
Protoporphyrin IX-modified chitosan-g-oligo(NiPAAm) polymeric micelles: from physical stabilization to permeability characterization in vitro†
Inbar
Schlachet
and
Alejandro
Sosnik
*
Laboratory of Pharmaceutical Nanomaterials Science, Department of Materials Science and Engineering, Technion-Israel Institute of Technology, Haifa, Israel. E-mail: alesosnik@gmail.com; sosnik@tx.technion.ac.il; Tel: +972-077-887-1971
First published on 1st December 2016
Abstract
Two main hurdles persist towards the more extensive bench-to-bed side translation of non-parenteral polymeric micelles. The first pertains to their thermodynamically-driven disassembly under uncontrolled dilution conditions in the biological milieu and upon interaction with biomacromolecules (e.g., proteins). The second is related to the relatively poor understanding of the pathways by which polymeric micelles improve the bioavailability of the payload by mucosal routes (e.g., intestinal). In this work, a chitosan-g-oligo(N-isopropylacrylamide) (CS-g-oligo(NiPAAm)) copolymer was modified with non-cytotoxic amounts of protoporphyrin IX (PP), a planar molecule of amphiphilic character that undergoes self-aggregation in water by forming π–π stacked supramolecular structures, to induce micellization under disfavored conditions and to serve as a fluorescent tracer for the measurement of the micelle permeability across a model of the intestinal epithelium in vitro. Findings indicated that the conjugation of PP amounts as low as 2% w/w induced the formation of micelles at temperatures below the lower critical solution temperature of oligo(NiPAAm) (30–32 °C). Moreover, permeability studies conducted at both 4 °C and 37 °C strongly suggested that despite the relatively large size of the micelles (200–300 nm), they cross the epithelial monolayer mainly by a paracellular pathway due to the opening of tight junctions. Complementary uptake studies by flow cytometry indicated that no endocytosis, though due to passive or facilitated diffusion, some internalization takes place.
1. Introduction
Polymeric micelles are nanostructures produced by the spontaneous self-aggregation of amphipathic block copolymers above the critical micellar concentration (CMC) and they exhibit a hydrophobic and a hydrophilic domain, namely a core and corona, respectively.1,2 The molecular structure and weight and the architecture of the copolymer govern the micellar nanostructure. Thus, amphiphilic diblocks and triblocks usually give place to well-defined core–corona configurations of relatively small size (<100 nm) and variable morphology,3 whereas in the case of graft copolymers, the formation of larger (several hundreds of nanometers) and more complex (e.g., multimicellar) structures could be anticipated.4 Polymeric micelles have become a gold-standard nanotechnology platform to attempt the encapsulation, release and passive and active targeting of drugs classified into Class II and IV of the Biopharmaceutics Classification System.5 Remarkably, in a very recent publication of the European Technology Platform for Nanomedicine, they appear as the second most popular nano-drug delivery system for the treatment of cancer, after the clinically relevant liposomes.6Despite the proven pharmacokinetic benefits by non-parenteral routes,7–10 only countable formulations based on polymeric micelles progressed from preclinical to Phase 2 and 3 clinical trials (and eventually reached the market), all of them for the intravenous chemotherapy of cancer.11 Recently, we elucubrated on possible strategies to design mucoadhesive polymeric micelles for mucosal drug delivery12 and developed a series of polysaccharide-based prototypes with high chemical flexibility and modularity that in water formed self-assembly systems in the 100–400 nm size-range and encapsulated hydrophobic molecules such as rifampicin and indinavir free base.13,14
Two main hurdles persist towards the more extensive bench-to-bed side translation of non-parenteral polymeric micelles. The first pertains to the disassembly under uncontrolled dilution conditions and upon interaction with biomacromolecules (e.g., proteins) in the biological milieu. Covalent crosslinking of the corona or the core is a classical approach to stabilize them.15,16 However, the use of unselective and aggressive chemistry compromises their applicability due to the possibility of cross-reactions with functional moieties in the cargo. To address this limitation, we engineered amphiphilic nanogels produced by the controlled ionotropic corona-crosslinking of thermo-sensitive polymeric micelles based on graft copolymers of chitosan (CS) and oligo(N-isopropylacrylamide) (oligo(NiPAAm)) as hydrophilic and hydrophobic components, respectively. PolyNiPAAm displays a reversible hydrophilic-to-hydrophobic thermal transition upon heating above its lower critical solution temperature (LCST) of 30–32 °C. Thus, before crosslinking, the micellization of the copolymers was reversibly “switched” on and off by applying heating–cooling cycles between room temperature (RT) and 37 °C.17 In this context, copolymers did not display CMC below the LCST of polyNiPAAm. Then, selective crosslinking of the hydrophilic backbone under mild conditions at 37 °C stabilized the micelles and prevented their disassembly upon cooling to RT.17 The second drawback for the clinical translation of polymeric micelles by mucosal routes (e.g., intestinal) is related to the relatively poor understanding of the pathways by which they improve the bioavailability of the payload. For instance, it is still unclear whether polymeric micelles cross epithelia in the apical-to-basal direction as fully assembled, partly or completely disassembled systems or if they only perform as drug reservoirs localized in the apical side of the mucosal tissue, facilitating the absorption of the free drug owing to the timeous release of the cargo in a molecularly dispersed form.18 Moreover, the description of a general mechanism for any kind of polymeric micelle is probably unfeasible owing to the fact that the size, the surface charge, the chemical composition and the nano-mechanical and nano-rheological properties influence the interaction with biological barriers such as the intestinal epithelium.
Protoporphyrin IX (PP) is the iron-free form of hemin, a planar molecule of amphiphilic character that undergoes self-aggregation in water forming π–π stacked supramolecular structures of different nature.19 In this context, the conjugation of PP is expected to confer self-assembly properties to hydrophilic polymers. In addition, PP also displays a series of strong fluorescence emission bands that make it a very appealing tracer for tracking the fate of PP-modified nanomaterials in different biological models in vitro and in vivo.20
We hypothesized that the modification of CS-g-oligo(NiPAAm) copolymers with non-cytotoxic amounts of PP would promote micellization even below the LCST and, at the same time, serve as a fluorescent probe for the measurement of the permeability of these polymeric micelles across a model of the intestinal epithelium in vitro.
In this framework, as a proof of concept, we initially demonstrated the self-assembly behavior of a conjugate of PP with a hydrophilic monofunctional poly(ethylene glycol) (MPEG) block. Then, growing amounts of PP (2–36% w/w) were conjugated to a CS-g-oligo(NiPAAm) derivative and the self-assembly and surface charge properties were characterized at 25 °C and 37 °C. Finally, comparative cell compatibility assays in a model of the intestinal epithelium, Caco2 cells, enabled the optimization of the experimental conditions for the characterization of the intestinal permeability in vitro employing cell monolayers cultured in cell culture inserts. Findings indicated that PP induces the formation of micelles at RT. Moreover, permeability studies strongly suggested that despite the relatively large size of the micelles (200–300 nm), they cross the epithelial monolayer mainly by paracellular transport.21
2. Experimental section
2.1 Materials
Low molecular weight CS (degree of deacetylation of 94%; viscosity of ≤100 mPa s, Glentham Life Sciences, Corsham, England), cerium(IV) ammonium nitrate (CAN, Strem Chemicals, Inc., Newburyport, MA, USA), nitric acid 70% (Bio-Lab, Jerusalem, Israel), hydroquinone (HQ, Merck, Hohenbrunn, Germany) and tetramethylethylenediamine (TEMED, Alfa Aesar, Heysham, England) were used as received. NiPAAm (99% purity) was supplied by J & K (Beijing, China) and recrystallized from hexane at 60 °C before use. Protoporphyrin IX (PP, Strem Chemicals, Inc.), dicyclohexylcarbodiimide (DCC, Alfa Aesar, Heysham, UK), 4-dimethylaminopyridine (DMAP, Alfa Aesar), and polyethylene glycol monomethyl ether of molecular weight 1900 g mol−1 (MPEG1900, Alfa Aesar) were used as received. All the solvents were of analytical or spectroscopic grade, purchased from Bio-Lab, Gadot (Netanya, Israel) and Cambridge Isotope Laboratories, Inc. (Tewksbury, MA, USA) and used without further purification.2.2 Synthetic methods
2.3 Characterization methods
The different PP-containing products were fully characterized and compared with the corresponding PP-free precursors.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 were used for molecular weight calibration.
