
Gemcitabine and chlorotoxin conjugated iron oxide nanoparticles for glioblastoma therapy†
Qingxin
Mu
ab,
Guanyou
Lin
c,
Victoria K.
Patton
d,
Kui
Wang
a,
Oliver W.
Press
b and
Miqin
Zhang
*a
aDepartment of Materials Science and Engineering, University of Washington, Seattle, Washington 98195, USA. E-mail: mzhang@uw.edu
bClinical Research Division, Fred Hutchinson Cancer Research Centre, Seattle, Washington 98109, USA
cDepartment of Bioengineering, University of Washington, Seattle, Washington 98195, USA
dDepartment of Chemical Engineering, University of Washington, Seattle, Washington 98195, USA
First published on 24th November 2015
Abstract
Many small-molecule anti-cancer drugs have short blood half-lives and toxicity issues due to non-specificity. Nanotechnology has shown great promise in addressing these issues. Here, we report the development of anti-cancer drug gemcitabine-conjugated iron oxide nanoparticles for glioblastoma therapy. A glioblastoma targeting peptide, chlorotoxin, was attached after drug conjugation. The nanoparticles have a small size (∼32 nm) and uniform size distribution (PDI ≈ 0.1), and are stable in biological medium. The nanoparticles effectively enter cancer cells without losing potency compared to the free drug. Significantly, the nanoparticles showed a prolonged blood half-life and the ability to cross the blood–brain barrier in wild type mice.
Many small molecule anti-cancer drugs encounter short blood half-life and off-target toxicity issues.1,2 Alternatively, drugs can be formulated into nanoparticles (NPs) to improve their overall pharmacokinetic profiles.3 One such example is gemcitabine (GEM). GEM is an FDA-approved anti-cancer drug for the treatment of pancreatic cancer, non-small cell lung cancer, breast cancer, ovarian cancer, bladder cancer, etc.4–9 GEM has also been tested for brain tumour therapy.10,11 The activity of GEM in brain tumour therapy is independent of methylguanine methyltransferase (MGMT) expression, an enzyme that is responsible for resistance to temozolomide (TMZ), a first-line brain tumour treatment drug.12 However, GEM has a half-life of ∼0.28 h in humans and mice and has several side effects.4,13 To address these issues, GEM has been modified and formulated into NPs. For example, GEM has been chemically modified with lipids (squalenoyl, stearoyl, etc.) to form lipid nanoparticles or loaded into liposomes to prolong blood circulation.14–17 GEM has been loaded onto polymeric NPs such as chitosan and polybutylcyanoacrylate NPs for targeted drug delivery.18,19 However, these NPs were either too large (>100 nm) to pass the BBB or too colloidally unstable in biological medium for brain tumour therapy.
Here, we report the development of a small and colloidally stable GEM-loaded nanocarrier to increase GEM's circulation time and overcome the blood–brain barrier (BBB) for targeted glioblastoma multiform (GBM) therapy. The nanocarrier is made of iron oxide-based NPs immobilized with GEM, chlorotoxin (CTX), and hyaluronic acid (HA). GBM is the most common and aggressive malignant primary brain tumour with very poor prognosis.20 Unlike liposomes or polymeric NPs, iron oxide NPs (IONPs) are small, stable and have superparamagnetic properties.21–23 IONPs are also biodegradable and sterilizable, making them a suitable candidate for an effective drug delivery carrier.21,24 CTX is a 39-mer peptide that is able to cross the BBB and target brain tumour cells.25 We have previously demonstrated brain tumour targeting of CTX-conjugated NPs.23,26,27 Tumour targeting ligands could be used to deliver drugs specifically to tumors and reduce systemic toxicity.28 A biodegradable cross linker, hyaluronic acid (HA), was used to bridge GEM and CTX with IONPs. HA is an anionic, non-sulfated glycosaminoglycan that is naturally present in human tissues. HA possesses repeated carboxyl groups on its glucuronic acid units that facilitate various chemical modifications and increase drug loading.29 A low molecular weight HA of ∼5 kDa was used in this study. The GEM and CTX conjugated IONPs (IONP-HA-GEM-CTX) (Scheme 1) were physicochemically characterized based on physical and hydrodynamic sizes, ζ-potential, drug loading and medium stability. The cellular uptake of the NPs and their effect on the potency in cell killing were evaluated. Furthermore, the pharmacokinetics, biodistribution, and BBB penetration of IONP-HA-GEM-CTX in wild type mice, were also studied.
