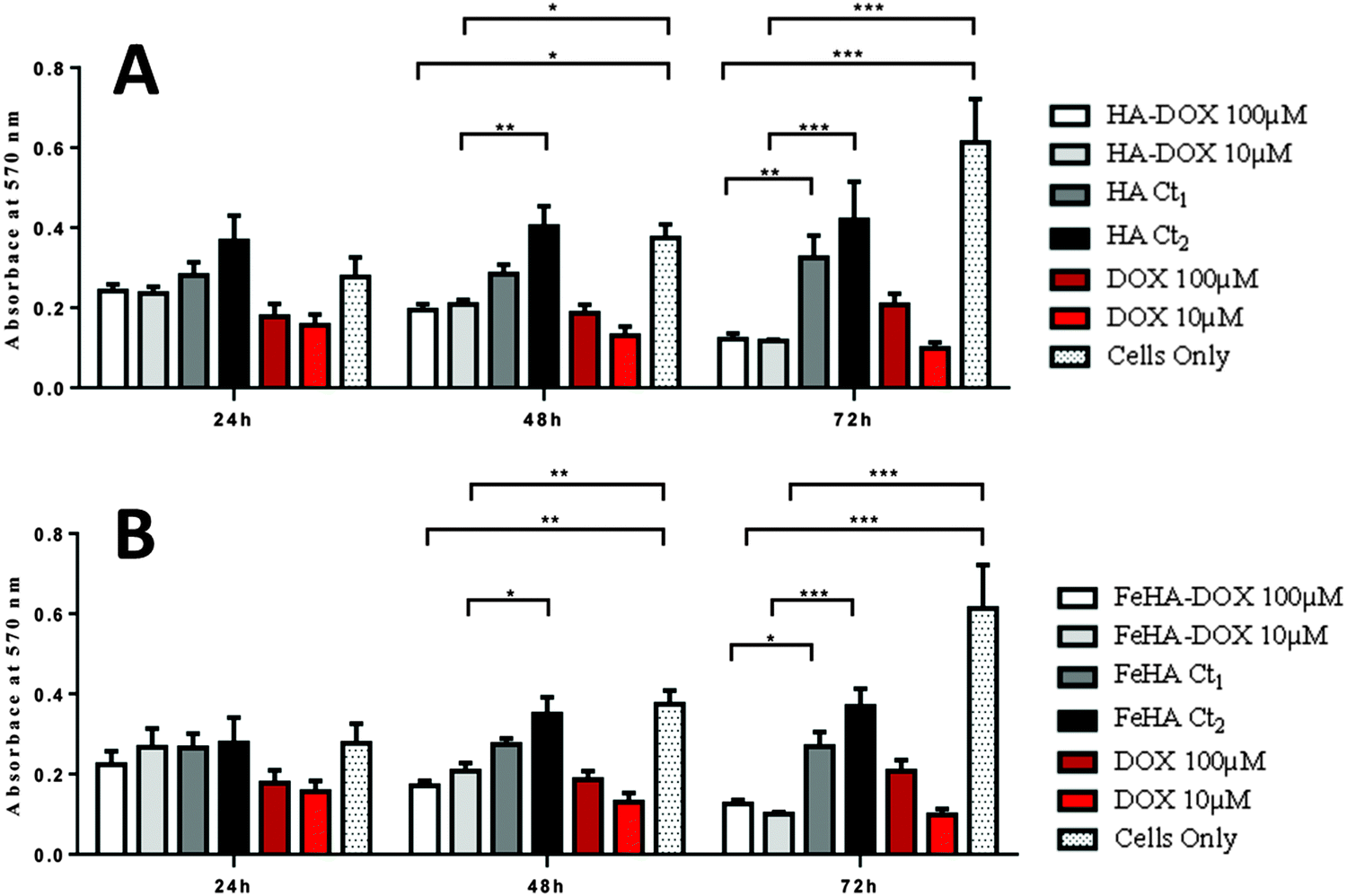 | ||
| Fig. 5 Analysis of SAOS-2 cell line viability by the MTT assay, after 24, 48 and 72 h of culture with 10 and 100 μM of DOX either free or loaded on (A) HA and (B) FeHA NPs, as well as with equivalent amounts of un-functionalized (A) HA and (B) FeHA. The sample with only cells was used as the control. The graph shows the average and standard deviation of 6 replicates; statistical significant differences among the samples are indicated in the graphs: *p ≤ 0.05, **p ≤ 0.01 and ***p ≤ 0.001. | ||
It is well reported that the calcium phosphate NPs can be internalized inside cells via clathrin- and caveolae-dependent endocytosis,77 while free DOX can diffuse through the plasma membrane due its lipophilic properties.78 Here, the nuclear localization of the different DOX-loaded HA and FeHA was evaluated by fluorescence microscopy. Fig. 6 and 7 show DOX internalized without any difference among the groups. It was also possible to observe a higher intensity of fluorescence in both HA-DOX and FeHA–DOX 100 μM groups (Fig. 6 and 7A–C) compared to 10 μM groups (Fig. 6 and 7D–F), indicating a dose-dependent drug internalization. Moreover, in agreement with the data previously reported,58,67 under all the tested conditions, after 24 hours DOX was mainly detected within the nuclei.