000 g mol−1 were used for molecular weight calibration.
Critical aggregation concentration (CAC) of MPEG–PP conjugates. To demonstrate that the conjugation of PP confers self-assembly properties to hydrophilic polymers, initially, the aggregation of the MPEG–PP conjugate was assessed. A stock aqueous solution of MPEG1900–PP (1% w/v in pure water) was prepared, serially diluted in the same medium to a final concentration in the 0.0001–0.1% w/v range and stabilized at two temperatures, 25 °C and 37 °C (24 h), to allow self-aggregation. Then, the intensity of the scattered light (DCR) expressed in kilo counts per second (kcps) was measured by Dynamic Light Scattering (DLS, Zetasizer Nano-ZS, Malvern Instruments, Malvern, UK) and plotted as a function of MPEG1900–PP concentration (% w/v). Measurements were conducted at a scattering angle of 173° to the incident beam and data were analyzed using CONTIN algorithms (Malvern Instruments). Data for each single specimen was the result of at least six runs. The aggregation was evidenced by a sharp increase in the scattering intensity and the intersection between the two straight lines was established as the CAC expressed in % w/v.
CMC of CS-N52 and CS-N52–PPX copolymers. The micellization of CS-g-oligo(NiPAAm) copolymers at 37 °C was already shown, while no aggregation was observed at RT.17 Stock aqueous solutions (1% w/v) of the different copolymers were prepared by direct dissolution in buffer acetate (pH 5), diluted (0.0001–0.1% w/v) in the same medium and stabilized at 25 °C and 37 °C (24 h). The DCR in kcps was measured by DLS (under the same conditions detailed above) and plotted as a function of the copolymer concentration (% w/v). Data for each single specimen were the result of at least six runs. The micellization was established as the sharp increase in the scattering intensity and the intersection between the two straight lines (see above).23
Size, size distribution and Z-potential. The hydrodynamic diameter (Dh), the size distribution (polydispersity index, PDI) and the Z-potential of 1% w/v systems were measured using the Zetasizer Nano-ZS in the same media detailed for CAC and CMC measurements (see above). For Z-potential, laser Doppler micro-electrophoresis was used. Values are expressed as mean ± S.D. of three independent samples prepared under identical conditions. Data for each single specimen were the result of at least six runs.
Nanoparticle tracking analysis (NTA). The quantification of the number of particles per volume unit (in mL) and the visualization of their Brownian movement were conducted by Nanoparticle Tracking Analysis (NTA, NanoSight® NS500-Zeta HSB system with a high sensitivity camera and 638 nm laser for fluorescence analysis, Malvern Instruments) under scattering and fluorescence mode. CS-N52 and CS-N52–PPX samples were prepared in the same medium used for the measurement of the CMC (see above).
Morphology of the micelles. The morphology of CS-N52–PP2 micelles was visualized by transmission electron microscopy (TEM). For this, a micellar suspension in buffer acetate of pH 5 (0.1% w/v, 25 μL) was cast on a carbon grid membrane, dried at RT and analyzed using an FEI Titan 80–300 kV S/TEM (Hillsboro, OR, USA) equipped with a Cs corrector at 300 kV in STEM and TEM modes, at RT. The diameter of the micelles was estimated using TEM AutoTune™ software (Gatan Digital Micrograph® software, Gatan, Inc., Pleasanton, CA, USA).
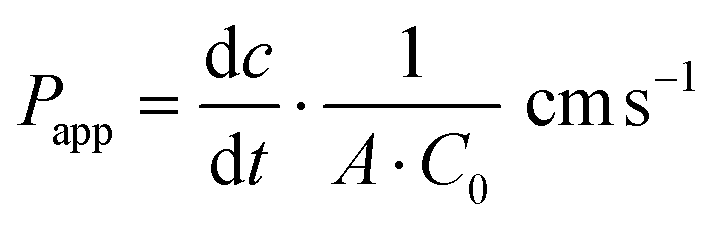 | (1) |
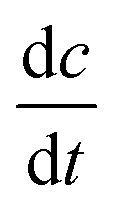 is the transport rate (μg s−1) across the monolayer, C0 is the initial concentration in the donor compartment (μg cm−3) and A is the surface area of the membrane (cm2).
is the transport rate (μg s−1) across the monolayer, C0 is the initial concentration in the donor compartment (μg cm−3) and A is the surface area of the membrane (cm2).
In addition, the size of the aggregates before and after the experiment in the donor and acceptor chambers was measured by DLS (see above).
To gain further insight into the possible permeation and cell uptake mechanisms, a similar experiment was conducted at both 4 °C and 37 °C, at different time points (1, 2 and 4 h). The cells were harvested and analyzed by using a BD LSR-II Analyzer Flow Cytometer (BD Biosciences, San Jose, CA, USA) equipped with a violet laser with a bandpass filter of 695/40 nm. Single cell populations of Caco2 cells were gated based upon forward and side scatter characteristics, and 5000 events from each experimental group were recorded and analyzed with BD FACSDiVa™ Software Version 6.1 (BD Biosciences). In addition, Caco2 cells were seeded in 96-well plates under the same conditions and exposed to the same micellar samples for 1, 2 and 4 h, at 4 and 37 °C. At each time point, the cells were washed with fresh culture medium and visualized with an Eclipse TS100 inverted fluorescent microscope equipped with a 1-FM Epi-Fluorescence Attachment (Nikon Instruments, Tokyo, Japan) under optical (OM) and fluorescence mode (FM).
3. Results and discussion
3.1 MPEG1900–PP conjugate
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN stretching vibration, 1430 and 1394 cm−1 of alkyl C–H antisymmetric deformations and 840 cm−1 of C–H bending.27 MPEG1900 presented bands at 1111 cm−1 (stretching vibration of C–O–C bonds) and 1346 cm−1 (vibration of the C–H bond of –CH3).28 Spectra of the MPEG–PP conjugate were the overlap of the bands of both precursors with a very weak signal at 1735 cm−1 that is characteristic of the newly formed ester bond (ESI Fig. S2†). GPC analysis was used to measure the molecular weight and compare it with the precursor. The results showed a monomodal distribution and a slight increase in the molecular weight from 3370 g mol−1 for pristine MPEG1900 to 3550 g mol−1 for MPEG1900–PP that was consistent with the conjugation of one MPEG residue per PP molecule. Finally, the PP content was measured by fluorescence spectrophotometry. The theoretical value expected for MPEG1900–PP according to the MPEG/PP feeding ratio and the experimental molecular weight of the polymer (see below) was 26% w/w. MPEG1900–PP contained 28% w/w of PP, supporting that one single MPEG block was conjugated to each PP molecule.
C and CN stretching vibration, 1430 and 1394 cm−1 of alkyl C–H antisymmetric deformations and 840 cm−1 of C–H bending.27 MPEG1900 presented bands at 1111 cm−1 (stretching vibration of C–O–C bonds) and 1346 cm−1 (vibration of the C–H bond of –CH3).28 Spectra of the MPEG–PP conjugate were the overlap of the bands of both precursors with a very weak signal at 1735 cm−1 that is characteristic of the newly formed ester bond (ESI Fig. S2†). GPC analysis was used to measure the molecular weight and compare it with the precursor. The results showed a monomodal distribution and a slight increase in the molecular weight from 3370 g mol−1 for pristine MPEG1900 to 3550 g mol−1 for MPEG1900–PP that was consistent with the conjugation of one MPEG residue per PP molecule. Finally, the PP content was measured by fluorescence spectrophotometry. The theoretical value expected for MPEG1900–PP according to the MPEG/PP feeding ratio and the experimental molecular weight of the polymer (see below) was 26% w/w. MPEG1900–PP contained 28% w/w of PP, supporting that one single MPEG block was conjugated to each PP molecule.
