
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzymatic site-selectivity enabled by structure-guided directed evolution
Jian-bo
Wang
ab,
Guangyue
Li
ab and
Manfred T.
Reetz
 *ab
*ab
aDepartment of Chemistry, Philipps-University Marburg, Hans-Meerwein Strasse 4, 35032, Marburg, Germany
bMax-Plank-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim, Germany. E-mail: reetz@mpi-muelheim.mpg.de
First published on 15th March 2017
Abstract
Biocatalytic site-selective (regioselective) organic transformations have been practiced for decades, but the traditional limitations of enzymes regarding narrow substrate acceptance and the often observed insufficient degree of selectivity have persisted until recently. With the advent of directed evolution, it is possible to engineer site-selectivity to suit the needs of organic chemists. This review features recent progress in this exciting research area, selected examples involving P450 monooxygenases, halogenases and Baeyer–Villiger monooxygenases being featured for illustrative purposes. The complementary nature of enzymes and man-made catalysts is emphasized.
 Jian-bo Wang | Jian-bo Wang obtained his bachelor's degree from NanKai University in 2004, then he pursued his doctoral research in Shanghai Institute Organic Chemistry (SIOC) from 2005, and graduated in 2010. After he worked in SIOC for about one and a half years, he went to Emory University, USA, to gain postdoctoral experience. After he finished his research in USA in 2014, he joined in Prof. M. T. Reetz's group as a postdoctoral fellow and now working in the field of biocatalysis, enzyme directed evolution and protein engineering. |
 Guangyue Li | Guangyue Li studied protein engineering at the Institute of Plant Protection, Chinese Academy of Agricultural Sciences (Laboratory of Prof. Dewen Qiu), obtaining his master degree in 2010. He then joined the group of Prof. Dunming Zhu at the Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, obtaining a doctoral degree in 2014 in the area of enzyme engineering. Currently he is a postdoctoral fellow in the group of M. T. Reetz focusing on directed evolution of epoxide hydrolase, alcohol dehydrogenase, Baeyer–Villiger monooxygenases and monoamine oxidase. |
 Manfred T. Reetz | Manfred T. Reetz is a synthetic organic chemist who proposed and developed the concept of directed evolution of stereoselective enzymes as catalysts in organic chemistry and biotechnology. Following two decades as Director at the Max-Planck-Institut für Kohlenforschung in Mülheim/Germany, he accepted in 2011 an emeritus position as Hans-Meerwein-Research Professor at Philipps-University in Marburg/Germany. |
Introduction
Site-selectivity and regioselectivity are terms that are often used interchangeably in synthetic organic chemistry. It means that a given reagent or catalyst induces a reaction selectively at one out of several possible positions in a compound. If the transformation is unselective, a mixture of products will arise, generally either constitutional isomers, enantiomers or diastereomers, depending upon the particular system. Since selectivity stands at the heart of modern organic chemistry, numerous selective reagents and catalysts have been developed and continue to be reported. Sometimes selectivity is possible only by resorting to protective group technology,1 but this requires additional steps. Indeed, as Gilbert Stork once said, “The best protective group is no protective group,” but this is generally not possible when using man-made reagents or catalysts. Introducing and removing protective groups site-selectively is in itself a fundamentally important task.1,2 The requirements as defined by the concepts of atom-economy,3a step-economy3b and redox-economy3c underscore the persisting challenges.Enzymes catalyze natural transformations generally with exquisite selectivity, including site-selectivity. Inspired by these well-known characteristics, chemists have long sought to harness wild type (WT) enzymes in isolated form, as lysates or as whole cells (strains) for catalyzing selective reactions of non-natural substrates.4,5 As in the case of synthetic reagents and catalysts,1–3 the ecological and economical aspects of enzyme catalysis in organic chemistry have been addressed.6 Several reviews focus on the use of enzymes in the production of pharmaceuticals.7
One of many practical examples of enzymes as catalysts in organic chemistry concerns the regioselective oxidation of cholic acid (1) with sole formation of the 12-keto product 2, catalyzed by 12α-hydroxysteroid dehydrogenase (12αHSDH),8 a specific alcohol dehydrogenase (ADH) (Scheme 1). Notice that the oxidation occurs at the sterically most hindered site, a result that is not possible using chemical reagents or catalysts without applying protective group methodology. Indeed, this transformation was also achieved by chemical means, albeit in four steps, three involving protection/deprotection and one requiring stoichiometric amounts of CrO3 as the oxidant.8
 | ||
| Scheme 1 Regioselective oxidation of cholic acid (1).8 | ||
Another early example of the complementarity of man-made catalysts/reagents and enzymes was reported in the chemoenzymatic synthesis of the important therapeutic compound cortisol (5) starting from readily available progesterone (3) by several pharmaceutical companies in the early 1950s, including Upjohn, Schering, Pfizer and Merck. In the Upjohn industrial process, an Aspergillus niger strain was used for oxidatively hydroxylating the desired C11-position regioselectively with formation of the C11-α-alcohol 4.9 This was the wrong diastereomer in an otherwise impressive site-selective CH-activating process. At the time the nature of the actual enzyme was not known, but it was most likely a cytochrome P450 monooxygenase (CYP), enzymes that have since been used in many selective oxidation reactions.10 Subsequent steps included chemical manipulation of the sidechain at the C17-position and inversion of configuration at position C11 (Scheme 2). The chemoenzymatic process was a practical alternative to the 40-step purely chemical synthesis of cortisone (as an entry to cortisol) by Woodward and coworkers at about the same time.11
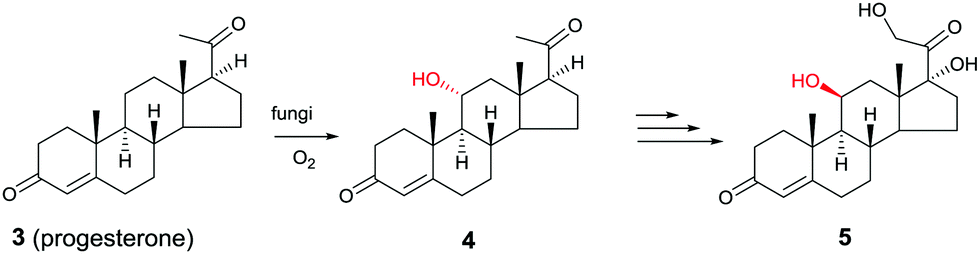 | ||
| Scheme 2 Chemoenzymatic synthesis of cortisol (5) by Upjohn.9 | ||
Perhaps even more impressive is the biocatalytic one-pot conversion of 5-aminolevulinic acid (6) into compound 7 in 20% overall yield, a known precursor of vitamin B12 (8), catalyzed by a mixture of 12 enzymes (Scheme 3).12 The cascade sequence involves regio- and stereoselective C-methylation, oxidation, reduction, ring contraction, decarboxylation and finally [1,5]-sigmatropic rearrangement. This stands in contrast to the purely chemical synthesis of vitamin B12 by Woodward, Eschenmoser and coworkers, a monumental feat which, of course, had many spinoffs that are of substantial benefit to synthetic organic chemistry.13 Neither approach is exploited in the industrial vitamin B12 production, which in reality is accomplished by the biocatalytic process of fermentation, a fascinating accomplishment in its own right.14
