
Characterization of lectin binding affinities via direct LC-MS profiling: implications for glycopeptide enrichment and separation strategies†
Feifei
Zhu
,
David E.
Clemmer
and
Jonathan C.
Trinidad
*
Department of Chemistry, Indiana University, 800 Kirkwood Ave., Bloomington, IN 47405, USA. E-mail: trinidad@indiana.edu
First published on 10th November 2016
Abstract
Determining the affinity between a lectin and its target glycans is an important goal, both for understanding the biological functions of a given lectin as well as enabling the use of that lectin for targeted enrichment of glycosylated species from complex samples. While the overall selectivities of many lectins have been characterized, such studies generally require individually purified lectins and glycans. From these analyses, it is clear that a given lectin does not bind all of its target glycans with the same affinity. Rather, lectins display a continuum of affinities for the range of glycan structures they may encounter. Because of this continuum, it is not straightforward in practice to determine which set of structures will be enriched using a lectin as an affinity reagent. Here we describe the development of glycan affinity chromatography coupled directly to electrospray mass spectrometry, which enables direct analysis of interactions of lectins with both glycans and glycoconjugates from complex mixtures. By observing the elution behavior of individual species, we are able to determine exactly which set of glycoconjugates would be enriched for a given lectin. Furthermore, this approach allows for the direct assessment of affinity constants between an individual lectin and a large number of glycans in a single experiment, which can be conducted using a complex mixture of unpurified glycans of varying concentrations.
Introduction
Glycosylation on membrane and secreted proteins regulates a range of critical functions in biological systems.1–3 Mapping of protein glycosylation patterns and determination of glycan structures are primarily accomplished through the use of mass spectrometry (MS).4–8 Analysis of protein glycosylation is particularly challenging because these modifications are frequently sub-stoichiometric and can display extensive structural microheterogeneity (i.e. a range of distinct glycans can modify exactly the same site on different versions of the protein).9,10 Additionally, glycopeptides ionize with lower efficiency than non-glycosylated peptides, which may result in suppression of glycopeptide signals in MS.11–13 As a consequence, when complex biological material is to be analyzed, enrichment of glycopeptides prior to MS analyses is necessary in order to maximize the number of species that can be characterized.14,15Multiple strategies exist to enrich glycosylated proteins/peptides, including lectin-based approaches,16–20 hydrophilic interaction chromatography (HILIC),21–24 and boronate-based chromatography.25,26 The use of immobilized lectin, either in a precipitation-type or chromatographic format, is perhaps the most widely employed glycan enrichment approach. In lectin affinity chromatography (LAC), lectins are immobilized onto a chromatographic matrix such as silica, agarose, or POROS.27 LAC typically employs high ionic strength buffer, and elution of tightly-bound glycosylated species is accomplished by injection of a specific saccharide to competitively displace these species. These procedures require additional washing and desalting steps before MS analysis, and therefore are typically accomplished offline. While offline fractionation is widely used, it can result in potential sample loss.28 In solving this issue, methods have been reported that allow on-line desalting and fractionation of glycopeptide mixtures subsequent to glyco-enrichment.16
We recently published a manuscript describing a WGA-based glycopeptide enrichment approach that allowed for the identification of 2500 unique glycopeptides,29 which was a significant advance in the number of glycopeptides identified in a single experiment. Nevertheless, there remains a substantial need for improved glycopeptide enrichment techniques. In those experiments, it was necessary to run the peptide digest over the lectin column three consecutive times to achieve sufficient separation from non-modified peptides, and the final purified fraction still contained approximately 1/3 non-glycosylated species. A limitation of agarose precipitation or column-based lectin affinity approaches is the difficulty in quantitatively determining the effect of varying wash and elution conditions.30–32 Individual wash or chromatographic fractions can be analyzed for the ratio of non-modified versus glycosylated species, but such an approach is laborious and semi-quantitative at best. This may partially explain the widespread presence of non-glycosylated proteins and peptides that are identified as background in such analyses.
In many cases, the binding affinities of a lectin for a panel of sugar structures have been determined. Such studies often focus on monosaccharides or simple oligosaccharides rather than the range of more complicated glycan structures produced by an organism. Many techniques exist for studying sugar–protein interactions, including surface plasmon resonance,33,34 titration calorimetry,35 frontal affinity chromatography,36–38 capillary affinity electrophoresis,39,40 and lectin microarray techniques.41,42 However, measuring the glycan–lectin binding interactions can be challenging because the interaction is often weak, with association constants less than 105 M−1. In addition, many of the above affinity measurement techniques require determination of precise concentrations of individual glycans, which can be difficult to achieve in a complex mixture.
This work reports the development of affinity chromatography directly coupled to MS for rapid screening of glycopeptides and evaluation of weak affinity glycan–lectin interactions. Determination of peak elution profiles allows for precise determination of binding affinities. Using this approach, we have compared a series of glycopeptides varying in the glycan and/or peptide moiety to investigate their relative contribution to glycopeptide binding. We evaluated several types of resin as suitable matrices for the enrichment and separation, and demonstrated that the methacrylate resin has fewer non-specific interactions than the polystyrene-based resin. This profiling approach allows for direct, quantitative optimization of parameters for glycopeptide enrichment strategies.
Results and discussion
Glycopeptide enrichment profiles can be directly measured by LC-MS
Fig. 1A shows the MS spectrum obtained via direct infusion of a tryptic digest mixture from several glycoproteins. As can be seen, non-glycosylated peptides dominated the spectrum and only a few low intensity glycopeptide peaks were observed. We have previously demonstrated that when WGA was immobilized in an HPLC column, both O-GlcNAcylated as well as more complex N- and O-linked glycopeptides can be successfully enriched and identified from digestions of complex protein mixtures. These peptides were not generally fully resolved from the unmodified peptide pool, rather they eluted at the tail end of that distribution.29,31 We wish to better define the separation of non-modified from glycosylated peptides by analyzing the eluant from a lectin column directly using MS with the goal of enhancing the overall enrichment and relative yield. Lectin based enrichment typically uses tris(hydroxymethyl)aminomethane (Tris) as a buffer component. However this salt is poorly compatible with ESI, and in addition has been shown to cause side reactions with sialic acids.43 To avoid these issues, we replaced Tris with ammonium acetate. A series of ammonia acetate concentrations was tested (1, 10, 25, 50, 100, and 150 mM), and no significant effect on glycopeptide retention times was observed as function of ammonia acetate concentration (data not shown). However, higher concentrations gave increased salt adduct peaks that complicated the MS spectra, and at 150 mM, protonated glycopeptide signals were completely suppressed. We therefore chose to use 25 mM ammonium acetate, since this was the highest buffering capacity for which significant salt adduct formation was not observed. | ||
| Fig. 1 MS spectra of a mixture of glycopeptides from a tryptic digest of ovomucoid, RNase B and α-crystallin. (A) shows the m/z distribution of this sample when directly infused into the mass spectrometer. Minor peaks can be seen for two glycoforms of SIEFGTNISK at m/z = 1299 and 1603. (B) shows the TIC of an online LAC using a WGA-coated POROS column at 0.1 ml min−1 flow rate. The survey scan was summed from 10–20 minutes (1C) and 20–40 minutes (1D). Peaks corresponding to glycopeptides listed in Table 1 are colored red, with the specific glycan structures listed for the most prominent peaks. | ||
When a column with WGA immobilized on POROS resin was used to separate the peptides, the bulk of the unbound, non-modified peptides eluted between 7 and 20 min (Fig. 1B). In contrast, most glycopeptides had much longer retention times, ranging from 20 to 40 min (Fig. 1C). Fig. 1D demonstrates that the lectin affinity chromatography effectively enriched the glycopeptides in the mixture, leading to significant increases in their MS signals, and the majority of the most intense peaks corresponded to glycopeptides.