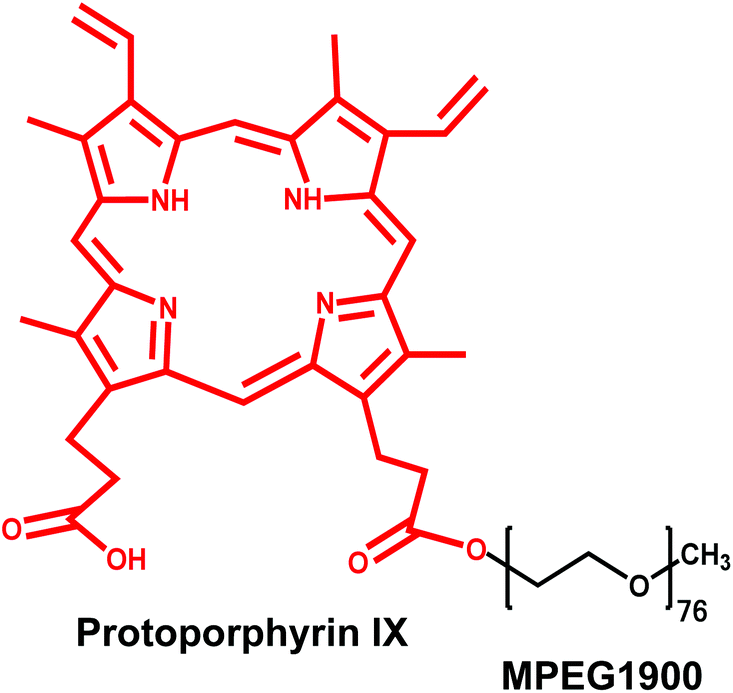 | ||
| Fig. 1 Chemical structure of the MPEG1900–PP conjugate. Molecules are not represented to scale. | ||
We hypothesized that the conjugation of PP to MPEG1900 would confer self-aggregation properties to the hydrophilic polymer. Since the formation of micellar structures could not be confirmed, the minimum concentration for the formation of aggregates is referred to as CAC. Regardless of the temperature, the results confirmed the formation of aggregates at a relatively low concentration. When the effect of the T was evaluated, MPEG1900–PP showed an increase of the CAC upon heating from 1.4 × 10−2% w/v at 25 °C to 2.3 × 10−2% w/v at 37 °C most probably due to the weakening of the hydrophobic interactions between PP residues (ESI Fig. S4†).31
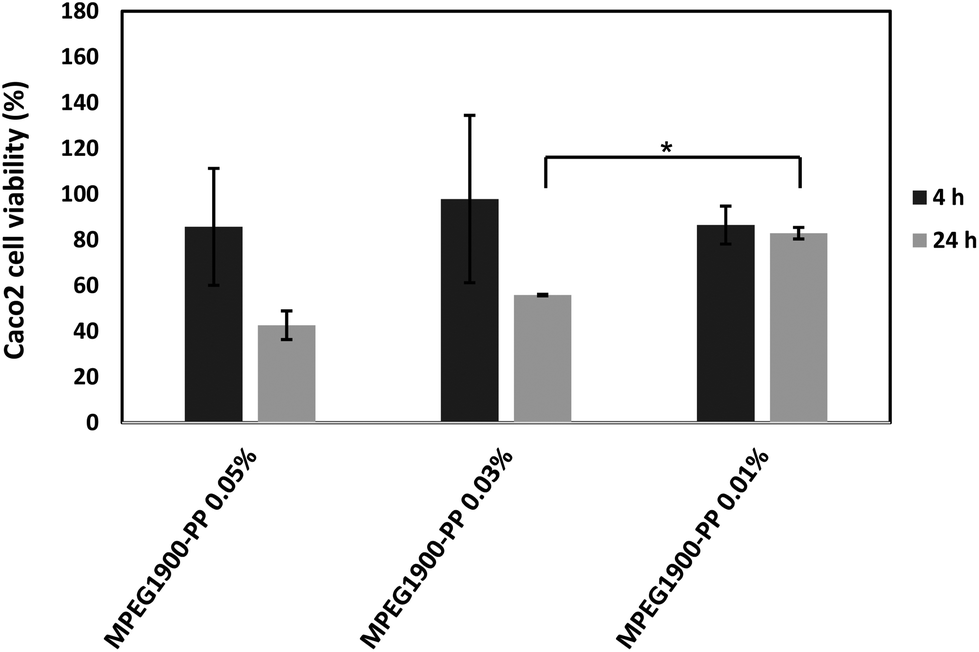 | ||
| Fig. 2 Caco2 cell viability upon exposure to different concentrations of MPEG1900–PP, as estimated by the MTT assay. *Statistically significant difference in cell viability (P < 0.05). | ||
3.2 CS-N52–PPX copolymers
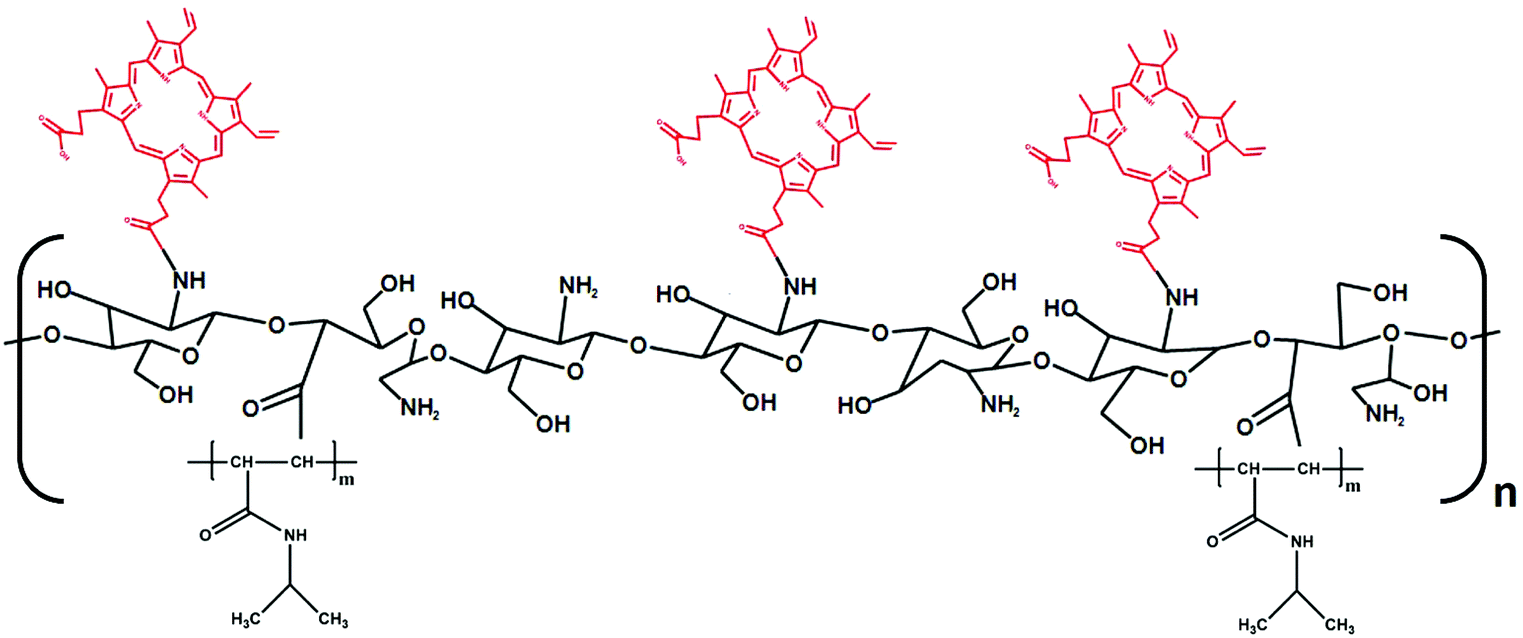 | ||
| Fig. 3 Chemical structure of CS-N52 modified with PP. Different PP contents were incorporated (2% to 36% w/w) to adjust the tracking and cytotoxicity properties of the micelles in the model of the intestinal epithelium in vitro. | ||
First, the PP content was quantified by fluorescence spectrophotometry. The gradual increase of the PP feed weight led to the expected increase of the PP content from 2% to 36% w/w (Table 1).