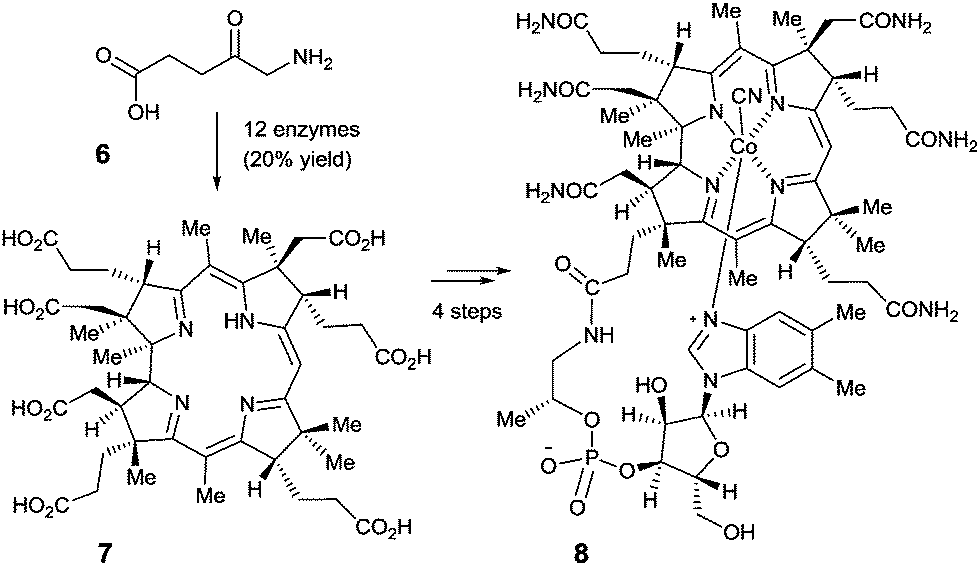 | ||
| Scheme 3 The Scott-synthesis of intermediate 7 as a precursor of vitamin B12 (8).12 | ||
When considering site-selectivity (regioselectivity) in general, essentially all of the enzyme types can be considered, including hydrolases, reductases, oxidases, transferases, lyases, isomerases and ligases, all in a variety of different kinds of reactions. Many of these have been exploited by academic and industrial researchers for the purpose of ensuring site-selectivity. For example, an important area is enzyme-catalyzed regioselective protection and deprotection of such functional moieties as:2,15
• Alcohol groups (e.g., in carbohydrates, poly-hydroxylated alkaloids and steroids, nucleosides)
• Thiol groups (e.g., in peptides)
• Amino groups (e.g., in polyhydroxylated alkaloids, aminosaccharides, β-lactam derivatives)
Hundreds of cases have been reported, one example being the site-selective deprotection of the fully O-acylated glucose derivative 9 with formation of either 10, catalyzed by porcine pancrease lipase (PPL),16a or 11, catalyzed by the esterase from Rhodosporidium toruloides (RTE)16b (Scheme 4). Further studies reporting enzymatic protection/deprotection continue to appear, as for example in reactions of monosaccharides.17 Sometimes the combination of chemical and enzymatic steps is especially effective.1,15
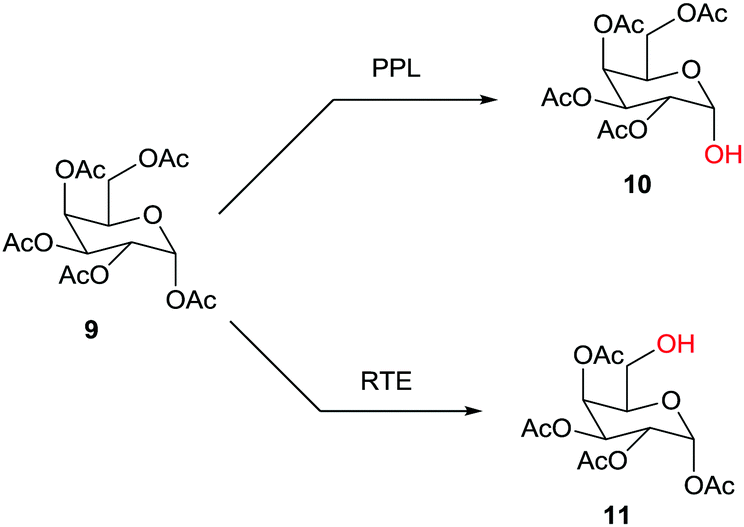 | ||
| Scheme 4 Site-selective deprotection of fully O-acylated glucose as a function of the enzyme (PPL: porcine pancreas lipase;16a RTE: esterase from Rhodosporium toruloides).16b | ||
In spite of these and other success stories, the traditional limitations of enzymes as catalysts in synthetic organic chemistry persisted for a long time, namely the often observed:4,5
• Wrong or poor enantio- or diastereoselectivity
• Wrong or insufficient regioselectivity (site-selectivity)
• Insufficient activity and limited substrate acceptance
• Insufficient robustness under operating conditions
All of these problems can be addressed and generally solved by applying the protein engineering technique of directed evolution.18 It involves recursive cycles of gene mutagenesis, expression and screening, the latter being the labor-intensive step. Because asymmetric catalysis is an integral part of synthetic organic chemistry, this parameter has been studied with great intensity to this day. This way of generating catalysts for stereoselective transformations was first reported in 199719 and has since been applied to essentially all enzyme types.18 Initially, efficiency was not of particular interest, since proof-of-principle was the primary goal.19 Different gene mutagenesis methods such as error-prone polymerase chain reaction (epPCR), saturation mutagenesis at the binding pocket, DNA shuffling or even mutator strains were applied.20 Some degree of selectivity improvement was generally observed irrespective of the mutagenesis technique, but soon it became clear that for real (industrial) applications, efficiency, speed and reliability are required. Consequently, the focus shifted to methodology development in directed evolution.20,21
In this ongoing endeavor, various strategies and techniques need be tested using one and the same enzyme and model reaction so that sound comparisons can be made, but unfortunately such comparative studies are rare.22 These and other studies18 indicate that saturation mutagenesis at sites lining the binding pocket of an enzyme is particularly effective.23 Based on precedence involving a saturation mutagenesis library generated by randomizing a 4-residue site lining the binding pocket of a lipase in kinetic resolution,24 systematization of this concept led to the technique of Combinatorial Active-Site Saturation Test (CAST) (Scheme 5).25 Thus, CAST is a convenient acronym for a process published earlier, readily distinguishing it from focused saturation mutagenesis at remote sites for other purposes. Since earlier protein engineering studies utilizing rationally designed mutations lining the binding pocket had resulted in changes in catalytic profiles (see discussion below), systematization by the CAST strategy appeared logical.
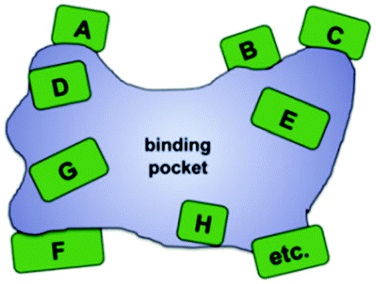 | ||
| Scheme 5 Systematization of saturation mutagenesis at sites lining the binding pocket of an enzyme (CASTing), a given site A, B, C, D, etc. comprising 1, 2, 3 or more residues.23,25 | ||
Statistical analysis based on the Patrick/Firth algorithm26 predicted that when using traditional NNK codon degeneracy encoding all 20 canonical amino acids as building blocks in the focused randomization process, the degree of oversampling for ensuring 95% library coverage increases astronomically as the size of a randomization site increases;23 for example, when randomizing a 5-residue CAST site, 108 transformants need to be screened, far beyond the capability of modern ee-screens.27 Therefore, reduced amino acid alphabets were introduced, and large randomization sites of 10 or more residues were split into smaller ones comprising two to four residues.23 Even when this was done, stereoselectivity sometimes remained at an insufficient level. Consequently, iterative saturation mutagenesis (ISM) was introduced.23,28 Accordingly, the best hit in one library is used as a template to perform saturation mutagenesis at another site, and the process is continued until the desired degree of stereoselectivity has been reached.
Rational criteria for choosing an appropriate set of amino acids as building blocks as part of the reduced amino acid alphabet were developed and guidelines for grouping residues into randomization sites were issued. These include guidance by X-ray structures, bioinformatics consensus data, computational techniques and exploratory NNK-based saturation mutagenesis at single positions requiring the screening of only 100 transformants.29 Thus, these strategies imply a rational process, away from “blind” directed evolution. When projecting such an approach to the directed evolution of site-selectivity (Scheme 6), it becomes clear that this catalytic parameter can be treated just like stereoselectivity. Indeed, saturation mutagenesis at sites lining the binding pocket (CASTing) can be expected to be a particularly viable approach to engineer both properties.
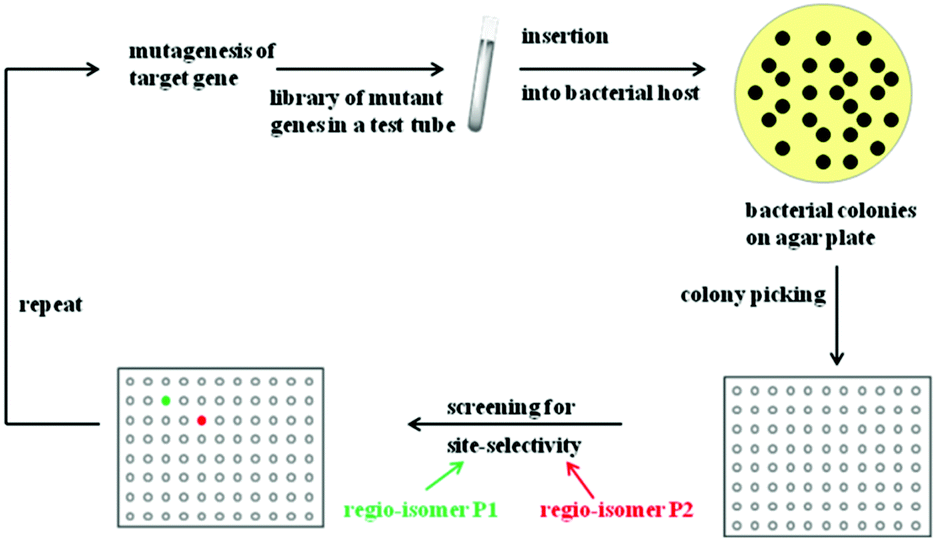 | ||
| Scheme 6 Directed evolution of site-selectivity featuring the case of two possible regioisomeric products P1 (green) and P2 (red). | ||
In the sections that follow, representative studies primarily utilizing focused library generation by saturation mutagenesis are analyzed, but selected cases of rational design, epPCR and DNA shuffling are also included for comparison. The three most intensively studied enzymes in terms of directed evolution of site-selectivity are featured, namely cytochrome P450 monooxygenases (CYPs), halogenases and Baeyer–Villiger monooxygenases (BVMOs). Rather than being comprehensive, the purpose of this article is to present selected studies for illustrative purposes. Literature citations referring to comprehensive accounts of these three enzyme types are included for the interested reader.