 | ||
| Scheme 1 Preparation of IONP-HA-GEM-CTX. (a) Conjugation of GEM onto IONP-NH2. (b) Conjugation of CTX onto IONP-HA-GEM. | ||
IONP-HA-GEM-CTX was synthesized by firstly conjugating GEM onto IONPs using HA as a bridging molecule (Scheme 1a) followed by the conjugation of CTX (Scheme 1b). 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and N-hydroxysuccinimide (NHS) were used for coupling of carboxyl and amine groups in these steps. CTX was conjugated onto the carboxyl groups of HA, which was conjugated onto the IONPs in the first step. According to the synthesis procedure of IONP-PEG-NH2, some PEG coatings on the IONP had amine groups (Scheme 1a and ESI†). IONP-HA-GEM-CTX was examined using transmission electron microscopy (TEM) with negative staining by uranyl acetate. The TEM images showed that these NPs had a core size of ∼12.5 nm with a uniform spherical shape (Fig. 1a). With negative staining, we were able to visualize surface coating of the NPs. The bright circles around the IONPs indicated that the IONPs had a surface coating with a thickness of ∼2.5 nm. However, when dispersed in aqueous solutions, the NPs had a hydrodynamic size of ∼32 nm (Fig. 1b and d) and a very narrow size distribution (PDI ≈ 0.1). The NPs with and without CTX conjugation had a slightly different ζ-potential while both were near neutral (Fig. 1c and d). As GEM has a primary amine and may interact with HA electrostatically during the conjugation process, we tested the drug loading with and without the presence of EDC and NHS. GEM was extracted from the NPs after conjugation and analysed by high-performance liquid chromatography (HPLC). It was observed that, with EDC and NHS, a clear and sharp GEM peak was observed. However, without EDC and NHS, no GEM could be detected indicating no physical absorption onto the NPs (Fig. 1e). The number of GEM molecules per NP was determined to be ∼20 (based on ∼0.64 nmol NP per mg Fe). The number of CTX molecules per particle on IONP-HA-GEM-CTX was estimated to be 5 ± 1.4 (based on free CTX after conjugation). The stability of the NPs in cell culture medium was also tested. IONP-HA-CTX and IONP-HA-GEM-CTX were incubated with complete DMEM supplemented with 10% FBS and antibiotics at 37 °C. The hydrodynamic sizes were monitored over a two-week period. The size of the NPs only increased slightly (Fig. 1f), likely due to serum protein adsorption.30 This indicates that the NPs had excellent stability in biological media which contain plenty of ions, proteins, amino acids, etc. To study the cellular uptake of NPs, IONP-HA-GEM-CTX was labeled with Cy3 for visualization in cells. HA was first reacted with Cy3-hydrazide through the carboxyl-hydrazide reaction before conjugation onto the IONPs. Two human GBM cell lines, SF-763 and U-118 MG, were used, representing different expressions of MGMT, which is responsible for the degree of TMZ resistance.31,32 The cells were incubated with NPs for 2 h, washed, fixed and stained with nucleus and membrane dyes. Cells were then mounted onto glass slides and imaged using a Leica SP8 confocal microscope. For easy visualization, the cell membranes and NPs were colored red and green, respectively. It was shown that IONP-HA-GEM-CTX-Cy3 entered cells effectively and distributed in the cytoplasm around nuclei (Fig. 2b and d).