Relative binding affinities of glycopeptides: effect of the glycan moiety
Because the lectin affinity column was coupled directly to the MS, we could use extracted ion chromatograms (XICs) at the masses of known peptides and glycopeptides to directly profile their interaction with WGA. Fig. 2A shows the normalized elution profiles of a series of non-modified and glycosylated peptides separated at a flow rate of 0.2 ml min−1. As can be seen, the peak elution times of the non-modified peptides ranged from 6.9 to 8.4 minutes, while those of the glycopeptides ranged from 9.1 to 15.8 minutes. Using this approach, a total of 25 unique glycosylated peptides derived from four standard glycoproteins were profiled, with the results listed in Table 1. In several instances, sets of glycopeptides were observed which contained the same peptide sequence, but a range of glycans. Included in this mixture was the O-GlcNAcylated protein, alpha crystallin. The GlcNAc-modified version of the peptide AIPVSREEKPSSAPSS had a retention time of 10.5 min, while the non-modified version eluted earlier, at 8.0 min, illustrating the notable binding affinity of WGA to peptides bearing as little as a single saccharide. Comparison of the glycopeptides 2, 4, 6, and 8, which contain the high-mannose glycans Man5GlcNAc2, Man6GlcNAc2, Man7GlcNAc2, and Man8GlcNAc2, respectively, indicates that sequentially increasing the number of Man residues has very little effect on the glycopeptide retention times on a WGA column (Table 1). We did not clearly observe peaks corresponding to the Man9GlcNAc2-modified version of peptides from RNase B, most likely due to the fact that it is present at very low relative abundance.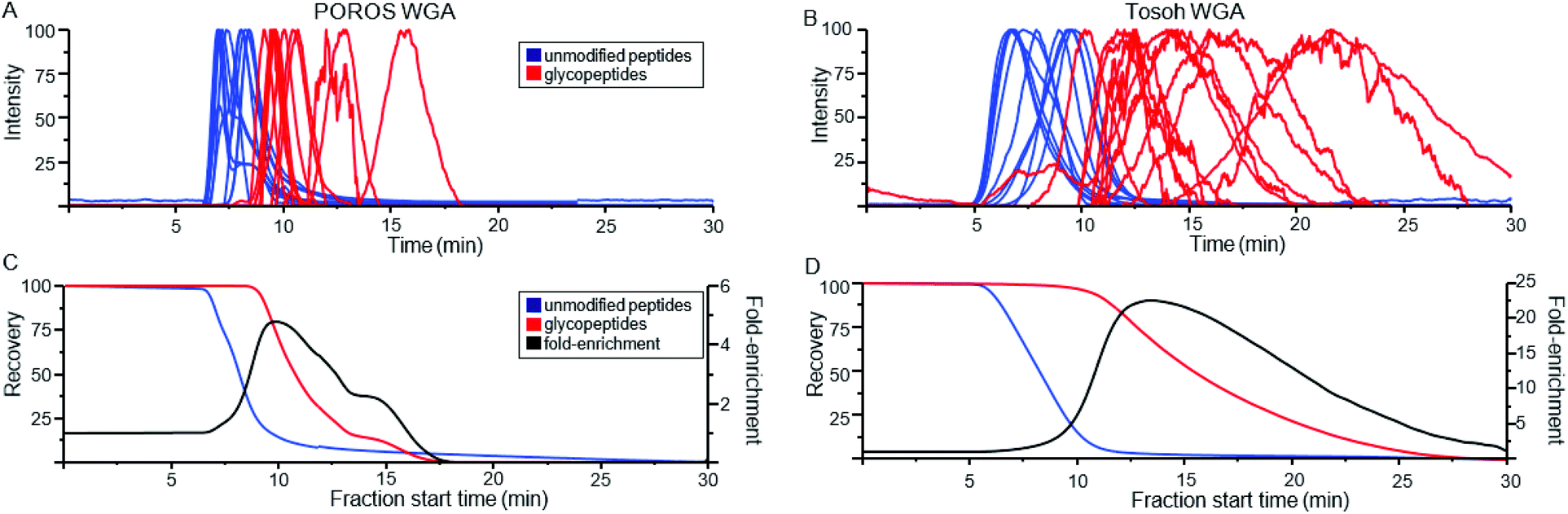 | ||
| Fig. 2 Normalized extracted ion chromatograms (XICs) of non-modified peptides (blue) and glycopeptides (red) obtained from WGA-coated POROS (A) and Tosoh (B) columns. The XICs have been smoothed by adjacent-averaging 30 points per window and background-subtracted based on the TIC. The peptide information is listed in Table 1. Recovery rate for non-modified and glycosylated peptides on the two columns were calculated as a function of fraction start time (C–D). Fold-recovery was also calculated by dividing the relative yield of glycopeptides at that time point by the non-modified peptide relative yield. | ||
| Peptide | z | Peptide sequenceb | Glycoprotein | Glycan type | Glycan formc | Elution time | |
|---|---|---|---|---|---|---|---|
| number | m/z | ||||||
| a A total of 24 glycopeptides and one non-modified peptide were analyzed at a 0.2 ml min−1 flow rate. b For each sequence, the site of glycosylation is indicated by an asterisk. The Uniprot IDs are as follows: α-crystallin (P02489); ovomucoid (P01005); RNase B (P61823); HRP (P00433); fetuin (P12763). c The observed glycans were annotated as follows: N-acetylglucosamine (GlcNAc); mannose (Man); galactose (Gal); N-acetylneuraminic acid (NeuAc); xylose (Xyl); fucose (Fuc). | |||||||
| 0 | 821.4 | 2 | AIPVSREEKPSSAPSS | α-Crystallin | NA | NA | 8.0 |
| 1 | 923.0 | 2 | AIPVS*REEKPSSAPSS | α-Crystallin | O-Linked | GlcNAc1 | 10.5 |
| 2 | 846.4 | 2 | N*LTK | RNAse B | High mannose | Man5GlcNAc2 | 10.8 |
| 3 | 967.9 | 2 | SRN*LTK | RNAse B | High mannose | Man5GlcNAc2 | 10.8 |
| 4 | 927.4 | 2 | N*LTK | RNAse B | High mannose | Man6GlcNAc2 | 11.1 |
| 5 | 1049.0 | 2 | SRN*LTK | RNAse B | High mannose | Man6GlcNAc2 | 11.2 |
| 6 | 1008.4 | 2 | N*LTK | RNAse B | High mannose | Man7GlcNAc2 | 11.0 |
| 7 | 1130.0 | 2 | SRN*LTK | RNAse B | High mannose | Man7GlcNAc2 | 11.0 |
| 8 | 1089.5 | 2 | N*LTK | RNAse B | High mannose | Man8GlcNAc2 | 11.5 |
| 9 | 1211.0 | 2 | SRN*LTK | RNAse B | High mannose | Man8GlcNAc2 | 11.5 |
| 10 | 994.4 | 2 | SIEFGTN*ISK | Ovomucoid | Core | Man3GlcNAc2 | 14.8 |
| 11 | 1299.1 | 2 | SIEFGTN*ISK | Ovomucoid | Hybrid | Man3GlcNAc5 | 18.5 |
| 12 | 1400.6 | 2 | SIEFGTN*ISK | Ovomucoid | Hybrid | Man3GlcNAc6 | 12.2 |
| 13 | 1481.6 | 2 | SIEFGTN*ISK | Ovomucoid | Hybrid | Gal1Man3GlcNAc6 | 12.4 |
| 14 | 1502.1 | 2 | SIEFGTN*ISK | Ovomucoid | Hybrid | Man3GlcNAc7 | 13.3 |
| 15 | 1603.7 | 2 | SIEFGTN*ISK | Ovomucoid | Hybrid | Man3GlcNAc8 | 12.4 |
| 16 | 1684.7 | 2 | SIEFGTN*ISK | Ovomucoid | Hybrid | Gal1Man3GlcNAc8 | 12.8 |
| 17 | 921.9 | 2 | NVGLN*R | HRP | Hybrid | Xyl1Man3Fuc1GlcNAc2 | 8.2 |
| 18 | 1306.1 | 2 | MGN*ITPLTGTQGQIR | HRP | Hybrid | Xyl1Man3GlcNAc2 | 8.8 |
| 19 | 1677.2 | 2 | SFAN*STQTFFNAFVEAMDR | HRP | Hybrid | Xyl1Man3Fuc1GlcNAc2 | 10.7 |
| 20 | 1698.2 | 2 | QLTPTFYDNSCPN*VSNIVR | HRP | Hybrid | Xyl1Man3Fuc1GlcNAc2 | 10.4 |
| 21 | 1836.4 | 2 | GLIQSDQELFSSPN*ATDTIPLVR | HRP | Hybrid | Xyl1Man3Fuc1GlcNAc2 | 8.2 |
| 22 | 1470.1 | 4 | VVHAVEVALATFNAESN*GSYLQLVEISR | Fetuin | Complex | NeuAc3Gal3Man3GlcNAc5 | 21.4 |
| 23 | 1542.9 | 4 | VVHAVEVALATFNAESN*GSYLQLVEISR | Fetuin | Complex | NeuAc4Gal3Man3GlcNAc5 | 30.6 |
| 24 | 1633.9 | 4 | RPTGEVYDIEIDTLETTCHVLDPTPLAN*CSVR | Fetuin | Complex | NeuAc3Gal3Man3GlcNAc5 | 25.1 |
| 25 | 1706.2 | 4 | RPTGEVYDIEIDTLETTCHVLDPTPLAN*CSVR | Fetuin | Complex | NeuAc4Gal3Man3GlcNAc5 | 35.5 |