Engineering site-selectivity of cytochrome P450 monooxygenases
As delineated in the Introduction, CYPs have been used industrially for a long time in the oxidative hydroxylation of organic compounds at specific sites.9,10 CYPs have large binding pockets, which leads to two basic problems: Small molecules such as C3–C5 alkanes are generally not accepted, while larger ones are often randomly attacked at different positions with formation of undesired mixtures of regio- and stereoisomers.10 Rational design utilizing site-specific mutagenesis continues to provide progress in this challenging research area, but ongoing studies relying on directed evolution indicate that this approach constitutes the more general and reliable strategy.10f,30 The fusion of the two approaches is underway, driven by the continuous accumulation of data revealing the effects of point mutations on regio- and stereoselectivity.Before discussing selected examples of the directed evolution approach, some comments regarding rational design are in order.31 Application of this form of protein engineering of CYPs dates back to the 1990s, the focus of interest in most cases being on mechanistic questions, metabolism of therapeutic drugs and natural products and sometimes activity enhancement.10,31 Mutations at residues lining the binding pocket were generally designed, guided by the size and electronic nature of the residue sidechains, but the control of regioselectivity for synthetic purposes was not the main purpose of most of these studies. An interesting early example concerns the oxidative hydroxylation of ethylbenzene with regioselective formation of (R)-2-phenylethanol (46% ee), catalyzed by P450-cam; triple mutant T101M/T185F/V247M increases coupling of reducing equivalents to product formation and also enhances (R)-selectivity (74% ee), while maintaining full regioselectivity.32
In another revealing study, P450-cam was subjected to rational re-design in order to manipulate site-selectivity in the oxidative hydroxylation of (+)-α-pinene,33 guided by the enzyme's crystal structure harboring camphor.34 WT P450-cam leads to a mixture of oxidation products comprising (+)-α-pinene epoxide (4%), (+)-cis-verbenol (31%), (+)-myrtenol (4%), (+)-verbenone (10%) and 51% of unidentified compounds. Reasonable choices for point mutations were made, including F87A, F87W, Y96F and V247L. Combinations thereof were also considered, variant F87W/Y96F/L247A proving to be the best catalyst for the formation of (+)-cis-verbenol with 86% regioselectivity.33 In other studies, both rational design and random mutagenesis were applied with varying degrees of success.10,31
Several early directed evolution studies employing P450-BM3 from Bacillus megaterium,10 a self-sufficient fusion protein composed of a P450 monooxygenase and an NADPH diflavin reductase, focused primarily on the use of sequential cycles of epPCR in order to increase activity of n-alkane hydroxylation, but shuffling techniques were also applied.35 Enhanced activity was evolved by epPCR-based random mutagenesis, leading to the introduction of point mutations mostly remote from the active site, but reasonable control of regioselectivity was not achieved until subsequent studies.36 For example, an industrially important goal was site-selectivity at terminal positions of n-alkanes. Accordingly, it was stated that36a “Changing substrate specificity and regioselectivity often requires multiple mutations within the active sites, not likely to be found by random mutagenesis of the full protein.”
This switch in mutagenesis strategy followed the experience at the time regarding directed evolution of enzyme stereoselectivity.20,24 Consequently, saturation mutagenesis at 11 residues around the binding pocket of P450-BM3 was performed, the mutations of some of the most active hits then being combined combinatorially by overlap extension PCR.36a This procedure is different from ISM.18a,28 In this way a small library of mutants was generated, one of the best variants leading to 52% terminal hydroxylation of n-octane. Other mutants favored the 2- and 3-positions of n-alkanes, but enantioselectivity, when measured, proved to be poor to moderate.36a Interestingly, by screening previous P450-BM3 mutants generated for other substrates, phenyl acetic acid esters were hydroxylated solely at the benzylic position with up to 93% ee (S).37 In other P450-BM3 work, a triple mutant but not the WT, was found to metabolize testosterone and several drug-like compounds such as amodiaquine with formation of product mixtures.36b
In another directed evolution study of P450-BM3, the combination of epPCR and DNA shuffling was applied once more.38 The most active variants were shown to be A330P, KT2 (A191T/N239H/I259V/A276T/L353I), KSK19 (F87A/H171L/Q307H/N319Y) and KT5 (F87A/A330P/E377A/D425N), which were tested in the oxidation of naphthalene and n-propylbenzene. Scheme 7 shows that WT P450-BM3 leads to exclusive hydroxylation at the most reactive benzylic position. In contrast, variant KT5 changes site-selectivity toward the neighboring less reactive C-atom.38 Activity increased substantially in all cases and in reactions of other aliphatic and aromatic substrates as well. Enantioselectivity was not reported.
 | ||
| Scheme 7 The effect of P450-BM3 mutations on the site-selectivity of the oxidative hydroxylation of n-propylbenzene.38 | ||
Subsequently the first case of directed evolution of high inverted stereoselectivity with maintained site-selectivity appeared in which P450-pyr was used as the catalyst in the oxidative hydroxylation of N-benzylpyrrolidine (12) (Scheme 8).39 WT P450-pyr shows complete regioselectivity favoring the 3-position, but enantioselectivity with preference for (S)-13 is poor (43% ee). Guided by the crystal structure of WT P450-pyr, 17 amino acid positions within 5 Å of the docked substrate were individually subjected to saturation mutagenesis. An improved (S)-selective variant was identified (65% ee), while inversion of enantioselectivity with maintained regioselectivity was also achieved, single mutant N100S showing 42% ee in favor of (R)-13. Iterative saturation mutagenesis (ISM) was then applied by using this mutant as a template for saturation mutagenesis at the other single CAST positions, leading to the final mutant N100S/T186I with 83% ee (R). In a later study, (S)-selectivity were boosted to 98% ee by applying several rounds of structure-guided ISM, an impressive achievement.40
 | ||
| Scheme 8 Directed evolution of enantioselectivity with maintained site-selectivity of P450-pyr as catalyst in the hydroxylation of N-benzylpyrrolidine (12) leading preferentially either to (R)-13 (83% ee)39 or (S)-13 (98% ee).40 | ||
The first reported example of enhancing site-selectivity of a CYP while evolving both (R)- and (S)-selectivity on an optional basis concerns oxidative hydroxylation of compound 14 catalyzed by P450-BM3 (Scheme 9).41 WT is only 84% regioselective with moderate preference for allylic position C3, while enantioselectivity is poor in slight favor of (R)-15 (34% ee). By applying CAST/ISM at 23 residues surrounding the binding pocket and using reduced amino acid alphabets, regioselectivity was boosted to >95%, while enantioselectivity was evolved to 95% ee (R) or to 96% ee (S).41 Following acylation at the hydroxy function of (R)-15 and applying Pd-catalyzed γ-regioselective Tsuji–Trost allylic amination with retention of configuration, GABA-analog (R)-16 was obtained. In a different stereoselective follow-up reaction, epoxidation using m-chloro per-benzoic acid provided an epoxide with three stereogenic centers as a single stereoisomer.41
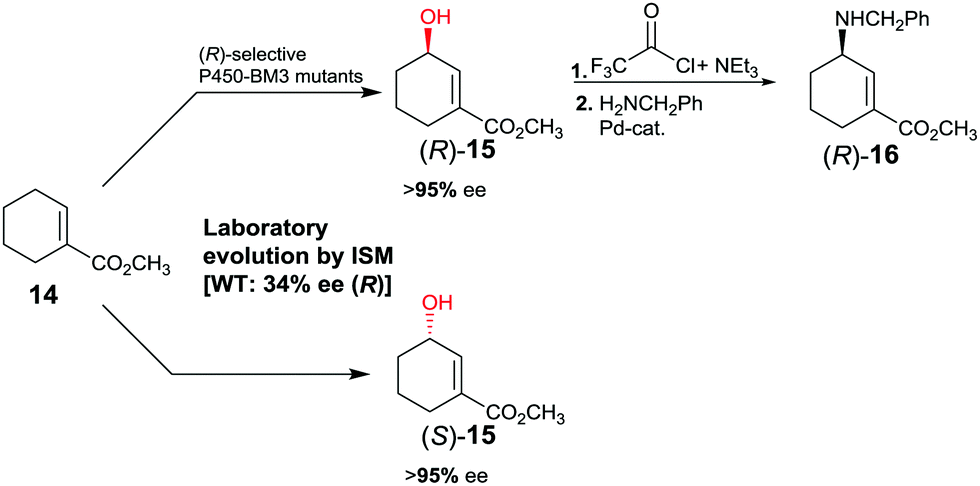 | ||
| Scheme 9 Directed evolution of P450-BM3 as catalyst in the regio- and enantioselective oxidative hydroxylation of ester 14.41 | ||
Late-stage CYP-catalyzed CH-activating oxidative hydroxylation of natural products or of other complex compounds with complete control of site-selectivity and stereoselectivity, enabled by directed evolution, is of considerable interest in organic chemistry as an alternative to synthetic reagents or catalysts. This is especially true when traditional methods fail. A number of further impressive examples of protein engineering of CYPs have been reported.10,42–45 An early example is the oxidative hydroxylation of testosterone (17) with regioselective formation of either 2β- or 15β-hydroxytestosterone (18 and 19, respectively) (Scheme 10).43a WT P450-BM3 does not accept this compound, but it was discovered that the known mutant F87A10 catalyzes hydroxylation with formation of a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixture of 18 and 19 in addition to trace amounts of other oxidation products. Phenylalanine at position 87 was known to partially shield the active site, therefore alanine was introduced in previous studies.10 Consequently, this mutant was used as the starting point in the steroid study, the goal being the evolution of a 2β- and a 15β-selective mutant.43a This was achieved by applying structure-guided ISM. The best 2β-selective variant ensured 97% site-selectivity with complete β-diastereoselectivity, and the best 15β-mutant allowed for 96% site-selectivity also with full β-diastereoselectivity.43a