| Copolymer | Theoretical PP content (% w/w) | Experimental PP content (% w/w) |
|---|---|---|
| CS-N52–PP2 | 2 | 2 |
| CS-N52–PP6 | 5 | 6 |
| CS-N52–PP9 | 10 | 9 |
| CS-N52–PP36 | 30 | 36 |
To support the conjugation of PP, the complete UV-Vis absorption pattern of the CS-N52 and CS-N52–PPX copolymers with growing PP contents was measured. CS-N52 did not show any absorption peak in the whole analysis range (ESI Fig. S5†). Conversely, PP-containing copolymers showed a clear peak at 408 nm that is characteristic of pure PP. It is important to remark that the absorption maximum did not undergo any shift upon conjugation. In addition, the increase of the PP content resulted in a linear increase of the absorbance. PP-content values were very similar to those obtained by fluorescence (data not shown). To confirm the conjugation of PP to the polymeric backbone, the characterization was complemented with 1H-NMR analysis where CS-N52–PPX derivatives showed the signals of CS, oligo(NiPAAm) and PP (Fig. 4). Moreover, the higher the PP content, the stronger the relative intensity of the peaks assigned to PP. The spectrum of a physical mixture of CS-N52:PP (98:2 weight ratio) is presented instead of CS-N52–PP2; PP contents below 6% w/w could not be detected by this method.
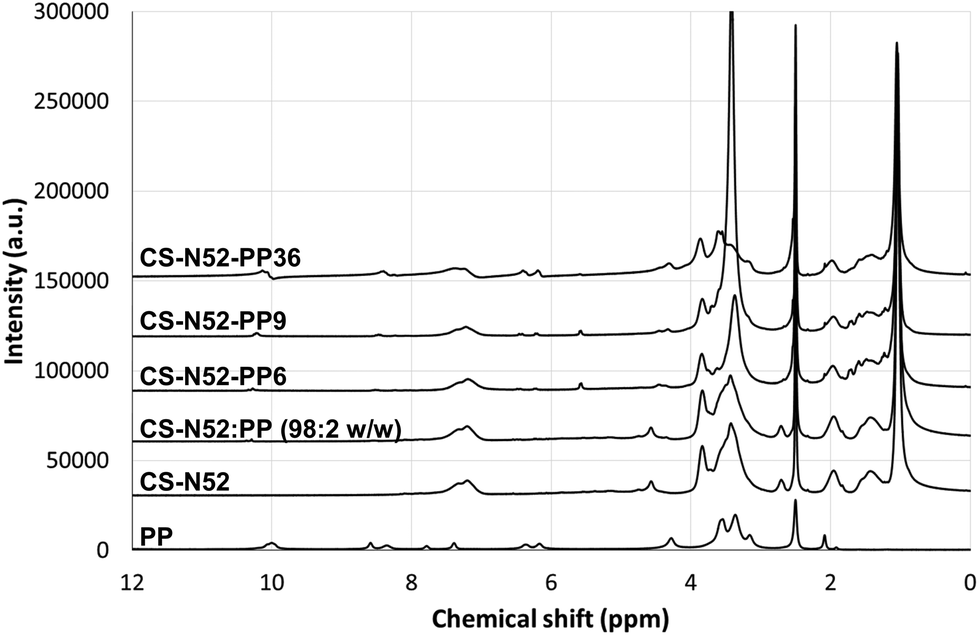 | ||
| Fig. 4
1H-NMR spectra of pure PP, CS-N52, CS-N52:PP (98:2 weight ratio) physical mixture and different CS-N52–PPX derivatives of growing PP content. | ||
Finally, FTIR characterization was in good agreement with the previous analyses, CS-N52–PPX displaying characteristic peaks of the CS-N52 copolymer17 and the sharp weakening of the band at 1700 cm−1 belonging to the –COOH group of PP, confirming that the porphyrin was efficiently conjugated to the copolymers and not in free form (ESI Fig. S6†).
The properties of the aggregates were characterized in terms of size, size distribution, Z-potential and morphology. All the derivatives showed a monomodal size distribution at both temperatures. In addition, the conjugation of greater PP amounts led to an increase of the size (Table 3). This behavior could rely on two mechanisms. The formation of larger hydrophobic domains is due to the increase of the PP content that leads to a moderate size growth or a substantial change of the aggregation pattern that more significantly affects the size. The very gradual size increase between 2% and 9% w/w PP suggested that the aggregation was not majorly affected. In addition, PDI values remained below 0.200. The incorporation of a substantially higher amount of porphyrin (36%) resulted in a more noticeable size and PDI increase (sizes were in the 1.5–2 μm range) that was in good agreement with secondary assembly phenomena (Table 3).
| Sample | Temperature (°C) | DLS analysis | NTA analysis | |||
|---|---|---|---|---|---|---|
| D h (nm) (±S.D.) | PDI (±S.D.) | Z-Potential (mV) | D h (nm) (±S.D.) | Concentration (109 particles per mL) | ||
| ND: Not determined due to limitation of the measurement range that is up to 1 μm. | ||||||
| CS-N52–PP2 | 25 | 205 (12) | 0.159 (0.087) | +7.04 | 133 (1) | 0.3 (0.02) |
| 37 | 244 (34) | 0.159 (0.087) | +16.5 | 158 (6) | 2.6 (0.1) | |
| CS-N52–PP6 | 25 | 215 (36) | 0.325 (0.038) | +12.4 | 150 (2) | 0.4 (0.1) |
| 37 | 250 (7) | 0.157 (0.021) | +14.6 | 154 (4) | 4.4 (0.2) | |
| CS-N52–PP9 | 25 | 246 (18) | 0.256 (0.019) | +12.2 | 152 (2) | 0.3 (0.03) |
| 37 | 285 (23) | 0.195 (0.018) | +15.2 | 158 (4) | 5.3 (0.6) | |
| CS-N52–PP36 | 25 | 1419 (247) | 0.867 (0.088) | +9.3 | ND | ND |
| 37 | 1966 (1629) | 0.919 (0.134) | +12.7 | ND | ND | |
The greater number of particles observed by NTA upon heating indicated the still fundamental contribution of the thermal transition of oligo(NiPAAm) to micellization, also in the PP-modified copolymers. On the other hand, it is remarkable that such small PP contents facilitated the formation of micelles even below the abovementioned LCST (Fig. 5).