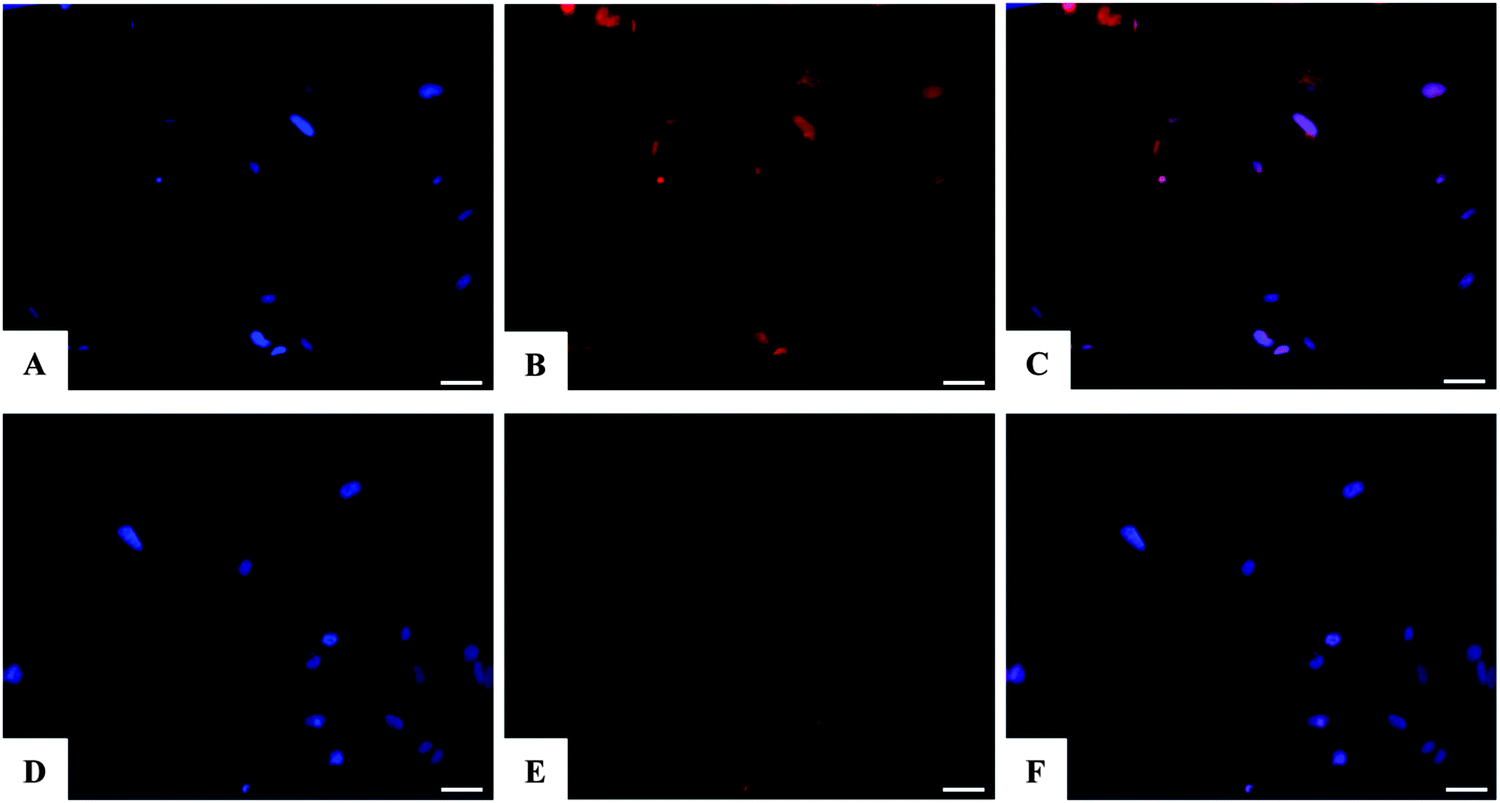 | ||
| Fig. 6 Analysis of the internalization of HA–DOX NPs in the SAOS-2 cell line after 24 h of culture by fluorescence microscopy. Images A, B and C show DAPI nuclear staining (blue), nuclear localization of DOX (red) and merge of the co-localization of DAPI and DOX for the sample HA–DOX 100 μM. Images D, E and F show DAPI nuclear staining (blue), nuclear localization of DOX (red) and merge of the co-localization of DAPI and DOX for the sample HA–DOX 10 μM. Scale bars 100 μm. | ||
 | ||
| Fig. 7 Analysis of the internalization of FeHA–DOX NPs in the SAOS-2 cell line after 24 h of culture by fluorescence microscopy. Images A, B and C show DAPI nuclear staining (blue), nuclear localization of DOX (red) and merge of the co-localization of DAPI and DOX for the sample FeHA–DOX 100 μM. Images D, E and F show DAPI nuclear staining (blue), nuclear localization of DOX (red) and merge of the co-localization of DAPI and DOX for the sample FeHA–DOX 10 μM. Scale bars 100 μm. | ||
On the basis of our previous data and those described in others studies about the internalization of calcium phosphate NPs inside cells,58,67,79,80 and taking into account that the bonding between DOX and HA/FeHA NPs was stable under physiological conditions, it is realistic to assume that DOX-loaded NPs were easily and rapidly internalized by cells and able to carry inside their bound DOX. Once inside the cells, DOX can be released from the NPs where the slightly acidic conditions of lysosomes allowed faster degradation of FeHA and HA NPs.58,67,68 After that, DOX diffused into cell nuclei and induced DNA damage exerting its cell apoptosis effect.
Several studies reported that at initial stages free DOX is more effective than that attached to the iron oxide NPs due to the different mechanism of cellular uptake.81,82 Free DOX passes from the extracellular to the intracellular matrix by simple passive diffusion and reaches the nuclei easily, while in the case of iron oxide NPs-bound DOX, the drug is released from the NPs surface in lysosomes due to the acidic pH environment and hence DOX can reach the nuclei and intercalate DNA. The fact that in our case DOX coupled to apatitic NPs displayed comparable cytotoxicity of free DOX at all the time points was correlated with the fast internalization process and the rapid degradation of FeHA and HA in the lysosomal compartments respect to iron oxide NPs.
