
Metal-free 18F-labeling of aryl-CF2H via nucleophilic radiofluorination and oxidative C–H activation†
Gengyang
Yuan
ab,
Feng
Wang
c,
Nickeisha A.
Stephenson‡
a,
Lu
Wang
a,
Benjamin H.
Rotstein§
a,
Neil
Vasdev
a,
Pingping
Tang
c and
Steven H.
Liang
*a
aGordon Center for Medical Imaging & Division of Nuclear Medicine and Molecular Imaging, Massachusetts General Hospital and Department of Radiology, Harvard Medical School, 55 Fruit St., Boston, MA 02114, USA. E-mail: liang.steven@mgh.harvard.edu; Tel: +1-617-726-6107
bDepartment of Chemistry & Chemical Biology, Northeastern University, 360 Huntington Ave., Boston, MA 02115, USA
cState Key Laboratory and Institute of Elemento-Organic Chemistry, Nankai University, Collaborative Innovation Center of Chemical Science and Engineering, Tianjin, 300071, China
First published on 5th December 2016
Abstract
A metal-free and selective method to form [18F]aryl-CF2H through nucleophilic radiofluorination of benzyl (pseudo)halides and oxidative C–H activation of benzylic C–H bonds has been developed. The method is operationally simple and tolerates a variety of electron-neutral/deficient arenes and heteroarenes.
Fluorine bearing functionalities are found in a wide range of materials and biologically active molecules.1 Incorporation of fluorine-18 radionuclide into these molecules facilitates the application of noninvasive positron emission tomography (PET) imaging in quantifying biochemical and pharmacological processes. As a result, continuous efforts have been made to develop novel radiofluorination methods that provide a diverse range of 18F-labeled vectors to support molecular imaging research and drug discovery.2 Isosteric replacement of phenolic hydroxyl group with an aromatic difluoromethyl functionality (Ar-CF2H) has been demonstrated as a useful medicinal chemistry approach, leading to the discovery of bioactive molecules, including thiazopyr, fluxapyroxad, deracoxib, ZSTK474 etc. (Scheme 1).1d,3
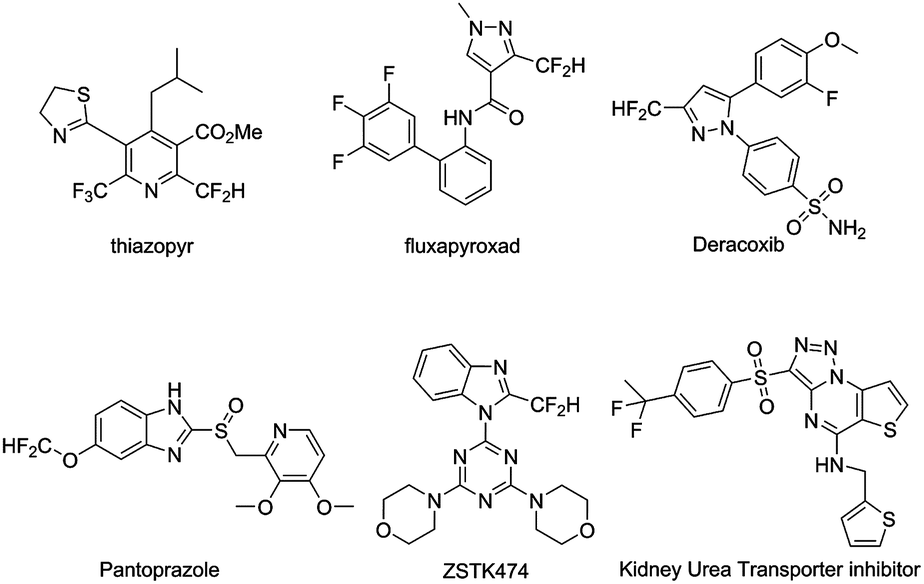 | ||
| Scheme 1 Aryl-CF2H containing bioactive molecules. | ||
The success of this substitution has been attributed to the similar acidic and isopolar characteristics of CF2H and OH. Moreover, CF2H functionality features hydrogen-bond-donating capability comparable to that of OH and NH groups, but much reduced hydrophilicity.4 Compared with recent advances in the preparation of non-radioactive Ar-CF2H,518F-labeling of this group has not yet been fully exploited. Initial methods relied on the silver(I)-mediated fluorodecarboxylation of α-fluorarylacetic acids with [18F]Selectfluor bis(triflate), an electrophilic fluorinating reagent derived from [18F]F2.6 An alternative 18F-incorporation method employed a silver(I)-mediated halogen exchange strategy with readily available [18F]fluoride under mild reaction conditions, but required aryl-CHFCl precursors prepared by multi-step synthesis.7 Our recent work disclosed a more practical method, starting from easily accessible aryl (pseudo)halides, via activation of a benzoyl auxiliary, to form [18F]Ar-CF2H functionality with an excellent substrate scope.8 However, to date, syntheses of [18F]aryl-CF2H have been restricted to low-to-moderate specific activity radiotracers, which may have limited their application in PET imaging studies, particularly for low density biological targets.