The elution behavior of peptides bearing hybrid glycans displayed a complex behavior. Glycopeptides 10–12, 14, and 15 contain increasing numbers of GlcNAc (Man3GlcNAcx, x = 2, 5, 6, 7, 8, respectively); however, their elution order does not increase directly with the number of GlcNAc residues (Table 1). Glycopeptide 11 containing the glycan Man3GlcNAc5 had the longest retention time of this set, at 18.5 min; either the addition or removal of a GlcNAc residue shortened this value (Table 1). These observations indicate that Man3GlcNAc5 (structures shown in Table S1†) has the highest affinity to WGA among these high-mannose and hybrid type glycans. Most lectins have carbohydrate-recognition domains that tend to form a shallow but relatively well-defined binding pocket to recognize specific glycan chains. It has previously been suggested that the tetrasaccharide GlcNAcβ1-4Manβ1-4GlcNAcβ1-4GlcNAc structural moiety can most efficiently fit into the binding pocket of WGA and that additional modifications to this structural moiety can reduce the binding affinity,44 which is consistent with our current observations. Glycopeptide 10, which contains the N-linked pentasaccharide core structure, does not have the full structural moiety due to the absence of the bisecting GlcNAc (Table S1†), and it possesses a decreased affinity compared to glycopeptide 11. In contrast, glycopeptides 12, 14, and 15 possess this tetrasaccharide structural moiety, but also contain additional GlcNAc residues that may sterically hinder this structure from fitting into the WGA binding pocket, which resulted in decreased binding affinities. These observations reinforce the notion that the binding affinity of a glycopeptide depends on the overall glycan structure that comes into contact with the lectin rather than simply the composition of the glycan. Experimental data regarding the relative binding affinities such as we can obtain may prove useful to evaluate the accuracy of in silico structural modeling of lectin binding proteins and their corresponding ligands.
The glycopeptides derived from HRP contain xylose (and fucose) modified versions of the N-linked pentasaccharide core (Table S1†). These glycopeptides eluted earlier from the WGA column relative to the other glycopeptides. The most significant difference between these and other glycopeptides tested is the presence of both fucose and xylose. Our previous manuscript analyzing glycopeptides from complex samples resulted in the identification of many glycopeptides which appeared to have fucose-modified core pentasaccharides.29 It is therefore most likely that the addition of xylose prevents the optimal binding between the pentasaccharide and WGA, thereby significantly reducing the glycan binding affinity.
The glycopeptides from fetuin, which contain complex-type glycans, show much higher retention times than the other glycopeptides (Table 1). These glycopeptides all contain terminal NeuAc residues, which, in addition to GlcNAc, have been demonstrated to interact relatively strongly with WGA.45,46 The better retention of these NeuAc-containing peptides relative to peptide 11 confirms that WGA can interact with multiple glycan structural elements.
Peptide sequence has a limited effect on lectin–glycopeptide interactions
While a lectin can bind to a glycopeptide containing specific sugar residues, non-specific interactions, such as peptide–lectin interactions, can sometimes impede the isolation and purification processes.47–49 Such non-specific interactions can become dominant in the case of intact glycoproteins,50 which may lead to unsuccessful or inefficient enrichment.51 To characterize possible non-specific interactions during the glycopeptide–lectin binding, the retention times of the glycopeptides bearing the same glycan but different peptide sequences were compared (Table 1). The set of glycopeptides 3, 5, 7, and 9 and glycopeptides 2, 4, 6, and 8 from RNase B differ slightly in the peptide sequence, but essentially the same retention profiles were obtained for pairs of glycopeptides with the same glycan.Glycopeptides 19 and 20 were also shown to have greater retention times than glycopeptide 21 despite the fact that they have the same glycan and have amino acid sequences of similar length. Comparison of the retention profiles for glycopeptides 22–25 from fetuin also suggests some degree of non-specific interactions. A close examination reveals that peptides containing aromatic residues, i.e., F, Y, and W, have longer retention times on the POROS resin material. The POROS resin is made of cross-linked polystyrene–divinylbenzene, which contains large arrays of aromatic rings that can interact with aromatic amino acid residues. We suspect that peptides containing aromatic residues show increased retention times, with multiple aromatic residues resulting in longer retention time shifts. Thus, it is important to consider these non-specific interactions when using POROS as the immobilization matrix.