 | ||
| Fig. 1 Characterization of NPs. (a) TEM micrograph of IONP-HA-GEM-CTX. Inset: Cartoon illustration of a coated IONP. (b and c) Size distribution of IONP-HA-GEM and IONP-HA-GEM-CTX weighted by intensity (b) and their ζ-potentials at pH 7.4 (c) as measured with DLS. (d) Properties of IONP-HA-CTX and IONP-HA-GEM-CTX. (e) HPLC analysis of GEM extracted from IONP-HA-GEM synthesized with or without the presence of EDC and NHS. (f) Stability of IONP-HA-CTX and IONP-HA-GEM-CTX in complete DMEM cell culture medium at 37 °C as determined by hydrodynamic size monitoring over 14 days. | ||
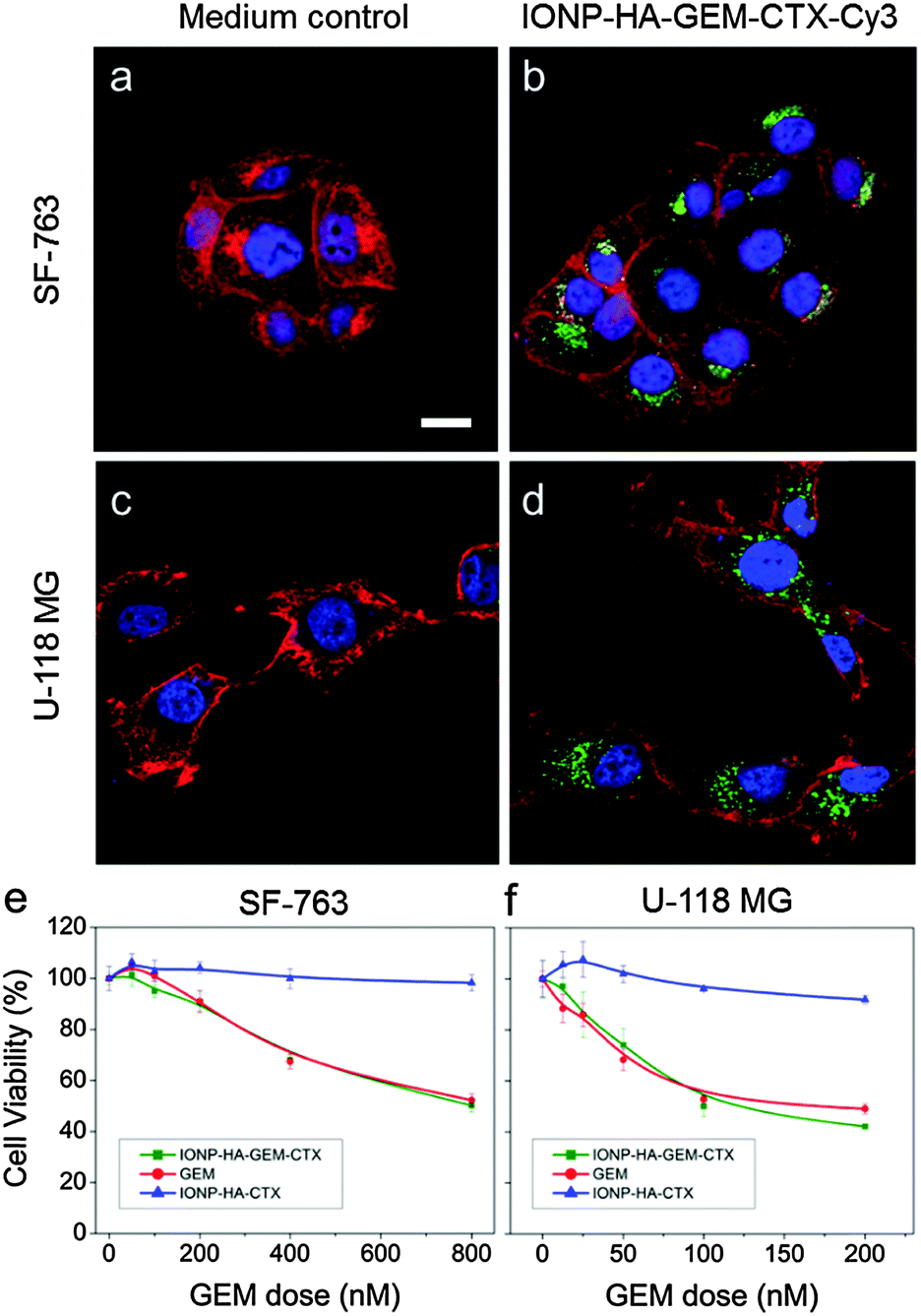 | ||
| Fig. 2 Cellular uptake of IONP-HA-GEM-CTX-Cy3 and viability after different treatments. (a and c) SF-763 (a) and U-118 MG (c) with medium control; (b and d) SF-763 (b) and U-118 MG (d) incubated with IONP-HA-GEM-CTX-Cy3 (40 μg mL−1 [Fe]) for 2 h. Blue, cell nucleus; red, cell membrane (false-coloured); green, NPs (false-coloured); (e) and (f), cell viability of SF-763 (e) and U-118 MG (f) after treatments of IONP-HA-GEM-CTX, IONP-HA-CTX or free GEM for 72 h. | ||
The cell viability was then tested by the Alamar Blue assay. Cells were treated with GEM, IONP-HA-GEM-CTX or drug-free IONP-HA-CTX for 3 days. The results showed that IONP-HA-CTX had no effect on cell viability. GEM and IONP-HA-GEM-CTX showed a similar cell kill profile to both cell lines (Fig. 2e and f). This suggests that after conjugating onto IONPs, GEM did not lose its potency as compared to free drug. The ultimate benefit of IONP-HA-GEM-CTX could be expected in vivo as the biodistribution of the drug plays an important role in determining in vivo efficacy.33