Recently we demonstrated the capability of silver-catalyzed oxidative activation of benzylic C–H bonds of aryl-CH3 for the synthesis of aryl-CF2H, which was further adapted to transition-metal-free conditions.9 Inspired by these discoveries, we speculated that a similar protocol could be applicable to the transformation of benzylic C–H to C–18F bond formation in the context of [18F]aryl-CFH2 intermediates, which could be easily obtained via nucleophilic radiofluorination of benzyl-(pseudo)halides with [18F]fluoride (Scheme 2). Herein, we report a general and metal-free method to radiolabel [18F]aryl-CF2H groups via a sequential nucleophilic radiofluorination of benzyl (pseudo)halides, followed by oxidative C–H activation.
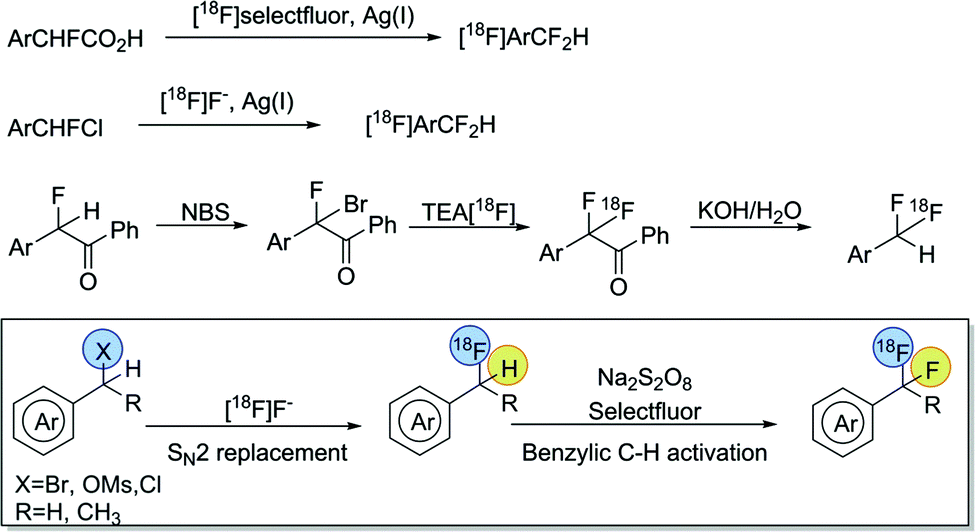 | ||
| Scheme 2 Radiolabeling of [18F]aryl-CF2H. | ||
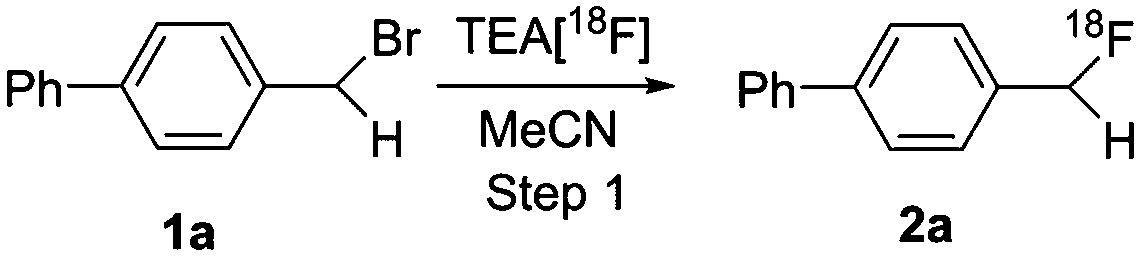
The feasibility of our method was experimentally confirmed, and further optimized by the radiosynthesis of 4-([18F]difluoromethyl)-1,1′-biphenyl ([18F]3a). An illustrative workflow is depicted in Scheme 3. Near quantitative radiochemical conversion (RCC; 98 ± 2%, n > 10) of 1a (8.09 μmol) to [18F]2a was achieved in 10 min in MeCN with tetraethylammonium bicarbonate (TEAB, 10.4 μmol) at 130 °C (Table 1). Under these conditions, the milder base used herein, compared with tetra-n-butylammonium hydroxide (TBAH),10 resulted in a 2-fold increase in RCC. Isolated [18F]2a was subsequently subjected to oxidative reaction conditions to install the benzyl 18F-fluoride. This step was optimized by varying the temperature and stoichiometry of reagents including AgNO3, Na2S2O8, and Selectfluor (Table 2). Under the initial optimal reaction conditions (Table 2, entry 3, RCC = 41 ± 6%), removal of AgNO3 gave superior RCC (50 ± 5%, entry 7), indicating that such C–H activation can be achieved under transition-metal-free conditions.9b Finally, compound [18F]3a was obtained in 50 ± 5% RCC with Na2S2O8 (33.6 μmol) and Selectfluor (56.5 μmol) at 120 °C for 10 min (Table 2, entry 7).