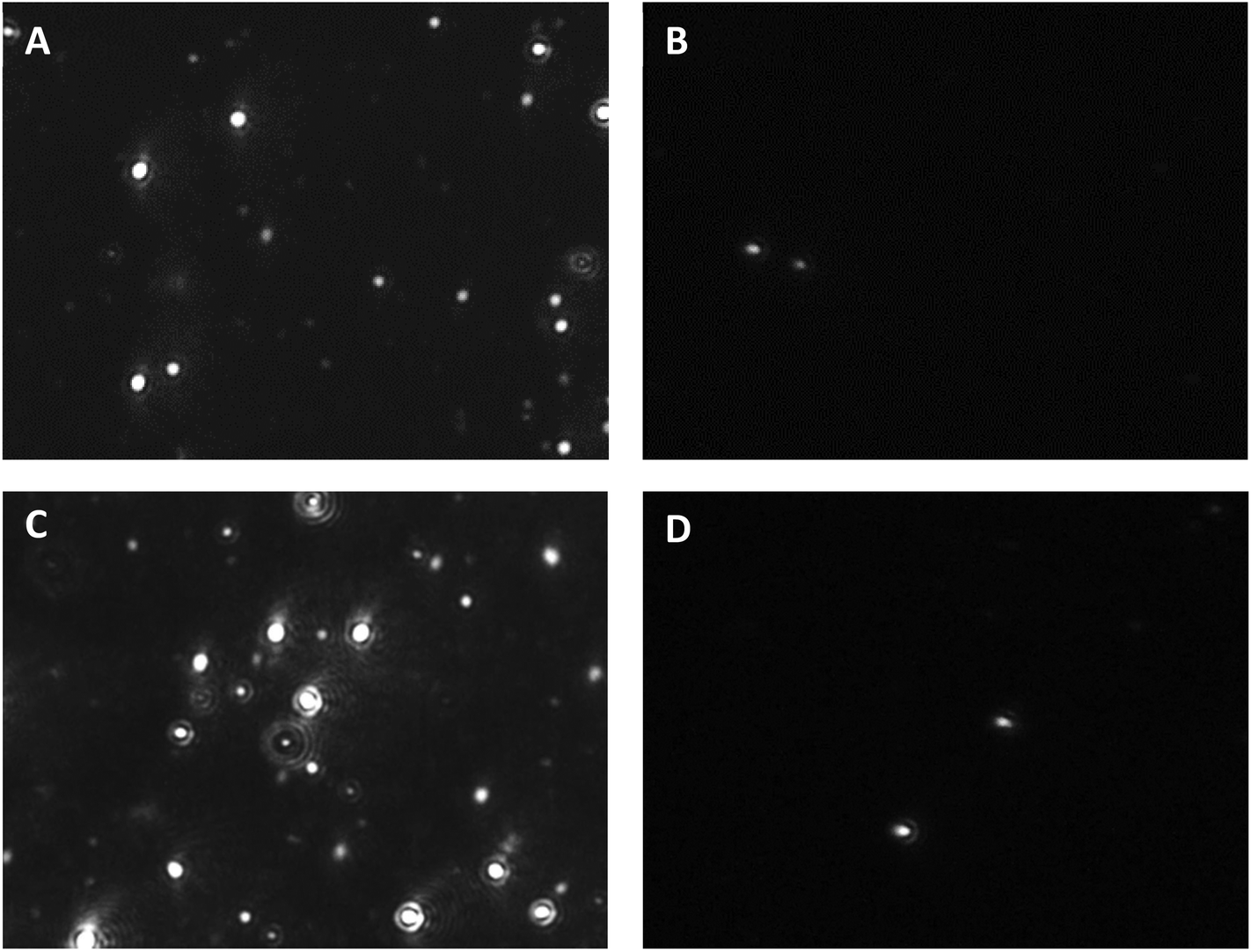 | ||
| Fig. 5 NTA snapshots of CS-N52–PP2 at (A, B) 25 and (C, D) 37 °C under (A, C) scattering and (B, D) fluorescence mode. The corresponding videos are provided as the ESI.† | ||
In addition, even though the fluorescence mode of the NTA instrument did not fit the strongest fluorescence emission of PP at 634 nm,20 the CS-N52–PP2 aggregates could still be visualized at both temperatures (Fig. 5B and D). Videos of CS-N52–PP2 micelles obtained under scattering and fluorescence mode are included as ESI videos.† In the case of CS-N52–PP36, the size of the aggregates was beyond the upper measurement limit of the NTA instrument (1 μm) and thus, they could not be quantified. In all the cases, the Z-potential remained positive due to the partial protonation of the amine side-groups of CS. Another intriguing aspect has to do with the increase of the size of the micelles upon heating that was probably associated with the change of the aggregation pattern below and above the LCST of poly(NiPAAm). Otherwise, a slight shrinkage would be expected. Finally, the spherical morphology of CS-N52–PPX micelles was visualized by TEM, as exemplified for CS-N52–PP2 in Fig. 6. The size range was in good agreement with the DLS and NTA data with some deviations due to the drying of the sample.
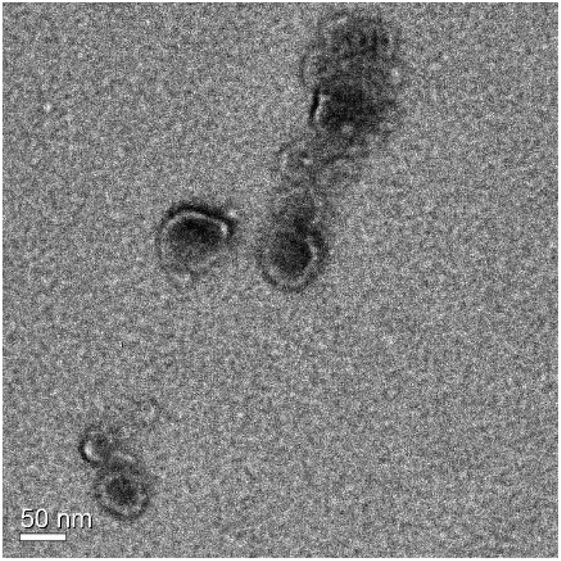 | ||
| Fig. 6 TEM micrograph of 0.1% w/v CS-N52–PP2 micelles at 37 °C, cast on carbon tape by TEM. | ||
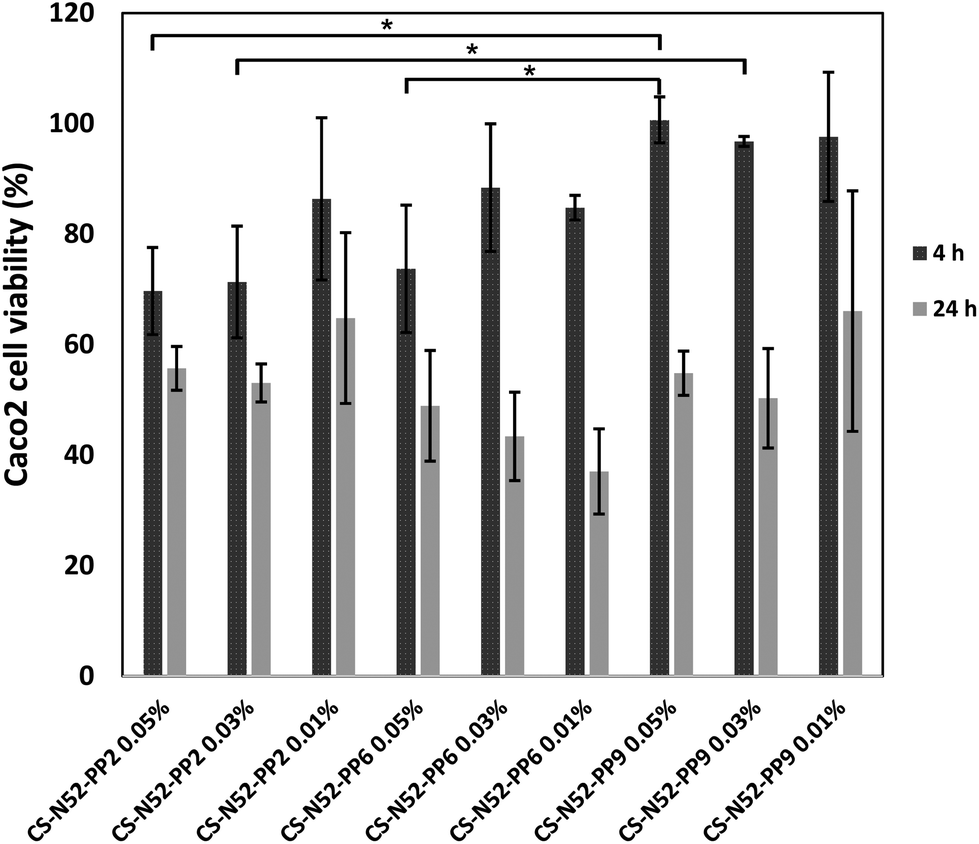 | ||
| Fig. 7 Caco2 cell viability upon exposure to different concentrations of CS-N52–PPX micelles with growing PP contents, as estimated by the MTT assay. *Statistically significant difference in cell viability (P < 0.05). | ||
This phenomenon could be observed when 0.05% and 0.03% w/v polymeric micelles containing 2% and 9% w/w PP were compared at 4 h. These findings were consistent with the decrease of the number of amine moieties in the side-chain of the polysaccharide owing to the conjugation of PP, a step that was previously shown to increase cell viability in MTT assays.17 In any event, the cell viability was high at 4 h, a condition that was critical to conduct the permeability studies (see below). Then, longer exposure reduced the viability for all the derivatives. Visualization of the cells under a fluorescence microscope did not reveal any significant uptake, as previously shown for CS-g-oligo(NiPAAm) polymeric micelles.17 However, since this observation was carried out at one specific time point, the involvement of passive or active transcellular transport mechanisms could not be ruled out.