In vitro tests with cells in the presence of a PEMF were not carried out because the time of the magnetic field to trigger an higher drug release from FeHA (3 days) was not comparable to that of cell internalization (few hours), therefore any effect would be further appreciated.
4. Conclusions
In this work, the effective ability of previously synthesized superparamagnetic FeHA29 to load DOX was demonstrated. FeHA displayed higher affinity for DOX in comparison to iron-free biomimetic HA NPs due to good affinity of the drug for the iron cations of the FeHA surface. The stability of the bonding between DOX and FeHA was stronger compared to HA; in fact, the quantity of DOX released at pH 7.4 from FeHA was lower than the drug released from HA. The release of DOX from FeHA was also assessed in the presence of a PEMF, and fascinatingly the extent of DOX released after 3 and 6 days under these conditions increased significantly in comparison to the drug release without a PEMF. This finding was explained by the mechanical shacking of superparamagnetic NPs breaking the bonding with DOX or allowing detachment of drug assemblies from the NPs surface, rather than by some higher degradation of NPs or a temperature increase. Finally, in vitro assays demonstrated that DOX loaded on HA and FeHA was able to exert its cytotoxic activity on SAOS-2 cells at the same level as free DOX, for all the concentrations and time points tested, because the functionalized NPs can be rapidly internalized within cells and release DOX, which accumulated in the nuclei and exerted cytotoxic activity.In light of our results and taking into account that the biocompatibility of FeHA particles, their pH-dependent biodegradability and ability to be moved by an external magnetic field were already demonstrated,29,30,68 and although further in vitro and in vivo studies are necessary, we can conclude that this new superparamagnetic nanosystem can represent a novel alternative to SPIONs to set up magnetic devices for stimulating personalized nanomedical applications such as targeted drug nano-carriers and scaffolds for bone tissue engineering with remotely controlled multi-functionalities.