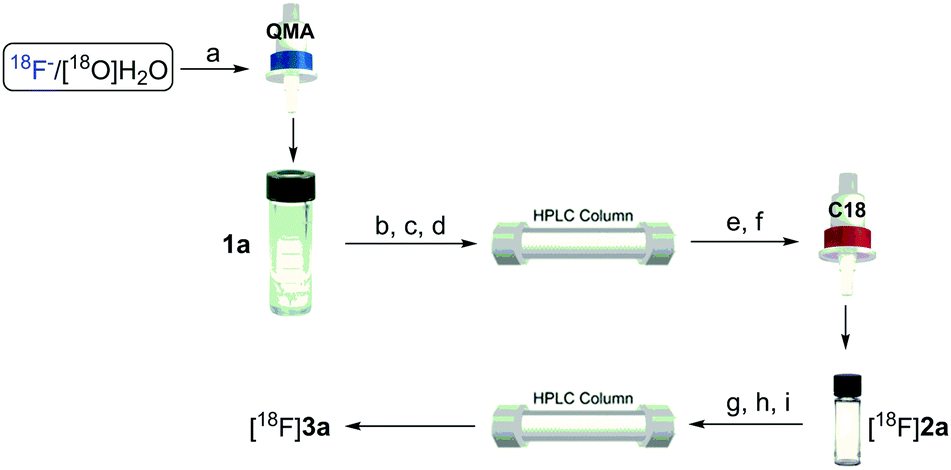 | ||
Scheme 3 Synthesis of [18F]2a and [18F]3a. Synthesis of [18F]2a: (a) trap and release 18F− into a V-vial via a solution of TEAB in 1 mL MeCN/H2O (v/v 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3); (b) azeotropic drying with anhydrous MeCN (3 × 1 mL, 3 min for each cycle); (c) addition of 1a, MeCN, 130 °C, 10 min; (d) dilution and subsequent purification of the reaction mixture (∼12 min); (e) concentration of the eluent via a C-18 cartridge (∼2 min); (f) release of [18F]2avia 1 mL MeCN (∼1 min). Synthesis of [18F]3a: (g) addition of Na2S2O8, Selectfluor and EtOH (∼1 min); (h) 120 °C, 10 min; (i) dilution and subsequent purification of the reaction mixture (∼12 min). :3); (b) azeotropic drying with anhydrous MeCN (3 × 1 mL, 3 min for each cycle); (c) addition of 1a, MeCN, 130 °C, 10 min; (d) dilution and subsequent purification of the reaction mixture (∼12 min); (e) concentration of the eluent via a C-18 cartridge (∼2 min); (f) release of [18F]2avia 1 mL MeCN (∼1 min). Synthesis of [18F]3a: (g) addition of Na2S2O8, Selectfluor and EtOH (∼1 min); (h) 120 °C, 10 min; (i) dilution and subsequent purification of the reaction mixture (∼12 min). | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | AgNO3 (mg) | Na2S2O8 (mg) | Selectfluor (mg) | Time (min) | Temp. (°C) | RCCa (%) |
| a Determined by radio-HPLC integration of product peaks, relative to [18F]fluoride and 18F-side products. | ||||||
| 1 | 8.0 | 4.0 | 20.0 | 10 | 120 | 20 ± 2 (n = 2) |
| 2 | 4.0 | 4.0 | 20.0 | 10 | 120 | 38 ± 5 (n = 2) |
| 3 | 4.0 | 8.0 | 20.0 | 10 | 120 | 41 ± 6 (n = 2) |
| 4 | 4.0 | 12.0 | 20.0 | 10 | 120 | 23 ± 3 (n = 2) |
| 5 | 4.0 | 8.0 | 5.0 | 10 | 120 | 0 (n = 2) |
| 6 | 4.0 | 8.0 | 15.0 | 10 | 120 | 33 ± 3 (n = 2) |
| 7 | 0.0 | 8.0 | 20.0 | 10 | 120 | 50 ± 5 (n = 2) |
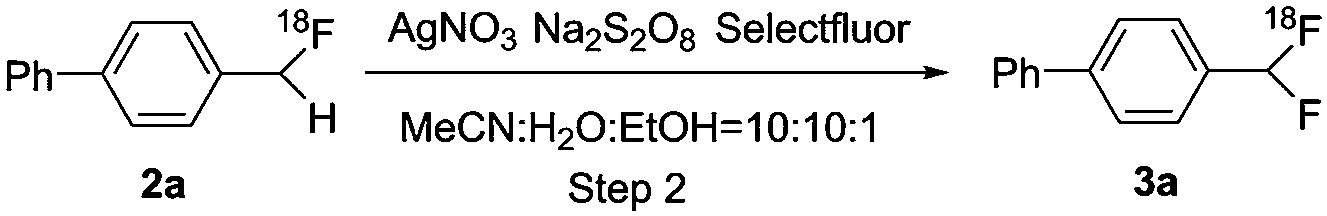