The rationale behind the design of polymeric micelles generated by the self-assembly of CS backbones hydrophobized with oligo(NiPAAm) is to extend the application of polymeric micelles to mucosal administration routes by exploiting the ability of CS to transiently open tight junctions between epithelial cells.21 At the same time, there are controversies about the ability of these nanocarriers to cross epithelia, in which form and by which pathway. In fact, it is unclear whether polymeric micelles can do so as assembled entities through a saturable (active transport) or non-saturable (passive diffusion) mechanism34 or, conversely, they are absorbed as high molecular weight unimers after gradual disassembly in the apical zone.34
The conjugation was envisioned to fulfill two complementary key roles, a physical stabilizer of the micelles and a fluorescent tracer for biological studies in vitro (this work) and eventually in vivo. In this framework, Caco2 cell monolayers were grown over approximately 3 weeks and the conductivity was followed up by measuring the TEER. On one hand, it is worth stressing that this model constitutes a simplification of the intestinal epithelium and thus, the results could only reveal part of the mechanisms involved in the permeability. On the other hand, Caco2 cells recapitulate very well one of the main hurdles for nanoparticle absorption in the intestinal epithelium, the presence of the tight junctions; the surface available for transcellular transport is substantially greater than that of the paracellular one due to them. This is a relevant aspect of the proposed experimental design because the polymeric micelles of this work are based on CS, a polysaccharide that was reported to open these junctions in a temporary manner. Once the conductivity was appropriate, the micelles were poured into the donor chamber, the fluorescence due to the permeability of CS-N52–PP2 measured over time in the acceptor one and the intensity interpolated in a calibration curve of the copolymer in HBSS (ESI Fig. S8†). The results indicated that the permeability coefficient for CS-N52–PP2 micelles in this model was 43 ± 9 × 10−7 cm s−1. This value was in good agreement with the literature.18,35,36 In addition, an experiment was carried out at 4 °C to verify the possible involvement of transcellular (energy-dependent) transport mechanisms. The permeability coefficient was 37 ± 4 × 10−7 cm s−1. Regardless of the decrease of Papp at 4 °C, the difference between these values was not statistically significant (P = 0.33). These results pointed out that the transport was mainly passive and stressed the fundamental role of the transient opening of tight junctions mediated by CS during the permeation process. Papp could be correlated with intestinal absorption in vivo.37 Values <1 × 10−6 cm s−1 indicate malabsorption, while above it good absorption. The polymeric micelles of this work showed permeability >1 × 10−6 cm s−1. In parallel, the size of the micelles before and after the experiment was measured by DLS. Findings showed that the initial size of the micelles (at time 0 h) and of the nanostructures in the acceptor chamber at the end of the experiment (4 h) was 357 ± 94 and 337 ± 98 nm, respectively, highlighting that the copolymer crossed the cell monolayer mainly in the form of assembled micelles and not as disassembled unimers. Otherwise, a change in the size distribution pattern should have been observed. Fluorescence spectrophotometry analysis confirmed that most of the PP conjugated to the polymeric backbone permeated across the monolayer. At 37 °C, 59% and 36% of the initial PP amount was detected in the acceptor and donor chambers, respectively, after 4 h. At 4 °C, values were similar: 46% (acceptor) and 44% (donor). The small difference to complete 100% of the initial PP, namely approximately 5–10% was probably internalized by the Caco2 cells (see below). Overall, these data confirmed that these nanocarriers are expected to increase the intestinal transport of an encapsulated hydrophobic cargo with respect to the free drug due to the capitalization of an enhanced paracellular pathway.
Aiming to gain additional insight into possible passive or active cell internalization mechanisms, Caco2 cells were initially incubated with CS-N52–PP2 polymeric micelles and at different time points visualized by optical and fluorescence microscopy. No fluorescence could be detected (Fig. 8A). Then, the cells were incubated in cell culture inserts, harvested at different time points and analyzed by flow cytometry. At 4 °C, the energy-dependent transcytosis mechanism (endocytosis, pinocytosis and macropinocytosis) is completely blocked and thus, no active uptake could take place.38 Thus, in the case that Caco2 cells actively take up the micelles, a significant drop in the percentage of PP-stained cells was expected at 4 °C with respect to 37 °C. Regardless of the T, the percentage of stained Caco2 cells after 1 and 4 h was almost identical (Fig. 8B and C). Conversely, a slight difference was measured at 2 h, values being 61% at 37 °C and 75% at 4 °C. This behavior was unexpected and probably stemmed from irreversible changes in the cell membrane and a consequent increase of the permeability at 4 °C.39 Moreover, these findings confirmed the involvement of a passive internalization process because otherwise, a substantial decrease in staining should have been obtained upon cooling.
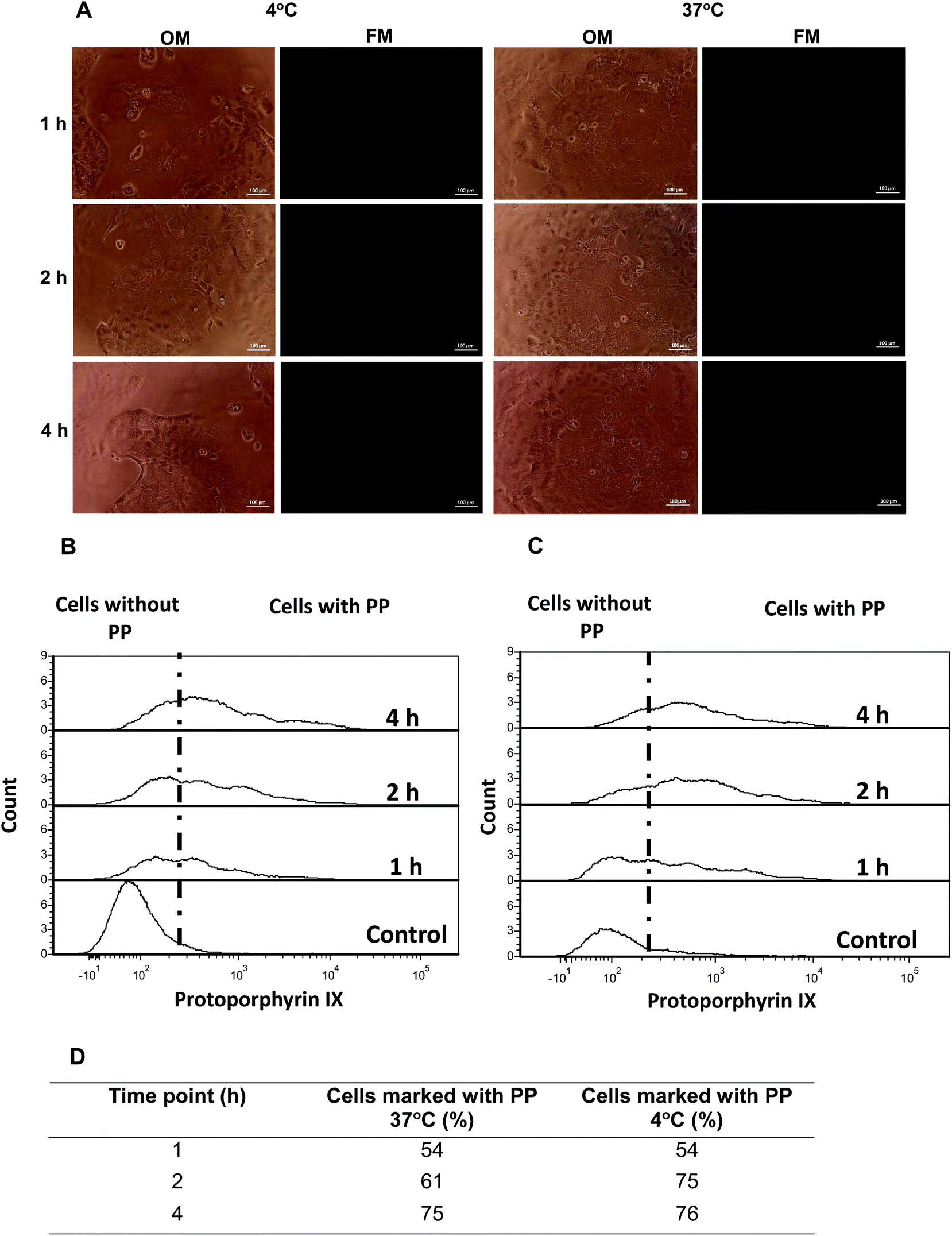 | ||
| Fig. 8 Caco2 cell uptake of CS-N52–PP2 polymeric micelles. (A) Micrographs of optical (OM) and fluorescence microscopy (FM) at different time points. (B, C) Flow cytometry results at (B) 37 °C and (C) 4 °C. (D) Summary of the percentage of cells stained with the fluorescent nanocarriers. | ||
Energy-independent diffusion of polymeric nanoparticles across cell membranes was reported elsewhere.40,41 This mechanism is more likely for positively-charged particles that disrupt the cell membrane than for neutral or negatively-charged counterparts.40 However, the claim that nanoparticles can passively diffuse across cell membranes remains controversial due to physicochemical and biological constraints,42 especially for nanoparticles with sizes >200 nm (this work). In addition, membrane disruption might result in extensive cell death, a phenomenon that was ruled out in MTT assays, after 4 h. Moreover, it is worth stressing that, as opposed to conventional polymeric nanoparticles or liposomes, polymeric micelles are self-assembly systems that exist in equilibrium with their non-self-assembled counterparts. Since our flow cytometry data showed identical PP staining at both T after 4 h, the internalization most likely stemmed from facilitated macromolecular diffusion of non-self-assembled polymeric molecules and not from the diffusion of assembled ones (micelles).43,44 These results were in good agreement with fluorescence microscopy assays.