Acknowledgements
We acknowledge support from the Italian Ministry for Education, University and Research (MIUR) in the framework of the Flagship Project NanoMax (PNR 201-2013), the French Egide association (Hubert-Curien “Galilée” program, project #28285UB), and the Università Italo-Francese (Programma Galileo 2012/2013). The authors also thank O. Marsan (CIRIMAT-Toulouse) and Yannick Coppel (LCC-Toulouse) for Raman and NMR technical support, respectively. The Biostim SPT pulse generator was kindly provided by IGEA (Carpi, Italy).References
- K. Fuhrmann, M. A. Gauthier and J.-C. Leroux, Mol. Pharmaceutics, 2014, 11, 1762–1771 CrossRef CAS PubMed.
- J. M. Coburn and D. L. Kaplan, Bioconjugate Chem., 2015, 26, 1212–1223 CrossRef CAS PubMed.
- S. Pietronave, M. Iafisco, D. Locarno, L. Rimondini and M. Prat, J. Appl. Biomater. Biomech., 2009, 7, 77–89 CAS.
- T. Sun, Y. S. Zhang, B. Pang, D. C. Hyun, M. Yang and Y. Xia, Angew. Chem., Int. Ed., 2014, 53, 12320–12364 CAS.
- K. Yan, P. Li, H. Zhu, Y. Zhou, J. Ding, J. Shen, Z. Li, Z. Xu and P. K. Chu, RSC Adv., 2013, 3, 10598–10618 RSC.
- L. H. Reddy, J. L. Arias, J. Nicolas and P. Couvreur, Chem. Rev., 2012, 112, 5818–5878 CrossRef CAS PubMed.
- M. Arruebo, R. Fernández-Pacheco, M. R. Ibarra and J. Santamaría, Nano Today, 2007, 2, 22–32 CrossRef.
- J. Kolosnjaj-Tabi, C. Wilhelm, O. Clement and F. Gazeau, J. Nanobiotechnol., 2013, 11, S7 CrossRef PubMed.
- A. Ranzoni, G. Sabatte, L. J. van Ijzendoorn and M. W. J. Prins, ACS Nano, 2012, 6, 3134–3141 CrossRef CAS PubMed.
- S. H. Lee, B. H. Kim, H. B. Na and T. Hyeon, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2014, 6, 196–209 CrossRef CAS PubMed.
- T. Kobayashi, Biotechnol. J., 2011, 6, 1342–1347 CrossRef CAS PubMed.
- J. Meng, B. Xiao, Y. Zhang, J. Liu, H. Xue, J. Lei, H. Kong, Y. Huang, Z. Jin, N. Gu and H. Xu, Sci. Rep., 2013, 3, 2655 Search PubMed.
- J.-J. Kim, R. K. Singh, S.-J. Seo, T.-H. Kim, J.-H. Kim, E.-J. Lee and H.-W. Kim, RSC Adv., 2014, 4, 17325–17336 RSC.
- H.-Y. Xu and N. Gu, Front. Mater. Sci., 2014, 8, 20–31 CrossRef.
- R. Sensenig, Y. Sapir, C. MacDonald, S. Cohen and B. Polyak, Nanomedicine, 2012, 7, 1425–1442 CrossRef CAS PubMed.
- T. Neuberger, B. Schöpf, H. Hofmann, M. Hofmann and B. von Rechenberg, J. Magn. Magn. Mater., 2005, 293, 483–496 CrossRef CAS.
- A. Singh and S. K. Sahoo, Drug Discovery Today, 2014, 19, 474–481 CrossRef CAS PubMed.
- C. Xu and S. Sun, Adv. Drug Delivery Rev., 2013, 65, 732–743 CrossRef CAS PubMed.
- F. M. Kievit and M. Zhang, Acc. Chem. Res., 2011, 44, 853–862 CrossRef CAS PubMed.
- M. Mahmoudi, S. Sant, B. Wang, S. Laurent and T. Sen, Adv. Drug Delivery Rev., 2011, 63, 24–46 CrossRef CAS PubMed.