:50 mixture of 18 and 19 in addition to trace amounts of other oxidation products. Phenylalanine at position 87 was known to partially shield the active site, therefore alanine was introduced in previous studies.10 Consequently, this mutant was used as the starting point in the steroid study, the goal being the evolution of a 2β- and a 15β-selective mutant.43a This was achieved by applying structure-guided ISM. The best 2β-selective variant ensured 97% site-selectivity with complete β-diastereoselectivity, and the best 15β-mutant allowed for 96% site-selectivity also with full β-diastereoselectivity.43a
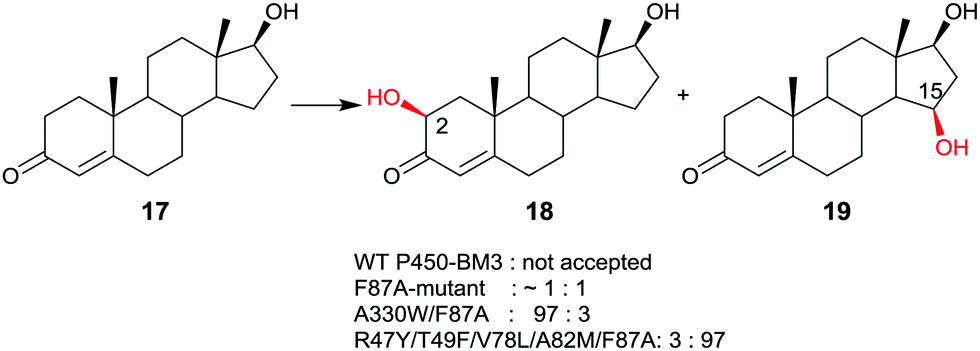 | ||
| Scheme 10 Regio- and stereoselective oxidative hydroxylation of testosterone (17) with formation of either 18 or 19 on an optional basis, catalyzed by mutants of P450-BM3.43a | ||
CYP-catalyzed hydroxylation is initiated by a radical mechanism in which the so-called Cpd I, a reactive high-spin heme-Fe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O intermediate, abstracts a hydrogen atom in the rate determining step with formation of a short-lived alkyl radical, followed by rapid alcohol formation by means of C–O bond formation.10 The optimal angle (C–H–O) in this abstraction step was calculated by QM to be about 130°.46 Hence, regio- and stereoselectivity depend on how the enzyme binds and places the substrate above the catalytically active FeO species. In order to explain the origin of regio- and diastereoselectivity of the two best mutants in steroid hydroxylation, extensive MD computations were carried out. Two very different substrate conformations in the binding pocket (poses) were predicted (Fig. 1), which are in line with the observed site-selectivity and diastereoselectivity.43a
O intermediate, abstracts a hydrogen atom in the rate determining step with formation of a short-lived alkyl radical, followed by rapid alcohol formation by means of C–O bond formation.10 The optimal angle (C–H–O) in this abstraction step was calculated by QM to be about 130°.46 Hence, regio- and stereoselectivity depend on how the enzyme binds and places the substrate above the catalytically active FeO species. In order to explain the origin of regio- and diastereoselectivity of the two best mutants in steroid hydroxylation, extensive MD computations were carried out. Two very different substrate conformations in the binding pocket (poses) were predicted (Fig. 1), which are in line with the observed site-selectivity and diastereoselectivity.43a
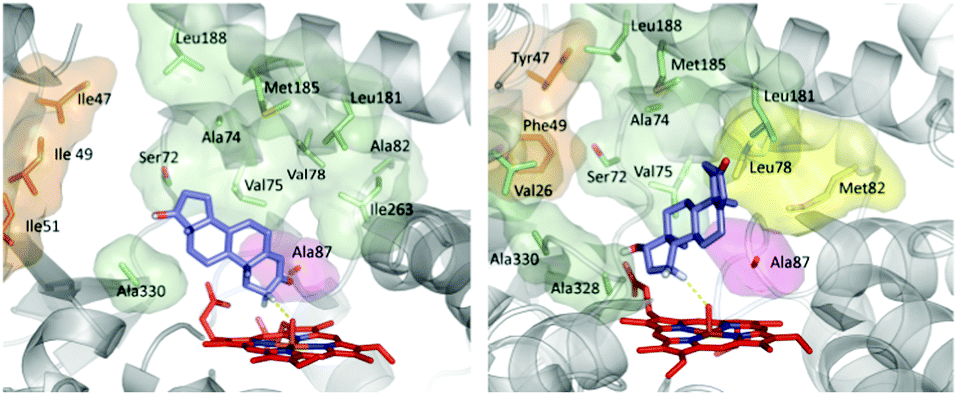 | ||
| Fig. 1 Calculated poses of testosterone (17) in P450-BM3 mutant R47I/T49I/F87A explaining 2β-selectivity (left) and in mutant R47Y/T49F/V78L/A82M/F87A shedding light on the origin of 15β-selectivity (right).43a | ||
The best mutants were then tested as catalysts in the hydroxylation of other steroids, likewise leading to synthetically interesting results.43a It should be mentioned that such a mutant library of about 300–400 members active in steroid hydroxylation provides many synthetically important opportunities. The “steroid library”43a as well as other small mutant collections with which different substrates were tested are (commercially) available.37,43,45 Moreover, structure(sequence)/selectivity trends have emerged,42–45 which taken together with extensive experience in the P450 area based on rational design of mutants by site-specific mutagenesis,31 constitutes the merging of the two protein engineering techniques. Nevertheless, it is currently still not possible to engineer specific mutants for targeting any CH-entity of steroids or of other natural products (or synthetic precursors) that a chemist might imagine.
As already pointed out, controlling P450-catalyzed regio- and stereoselectivity in the oxidative hydroxylation of non-functionalized alkanes is a daunting challenge.35 Using P450-pyr and n-octane as the substrate and CAST/ISM as the structure-guided mutagenesis technique, this was recently accomplished for the first time.44a Using n-octane (20) as the model substrate, mutant A77Q/I83F/N100S/F403I/T186I/L302V was found to ensure complete site-selectivity at position C2 and excellent enantioselectivity in favor of (S)-21 (98% ee) (Scheme 11).44a
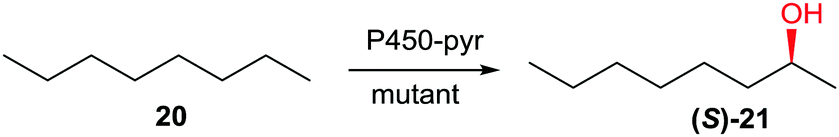 | ||
| Scheme 11 Complete regio- and enantioselective oxidative hydroxylation of n-octane (20) catalyzed by mutants of P450-pyr evolved by CAST/ISM.44a | ||
Other compounds such as n-propylbenzene likewise reacted with high selectivity. It would be interesting to extend this “dream reaction” by inverting the sense of enantioselectivity in favor of (R)-2-octanol and to target positions C3 and C4 in n-octane as well.
In another study, late-stage site-selective hydroxylation of the anti-malaria agent artemisinin (22) catalyzed by P450-BM3 mutants was accomplished, a compound that is not accepted by the WT enzyme (Scheme 12).44b Rather than adhering to the structure-guided approach employed earlier in the case of testosterone by starting with the known mutant F87A, a different strategy was chosen. First, a fingerprint technique was developed using and analyzing a collection of P450-BM3 mutants, which led to the identification of variant FL‡62 having 16 point mutations.44b This mutant catalyzes the formation of three main products 23, 24 and 25 in a ratio of 73:17:7 (Scheme 12). Then a number of saturation mutagenesis experiments were performed at defined CAST-type randomization sites 78/87, 78/81/87, 78/87/181, 78/87/184, 78/81/82/87, 81/82/87/184, 78/81/82/87/181/184, and 74/81/82/87/181/184. In earlier studies, some of these residues had been defined as “hot spots”.10,36,37,42–45 In a further step, 12500 transformants were assayed for activity using five semisynthetic chromogenic probes for easy detection, leading to 1950 active variants (criterion: >10% activity), and 522 functionally unique variants (criterion: >20% variation on at least one of the 5 fingerprint components relative to parent). This was followed by HPLC screening of several hundred variants using the real substrate 22. The reader is referred to details in the original publication.44b Suffice it to say that three variants were identified that are site-specific for positions C7(S), C7(R) and C6 (Scheme 12).
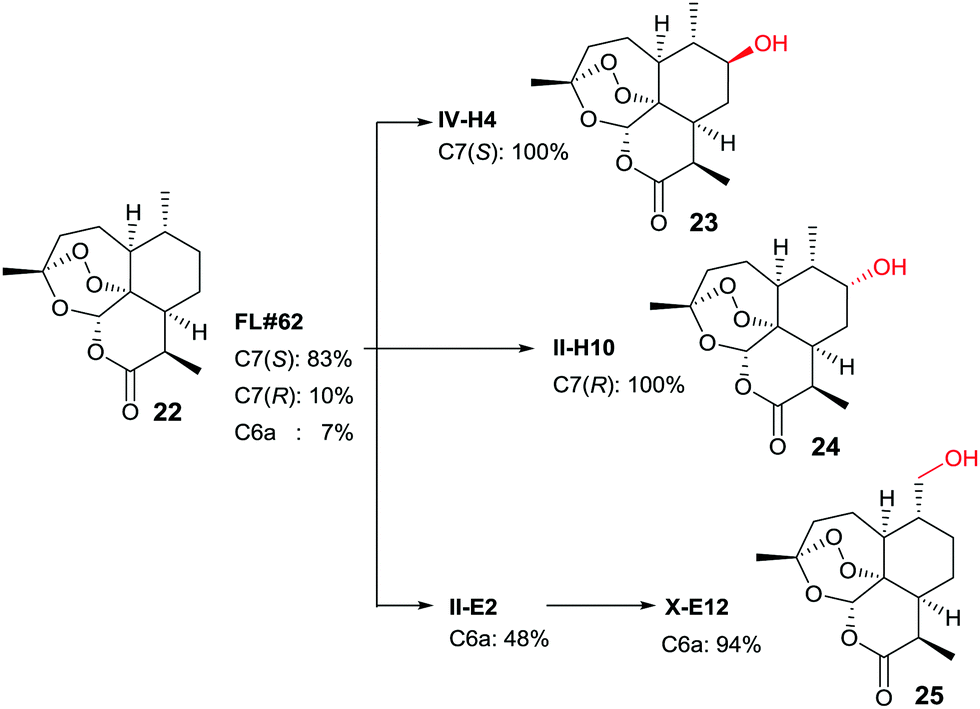 | ||
| Scheme 12 Late-stage oxidative hydroxylation of artemisinin (22) catalyzed by evolved mutants of P450-BM3.44b | ||
Here as in many other late-stage CYP-catalyzed oxidations, the particular “target positions” found following mutagenesis was not specified as a defined goal at the outset of the project. An exception is a study reporting bioorthogonal P450-BM3 catalyzed activation of caged compounds in living cells.43b A biologically active (anti-cancer) compound was first protected at a phenolic function in the form of a propargyl ether, this pro-drug compound then being deprotected with release of the biologically active substance by site-selective hydroxylation solely at the propargylic methylene-moiety. Nevertheless, the general problem of targeting any hydroxylation position in a given compound is one of the remaining challenges in this research area, especially if the technique should become an integral part of modern synthetic (natural products) chemistry. A logical strategy would be to subject a given compound to CYP-catalyzed oxidative hydroxylation using an appropriate library of mutants, to identify a mutant that produces at least a small amount of the desired hydroxylation product, and then to apply structure-driven directed evolution to increase the respective regio- and stereoselectivity. Aside from this issue, numerous other studies reporting CYP-catalyzed regio- and stereoselective hydroxylation have appeared recently, most of them exploiting structure-guided directed evolution by focusing saturation mutagenesis on sites lining the binding pocket.43–45
Decades ago, a P450 monooxygenase was shown to be a catalyst in nitrene mediated regioselective C–H insertion using azides with formation of amines analogous to hydroxylation leading to alcohols.47a More recently this has been exploited in the manipulation of site-selectivity and stereoselectivity, the controlling element being directed evolution (usually saturation mutagenesis).47b–d More work is necessary in this intriguing area so that CYP-catalyzed amine synthesis becomes a truly useful tool in organic chemistry.