Glycopeptides display better retention to WGA immobilized on methacrylic polymer beads compared to polystyrenedivinylbenzene beads
In addition to the POROS polystyrenedivinylbenzene-based resin, WGA was immobilized on two other matrices. The first was a Tosoh Toyopearl resin, which is composed of methacrylic polymer beads. The second was activated silica beads. Fig. 2B shows the elution profile of peptides run over the WGA-Tosoh resin using XICs from the same set of peptides as shown in Fig. 2A for the POROS resin. The results from the silica resin were very poor (data not shown), with almost all peptides eluting broadly, with low recovery, or not at all. We suspect that the complex behavior of the peptides and glycopeptides on the resin may be due to hydrophilic interactions with the silica. The use of organic solvents in the mobile phase may minimize this effect, but high levels of organic buffer may denature the lectin or interfere with its glycan-binding activity. As such, the silica resin was not investigated further.Overall, the Tosoh resin provided the best separation between non-modified peptides and glycosylated peptides. Non-glycosylated peptides had peak elution times between 6.6 and 9.7 minutes. Glycosylated peptides had peak elution times between 10.2 and 21.7 minutes. To estimate the relative enrichment capabilities of the POROS and Tosoh resins, we calculated the overall recoveries that would be obtained, from having collected the remaining column eluant at various start times (Fig. 2C and D). We estimated overall non-modified and glycosylated peptide recoveries by averaging the respective XICs in Fig. 2A and B. To estimate the fold-recovery at any time point, we divided the relative yield of glycopeptides at that time point by the non-modified peptide relative yield. These estimates will depend partly on the set of peptide elution profiles chosen, but using a range of distinct peptides and glycopeptides should give a relative approximation of the enrichment efficiency.
For the POROS resin, achieving a 95% yield of the 13 glycopeptides would require collecting the eluant starting at 9.1 minutes. This would also capture 22% of the non-modified peptides, corresponding to 4.3-fold enrichment. For the Tosoh resin, achieving a 95% glycopeptide yield would require collecting starting at 10.6 minutes, which would also capture 9.4% of the non-glycopeptides, corresponding to a 10-fold enrichment. The estimated recovery rates of these peptides in different retention time windows (to mimic offline fractionation) are shown in Table S2.† The Tosoh resin provided better overall separation between the non-modified and glycosylated peptides, which is a key factor to consider in offline fractionations. Despite the lower overall enrichment, the POROS resin was judged better for on-line LAC-MS because the narrow elution profiles gave more intense MS signals, and the samples being analyzed were not of high enough complexity such that the glycopeptides suffered significant ion suppression.
Isolated glycans display similar elution behaviors relative to glycopeptides and association constant is proportional to the adjusted retention time
While the above results indicate that the peptide structure plays a fairly small role in the affinity between lectins and glycopeptides, to directly measure the affinity of glycans, we isolated glycans by enzymatic hydrolysis using PNGase F. Fig. 3 shows the retention time profiles of a mixture of isolated glycans from ovomucoid and GlcNAc monosaccharide on a WGA-coated POROS column. The observed free glycans show a similar elution order as their corresponding glycopeptides (Table 1), confirming that the glycan moiety has the dominant contribution to the binding affinity. The two peaks observed for GlcNAc resulted from its α and β anomers. The α anomer was observed to have higher binding affinity than the β anomer by a factor of ∼1.7, which was close to the previously reported factor of 2.1.52,53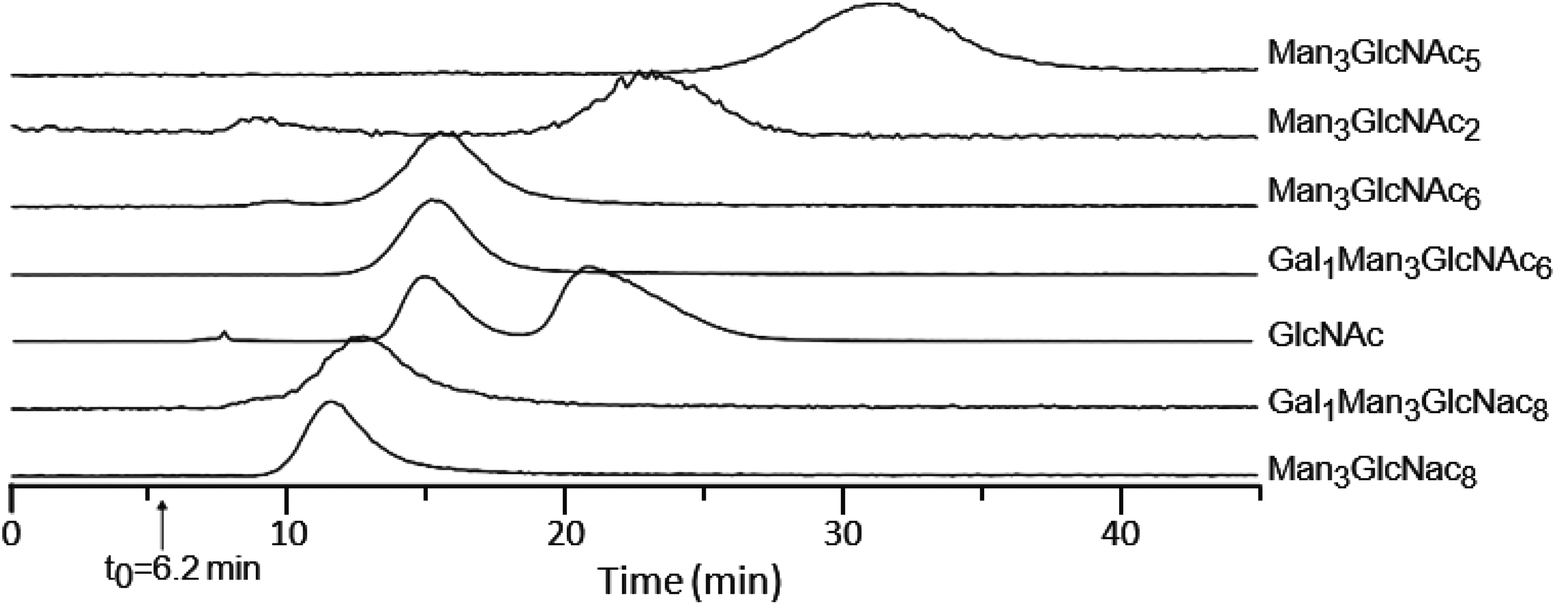 | ||
| Fig. 3 Retention time profiles of glycans and GlcNAc on the WGA-coated POROS column at a flow rate of 0.2 ml min−1. Glycans were released from ovomucoid by PNGase F and analyzed by LC-MS. GlcNAc elutes as a doublet, corresponding to its β and α anomers (at retention times of 15 and 20.8 minutes, respectively). See text for details. | ||
Our LAC-MS approach can also be used to calculate association constants, without the requirement that other approaches have to obtain individual glycans in their purified forms and/or at known concentrations. For lectin–glycan binding, the Ka value can be expressed as Ka = (tR − t0)/(t0C[L]). In this equation, tR and t0 refer to the analyte retention time and void volume time, respectively; C is a constant reflecting the physical characteristics of the matrix including the porosity; and [L] is the effective lectin concentration. The product C[L] can be empirically determined for a given lectin column using a reference standard with a known Ka. The Ka value for WGA interacting with β-GlcNAc is 2 × 102 M−1.52,53 For species measured on the same lectin column, Ka is proportional to the adjusted retention time (tR − t0)/t0. This allows us to calculate Ka values for the isolated glycan and demonstrate them to be in the range from 1 × 102 to 6 × 102 M−1 (Table 2). It is worthwhile to note that the equation is valid provided that the analyte has a concentration c0 (at injection) such that c0Ka ≪ 1.54 This condition is satisfied for our experiments since the Ka value for the glycan–lectin binding is usually on the order of 102–104 M−1,35,38,39 and the glycans used in this study were estimated to be on the μM range. While the Ka values for complex glycans reported in Table 2 have not been reported using alternative methods to calculate affinities, our observation that the Ka of α GlcNAc is approximately twice that of β GlcNAc is consistent with previously reported literature,52,53 thereby providing independent support for the accuracy of our calculations.