- N. Singh, G. J. S. Jenkins, R. Asadi and S. H. Doak, Nano Rev., 2010, 1, 5358 Search PubMed.
- J.-E. Kim, J.-Y. Shin and M.-H. Cho, Arch. Toxicol., 2012, 86, 685–700 CrossRef CAS PubMed.
- T. K. Jain, M. K. Reddy, M. A. Morales, D. L. Leslie-Pelecky and V. Labhasetwar, Mol. Pharmaceutics, 2008, 5, 316–327 CrossRef CAS PubMed.
- J. Huang, L. Bu, J. Xie, K. Chen, Z. Cheng, X. Li and X. Chen, ACS Nano, 2010, 4, 7151–7160 CrossRef CAS PubMed.
- M.-T. Zhu, W.-Y. Feng, Y. Wang, B. Wang, M. Wang, H. Ouyang, Y.-L. Zhao and Z.-F. Chai, Toxicol. Sci., 2009, 107, 342–351 CrossRef CAS PubMed.
- J. Gómez-Morales, M. Iafisco, J. M. Delgado-López, S. Sarda and C. Drouet, Prog. Cryst. Growth Charact. Mater., 2013, 59, 1–46 CrossRef.
- E. Boanini, M. Gazzano and A. Bigi, Acta Biomater., 2010, 6, 1882–1894 CrossRef CAS PubMed.
- M. Šupová, Ceram. Int., 2015, 41, 9203–9231 CrossRef.
- A. Tampieri, T. D'Alessandro, M. Sandri, S. Sprio, E. Landi, L. Bertinetti, S. Panseri, G. Pepponi, J. Goettlicher, M. Bañobre-López and J. Rivas, Acta Biomater., 2012, 8, 843–851 CrossRef CAS PubMed.
- S. Panseri, C. Cunha, T. D'Alessandro, M. Sandri, G. Giavaresi, M. Marcacci, C. T. Hung and A. Tampieri, J. Nanobiotechnol., 2012, 10, 32 CrossRef CAS PubMed.
- M. Iafisco, M. Sandri, S. Panseri, J. M. Delgado-López, J. Gómez-Morales and A. Tampieri, Chem. Mater., 2013, 25, 2610–2617 CrossRef CAS.
- A. Gloria, T. Russo, U. D'Amora, S. Zeppetelli, T. D'Alessandro, M. Sandri, M. Bañobre-López, Y. Piñeiro-Redondo, M. Uhlarz, A. Tampieri, J. Rivas, T. Herrmannsdörfer, V. A. Dediu, L. Ambrosio and R. De Santis, J. R. Soc., Interface, 2013, 10, 20120833 CrossRef CAS PubMed.
- O. Tacar, P. Sriamornsak and C. R. Dass, J. Pharm. Pharmacol., 2013, 65, 157–170 CrossRef CAS PubMed.
- Y. Octavia, C. G. Tocchetti, K. L. Gabrielson, S. Janssens, H. J. Crijns and A. L. Moens, J. Mol. Cell. Cardiol., 2012, 52, 1213–1225 CrossRef CAS PubMed.
- G. Aubel-Sadron and D. Londos-Gagliardi, Biochimie, 1984, 66, 333–352 CrossRef CAS PubMed.
- S. Cazalbou, G. Bertrand and C. Drouet, J. Phys. Chem. B, 2015, 119, 3014–3024 CrossRef CAS PubMed.
- P. Pascaud, P. Gras, Y. Coppel, C. Rey and S. Sarda, Langmuir, 2013, 29, 2224–2232 CrossRef CAS PubMed.
- H. Autefage, F. Briand-Mesange, S. Cazalbou, C. Drouet, D. Fourmy, S. Goncalves, J. P. Salles, C. Combes, P. Swider and C. Rey, J. Biomed. Mater. Res., Part B, 2009, 91B, 706–715 CrossRef CAS PubMed.
- A. L. Patterson, Phys. Rev., 1939, 56, 978–982 CrossRef CAS.
- L. Fassina, L. Visai, F. Benazzo, L. Benedetti, A. Calligaro, M. G. Cusella De Angelis, A. Farina, V. Maliardi and G. Magenes, Tissue Eng., 2006, 12, 1985–1999 CrossRef CAS PubMed.