Engineering site-selectivity of halogenases
Halogenation of aliphatic and aromatic compounds constitutes a fundamentally important transformation in synthetic organic and pharmaceutical chemistry. Of particular importance are those transformations that involve the CH-activating substitution of an H-atom by halogen in aliphatic or aromatic compounds. In addition to the standard synthetic methods, which do not routinely ensure high site-selectivity, biocatalysts of the type halogenases offer a fascinating alternative for controlling this catalytic parameter.48 Different types of halogenases have been identified which function by different mechanisms, including heme- and vanadium-dependent haloperoxidases as well as Fe2+/α-ketoglutarate-dependent and flavin-mediated halogenases (FDHs). During the last few decades the mechanism of these enzymes has been largely elucidated, although some uncertainties persist. Haloperoxidases oxidize halides such as Cl− or Br− with formation of hypohalites HOX which diffuse freely out of the enzyme. These reactive species then induce halogenation of electron-rich aromatic compounds,48 which means that protein engineering aimed at manipulating regioselectivity is difficult if not impossible. FDHs also utilize halide salts such as NaCl or NaBr. In these halogenases, flavin in the oxidized state is first reduced by NAD(P)H followed by reaction with oxygen and formation of the intermediate FAD-OOH. This peroxide then oxidizes halides to the electrophilic halogenating agent HOX which remains within the enzyme.48,49 At this point some mechanistic uncertainty remains. Some researchers contend that it is not HOX which halogenates the aromatic substrate. Rather, it was postulated that a lysine is first halogenated at the amino group leading to a reactive intermediate which then functions as the actual halogenation species.48 Recently novel halogenases have been shown to follow a nucleophilic mechanism.48 Fluorinases were discovered some time ago,50 but have not (yet) been applied in synthetic organic chemistry. In this review we focus only on FDHs.The finding that FDHs from different species show different site-selectivity in the reaction of a given natural or non-natural compound is of significant synthetic value. An impressive example is illustrated in Scheme 13, showing site-selective transformations of tryptophan (26) that would be difficult if not impossible using modern synthetic chlorination techniques.51 Other examples have been described,48 including the non-heme halogenase Amb05 which catalyzes the regio- and stereoselective halogenation of an amazingly wide range of alkaloids and other compounds at aliphatic CH-positions.52 Nevertheless, screening many enzymes of a given type does not routinely lead to the regioselectivity that the researcher may need, which means that alternative approaches are required.
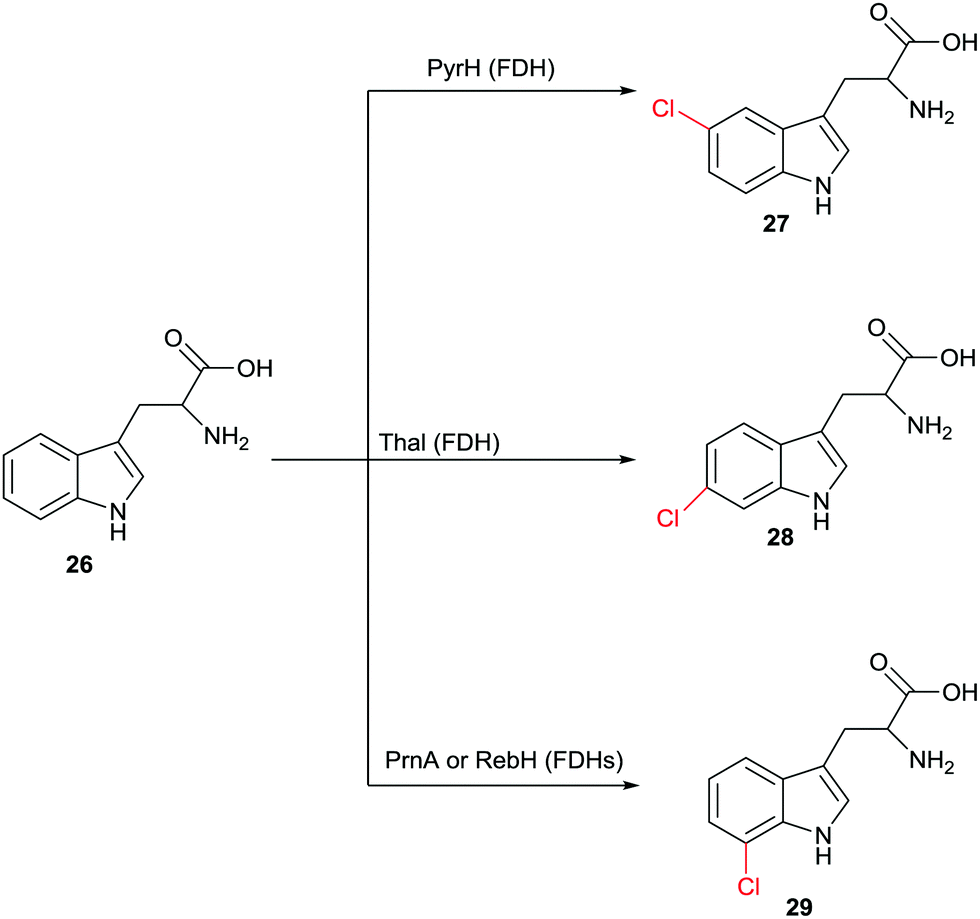 | ||
| Scheme 13 Regioselective chlorination of (R)- or (S)-tryptophan (26) at three different sites catalyzed by different halogenases.51 | ||
Protein engineering of an FDH by rational site-specific mutagenesis was first reported in 2011 in which mutant F103A of the halogenase PrnA was found to change the strict 7-selectivity of WT to a 2:1 mixture of the 7- and 5-chlorinated products.53 In other early work, rational design on the basis of structure-guided mutagenesis was applied to other FDHs with notable changes in site-selectivity in reactions of structurally different substrates, although a 100% shift in selectivity remains to be achieved.54,55
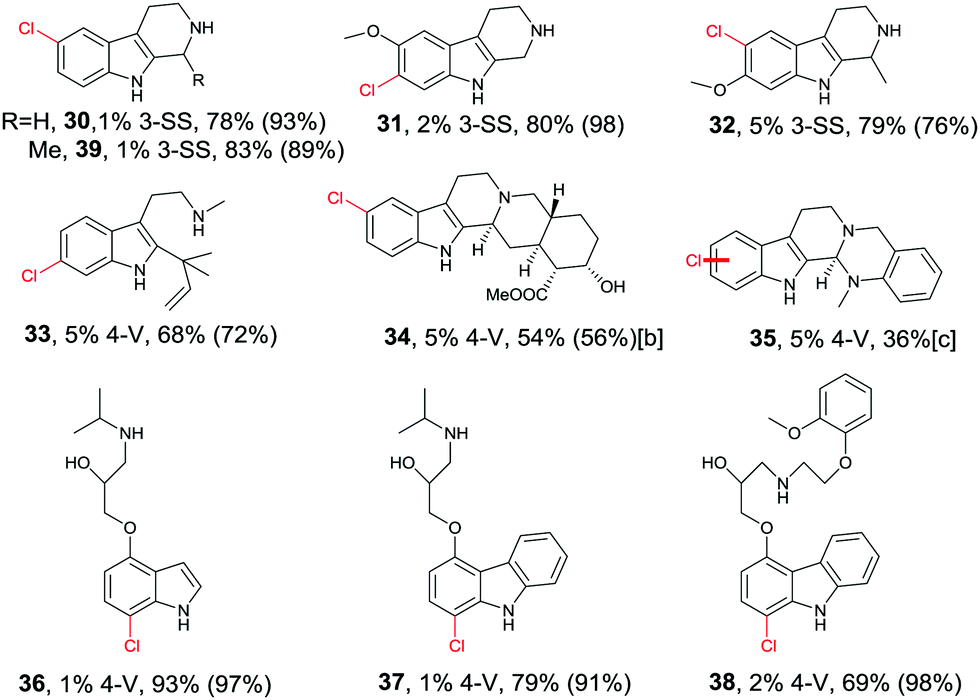
Directed evolution of halogenases offers a more general and reliable solution to the problem of controlling the regioselectivity of halogenation. In one study the well-known tryptophan 7-halogenase RebH was first subjected to directed evolution in order to enhance its thermostability.56 Two rounds of epPCR and combining some of the point mutations led to several improved variants, the best one showing an 18 °C increase in Topt relative to WT RebH with only a small tradeoff in activity. This required the screening of only 1600 transformants.56 In a subsequent study aimed at increasing activity and expanding substrate range, one of the stabilized mutants was used as a template for further mutagenesis, several cycles epPCR again being applied.57 Following the screening of about 2200 transformants, two prominent variants were identified: 3-SS which is ideal for site-selective chlorination of tricyclic tryptoline derivatives, and 4-V which shows broad substrate range of indole and carbazole derivatives, likewise with high site-selectivity (Table 1).57 All transformations are synthetically interesting, the meta-products such as 31 and 33 deserving special attention.