| Glycan | t R | t R − t0 | K a (M−1) |
|---|---|---|---|
| a A Ka value of 2 × 102 M−1 was used for the interaction between WGA and β-GlcNAc.52,53 | |||
| β-GlcNAc | 15.1 | 8.9 | 2 × 102![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
| α-GlcNAc | 21.2 | 15.0 | 3 × 102 |
| Man3GlcNAc2 | 23.0 | 16.8 | 4 × 102 |
| Man3GlcNAc5 | 31.3 | 25.1 | 6 × 102 |
| Gal1Man3GlcNAc6 | 15.6 | 9.4 | 2 × 102 |
| Man3GlcNAc8 | 11.5 | 5.3 | 1 × 102 |
| Gal1Man3GlcNAc8 | 12.6 | 6.4 | 1 × 102 |
Glycopeptide binding affinities are evaluated against different lectins
While the binding properties of a few lectins, such as WGA and Con A, have been extensively studied, many lectins remain only partially characterized with respect to their glycan-binding specificities.15 We therefore evaluated online LAC as an efficient approach to evaluate lectin binding specificities. In addition to WGA, Con A, LCA, APA and SBA lectins were also immobilized on individual POROS columns and their abilities to retain a range of glycopeptides were evaluated. Fig. 4 shows the adjusted retention times of glycopeptides on these five different columns. WGA, which has been discussed above, shows a relatively wide range of specificity towards different types of glycopeptides. As a lectin known to be specific to GlcNAc and NeuAc residues,45,46,55,56 our data indicate that its affinity to glycans decreases in the following order: the NeuAc-terminated complex type, the GlcNAc-rich hybrid type, and the high-mannose type. It appears that some of the HRP glycopeptides are retained slightly; however, most of the additional retention is likely caused by the non-specific interaction between the POROS resin and the peptide moiety as described above. It seems that the poor affinity of these glycopeptides is due to the xylose residue modifying the core pentasaccharide.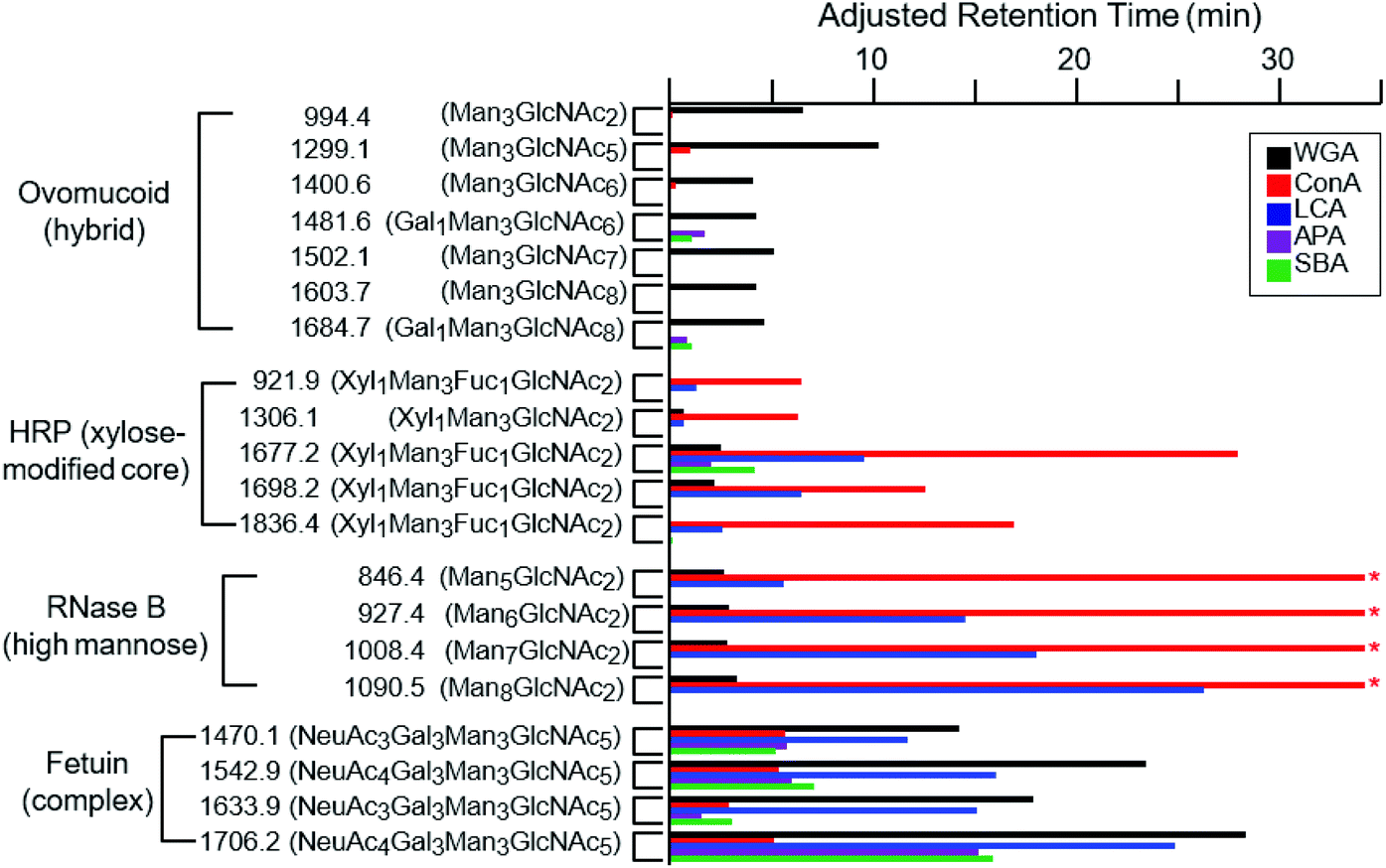 | ||
| Fig. 4 The adjusted retention times of the glycopeptides on different lectin-coated POROS columns at the flow rate of 0.2 ml min−1. These glycopeptides are from four proteins that contain distinct types of glycan structures: high-mannose glycans from RNase B, xylose (and fucose) modified core pentasaccharide from HRP, hybrid glycans from ovomucoid, and complex glycans from fetuin (Table 1). Glycopeptides are shown with m/z labels and associated glycan structures. The * symbols for the high-mannose glycopeptides from RNase B indicate that the retention times of these glycopeptides on the Con A-coated column exceeded the time course of the experiment. | ||
Con A and LCA are known to capture mannose-rich carbohydrates.57,58 The high-mannose glycopeptides from RNase B were well retained by the LCA lectins (Fig. 4). In fact, the RNase B high-mannose glycopeptides were not eluted off the Con A column during the one hour gradient due to their tight binding affinities, with Ka values around 105 M−1.59,60 These glycopeptides were subsequently recovered by injection of methyl-α-mannose. The relative affinities of the different types of glycopeptides to the Con A lectin are in agreement with previously published values.34,47,61,62 LCA was found to bind complex type glycopeptides, in addition to its reported specificity for the chitobiose core structure.63,64 The xylose-modified core glycopeptides from HRP were well retained by both Con A and LCA, with slight variation in the relative affinities for individual species, suggesting the existence of somewhat minor lectin–peptide interactions. Neither lectin shows significant affinity for the hybrid-type glycopeptides from ovomucoid.