GEM has a short blood half-life (∼0.28 h).4,13 As we have previously demonstrated long-lasting blood circulation with O6-benzylguanine conjugated IONPs,23 we expected that the GEM-conjugated IONPs would show prolonged blood circulation as well. To verify this, we labelled IONP-HA-GEM-CTX with a near infrared dye Cy5.5 similar to the aforementioned Cy3 labelling for in vivo detection purposes. The resultant IONP-HA-GEM-CTX-Cy5.5 was injected into wild type mice intravenously with 0.2 mg Fe equivalents per mouse. Blood samples were collected at various time points and the NP fluorescence from the blood sample was measured by a microplate reader (Ex: 673 nm; Em: 727 nm). In the meantime, various organs were collected at 3 h and 48 h after NP injection for biodistribution study. Note that the mice behave normally throughout this experiment and 4 weeks after the experiment, and there was no body weight loss, indicating the non-toxicity of the IONPs. From the pharmacokinetic profile of IONP-HA-GEM-CTX-Cy5.5, it can be seen that the clearance of NPs from blood circulation fits a power law distribution curve (Fig. 3a). The blood half-life was estimated to be ∼2.8 h, which is 10 fold longer than that of free GEM.13 NP localization in various organs was quantified by an IVIS 200 imaging system. To avoid the autofluorescence of tissues, the excitation was set to be 710 nm and the emission was set to be 810–875 nm. The distribution of IONP-HA-GEM-CTX-Cy5.5 in wild type mice is shown in Fig. 3b. IONP-HA-GEM-CTX-Cy5.5 had the highest accumulation in the liver at 3 h but more than 50% was eliminated at 48 h. The spleen also showed an NP signal at 3 h with a slight increase at 48 h.