|
Although recursive epPCR proved to be successful in this study, it can be anticipated that saturation mutagenesis at active site residues and possibly ISM are likely to be more efficient, probably providing a high diversity of mutants showing different site-selectivity. Indeed, this was demonstrated in a subsequent study in which tryptamine (40) was used as the model substrate (Scheme 14).58 At the outset of the project, the goal was defined as the search for three different RebH mutants leading to the ortho-, meta- and para-products 41, 42 and 43, respectively. In the case of chlorination product 41, epPCR sufficed, but in order to evolve meta- and para-selectivity, it was more productive to apply saturation mutagenesis and iterative saturation mutagenesis at rationally chosen sites lining the binding pocket (CAST/ISM).58
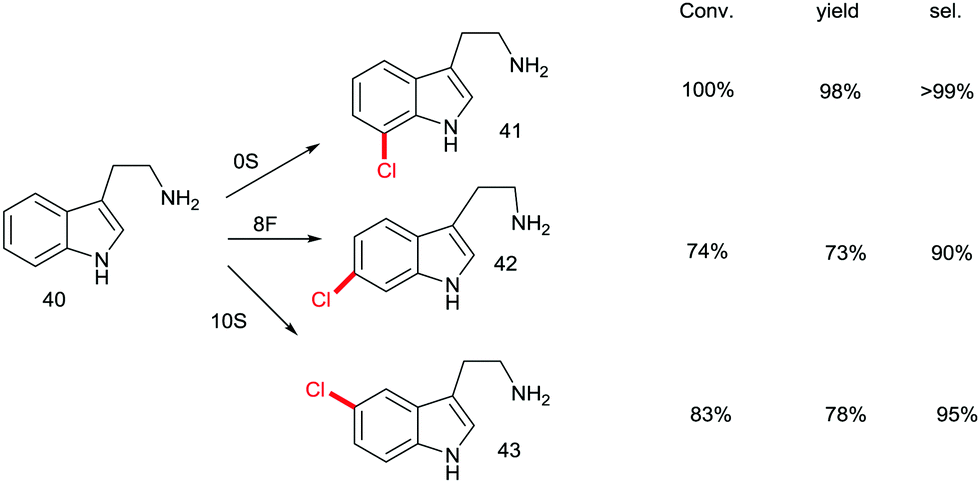 | ||
| Scheme 14 Site-selective chlorination of tryptamine (40) catalyzed by RebH mutants OS, 8F and 10S evolved by epPCR, saturation mutagenesis and iterative saturation mutagenesis (ISM), respectively.58 | ||
Directed evolution of flavin-dependent regioselective halogenases is a relatively new research area from which more advances can be expected. It is likely that structure-guided saturation mutagenesis will play the dominant role in future studies. In view of the fact that many therapeutic drugs contain halogens on aromatic residues, this research will continue to play an important role. Hopefully, certain non-flavin-dependent halogenases can also be engineered for site-selectivity. The site-selective introduction of halogen in aromatic compounds provides pivotal intermediates for subsequent elaboration by Pd-catalyzed C–C, C–O, or C–N bond formation with the emergence of a wide range of new pharmaceutically interesting derivatives.55c In a recently reported technique, this concept was nicely implemented by developing membrane compartmentalization in a two-step cascade sequence requiring no intermediate workup.55c Directed evolution of fluorinases50 for manipulating site-selectivity remains to be studied.
Engineering site-selectivity of Baeyer–Villiger monooxygenases
The Baeyer–Villiger reaction of an unsymmetrical ketone 44 with a peroxy reagent of the type ROOH or RCO3H can produce two different constitutionally isomeric esters 46 and/or 48, site-selective σ-bond migration being determined by the relative migratory tendency of the groups R1 and R2 at the stage of the Criegee-intermediates 45/47 (Scheme 15).59 For maximal activity the well-known stereoelectronic effect requiring an anti-periplanar arrangement must be ensured, as underscored by several QM studies.60 Those groups that stabilize partial positive charge best in the fragmentation of the Criegee intermediate define the σ-bond that has the highest migratory tendency, e.g., a benzyl group is favored over a methyl group.59,60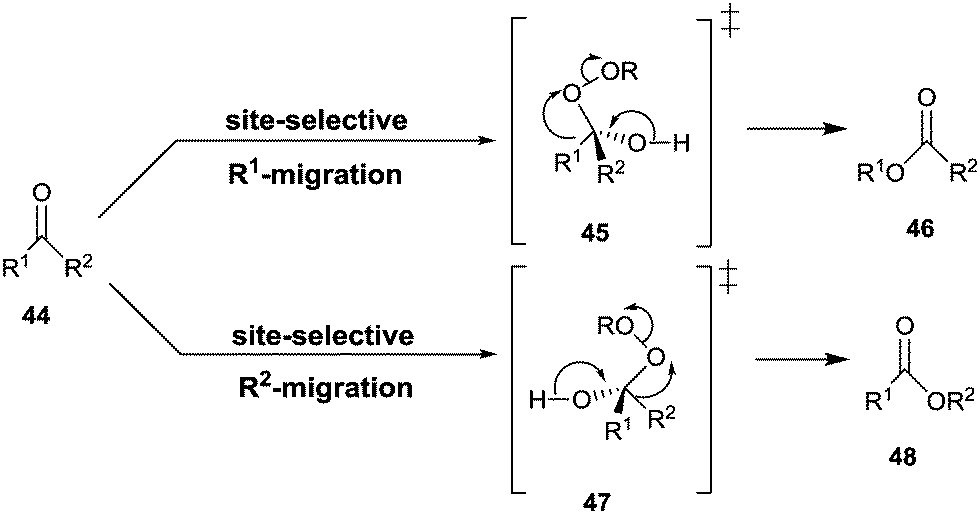 | ||
| Scheme 15 Regioselective σ-bond migration in the Baeyer–Villiger reaction of ketones 44. | ||
In synthetic organic chemistry many reagents and catalysts have been developed for Baeyer–Villiger reactions,59 including chiral catalysts that lead to enantiomerically pure or enriched products in the desymmetrization of prochiral ketones such as 3-substituted cyclobutanones or 4-substituted cyclohexanones.61 In such processes a special case of site-selectivity is involved. The two migratory groups, although identical in the isolated ketone, are different in the chiral environment of the catalyst at the stage of the Criegee-intermediate. Oxidative kinetic resolution of racemic ketones is also possible. Currently, the Feng-system using man-made catalysts appears to be the most efficient approach in both types of stereoselective transformations.61g,h
While this research is ongoing, Baeyer–Villiger monooxygenases (BVMOs) constitute an attractive enzymatic alternative.62 Indeed, they have a notably broader substrate range than synthetic catalysts. BVMOs are flavin-dependent, dioxygen in air reacting to form the actual oxidant FAD-OOH in close vicinity to the binding pocket of the enzyme. Directed evolution has been exploited a number of times in order to enhance or invert enantioselectivity of BVMOs.62–64 In the first reported case, desymmetrization of 4-hydroxycyclohexanone by WT cyclohexanone monooxygenase (CHMO) from Acinetobacter sp. NCIMB 9871 was found to proceed with poor enantioselectivity amounting to only 9% (R), while epPCR provided a mutant F432S which reversed stereoselectivity to 79% ee (S).63a Notice once more that the two possible migrating groups are sterically and electronically identical, but this changes in the chiral environment of the enzyme's binding pocket so that site-selective σ-bond migration becomes possible (Scheme 16). At the time, this was a formal conjecture, which was later studied in detail by theoretical analyses. Accordingly, QM/MM studies of 4-methyl- and 4-hydroxy-cyclohexanone provided mechanistic details of BVMOs, showing that deprotonated flavin hydroperoxide (FAD-OO−) and the anionic form of Criegee intermediates are involved.65 Importantly, mutant F432S proved to be effective in the desymmetrization of a number of structurally different ketones without the need to perform additional mutagenesis experiments (Table 2).66 In the kinetic resolution of a racemic ketone reported in an earlier study, epPCR was also applied,63b although subsequently researchers turned away from “blind” directed evolution and opted for structure-guided generation of focused saturation mutagenesis libraries.64
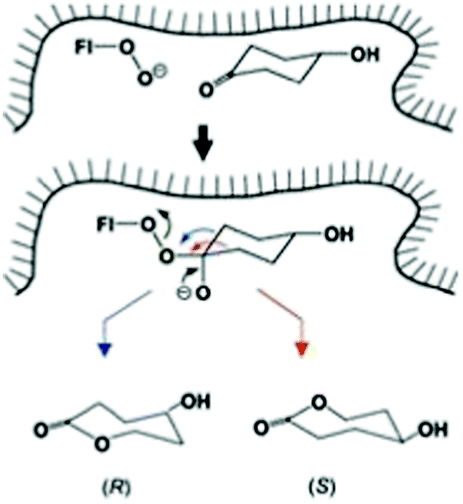 | ||
| Scheme 16 Schematic representation of site-selective migration of two enantiotopic alkyl groups in 4-hydroxycyclohexanone in the binding pocket of CHMO leading to enantiomeric products.63a Blue arrow correlates with the (R)-enantiomer, red arrow with the (S)-antipode. | ||