SBA and APA lectins show very similar binding specificities. As galactose-specific lectins, they have been shown to bind Gal1Man3GlcNAc6 and Gal1Man3GlcNAc8, respectively.65–67 We demonstrate that these lectins bind galactose-containing complex glycans from fetuin. In fact, these fetuin glycopeptides are more strongly retained than either Gal1Man3GlcNAc6 or Gal1Man3GlcNAc8.
Experimental
Materials and chemicals
POROS AL resin and Toyopearl AF-formyl-650M resin were obtained from Applied Biosystems (Foster City, CA, USA) and Tosoh Bioscience (King of Prussia, PA, USA), respectively. Luna 5μ silica (2) resin and stainless steel columns (2 × 250 mm) were obtained from Phenomenex (Torrance, CA, USA). Wheat germ agglutinin (WGA), concanavalin A (Con A), lens culinaris agglutinin (LCA), and soybean agglutinin (SBA) were from Vector Labs (Burlingame, CA, USA). PNGase F, 2-mercaptoethanol, abrus precatorius agglutinin (APA), ovomucoid, α-crystallin (from bovine eye), ribonuclease B (RNase B), horseradish peroxidase (HRP), bovine fetuin, trypsin (TPCK-treated), dithiothreitol (DTT), iodoacetamide, trizma base, HEPES sodium salt, methyl-α-mannose, N-acetylglucosamine (GlcNAc), galactose (Gal), manganese acetate and sodium cyanoborohydride (NaCNBH3) were all purchased from Sigma-Aldrich (St Louis, MO, USA). Urea, ammonium bicarbonate, sodium chloride, and calcium chloride were all obtained from Mallinckrodt Pharmaceuticals (St Louis, MO, USA). C18 Sep-Pak cartridges were from Waters (Milford, MA, USA). Trifluoroacetic acid (TFA), water and acetonitrile were obtained from EMD Chemicals (Darmstadt, Germany). Formic acid was from Alfa Aesar (Ward Mill, MA, USA) and ethanol (200 proof, absolute, anhydrous) was from Pharmco-AAPER (Brookfield, CT, USA).Activation of silica resin
One ml of silica resin was suspended in two ml of ethanol. While stirring the mixture, 30 μl of triethoxysilane was added and the reaction was allowed to proceed for ∼14 hours under anhydrous conditions. The resulting silica product was filtered and washed with 10 ml of hexane, 10 ml of water and 10 ml of hexane before drying under vacuum.Lectin immobilization
Lectins were attached to the aldehyde-activated resin using reductive amination. Briefly, 25 mg of lectin was dissolved in 2.5 ml of 10 mM HEPES buffer containing 150 mM NaCl, 1 mM CaCl2 and 1 mM Mn(CH3COO)2. The solution was transferred to a 5 ml tube containing 1 ml of the aldehyde-activated resin, followed by the addition of 12.5 mg NaCNBH3. To shield the lectin carbohydrate-binding region from reacting with aldehyde groups on the resin, a monosaccharide (i.e., methyl-α-mannose for Con A and LCA, GlcNAc for WGA, and Gal for SBA and APA) was added to the reaction mixture at a final concentration of 5 mM. The reaction mixture was rotated at 4 °C for 48 hours, spun down and the supernatant was removed. To quench the remaining aldehyde groups, 1 ml of quenching solution (200 mM Tris, 150 mM NaCl, 1 mM CaCl2, and 1 mM Mn(CH3COOH)2) and 5 mg NaCNBH3 were added to the resin. The reaction mixture was rotated at room temperature for three hours. The supernatant was removed and the resin was washed with the quenching buffer and packed into a 2 × 250 mm stainless steel column.Tryptic digestion
Individual glycoproteins (∼2 mg) were dissolved in 100 μl of 100 mM ammonium acetate containing 8 M urea. After the addition of 5 μl of 20 mM DTT, the solution was incubated at 56 °C for 1 hour. The solution was then mixed with 5 μl of 25 mM ammonium acetate solution containing 84 mM iodoacetamide and incubated in dark at room temperature for 45 min. The reaction mixture was diluted with 100 mM ammonium acetate to a final volume of 400 μl. Trypsin was added at a 50:1 (w:w) protein:trypsin ratio and incubated at 37 °C for 14 hours. The tryptic digest was desalted using a C18 Sep-Pak cartridge and dried under vacuum.
Isolation of glycans
The glycans from ovomucoid were enzymatically cleaved using PNGase F. The glycoprotein (∼2 mg) was dissolved in 50 mM phosphate buffer containing 0.1% 2-mercaptoethanol (pH = 7.5) at a concentration 2 mg ml−1. The solution was heated at 100 °C for 10 min and allowed to cool to room temperature before the addition of 2 μl of 500 unit per ml PNGase F. The mixture was then incubated at 37 °C for 14 hours. The glycans were cleaned by removing the peptides using a C18 Sep-Pak cartridge. The cartridge was pre-conditioned with 5 ml of 85% ACN and 0.1% TFA solution followed by 5 ml of 5% ACN and 0.1% TFA solution. The tryptic digest was diluted with 1 ml of 5% ACN and 0.1% TFA solution and loaded three times onto the cartridge. The resulting solution containing unbound glycans was collected and dried under vacuum.Lectin affinity chromatography-mass spectrometry (LAC-MS)
The LAC-MS experiments were performed on a Waters Acquity UPLC coupled to a Synapt G2S HDMS instrument (Milford, MA, USA). The tryptic digests or isolated glycans were dissolved in buffer A (25 mM ammonium acetate and 0.2 mM metal acetate, depending on which metals are required for the glycan–lectin binding) at a concentration of 10 mg ml−1 glycoprotein, and 4 μl of the sample was injected on to the column. Buffer B, containing 99% ACN and 1% formic acid, was infused post-column using a static three-way union. An isocratic flow composed of 50% buffer A and 50% buffer B was used, and four different flow rates (0.05, 0.1, 0.2, and 0.3 ml min−1) were tested. The lectin column was kept in an ice bath during all experiments. Preliminary experiments running the lectin column at room temperature showed poor retention and/or peak shape for both free GlcNAc and glycopeptides and we did not examine the effect of temperature in more detail. The electrospray ionization (ESI) voltage of the source capillary was set at 3.0 kV, and the source and desolvation temperature were maintained at 140 and 500 °C, respectively. When not in use, columns should be stored at 4 °C with 0.1% sodium azide and 10 mM GlcNAc. We have successfully used individual columns for over 100 runs over 12 months when stored in this fashion. To monitor possible lectin degradation, retention times of a standard glycoprotein digest can be measured over time using online-LWAC. Alternatively, the column could be connected to a UV detector, and the binding capacity/retention can be determined by injecting increasing amounts of GlcNAc.Conclusions
An online LAC-MS platform has been developed that can be used to characterize biomolecular bindings of weak affinity. It allows characterization of glycan–lectin binding affinities and helps effectively capture species of interest in a glycome. By using a reference standard, association constants for glycan–lectin interactions can be calculated from complex mixtures containing multiple glycans of unknown concentrations. This platform can be easily adapted from regular LC-MS systems, and does not require a series of conditioning, washing, and elution steps used in conventional LAC. Several chromatographic matrices have been compared based on their separation efficiencies and non-specific interactions with the glycopeptides. While non-specific interactions cannot be completely eliminated, this approach can measure their contribution to glycopeptide retention.Immobilization of WGA on Tosoh resin allowed better separation between glycosylated and non-glycosylated peptides. This could be due to higher overall protein binding and/or a higher percentage of bound WGA remaining functional with respect to glycopeptide binding. Initial lectin immobilization was performed in the presence of the respective target glycan in an attempt to limit covalent modification of the substrate recognition region. Over the course of these and previous glycopeptide experiments, we have constructed four different columns using WGA immobilized on POROS. We have assayed the binding ability of these columns by measuring their retention of GlcNAc as measured by UV and seen vary little column-to-column variability in binding ability.52 While equivalent binding conditions were used for both resins, it is possible that a higher effective WGA surface concentration was immobilized on the Tosoh resin or that effects such as resin particle size and pore size allow more interaction events between the glycopeptides and immobilized WGA. For both the Tosoh and POROS columns, the elution peaks for peptides and glycopeptides are significantly broader than would be obtained by reverse phase LC-MS. Therefore, for deep glycopeptide profiling from complex mixtures, offline LAC (using conditions determined by online LAC) followed by orthogonal LC-MS of enriched fractions would likely provide much higher depth of coverage compared to online LAC. For analysis of single proteins or simple mixtures, online LAC may be a more straightforward approach that allows determination of a protein's glycoforms as well as providing information regarding the glycan–substrate interaction.