- C. Rey, C. Combes, C. Drouet, H. Sfihi and A. Barroug, Mater. Sci. Eng., C, 2007, 27, 198–205 CrossRef CAS.
- R. Z. LeGeros, R. Kijkowska, C. Bautista and J. P. Legeros, Connect. Tissue Res., 1995, 33, 203–209 CrossRef CAS PubMed.
- M. Iafisco, M. Marchetti, J. Gómez Morales, M. a. A. Hernández-Hernández, J. M. García Ruiz and N. Roveri, Cryst. Growth Des., 2009, 9, 4912–4921 CAS.
- J. M. Delgado-López, M. Iafisco, I. Rodríguez, A. Tampieri, M. Prat and J. Gómez-Morales, Acta Biomater., 2012, 8, 3491–3499 CrossRef PubMed.
- S. Koutsopoulos, J. Biomed. Mater. Res., 2002, 62, 600–612 CrossRef CAS PubMed.
- X. Ma and D. E. Ellis, Biomaterials, 2008, 29, 257–265 CrossRef CAS PubMed.
- C. Rey, C. Combes, C. Drouet, S. Cazalbou, D. Grossin, F. Brouillet and S. Sarda, Prog. Cryst. Growth Charact. Mater., 2014, 60, 63–73 CrossRef CAS.
- T. Ito, L. Sun, M. A. Bevan and R. M. Crooks, Langmuir, 2004, 20, 6940–6945 CrossRef CAS PubMed.
- F.-C. Wu, R.-L. Tseng and R.-S. Juang, Chem. Eng. J., 2009, 150, 366–373 CrossRef CAS.
- C. Aharoni and F. C. Tompkins, in Advances in Catalysis and Related Subjects, ed. D. D. Eley, H. Pines and P. B. Weisz, Academic Press, New York, 1970, vol. 21, pp. 1–49 Search PubMed.
- L. E. Gerweck, S. V. Kozin and S. J. Stocks, Br. J. Cancer, 1999, 79, 838–842 CrossRef CAS PubMed.
- Z. Fülöp, R. Gref and T. Loftsson, Int. J. Pharm., 2013, 454, 559–561 CrossRef PubMed.
- L. Benaziz, A. Barroug, A. Legrouri, C. Rey and A. Lebugle, J. Colloid Interface Sci., 2001, 238, 48–53 CrossRef CAS PubMed.
- K. Achelhi, S. Masse, G. Laurent, A. Saoiabi, A. Laghzizil and T. Coradin, Dalton Trans., 2010, 39, 10644–10651 RSC.
- L. K. Koopal, W. H. van Riemsdijk, J. C. M. de Wit and M. F. Benedetti, J. Colloid Interface Sci., 1994, 166, 51–60 CrossRef CAS.
- Q. Luo and J. D. Andrade, J. Colloid Interface Sci., 1998, 200, 104–113 CrossRef CAS.
- M. Choimet, A. Tourrette and C. Drouet, J. Colloid Interface Sci., 2015, 456, 132–137 CrossRef CAS PubMed.
- I. Rodríguez-Ruiz, J. M. Delgado-López, M. A. Durán-Olivencia, M. Iafisco, A. Tampieri, D. Colangelo, M. Prat and J. Gómez-Morales, Langmuir, 2013, 29, 8213–8221 CrossRef PubMed.
- A. Al-Kattan, F. Errassifi, A. M. Sautereau, S. Sarda, P. Dufour, A. Barroug, I. D. Santos, C. Combes, D. Grossin, C. Rey and C. Drouet, Adv. Eng. Mater., 2010, 12, B224–B233 CrossRef.
- M. Iafisco, B. Palazzo, G. Martra, N. Margiotta, S. Piccinonna, G. Natile, V. Gandin, C. Marzano and N. Roveri, Nanoscale, 2012, 4, 206–217 RSC.
- E. Hayakawa, K. Furuya, T. Kuroda, M. Moriyama and A. Kondo, Chem. Pharm. Bull., 1991, 39, 1282–1286 CrossRef CAS.
- R. Anand, S. Ottani, F. Manoli, I. Manet and S. Monti, RSC Adv., 2012, 2, 2346–2357 RSC.
- H. Cortes-Funes, M. Gosalvez, A. Moyano, A. Mañas and C. Mendiola, Cancer Treat. Rep., 1979, 63, 903–908 CAS.