| Substrate | % ee |
|---|---|
|
94 |
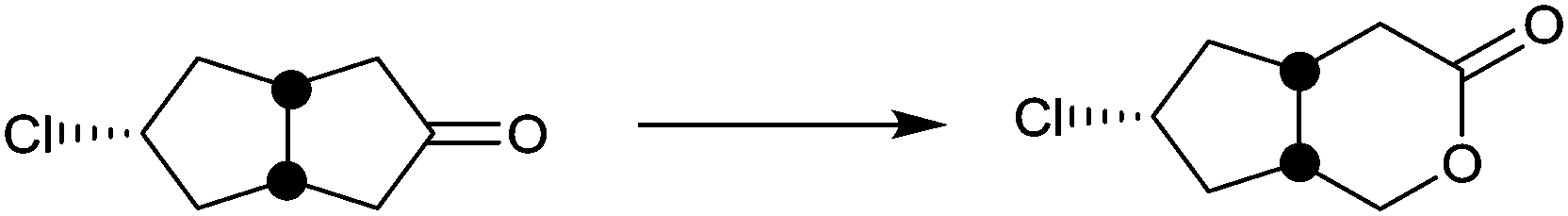
|
99 |
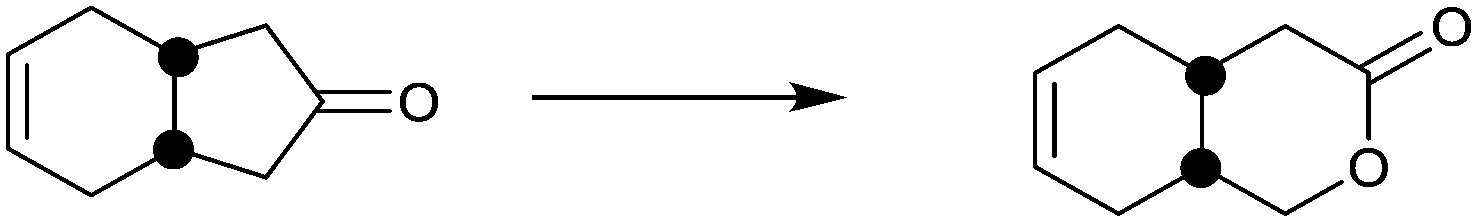
|
91 |

|
97 |
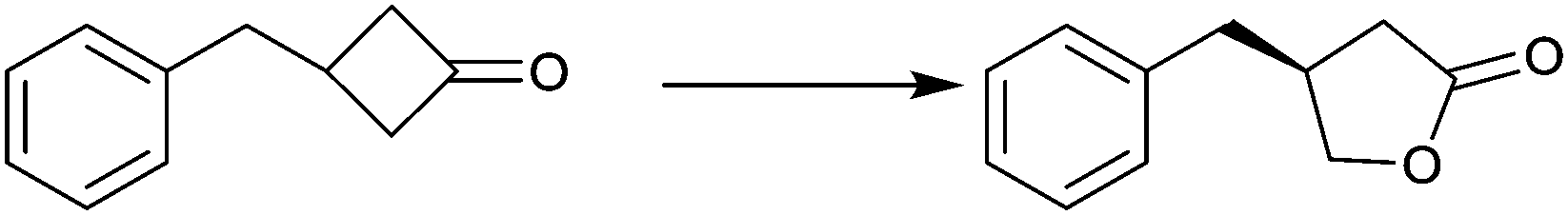
|
78 |

|
96 |
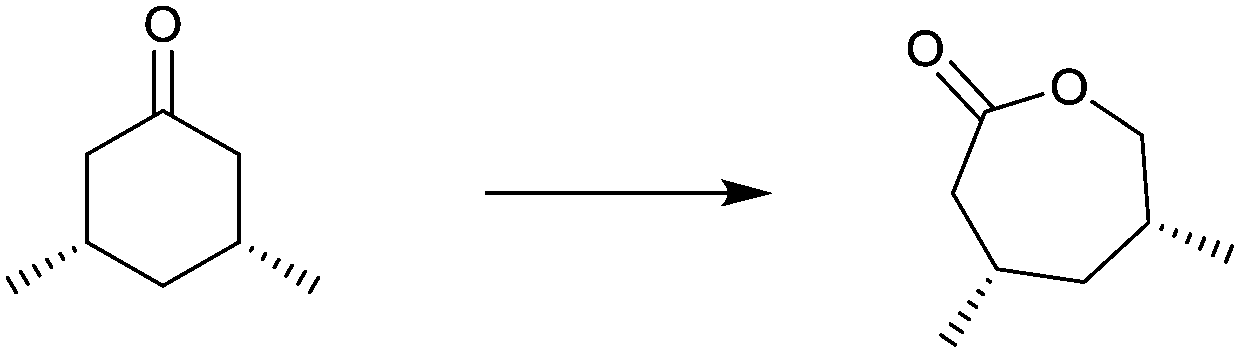
|
>99 |
|
>99 |

|
>99 |

|
99 |
In spite of the impressive performance of CHMO variant F432S (Table 2), the system has limitations, including the insufficient robustness of this BVMO. Moreover, a more rational approach to directed evolution based on crystal structural data was necessary. The first step in this direction was undertaken by a structure-guided approach based on the use of CASTing in combination with a reduced amino acid alphabet, the BVMO in this case being cyclopentanone monooxgenase (CPMO).67 A homology model was first generated using the X-ray data of phenyl acetone monooxygenase (PAMO), a thermostable BVMO.68 On this basis, four CAST residues F156, G157, G449 and F450 in direct neighborhood to the modeled Criegee intermediate in CPMO were identified as putative hot spots. Two saturation mutagenesis libraries at randomization sites F156/G157 (site A) and G449/F450 (site B) were generated using NDT codon degeneracy, which encodes 12 amino acids as a structurally balanced set of combinatorial building blocks (Phe, Leu, Ile, Val, Tyr, His, Asn, Asp, Cys, Arg, Ser, and Gly).67 This was the first case of using a reduced amino acid alphabet in directed evolution of stereoselective enzymes. At the time only mini-libraries were screened amounting to only 150 transformants, three substrates being considered, namely 4-methyl-, 4-acetoxy- and 4-tert-butylcyclohexanone. Active mutants were found only for 4-methyl- and 4-acetoxycyclohexanone, the best enantioselectivities amounting to 92% ee (R) and 90% ee (S) for these two substrates, respectively. This compares well with the poor performance of WT CPMO 46% ee (R) and 5% ee (S), respectively.67
These early results suggested that structure-guided CASTing constitutes the preferred mutagenesis strategy in the directed evolution of site-selective BVMOs leading to high stereoselectivity, a conclusion that was supported by subsequent studies using the thermostable PAMO.69 In some cases CASTing was supported by a bioinformatics technique by aligning the DNA sequence of eight BVMOs in a loop region near the active site in order to identify amino acids to be used as building blocks in focused saturation mutagenesis.69a,b In a different study, an alternative strategy was successfully tested, namely structure-inspired subdomain exchanges,69c but this interesting approach has not been generalized to date.
Site-selective σ-bond migration in Baeyer–Villiger reactions can also lead to cis/trans-isomers in a new type of selective olefin synthesis, an unusual phenomenon observed using CHMO as the catalyst in the reaction of ketone 49 (Scheme 17).70 This kind of selectivity had not been addressed previously in Baeyer–Villiger reactions catalyzed by synthetic reagents or catalysts. The trans- and cis-isomers of 51 can be expected to be thermodynamically essentially identical, which underscores the challenge in inverting site-selectivity.
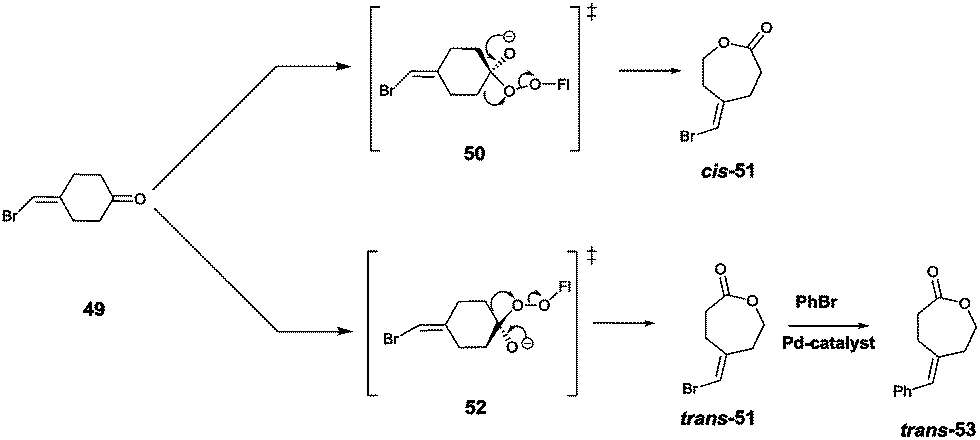 | ||
| Scheme 17 Site-selective σ-bond migration in the Baeyer–Villiger reaction of ketone 49 leading to cis- and trans-configurated olefins Z-51 and E-51via transition states 50 and 52, respectively.70 | ||
The discovery that WT CHMO is 99% trans-selective in favor of olefin trans-51 was somewhat surprising.70 Transition state 52 rather than 50 is involved. This result was exploited synthetically by performing a Pd-catalyzed Suzuki reaction of trans-51 with exclusive formation of the respective phenyl derivative trans-53 (Scheme 17). The difficult problem of inverting trans-selectivity was solved by applying CAST/ISM.70 Three CAST randomization sites A (F432/T433), B (L143) and C (F505) were chosen for saturation mutagenesis guided by the crystal structure of a homologous CHMO which had just become available at the time.71 These constitute rough guesses where steric clashes may occur, the limited number of chosen CAST sites minimizing the screening effort. In the case of 2-residue site A, NDT codon degeneracy encoding 12 amino acids was chosen, while the standard NNK (encoding 20 amino acids) was used for libraries B and C. Pathway A → B → C provided the quadruple mutant F432I/T433G/L143M/F505C showing the best reversed product ratio trans-51:cis-51 = 18:82. In separate follow-up reactions, both trans-51 and cis-51 were subjected to Pd-catalyzed carbonylation with selective formation of new trans- and cis-alkenes.70
An even more challenging task is to evolve BVMO mutants that invert the natural migratory tendency of σ-bonds in Baeyer–Villiger reactions with formation of so-called “abnormal” products. Very few exceptions to the general rule have been noted using synthetic reagents or catalysts, these examples generally involving unusual structural features of the starting ketones.59,72 In the case of BVMOs, exceptions are also known, but here again built-in bias due to the structural characteristics of the ketone generally plays the dominant role, as in the case of some strained bicyclic substrates62 or certain β-hydroxy- or β-aminoketones.73 Thus, it would be an outstanding achievement if the abnormal product were to be formed when reacting such substrates as phenyl acetone or 2-alkylcyclohexanones. To date this ambitious goal has not been reached. However, by screening a collection of different BVMOs, it was discovered that the reaction of 2-butanone provided mainly methyl propanoate as the abnormal product,74 an encouraging sign that general reversal by means of directed evolution could become reality in future studies.