In any event, it is worth indicating that the percentage of intracellular PP was always <10% of the initial amount and that most of it was detected in the acceptor chamber in the form of polymeric micelles, while the rest remained in the donor one. Future work will be dedicated to investigate the effect of micellar concentration and loading on the Papp.
4. Conclusions
In this study, PP was conjugated to a CS-g-oligo(NiPAAm) copolymer to achieve two goals, higher micellization capacity and tracking properties in permeability studies in vitro. Overall, the results point out the beneficial effect of the modification with CMC values at RT that resemble those of the unmodified copolymer at 37 °C, even for the derivative with the lowest PP content of 2% w/w. Furthermore, this modification enabled the measurement of the permeability coefficient using a standardized method, Caco-2 monolayers, where the ability of the micelles to cross the cell membrane in the assembled form by a passive paracellular pathway was confirmed. Conversely, the transcellular pathway contributed in a much less substantial way. Next studies will be oriented to evaluate the performance of PP-modified micelles in other models of mucosal tissues in vitro and the pharmacokinetics in vivo.Acknowledgements
The authors acknowledge the financial support of Technion (internal grant #2019300), the European Union's – Seventh Framework Programme under grant agreement #612765-MC-NANOTAR, the Israel Science Foundation (ISF, grant #269/15), the Teva National Network of Excellence in Neuroscience Research and the Russel Berrie Nanotechnology Institute (RBNI) (Technion). IS acknowledges the partial scholarship support of RBNI.References
- G. Gaucher, M. Dufresne, V. Sant, N. Kang, D. Maysinger and J. C. Leroux, Block copolymer micelles: preparation, characterization and application in drug delivery, J. Controlled Release, 2005, 109, 169–188 CrossRef CAS PubMed.
- K. Kataoka, A. Harada and Y. Nagasaki, Block copolymer micelles for drug delivery: design, characterization and biological significance, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Selective Localization of Preformed Nanoparticles in Morphologically Controllable Block Copolymer Aggregates in Solution, Acc. Chem. Res., 2012, 45, 1657–1666 CrossRef CAS PubMed.
- X. Chen, S. Ding, G. Qu and C. Zhang, Synthesis of Novel Chitosan Derivatives for Micellar Solubilization of Cyclosporine A, J. Bioact. Compat. Polym., 2008, 23, 563–578 CrossRef CAS.
- G. L. Amidon, H. Lennernas, V. P. Shah and J. R. Crison, A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability, Pharm. Res., 1995, 12, 413–420 CrossRef CAS.
- The ETP Nanomedicine. Strategic Research and Innovation Agenda: a vision for Nanomedicine in Europe for 2016–2030. http://www.etp-nanomedicine.eu/public/news-events/newsletter/articles/available-update-of-the-strategic-research-and-innovation-agenda-a-vision-of-nanomedicine-in-europe-for-2016-2030 (accessed in June 2016).
- X. Hu, F. F. Yang, L. H. Quan, C. Y. Liu, X. M. Liu, C. Ehrhardt and Y. H. Liao, Pulmonary delivered polymeric micelles–pharmacokinetic evaluation and biodistribution studies, Eur. J. Pharm. Biopharm., 2014, 88, 1064–1075 CrossRef CAS PubMed.
- D. A. Chiappetta, C. Hocht, J. A. W. Opezzo and A. Sosnik, Intranasal administration of antiretroviral-loaded micelles for anatomical targeting to the brain in HIV, Nanomedicine, 2013, 8, 223–237 CrossRef CAS PubMed.
- N. Akhtar and K. Pathak, Polymeric Micelles for Ocular Delivery: Progress and Issues, Pharm. Nanotechnol., 2015, 3, 171–187 CrossRef CAS.
- G. Gaucher, P. Satturwar, M. C. Jones, A. Furtos and J. C. Leroux, Polymeric micelles for oral drug delivery, Eur. J. Pharm. Biopharm., 2010, 76, 147–158 CrossRef CAS PubMed.
- H. Cabral and K. Kataoka, Progress of drug-loaded polymeric micelles into clinical studies, J. Controlled Release, 2014, 190, 465–476 CrossRef CAS PubMed.
- A. Sosnik and M. Menaker Raskin, Polymeric micelles in mucosal drug delivery: Challenges towards clinical translation, Biotechnol. Adv., 2015, 33, 1380–1392 CrossRef CAS PubMed.
- A. Sosnik, J. C. Imperiale, B. Vázquez-González, M. Menaker Raskin, F. Muñoz-Muñoz, G. Burillo, G. Cedillo and E. Bucio, Mucoadhesive thermo-responsive chitosan-g-poly(N-isopropylacrylamide) polymeric micelles via a one-pot gamma-radiation-assisted pathway, Colloids Surf., B, 2015, 136, 900–907 CrossRef CAS PubMed.
- R. J. Glisoni, S. S. Quintana, L. M. Molina, M. Calderon, A. G. Moglioni and A. Sosnik, Chitosan-g-oligo(epsilon-caprolactone) polymeric micelles: microwave-assisted synthesis and physicochemical and cytocompatibility characterization, J. Mater. Chem. B, 2015, 3, 4853–4864 RSC.
- X. Jiang, Z. Ge, J. Xu, H. Liu and S. Liu, Fabrication of multiresponsive shell cross-linked micelles possessing pH-controllable core swellability and thermo-tunable corona permeability, Biomacromolecules, 2007, 8, 3184–3192 CrossRef CAS PubMed.
- M. Talelli, M. Barz, C. J. Rijcken, F. Kiessling, W. E. Hennink and T. Lammers, Core-crosslinked polymeric micelles: Principles, preparation, biomedical applications and clinical translation, Nano Today, 2015, 10, 93–117 CrossRef CAS PubMed.