The scope of this methodology was then explored with more challenging substrates (Scheme 4). Under the optimized reaction conditions, all 18F-monofluorinated products were synthesized quantitatively, except [18F]2e (85 ± 5%) and [18F]2f (70 ± 3%). The isolated 18F-labeled compounds ([18F]2b–2i) were then subjected to the second step under optimized reaction conditions as described above. The final products [18F]3b–3h were obtained with moderate-to-good RCCs, ranging from 10% to 45%. As shown in Scheme 4, para-substituted electron-deficient ketone substrates [18F]2b and [18F]2c gave 10% and 15% RCCs, respectively. Ester substitution in the para-position of [18F]2d also yielded 12% RCC. Substrate [18F]2e exemplified the applicability of this method for a multi-substituted, sterically hindered compound with 28% RCC. In addition, a bromide motif in the product [18F]3e could allow further coupling reactions with various counterparts to provide more complex molecules. This method was also found to give good conversion for substrate [18F]2f (RCC = 45%) possessing a primary amide. The success of 2-arylpyridine substrate [18F]2g showed the compatibility of this labeling strategy for N-heteroatom bearing compounds. It is also noteworthy that substrate [18F]2h demonstrated the introduction of a second fluorine atom onto a tertiary carbon. Furthermore, product [18F]3h, bearing a (1,1-difluoroethyl)benzene fragment, is a key structural motif in a series of potent kidney urea transporter inhibitors,11 analogous to the compound shown in Scheme 1. The electron-rich arene 1i led to no desired product [18F]3i, as [18F]2i immediately decomposed to [18F]fluoride under our standard oxidative conditions as well as alternative oxidation reagents, for example 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo [2.2.2] octane bis(hexafluorophosphate) (F-TEDA-PF6).12 It is possible that the increased reactivity brought by the electron donating groups makes [18F]2i more vulnerable to the fluorinating reagents and/or radical defluorination.