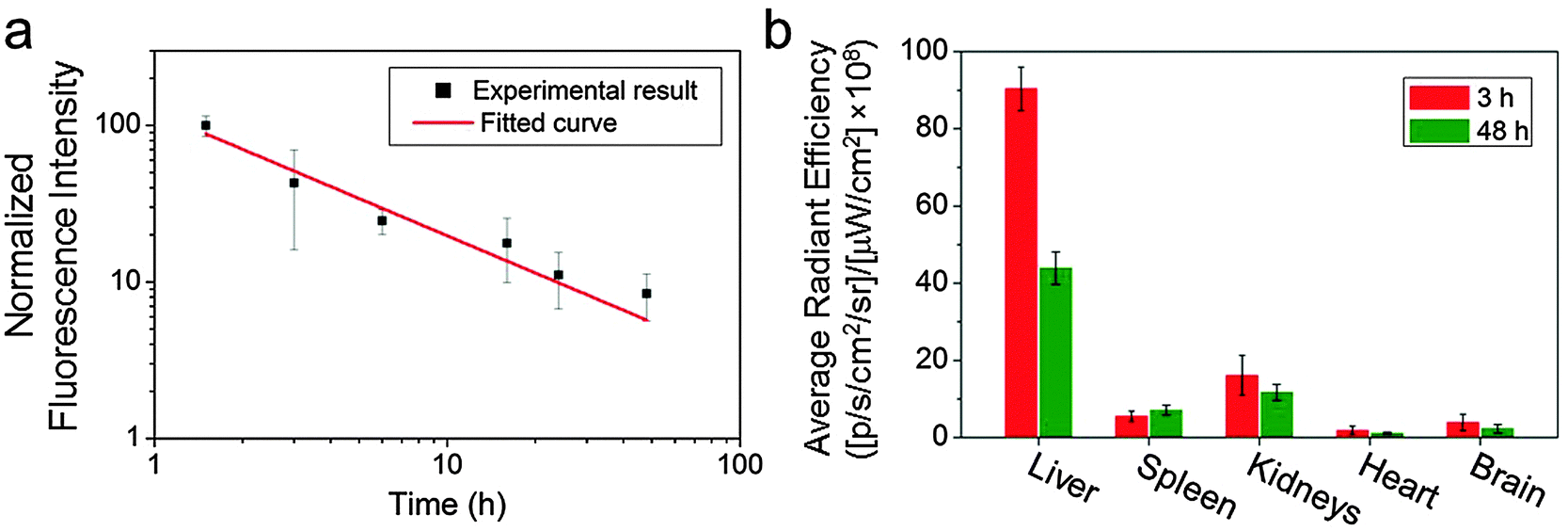 | ||
| Fig. 3 Pharmacokinetics and biodistribution of IONP-HA-GEM-CTX-Cy5.5 in wild type mice. (a) Blood clearance profile of IONP-HA-GEM-CTX-Cy5.5 determined using fluorescence measurements. The curve indicates a power law distribution fit to the data (n = 3 mice per time point); (b) Ex vivo measurement of the fluorescence intensity from various organs at 3 h and 48 h post injection using the IVIS 200 imaging system. | ||
The kidney had the second strongest signal at 3 h among all organs with a moderate decrease at 48 h. Based on this information, it can be expected that IONP-HA-GEM-CTX-Cy5.5 is mainly degraded in liver and is excreted through the renal system. There was nearly no signal from the heart and a low signal from the brain. Since these wild type mice had no tumours in the brain, it is expected that not many NPs would accumulate in the brain. However, it is imperative to know whether IONP-HA-GEM-CTX-Cy5.5 is able to cross the BBB in vivo and thus serve as a potential drug delivery carrier for GBM treatment.
To evaluate the BBB permeability of IONP-HA-GEM-CTX-Cy5.5, we analyzed brain sections of mice at 2 h and 6 h after intravenous injections of NPs (Fig. 4). Tissue sections were stained with the anti-CD31 antibody for visualization of endothelial blood vessels and DAPI for nuclei. The brain tissues from untreated mice showed no signal from NPs (top row). The images from the mouse at 2 h after NP administration showed several red dots near blood vessels indicating extravasation of NPs from blood vessels (middle row). There were some NPs around blood vessels at 6 h (bottom row) but the amount was much less than those at 2 h. This result and the biodistribution result