- M. Menaker Raskin, I. Schlachet and A. Sosnik, Mucoadhesive nanogels by ionotropic crosslinking of chitosan-g-oligo(NiPAam) polymeric micelles as novel drug nanocarriers, Nanomedicine, 2016, 11, 217–233 CrossRef PubMed.
- F. Mathot, A. des Rieux, A. Ariën, Y.-J. J. Schneider, M. Brewster and V. Préat, Transport mechanisms of mmePEG750P(CL-co-TMC) polymeric micelles across the intestinal barrier, J. Controlled Release, 2007, 124, 134–143 CrossRef CAS PubMed.
- L. M. Scolaro, M. Castriciano, A. Romeo, S. Patanè, E. Cefalì and M. Allegrini, Aggregation behavior of protoporphyrin IX in aqueous solutions: Clear evidence of vesicle formation, J. Phys. Chem. B, 2002, 106, 2453–2459 CrossRef CAS.
- H. Schneckenburger, M. Lang, T. Köllner, A. Rück, M. Herzog and R. Steiner, Fluorescence spectra and microscopic imaging of porphyrins in single cells and tissues, Lasers Med. Sci., 1989, 4, 159–166 CrossRef.
- K. Sonaje, E. R. Chuang, K. J. Lin, T. C. Yen, F. Y. Su, M. T. Tseng and H. W. Sung, Opening of epithelial tight junctions and enhancement of paracellular permeation by chitosan: microscopic, ultrastructural, and computed-tomographic observations, Mol. Pharmaceutics, 2012, 9, 1271–1279 CrossRef CAS PubMed.
- B. Neises and W. Steglich, Simple Method for the Esterification of Carboxylic Acids, Angew. Chem., Int. Ed. Engl., 1978, 17, 522–524 CrossRef.
- Ö. Topel, B. A. Çakır, L. Budama and N. Hoda, Determination of critical micelle concentration of polybutadiene-block-poly(ethyleneoxide) diblock copolymer by fluorescence spectroscopy and dynamic light scattering, J. Mol. Liq., 2013, 177, 40–43 CrossRef.
- I. Hubatsch, E. G. E. Ragnarsson and P. Artursson, Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers, Nat. Protoc., 2007, 2, 2111–2119 CrossRef CAS PubMed.
- R. J. Abraham, S. C. M. Fell, H. Pearson and K. M. Smith, The nmr spectra of porphyrins—15: Self-aggregation in zinc(II) protoporphyrin-IX dimethyl ester, Tetrahedron, 1979, 35, 1759–1766 CrossRef CAS.
- J. Zhang, Y. J. Zhou, Z. G. Su and M. Guang-Hui, Synthesis of monomethoxy poly(ethylene glycol) without diol poly(ethylene glycol), J. Controlled Release, 2007, 105, 3780–3786 CAS.
- L. J. Boucher, Manganese porphyrin complexes. I. Synthesis and spectroscopy of manganese(III) protoporphyrin IX dimethyl ester halides, J. Am. Chem. Soc., 1968, 90, 6640–6645 CrossRef CAS.
- H. Sun, L. Zhang, X. Zhang, C. Zhang, Z. Wei and S. Yao, 188Re-labeled MPEG-modified superparamagnetic nanogels: Preparation and targeting application in rabbits, Biomed. Microdevices, 2008, 10, 281–287 CrossRef CAS PubMed.
- J. Gonzalez-Lopez, C. Alvarez-Lorenzo, P. Taboada, A. Sosnik, I. Sandez-Macho and A. Concheiro, Self-associative behavior and drug-solubilizing ability of poloxamine (tetronic) block copolymers, Langmuir, 2008, 24, 10688–10697 CrossRef CAS PubMed.
- V. E. Diz, G. a. Gauna, C. A. Strassert, J. Awruch and L. E. Dicelio, Photophysical properties of microencapsulated phthalocyanines, J. Porphyrins Phthalocyanines, 2010, 14, 278–283 CrossRef CAS.
- R. Rai, V. Kumar and S. Pandey, Aggregation of a model porphyrin within poly(ethylene glycol) (PEG): effect of water, PEG molecular weight, ionic liquids, salts, and temperature, Phys. Chem. Chem. Phys., 2014, 16, 7263–7273 RSC.
- B. Zhu, Q. Liu, Y. Wang, X. Wang, P. Wang, L. Zhang and S. Su, Comparison of accumulation, subcellular location, and sonodynamic cytotoxicity between hematoporphyrin and protoporphyrin IX in L1210 cells, Chemotherapy, 2010, 56, 403–410 CrossRef CAS PubMed.
- C. Pereira, J. Costa and B. Sarmento, Cell-based in vitro models for intestinal permeability studies, in Concepts and Models for Drug Permeability Studies, ed. B. Sarmento, Woodhead Publishing, Cambridge, UK, 2015, pp. 57–81 Search PubMed.
- F. Mathot, L. van Beijsterveldt, V. Preat, M. Brewster and A. Arien, Intestinal uptake and biodistribution of novel polymeric micelles after oral administration, J. Controlled Release, 2006, 111, 47–55 CrossRef CAS PubMed.
- M. M. Thanou, A. F. Kotzé, T. Scharringhausen, H. L. Lueßen, A. G. De Boer, J. C. Verhoef and H. E. Junginger, Effect of degree of quaternization of N-trimethyl chitosan chloride for enhanced transport of hydrophilic compounds across intestinal Caco-2 cell monolayers, J. Controlled Release, 2000, 64, 15–25 CrossRef CAS PubMed.
- A. F. Kotzé, H. L. Lueßen, B. J. de Leeuw, A. G. de Boer, J. C. Verhoef and H. E. Junginger, Comparison of the effect of different chitosan salts and N-trimethylchitosan chloride on the permeability of intestinal epithelial cells (Caco-2), J. Controlled Release, 1998, 51, 35–46 CrossRef.
- S. Yee, In vitro permeability across Caco-2 cells (colonic) can predict in vivo (small intestinal) absorption in man–fact or myth, Pharm. Res., 1997, 14, 763–766 CrossRef CAS.
- D. Vercauteren, R. E. Vandenbroucke, A. T. Jones, J. Rejman, J. Demeester, S. C. De Smedt, N. N. Sanders and K. Braeckmans, The use of inhibitors to study endocytic pathways of gene carriers: Optimization and pitfalls, Mol. Ther., 2010, 18, 561–569 CrossRef CAS PubMed.
- M. Edidin and V. A. Petit, The effect of temperature on the lateral diffusion of plasma membrane proteins, in Ciba Foundation Symposium 52 - The Freezing of Mammalian Embryos, ed. K. Elliott and J. Whelan, John Wiley & Sons, Ltd., Chichester, UK, 1977, pp. 155–174 Search PubMed.
- T. Wang, J. Bai, X. Jiang and G. U. Nienhaus, Cellular uptake of nanoparticles by membrane penetration: A study combining confocal microscopy with FTIR spectroelectrochemistry, ACS Nano, 2012, 6, 1251–1259 CrossRef CAS PubMed.
- L. Treuel, X. Jiang and G. U. Nienhaus, New views on cellular uptake and trafficking of manufactured nanoparticles, J. R. Soc., Interface, 2013, 10, 20120939 CrossRef PubMed.
- L. K. Bogart, G. Pourroy, C. J. Murphy, V. Puntes, T. Pellegrino, D. Rosenblum, D. Peer and R. Lévy, Nanoparticles for imaging, sensing, and therapeutic intervention, ACS Nano, 2014, 8, 3107–3122 CrossRef CAS PubMed.
- R. Gaur, L. Mishra and S. K. Sen Gupta, Diffusion and transport of molecules in living cells, in Modelling and Simulation of Diffusive Processes: Methods and Applications, ed. S. K. Basu and N. Kumar, Springer International Publishing, Switzerland, 2014, pp. 27–49 Search PubMed.
- P. Gribbon and T. E. Hardingham, Macromolecular diffusion of biological polymers measured by confocal fluorescence recovery after photobleaching, Biophys. J., 1998, 75, 1032–1039 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6bm00667a |
| This journal is © The Royal Society of Chemistry 2017 |