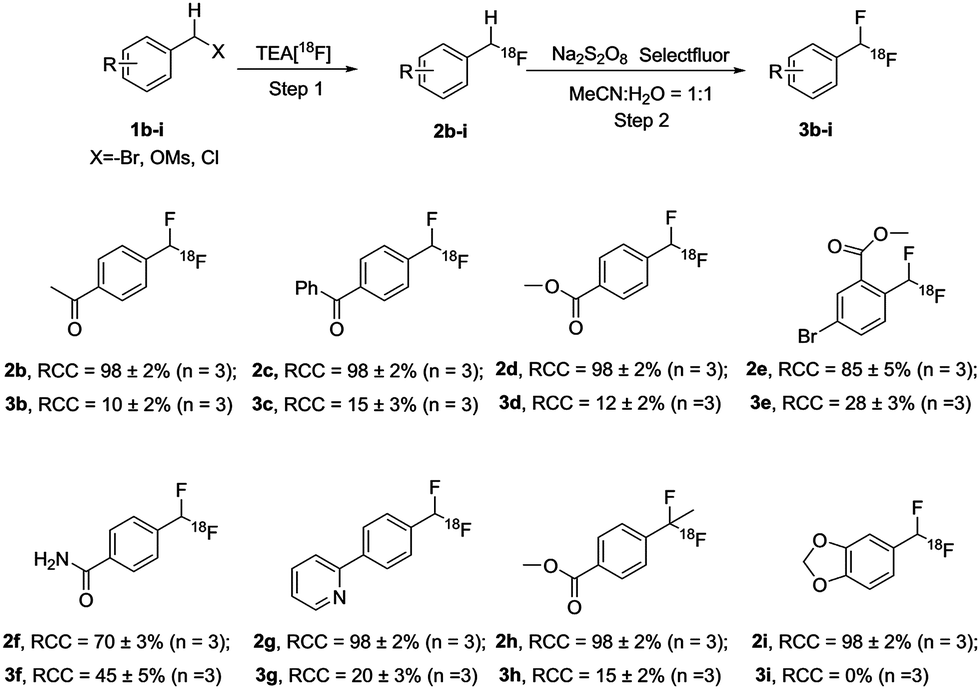 | ||
| Scheme 4 Scope of oxidative benzylic C–H activation in preparing the [18F]aryl-CF2H products. | ||
A plausible reaction mechanism is shown in Scheme 5. Homolytic cleavage of a peroxydisulfate anion produces two sulfate radical anions at elevated temperature, which oxidizes the benzylic C–H bond of [18F]aryl-CFH2 (A) to a benzylic radical (B). Then, Selectfluor reacts through a radical mechanism,13 providing a fluorine radical for bond formation with the benzylic radical (B) to produce the desired [18F]aryl-CF2H (C).