shown in Fig. 3b suggest that IONP-HA-GEM-CTX-Cy5.5 was able to pass the BBB in live mice and accumulate in brains, although only a small amount of NPs were observed, because these mice did not bear brain tumours. A higher NP amount in the brains of tumour-bearing mice is expected because of the active targeting mediated by CTX.23,26,34
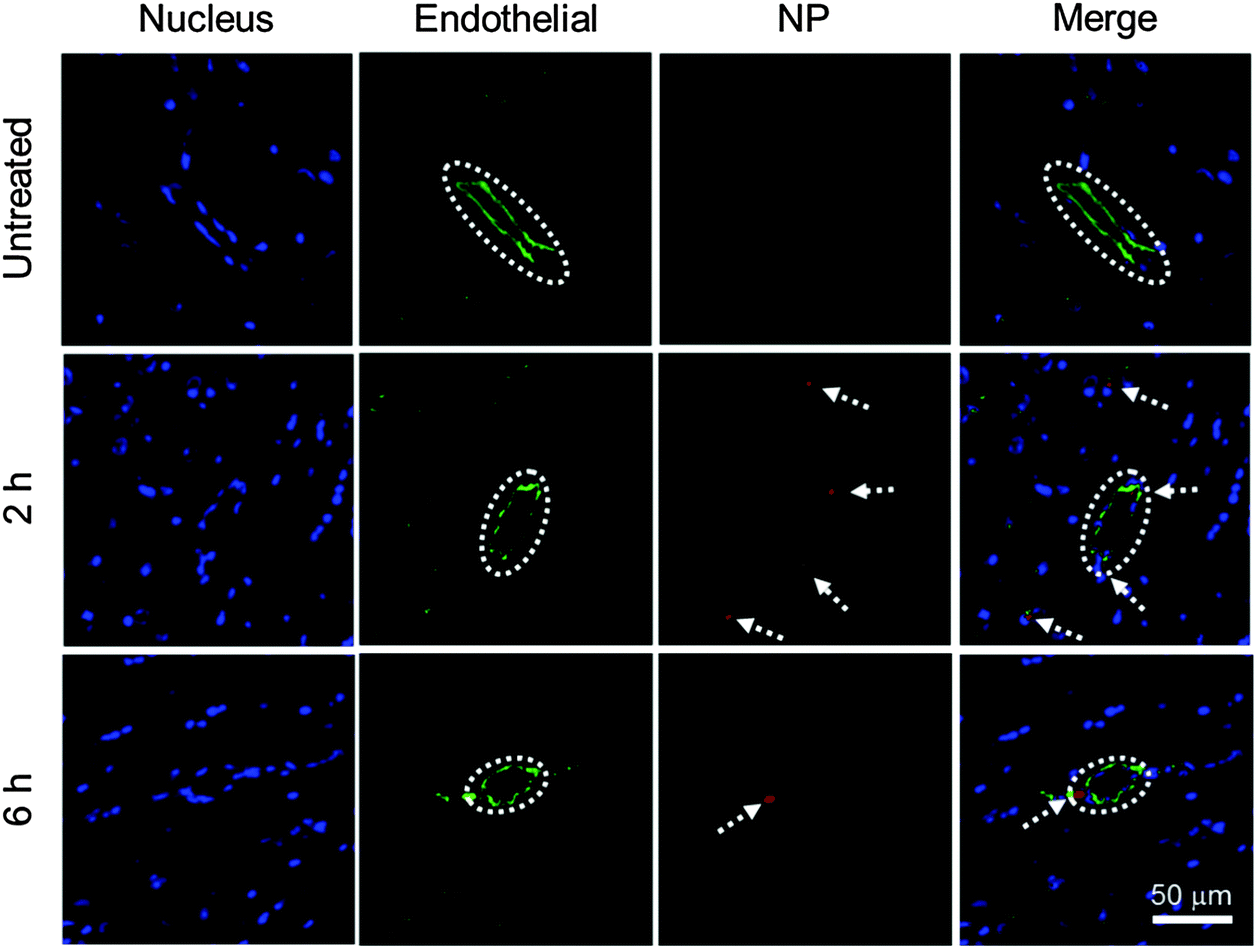 | ||
| Fig. 4 In vivo BBB permeability study. Confocal fluorescence microscopy images of mouse brain tissue sections with one of three treatment conditions: untreated (top), at 2 h (middle) and at 6 h (bottom) after injection of IONP-HA-GEM-CTX-Cy5.5. Cell nucleus was stained with DAPI (blue); endothelial cells were stained with anti-CD31 antibody (green) and NPs were red (false-coloured). Blood vessels are highlighted in dashed circles. The scale bar corresponds to 100 μm and applies to all photos. | ||
Conclusions
IONP-HA-GEM-CTX produced in this study has small size, uniform shape, and great stability in biological medium. Significantly, IONP-HA-GEM-CTX effectively entered and killed GBM cells, had prolonged blood circulation, and was excreted from the renal system. Furthermore, the NPs demonstrated the ability to cross the BBB in live mice. Our experimental results suggest that IONP-HA-GEM-CTX has the potential to improve the in vivo performance of GEM.Acknowledgements
The work is supported by NIH grant R01CA161953. Q. M. acknowledges support from an NIH Ruth L. Kirschstein T32 Fellowship (T32CA138312). We also acknowledge the support from NIH to the UW Keck Microscopy Facility (S10OD016240).Notes and references
- S. Hoelder, P. A. Clarke and P. Workman, Mol. Oncol., 2012, 6, 155–176 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS PubMed.