Multidimensional lectin chromatography or serial lectin chromatography is often used in order to cover a broad range of glycomic features. However, the choice of the lectin combination in these approaches is mainly qualitative and empirical. Using online LAC-MS, the global affinities of the lectin combination towards different glycan structures can be measured in a systematic manner. Importantly, these measurements can be obtained in a multiplexed fashion, with a single LAC-MS analysis capable of providing affinity data on many individual glycans in a complex mixture. In addition to plant-derived lectins, our approach can characterize binding specificities for other lectins or glycan-binding proteins in general. We also envision its general utility for characterizing interactions between pairs of proteins or between proteins and small molecules.
Our comparison of the glycan-binding affinities of five different lectins was particularly informative. A key step in most glycoproteomic experiments is the initial enrichment of a broad pool of glycopeptides. To account for the fact that individual lectins are relatively specific for specific saccharides, multi-lectin enrichment approaches have been adopted.19,20 Here we provide direct evidence that WGA is able to enrich a wide range of glycan structures, including complex, hybrid and high-mannose N-glycans. This is likely due to that fact that not only can WGA bind GlcNAc and NeuAc, but it can interact with the core pentasaccharide which is common to all N-glycans. However, the presence of xylose appears to inhibit recognition of WGA for the core pentasaccharide. As such, while WGA has the broadest glycan affinity of the five lectins tested, it cannot be used as a universal affinity reagent for any type of glycopeptide. For those peptides, a mannose or galactose-specific lectin would be more appropriate.
Acknowledgements
We gratefully acknowledge the funding support of this research by the Indiana University METACyt Initiative.References
- J. N. Arnold, M. R. Wormald, R. B. Sim, P. M. Rudd and R. A. Dwek, Annu. Rev. Immunol., 2007, 25, 21–50 CrossRef PubMed.
- P. M. Rudd, T. Elliott, P. Cresswell, I. A. Wilson and R. A. Dwek, Science, 2001, 291, 2370–2376 CrossRef PubMed.
- A. Helenius and M. Aebi, Science, 2001, 291, 2364–2369 CrossRef PubMed.
- L. Han and C. E. Costello, Biochemistry, 2013, 78, 710–720 Search PubMed.
- C. S. Chu, M. R. Ninonuevo, B. H. Clowers, P. D. Perkins, H. J. An, H. Yin, K. Killeen, S. Miyamoto, R. Grimm and C. B. Lebrilla, Proteomics, 2009, 9, 1939–1951 CrossRef PubMed.
- C. E. Costello, J. M. Contado-Miller and J. F. Cipollo, J. Am. Soc. Mass Spectrom., 2007, 18, 1799–1812 CrossRef PubMed.
- N. Leymarie and J. Zaia, Anal. Chem., 2012, 84, 3040–3048 CrossRef PubMed.
- W. Morelle and J. C. Michalski, Nat. Protoc., 2007, 2, 1585–1602 CrossRef PubMed.
- A. Dell and H. R. Morris, Science, 2001, 291, 2351–2356 CrossRef PubMed.
- K. Khatri, G. O. Staples, N. Leymarie, D. R. Leon, L. Turiak, Y. Huang, S. Yip, H. Hu, C. F. Heckendorf and J. Zaia, J. Proteome Res., 2014, 13, 4347–4355 CrossRef PubMed.
- Y. Makino, K. Omichi and S. Hase, Anal. Biochem., 1998, 264, 172–179 CrossRef PubMed.
- E. Lattova, S. Snovida, H. Perreault and O. Krokhin, J. Am. Soc. Mass Spectrom., 2005, 16, 683–696 CrossRef PubMed.
- P. Kang, Y. Mechref, Z. Kyselova, J. A. Goetz and M. V. Novotny, Anal. Chem., 2007, 79, 6064–6073 CrossRef PubMed.
- Y. Mechref and M. V. Novotny, Chem. Rev., 2002, 102, 321–369 CrossRef PubMed.
- W. R. Alley Jr., B. F. Mann and M. V. Novotny, Chem. Rev., 2013, 113, 2668–2732 CrossRef PubMed.
- M. Madera, Y. Mechref and M. V. Novotny, Anal. Chem., 2005, 77, 4081–4090 CrossRef PubMed.
- M. Bedair and Z. El Rassi, J. Chromatogr., A, 2005, 1079, 236–245 CrossRef PubMed.
- M. Madera, Y. Mechref, I. Klouckova and M. V. Novotny, J. Proteome Res., 2006, 5, 2348–2363 CrossRef PubMed.
- R. Qiu and F. E. Regnier, Anal. Chem., 2005, 77, 2802–2809 CrossRef PubMed.
- Z. Zeng, M. Hincapie, S. J. Pitteri, S. Hanash, J. Schalkwijk, J. M. Hogan, H. Wang and W. S. Hancock, Anal. Chem., 2011, 83, 4845–4854 CrossRef PubMed.
- G. Palmisano, S. E. Lendal, K. Engholm-Keller, R. Leth-Larsen, B. L. Parker and M. R. Larsen, Nat. Protoc., 2010, 5, 1974–1982 CrossRef PubMed.
- Z. L. Nikolov and P. J. Reilly, J. Chromatogr., 1985, 325, 287–293 CrossRef.
- A. J. Alpert, J. Chromatogr., 1990, 499, 177–196 CrossRef PubMed.
- Y. Takegawa, K. Deguchi, T. Keira, H. Ito, H. Nakagawa and S. Nishimura, J. Chromatogr., A, 2006, 1113, 177–181 CrossRef PubMed.
- M. Glad, S. Ohlson, L. Hansson, M. O. Mansson and K. Mosbach, J. Chromatogr., 1980, 200, 254–260 CrossRef.