- M. Gosálvez, M. F. Blanco, C. Vivero and F. Vallés, Eur. J. Cancer, 1978, 14, 1185–1190 CrossRef.
- P. M. May, G. K. Williams and D. R. Williams, Inorg. Chim. Acta, 1980, 46, 221–228 CrossRef CAS.
- S. Kayal and R. V. Ramanujan, Mater. Sci. Eng., C, 2010, 30, 484–490 CrossRef CAS.
- M. Iafisco, J. M. Delgado-Lopez, E. M. Varoni, A. Tampieri, L. Rimondini, J. Gomez-Morales and M. Prat, Small, 2013, 9, 3834–3844 CrossRef CAS PubMed.
- S. Panseri, M. Montesi, M. Sandri, M. Iafisco, A. Adamiano, M. Ghetti, G. Cenacchi and A. Tampieri, J. Biomed. Nanotechnol. Search PubMed , accepted.
- C. R. Thomas, D. P. Ferris, J.-H. Lee, E. Choi, M. H. Cho, E. S. Kim, J. F. Stoddart, J.-S. Shin, J. Cheon and J. I. Zink, J. Am. Chem. Soc., 2010, 132, 10623–10625 CrossRef CAS PubMed.
- S. Brulé, M. Levy, C. Wilhelm, D. Letourneur, F. Gazeau, C. Ménager and C. Le Visage, Adv. Mater., 2011, 23, 787–790 CrossRef PubMed.
- L. Massari, G. Caruso, V. Sollazzo and S. Setti, Clin. Cases Miner. Bone Metab., 2009, 6, 149–154 Search PubMed.
- M. Barzegar-Jalali, K. Adibkia, H. Valizadeh, M. R. S. Shadbad, A. Nokhodchi, Y. Omidi, G. Mohammadi, S. H. Nezhadi and M. Hasan, J. Pharm. Pharm. Sci., 2008, 11, 167–177 CAS.
- C. G. Weber, M. Mueller, N. Vandecandelaere, I. Trick, A. Burger-Kentischer, T. Maucher and C. Drouet, J. Mater. Sci.: Mater. Med., 2014, 25, 595–606 CrossRef CAS PubMed.
- N. L. Ignjatovic, P. Ninkov, R. Sabetrasekh and D. P. Uskokovic, J. Mater. Sci.: Mater. Med., 2010, 21, 231–239 CrossRef CAS PubMed.
- U. Gbureck, E. Vorndran and J. E. Barralet, Acta Biomater., 2008, 4, 1480–1486 CrossRef CAS PubMed.
- Y. Liu, D. A. Peterson, H. Kimura and D. Schubert, J. Neurochem., 1997, 69, 581–593 CrossRef CAS PubMed.
- D. Y. E. Olton, J. M. Close, C. S. Sfeir and P. N. Kumta, Biomaterials, 2011, 32, 7662–7670 CrossRef CAS PubMed.
- A. Beljebbar, G. D. Sockalingum, J. F. Angiboust and M. Manfait, Spectrochim. Acta, Part A, 1995, 51, 2083–2090 CrossRef.
- S. P. Victor, W. Paul, M. Jayabalan and C. P. Sharma, CrystEngComm, 2014, 16, 9033–9042 RSC.
- Y.-H. Yang, C.-H. Liu, Y.-H. Liang, F.-H. Lin and K. C. W. Wu, J. Mater. Chem. B, 2013, 1, 2447–2450 RSC.
- K. Tomankova, K. Polakova, K. Pizova, S. Binder, M. Havrdova, M. Kolarova, E. Kriegova, J. Zapletalova, L. Malina, J. Horakova, J. Malohlava, A. Kolokithas-Ntoukas, A. Bakandritsos, H. Kolarova and R. Zboril, Int. J. Nanomed., 2015, 10, 949–961 CrossRef PubMed.
- M. I. Majeed, Q. Lu, W. Yan, Z. Li, I. Hussain, M. N. Tahir, W. Tremel and B. Tan, J. Mater. Chem. B, 2013, 1, 2874–2884 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tb01524c |
| ‡ Present address: University of Limoges, CNRS, SPCTS UMR 7315, CEC, 12 Rue Atlantis, 87068 Limoges Cedex, France. |
| This journal is © The Royal Society of Chemistry 2016 |