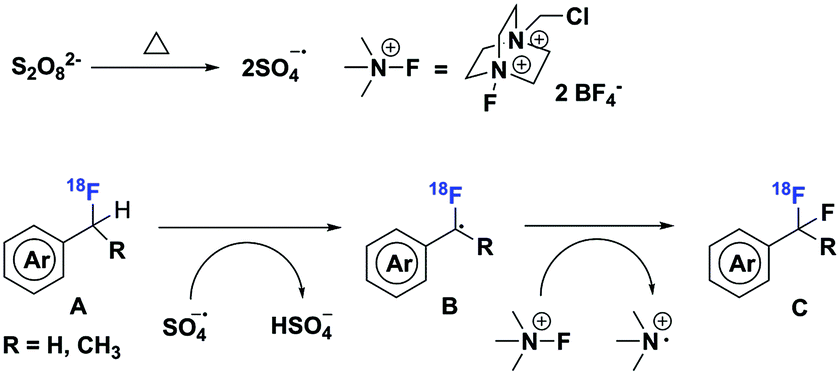 | ||
| Scheme 5 Proposed mechanism for the radiosynthesis of [18F]aryl-CF2H. | ||
Specific activity is a very critical parameter which should be considered in both radiolabeling methods and PET imaging studies.14 Obtaining high specific activity is still a challenge for 18F-labeling of many multi-fluorinated compounds.6a,7,8 As shown in Scheme 6, we measured the specific activity of the aryl-CF2H product [18F]3a by our protocol (see the ESI† for details). Briefly, compound [18F]2a was synthesized and isolated using a commercial automated radiofluorination unit (GE TRACERlab™ FXFN; Fig. S1, ESI†), starting from 1.7 GBq of [18F]fluoride, in 61% radiochemical yield (non-decay corrected) with >99% radiochemical purity and 51.8 GBq μmol−1 specific activity at the end-of-synthesis (∼60 min synthesis time from [18F]fluoride). The isolated intermediate [18F]2a was formulated and eluted from a C-18 cartridge with 1 mL of MeCN. It was diluted with 1 mL of sterile water, from which 0.4 mL was used for the second step. Under the optimized conditions, the mixture was heated for 10 min, cooled and diluted using a mobile phase. The reaction mixture was injected into semi-preparative HPLC for purification. The purified [18F]3a was isolated in 38% radiochemical yield (non-decay corrected, based on the added activity of [18F]2a) with a specific activity of 22.2 GBq μmol−1 at the end-of-synthesis (∼25 min synthesis time from the addition of [18F]2a). As shown in Table 3, the current method gives superior specific activity over other reported syntheses of [18F]aryl-CF2H.6a,7,8 Efforts were also made to further improve the specific activity by changing the counterion of the current fluorinating reagents to PF6− (F-TEDA-PF6) and OTf− (1-chloromethyl-4-fluoro-1,4-diazoniabicyclo [2.2.2]octane bis(trifluoromethane sulfonate; F-TEDA-OTf; see the ESI† for its preparation), though none of them gave the desired product [18F]3a. Since the tetrafluoroborate anion (BF4−) has been proven to give high specific activity comparable to that of OTf− in an analogous radiosynthesis of aryl-[18F]fluoride from (mesityl)(aryl)iodonium salt precursors,15 it is reasonable to postulate that there were limited exchange reactions between 19F and 18F under the present radiolabeling conditions.