- R. A. Petros and J. M. DeSimone, Nat. Rev. Drug Discovery, 2010, 9, 615–627 CrossRef CAS PubMed.
- S. Noble and K. L. Goa, Drugs, 1997, 54, 447–472 CrossRef CAS PubMed.
- C. Nabhan, N. Krett, V. Gandhi and S. Rosen, Curr. Opin. Oncol., 2001, 13, 514–521 CrossRef CAS PubMed.
- V. Heinemann, Oncology, 2001, 60, 8–18 CrossRef CAS PubMed.
- M. D. Shelley, G. Jones, A. Cleves, T. J. Wilt, M. D. Mason and H. G. Kynaston, BJU Int., 2012, 109, 496–505 CrossRef CAS PubMed.
- V. Heinemann, Oncology, 2003, 64, 191–206 CrossRef CAS PubMed.
- D. Lorusso, A. Di Stefano, F. Fanfani and G. Scambia, Ann. Oncol., 2006, 17, v188–v194 CrossRef PubMed.
- B. Geoerger, J. Chisholm, M.-C. Le Deley, J.-C. Gentet, C. M. Zwaan, N. Dias, T. Jaspan, K. Mc Hugh, D. Couanet, S. Hain, A. Devos, R. Riccardi, C. Cesare, J. Boos, D. Frappaz, P. Leblond, I. Aerts and G. Vassal, Eur. J. Cancer, 2011, 47, 230–238 CrossRef CAS PubMed.
- M. Morfouace, A. Shelat, M. Jacus, B. B. Freeman Iii, D. Turner, S. Robinson, F. Zindy, Y.-D. Wang, D. Finkelstein, O. Ayrault, L. Bihannic, S. Puget, X.-N. Li, J. M. Olson, G. W. Robinson, R. K. Guy, C. F. Stewart, A. Gajjar and M. F. Roussel, Cancer Cell, 2014, 25, 516–529 CrossRef CAS PubMed.
- G. Metro, A. Fabi, M. Mirri, A. Vidiri, A. Pace, M. Carosi, M. Russillo, M. Maschio, D. Giannarelli, D. Pellegrini, A. Pompili, F. Cognetti and C. Carapella, Cancer Chemother. Pharmacol., 2010, 65, 391–397 CrossRef CAS PubMed.
- L. A. Shipley, T. J. Brown, J. D. Cornpropst, M. Hamilton, W. D. Daniels and H. W. Culp, Drug Metab. Dispos., 1992, 20, 849–855 CAS.
- B. R. Sloat, M. A. Sandoval, D. Li, W.-G. Chung, D. S. P. Lansakara-P, P. J. Proteau, K. Kiguchi, J. DiGiovanni and Z. Cui, Int. J. Pharm., 2011, 409, 278–288 CrossRef CAS PubMed.
- J. L. Arias, L. H. Reddy and P. Couvreur, Langmuir, 2008, 24, 7512–7519 CrossRef CAS PubMed.
- S. Rejiba, L. H. Reddy, C. Bigand, C. Parmentier, P. Couvreur and A. Hajri, Nanomed.: Nanotechnol., Biol., Med., 2011, 7, 841–849 CrossRef CAS PubMed.