- T. Suksrichavalit, K. Yoshimatsu, V. Prachayasittikul, L. Bulow and L. Ye, J. Chromatogr., A, 2010, 1217, 3635–3641 CrossRef PubMed.
- A. Monzo, G. K. Bonn and A. Guttman, TrAC, Trends Anal. Chem., 2007, 26, 423–432 CrossRef.
- S. Magdeldin, J. J. Moresco, T. Yamamoto and J. R. Yates 3rd, J. Proteome Res., 2014, 13, 3826 CrossRef PubMed.
- J. C. Trinidad, R. Schoepfer, A. L. Burlingame and K. F. Medzihradszky, Mol. Cell. Proteomics, 2013, 12, 3474 Search PubMed.
- K. Vosseller, J. C. Trinidad, R. J. Chalkley, C. G. Specht, A. Thalhammer, A. J. Lynn, J. O. Snedecor, S. Guan, K. F. Medzihradszky, D. A. Maltby, R. Schoepfer and A. L. Burlingame, Mol. Cell. Proteomics, 2006, 5, 923–934 Search PubMed.
- J. C. Trinidad, D. T. Barkan, B. F. Gulledge, A. Thalhammer, A. Sali, R. Schoepfer and A. L. Burlingame, Mol. Cell. Proteomics, 2012, 11, 215–229 Search PubMed.
- J. C. Trinidad, A. Thalhammer, A. L. Burlingame and R. Schoepfer, Mol. Cell. Proteomics, 2013, 12, 29–41 Search PubMed.
- P. Mehta, R. D. Cummings and R. P. McEver, J. Biol. Chem., 1998, 273, 32506–32513 CrossRef PubMed.
- D. A. Mann, M. Kanai, D. J. Maly and L. L. Kiessling, J. Am. Chem. Soc., 1998, 120, 10575–10582 CrossRef.
- T. K. Dam and C. F. Brewer, Chem. Rev., 2002, 102, 387–429 CrossRef PubMed.
- B. Zhang, M. M. Palcic, H. Mo, I. J. Goldstein and O. Hindsgaul, Glycobiology, 2001, 11, 141–147 CrossRef PubMed.
- D. C. Schriemer and O. Hindsgaul, Comb. Chem. High Throughput Screening, 1998, 1, 155–170 Search PubMed.
- H. Tateno, S. Nakamura-Tsuruta and J. Hirabayashi, Nat. Protoc., 2007, 2, 2529–2537 CrossRef PubMed.
- Y. H. Chu, L. Z. Avila, H. A. Biebuyck and G. M. Whitesides, J. Med. Chem., 1992, 35, 2915–2917 CrossRef PubMed.
- K. Nakajima, Y. Oda, M. Kinoshita and K. Kakehi, J. Proteome Res., 2003, 2, 81–88 CrossRef CAS PubMed.
- O. Blixt, S. Head, T. Mondala, C. Scanlan, M. E. Huflejt, R. Alvarez, M. C. Bryan, F. Fazio, D. Calarese, J. Stevens, N. Razi, D. J. Stevens, J. J. Skehel, I. van Die, D. R. Burton, I. A. Wilson, R. Cummings, N. Bovin, C. H. Wong and J. C. Paulson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17033–17038 CrossRef CAS PubMed.
- A. Kuno, N. Uchiyama, S. Koseki-Kuno, Y. Ebe, S. Takashima, M. Yamada and J. Hirabayashi, Nat. Methods, 2005, 2, 851–856 CrossRef CAS PubMed.
- Z. Darula and K. F. Medzihradszky, J. Am. Soc. Mass Spectrom., 2014, 25, 977–987 CrossRef CAS PubMed.
- K. Yamamoto, T. Tsuji, I. Matsumoto and T. Osawa, Biochemistry, 1981, 20, 5894–5899 CrossRef CAS PubMed.
- M. Monsigny, C. Sene, A. Obrenovitch, A. C. Roche, F. Delmotte and E. Boschetti, Eur. J. Biochem., 1979, 98, 39–45 CrossRef CAS PubMed.
- K. A. Kronis and J. P. Carver, Biochemistry, 1982, 21, 3050–3057 CrossRef CAS PubMed.
- J. K. Scott, D. Loganathan, R. B. Easley, X. Gong and I. J. Goldstein, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 5398–5402 CrossRef CAS.
- T. Manabe, N. Higuchi, T. Okuyama and Y. Mukaiyama, J. Chromatogr., 1988, 431, 45–54 CrossRef CAS PubMed.
- M. A. Leon, Science, 1967, 158, 1325–1326 CAS.
- E. M. M. del Valle and M. A. Galan, Ind. Eng. Chem. Res., 2002, 41, 2296–2304 CrossRef.
- A. Lee, M. Nakano, M. Hincapie, D. Kolarich, M. S. Baker, W. S. Hancock and N. H. Packer, OMICS, 2010, 14, 487–499 CrossRef CAS PubMed.
- S. Ohlson, M. Bergstrom, L. Leickt and D. Zopf, Bioseparation, 1998, 7, 101–105 CrossRef CAS PubMed.
- L. Leickt, M. Bergstrom, D. Zopf and S. Ohlson, Anal. Biochem., 1997, 253, 135–136 CrossRef CAS PubMed.
- D. Zopf and S. Ohlson, Nature, 1990, 346, 87–88 CrossRef.
- A. K. Allen, A. Neuberger and N. Sharon, Biochem. J., 1973, 131, 155–162 CrossRef CAS PubMed.
- Y. Nagata and M. M. Burger, J. Biol. Chem., 1974, 249, 3116–3122 CAS.
- J. W. Becker, G. N. Reeke Jr., J. L. Wang, B. A. Cunningham and G. M. Edelman, J. Biol. Chem., 1975, 250, 1513–1524 CAS.
- N. M. Young, M. A. Leon, T. Takahashi, I. K. Howard and H. J. Sage, J. Biol. Chem., 1971, 246, 1596–1601 CAS.
- P. H. Liang, S. K. Wang and C. H. Wong, J. Am. Chem. Soc., 2007, 129, 11177–11184 CrossRef CAS PubMed.
- R. Gutierrez Gallego, S. R. Haseley, V. F. van Miegem, J. F. Vliegenthart and J. P. Kamerling, Glycobiology, 2004, 14, 373–386 CrossRef PubMed.
- C. F. Brewer and L. Bhattacharyya, J. Biol. Chem., 1986, 261, 7306–7310 CAS.
- J. U. Baenziger and D. Fiete, J. Biol. Chem., 1979, 254, 2400–2407 CAS.
- K. Yamamoto, T. Tsuji and T. Osawa, Carbohydr. Res., 1982, 110, 283–289 CrossRef CAS PubMed.
- K. A. Maupin, D. Liden and B. B. Haab, Glycobiology, 2012, 22, 160–169 CrossRef CAS PubMed.
- T. Aoki, T. Araki and M. Kitamikado, Eur. J. Biochem., 1990, 187, 461–465 CrossRef CAS PubMed.
- S. Sueyoshi, T. Tsuji and T. Osawa, Carbohydr. Res., 1988, 178, 213–224 CrossRef CAS PubMed.
- S. Olsnes, E. Saltvedt and A. Pihl, J. Biol. Chem., 1974, 249, 803–810 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6an02043g |
| This journal is © The Royal Society of Chemistry 2017 |