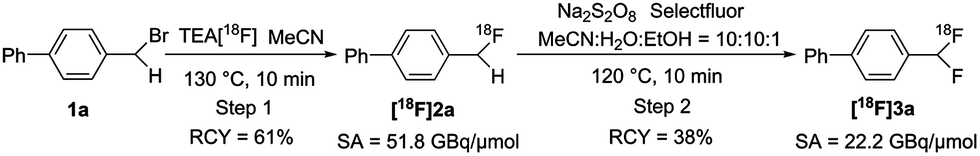 | ||
| Scheme 6 Specific activity determination for 4-([18F]difluoromethyl)-1,1′-biphenyl ([18F]3a). | ||
| Ref. | [18F]aryl-CF2H | Reaction type | Starting radioactivity (GBq) | Specific activity (GBq μmol−) |
|---|---|---|---|---|
| a Specific activity after radioactive decay correction. | ||||
| Gouverneur et al.6a |
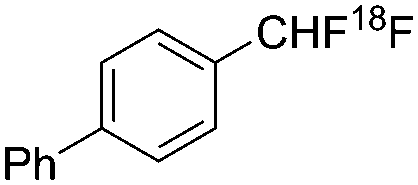
|
Electrophilic | — | 2.5a |
| Gouverneur et al.7 |
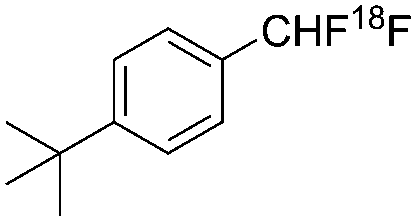
|
Nucleophilic | 3.5 | 0.03 |
| Liang, Vasdev, Ritter et al.8 |
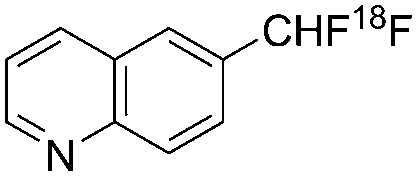
|
Nucleophilic | 1.4 | 3.0 |
| This work |
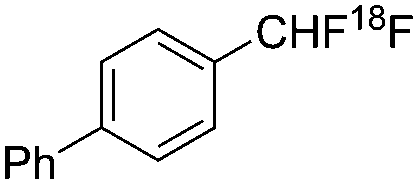
|
Radical | 1.7 | 22.2 |
In conclusion, we have investigated a metal-free oxidative C–H activation strategy for the synthesis of [18F]aryl-CF2H with good radiochemical yields and reasonably high specific activity. This method was successfully applied to mono- or multi-substituted electron-neutral and electron-deficient substrates, among which the amide group, N-containing heterocycle, and tertiary benzylic C–H bond were well tolerated. Further ongoing improvement resides in applying this method to electron-rich substituted arenes.
We would like to thank the staff at the radiochemistry program, Gordon Center for Medical Imaging, Nuclear Medicine and Molecular Imaging, Massachusetts General Hospital, MA, and Department of Chemistry & Chemical Biology, Northeastern University for their generous support. We also thank Drs Lee Collier and Hema Krishnan for helpful discussion. S. H. L is a recipient of NIH career development award from the National Institute on Drug Abuse (DA038000).
Notes and references
- (a) P. Jeschke, ChemBioChem, 2004, 5, 571 CrossRef PubMed; (b) K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (c) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed; (d) J. Wang, M. Sanchez-Rosello, J. L. Acena, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; (e) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- (a) S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719 CrossRef CAS PubMed; (b) M. Tredwell and V. Gouverneur, Angew. Chem., Int. Ed., 2012, 51, 11426 CrossRef CAS PubMed; (c) A. F. Brooks, J. J. Topczewski, N. Ichiishi, M. S. Sanford and P. J. Scott, Chem. Sci., 2014, 5, 4545 RSC; (d) M. G. Campbell and T. Ritter, Chem. Rev., 2015, 115, 612 CrossRef CAS PubMed; (e) F. Buckingham and V. Gouverneur, Chem. Rev., 2016, 7 Search PubMed; (f) O. Jacobson, D. O. Kiesewetter and X. Chen, Bioconjugate Chem., 2015, 26, 1 CrossRef CAS PubMed.
- (a) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef CAS PubMed; (b) S. Kaneko, T. Yamazaki and T. Kitazume, J. Org. Chem., 1993, 58, 2302 CrossRef CAS; (c) W. F. Goure, K. L. Leschinsky, S. J. Wratten and J. P. Chupp, J. Agric. Food Chem., 1991, 39, 981 CrossRef CAS; (d) G. W. Rewcastle, S. A. Gamage, J. U. Flanagan, R. Frederick, W. A. Denny, B. C. Baguley, P. Kestell, R. Singh, J. D. Kendall, E. S. Marshall, C. L. Lill, W. J. Lee, S. Kolekar, C. M. Buchanan, S. M. Jamieson and P. R. Shepherd, J. Med. Chem., 2011, 54, 7105 CrossRef CAS PubMed; (e) M. O. Anderson, J. Zhang, Y. Liu, C. Yao, P. W. Phuan and A. S. Verkman, J. Med. Chem., 2012, 55, 5942 CrossRef CAS PubMed.