- E. Dalla Pozza, C. Lerda, C. Costanzo, M. Donadelli, I. Dando, E. Zoratti, M. T. Scupoli, S. Beghelli, A. Scarpa, E. Fattal, S. Arpicco and M. Palmieri, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 1396–1404 CrossRef CAS PubMed.
- G. Arya, M. Vandana, S. Acharya and S. K. Sahoo, Nanomed.: Nanotechnol., Biol., Med., 2011, 7, 859–870 CrossRef CAS PubMed.
- C.-X. Wang, L.-S. Huang, L.-B. Hou, L. Jiang, Z.-T. Yan, Y.-L. Wang and Z.-L. Chen, Brain Res., 2009, 1261, 91–99 CrossRef CAS PubMed.
- P. Y. Wen and S. Kesari, N. Engl. J. Med., 2008, 359, 492–507 CrossRef CAS PubMed.
- C. Fang, N. Bhattarai, C. Sun and M. Q. Zhang, Small, 2009, 5, 1637–1641 CrossRef CAS PubMed.
- C. Sun, J. S. H. Lee and M. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1252–1265 CrossRef CAS PubMed.
- Z. R. Stephen, F. M. Kievit, O. Veiseh, P. A. Chiarelli, C. Fang, K. Wang, S. J. Hatzinger, R. G. Ellenbogen, J. R. Silber and M. Zhang, ACS Nano, 2014, 8, 10383–10395 CrossRef CAS PubMed.
- N. Kohler, G. E. Fryxell and M. Zhang, J. Am. Chem. Soc., 2004, 126, 7206–7211 CrossRef CAS PubMed.
- J. Deshane, C. C. Garner and H. Sontheimer, J. Biol. Chem., 2003, 278, 4135–4144 CrossRef CAS PubMed.
- O. Veiseh, C. Sun, C. Fang, N. Bhattarai, J. Gunn, F. Kievit, K. Du, B. Pullar, D. Lee, R. G. Ellenbogen, J. Olson and M. Zhang, Cancer Res., 2009, 69, 6200–6207 CrossRef CAS PubMed.
- C. Fang, O. Veiseh, F. Kievit, N. Bhattarai, F. Wang, Z. Stephen, C. Li, D. Lee, R. G. Ellenbogen and M. Zhang, Nanomedicine, 2010, 5, 1357–1369 CrossRef CAS PubMed.
- L. Brannon-Peppas and J. O. Blanchette, Adv. Drug Delivery Rev., 2012, 64(Supplement), 206–212 CrossRef.
- G. D. Prestwich, D. M. Marecak, J. F. Marecek, K. P. Vercruysse and M. R. Ziebell, J. Controlled Release, 1998, 53, 93–103 CrossRef CAS PubMed.
- M. M. Yallapu, N. Chauhan, S. F. Othman, V. Khalilzad-Sharghi, M. C. Ebeling, S. Khan, M. Jaggi and S. C. Chauhan, Biomaterials, 2015, 46, 1–12 CrossRef CAS PubMed.
- M. S. Bobola, S. Varadarajan, N. W. Smith, R. D. Goff, D. D. Kolstoe, A. Blank, B. Gold and J. R. Silber, Clin. Cancer Res., 2007, 13, 612–620 CrossRef CAS PubMed.
- N. Gaspar, L. Marshall, L. Perryman, D. A. Bax, S. E. Little, M. Viana-Pereira, S. Y. Sharp, G. Vassal, A. D. J. Pearson, R. M. Reis, D. Hargrave, P. Workman and C. Jones, Cancer Res., 2010, 70, 9243–9252 CrossRef CAS PubMed.
- C. Fang, F. M. Kievit, Y. C. Cho, H. Mok, O. W. Press and M. Zhang, Nanoscale, 2012, 4, 7012–7020 RSC.
- F. M. Kievit, O. Veiseh, C. Fang, N. Bhattarai, D. Lee, R. G. Ellenbogen and M. Q. Zhang, ACS Nano, 2010, 4, 4587–4594 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tb02123e |
| This journal is © The Royal Society of Chemistry 2016 |