- J. A. Erickson and J. I. McLoughlin, J. Org. Chem., 1995, 60, 1626 CrossRef CAS.
- L. Xu and D. A. Vicic, J. Am. Chem. Soc., 2016, 138, 2536 CrossRef CAS PubMed and ref. 3–12 therein.
- (a) S. Mizuta, I. S. Stenhagen, M. O'Duill, J. Wolstenhulme, A. K. Kirjavainen, S. J. Forsback, M. Tredwell, G. Sandford, P. R. Moore, M. Huiban, S. K. Luthra, J. Passchier, O. Solin and V. Gouverneur, Org. Lett., 2013, 15, 2648 CrossRef CAS PubMed; (b) F. Yin, Z. Wang, Z. Li and C. Li, J. Am. Chem. Soc., 2012, 134, 10401 CrossRef CAS PubMed; (c) H. Teare, E. G. Robins, A. Kirjavainen, S. Forsback, G. Sandford, O. Solin, S. K. Luthra and V. Gouverneur, Angew. Chem., Int. Ed., 2010, 49, 6821 CrossRef CAS PubMed.
- S. Verhoog, L. Pfeifer, T. Khotavivattana, S. Calderwood, T. L. Collier, K. Wheelhouse, M. Tredwell and V. Gouverneur, Synlett, 2016, 25 CAS.
- H. Shi, A. Braun, L. Wang, S. H. Liang, N. Vasdev and T. Ritter, Angew. Chem., Int. Ed., 2016, 55, 10786 CrossRef CAS PubMed.
- (a) P. Xu, S. Guo, L. Wang and P. Tang, Angew. Chem., Int. Ed., 2014, 53, 5955 CrossRef CAS PubMed; (b) J. J. Ma, W. B. Yi, G. P. Lu and C. Cai, Org. Biomol. Chem., 2015, 13, 2890 RSC.
- K. C. Lee, S.-Y. Lee, Y. S. Choe and D. Y. Chi, Bull. Korean Chem. Soc., 2004, 25, 1225 CrossRef CAS.
- Y. Liu, C. Esteva-Font, C. Yao, P. W. Phuan, A. S. Verkman and M. O. Anderson, Bioorg. Med. Chem. Lett., 2013, 23, 3338 CrossRef CAS PubMed.
- T. Furuya, A. E. Strom and T. Ritter, J. Am. Chem. Soc., 2009, 131, 1662 CrossRef CAS PubMed.
- (a) P. T. Nyffeler, S. G. Duron, M. D. Burkart, S. P. Vincent and C. H. Wong, Angew. Chem., Int. Ed., 2004, 44, 192 CrossRef PubMed; (b) Y. A. Serguchev, M. V. Ponomarenko, L. F. Lourie and A. A. Fokin, J. Phys. Org. Chem., 2011, 24, 407 CrossRef CAS.
- J. J. M. de Goeij and M. L. Bonardi, J. Radioanal. Nucl. Chem., 2005, 263, 13 CrossRef CAS.
- N. Ichiishi, A. F. Brooks, J. J. Topczewski, M. E. Rodnick, M. S. Sanford and P. J. Scott, Org. Lett., 2014, 16, 3224 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc07913j |
| ‡ Current address: Department of Chemistry, University of the West Indies, Mona, Kingston 7, Jamaica. |
| § Current address: Department of Biochemistry, Microbiology and Immunology, University of Ottawa & University of Ottawa Heart Institute, 40 Ruskin Street, Ottawa, Ontario, USA. |
| This journal is © The Royal Society of Chemistry 2017 |