An unusual switch in site-selective σ-bond migration was recently achieved by applying protein engineering to the CHMO from Arthrobacter sp. as catalyst in the Baeyer–Villiger oxidation of (+)-trans-dihydrocarvone (54) (Scheme 18).75 Previously it had been reported that WT CHMO-Arthro delivers not the expected normal product 56, but exclusively the abnormal lactone 55, while the enantiomeric substrate (−)-trans-dihydrocarvone leads solely to the normal product.76 It should be noted that reactions of both enantiomers catalyzed by a synthetic catalyst comprising a Sn-zeolite lead solely to the normal product.77 The goal of the recent biocatalysis study was to induce the BVMO to catalyze formation of the normal product 56, which was accomplished by a combination of directed evolution and rational design. X-ray structural data of open and closed CHMOs,78 homology modeling of the closed and rotated enzyme conformations, and consideration of the BVMO mechanism62,79 were used as a guide in this endeavor.75 Then 16 residues most likely to be responsible for placing the substrate in the binding pocket for smooth reactions to occur were considered for alanine scanning. Formation and screening of 13 mutants led to the identification of variant F299A, which was 59% selective for the normal product 56 (Scheme 18). Instead of performing saturation mutagenesis at this obvious hot spot, the authors preferred to pursue the search for better mutants by rational design. One of the best variants proved to be F299A/F330A/F485A, which is 99% selective in favor of the normal product 56.75
 | ||
| Scheme 18 CHMO-Arthro catalyzed Baeyer–Villiger oxidation of (+)-trans-dihydrocarvone (54) with formation of the formal “abnormal” product 55 generated by the WT enzyme and normal product 56 provided by an evolved mutant thereof.75 | ||
This study nicely demonstrates that the combination of directed evolution and rational design, both guided by X-ray structural data and mechanistic knowledge, can lead to a complete switch in regioselectivity of a BVMO.75 Although in this particular case the WT enzyme favors the abnormal product, for reasons that are not well understood, and the engineered variant delivers the normal lactone, important mechanistic lessons were learned.
Conclusions
Enzymes are ideally suited for achieving high site-selectivity in many types of transformations of interest to organic chemists, but unfortunately a given wild-type (WT) often does not show the catalytic profile that the researcher aims for. In these cases directed evolution offers an optimal means to solve such problems. It has been shown that essentially any mutagenesis technique can be successful, including epPCR, DNA shuffling and saturation mutagenesis.18 However, since the labor-intensive bottleneck is the screening step, much effort has gone into methodology development aimed at ensuring small and highest-quality mutant libraries which require minimal screening.18,20,21,23,29 The last 10 years have shown that structure-guided saturation mutagenesis at sites lining the binding pocket constitutes the method of choice, often in an iterative manner (CAST/ISM). However, epPCR-based random mutagenesis can also lead to mutations near the binding pocket, but also at remote sites which likewise influence activity as well as regio- and stereoselectivity.18 For this reason a new technique was recently developed in which ePCR and CASTing are performed simultaneously in a single one-step molecular biological process.80 Applying epPCR and saturation mutagenesis sequentially is well known,18 but the simultaneous technique may prove to be superior. It remains to be seen how beneficial this approach is in evolving site-selectivity of enzymes in general.Structure guided saturation mutagenesis is illustrated and analyzed in the present review by featuring representative examples encompassing cytochrome P450 monooxygenases, halogenases and Baeyer–Villiger monooxygenases as catalysts in regioselective (site-selective) transformations. Site-selectivity has also been engineered using other types of enzymes;18 recent publications reporting regioselectivity include glycoside hydrolase in potential universal blood formation,81a glycosidase in vaccine production81b or in 2-O-D-glucopyranosyl-L-ascorbic acid formation,81c tetralose phosphorylase in β-galactose-1-phosphate formation,81d sugar invertase in the production of D-psicose from sucrose,81e sialyltransferase in the formation of α-2,3- and α-2,6-sialyl human milk oligosaccharides,81f nitroreductase in mono-reduction of dinitro compounds,81g P450-induced nitration81h and desaturase for introducing olefinic functions in fatty acids.81i Researchers wanting to engineer biocatalytic site-selectivity in future work, possibly involving simultaneous stereoselectivity, can profit from computational techniques82 and from the experience gained in the directed evolution of the three types of enzymes discussed in the present review.
Acknowledgements
We thank the Max-Planck-Society for generous financial support. Open Access funding provided by the Max Planck Society.References
- (a) P. G. M. Wuts, Greene's Protective Groups in Organic Synthesis, Wiley-VCH, Weinheim, 5th edn, 2014 Search PubMed; (b) P. J. Kocienski, Protecting Groups, Thieme, 3rd edn, 2005 Search PubMed.
- T. Pathak and H. Waldmann, Curr. Opin. Chem. Biol., 1998, 2, 112–120 CrossRef CAS PubMed.
- (a) B. M. Trost, Science, 1991, 254, 1471–1477 CAS; (b) P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40–49 CrossRef CAS PubMed; (c) N. Z. Burns, P. S. Baran and R. W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 2854–2867 CrossRef CAS PubMed.
- (a) A. Goswami and J. D. Stewart, Organic Synthesis Using Biocatalysis, Elsevier, 1st edn, 2015 Search PubMed; (b) K. Drauz, H. Gröger and O. May, Enzyme Catalysis in Organic Synthesis, John Wiley-VCH, Weinheim, 3rd edn, 2012 Search PubMed; (c) K. Faber, Biotransformations in Organic Chemistry, Springer, Heidelberg, 6th edn, 2011 Search PubMed; (d) A. Liese, K. Seelbach and C. Wandrey, Industrial Biotransformations, Wiley-VCH, Weinheim, 2006 Search PubMed.
- (a) M. T. Reetz, J. Am. Chem. Soc., 2013, 135, 12480–12496 CrossRef CAS PubMed; (b) N. J. Turner and E. O'Reilly, Nat. Chem. Biol., 2013, 9, 285–288 CrossRef CAS PubMed.
- (a) R. Sheldon, Green Chem., 2017, 19, 18–43 RSC; (b) Y. Ni, D. H. Holtmann and F. Hollmann, ChemCatChem, 2014, 6, 930–943 CrossRef CAS.
- (a) Biocatalysis in the Pharmaceutical and Biotechnology Industries, ed. R. N. Patel, CRC Press, 2006 Search PubMed; (b) R. N. Patel, Expert Opin. Drug Discovery, 2008, 2, 187–245 CrossRef PubMed; (c) A. Bezborodov and N. Zagustina, Appl. Biochem. Microbiol., 2016, 52, 237–249 CrossRef CAS.
- G. Carrea, R. Bovara, R. Longhi and S. Riva, Enzyme Microb. Technol., 1985, 7, 597–600 CrossRef CAS.
- J. A. Hogg, Steroids, 1992, 57, 593–616 CrossRef CAS PubMed.
- Reviews of CYPs: (a) P. R. Ortiz de Montellano, Cytochrome P450: Structure, Mechanism, and Biochemistry, Springer, Berlin, 3rd edn, 2005 Search PubMed; (b) E. M. Isin and F. P. Guengerich, Biochim. Biophys. Acta, Gen. Subj., 2007, 1770, 314–329 CrossRef CAS PubMed; (c) A. W. Munro, H. M. Girvan and K. J. McLean, Nat. Prod. Rep., 2007, 24, 585–609 RSC; (d) P. R. Ortiz de Montellano, Chem. Rev., 2010, 110, 932–948 CrossRef CAS PubMed; (e) S. Shaik, S. Cohen, Y. Wang, H. Chen, D. Kumar and W. Thiel, Chem. Rev., 2010, 110, 949–1017 CrossRef CAS PubMed; (f) C. J. C. Whitehouse, S. G. Bell and L.-L. Wong, Chem. Soc. Rev., 2012, 41, 1218–1260 RSC; (g) F. P. Guengrich, M. Egli and M. R. Waterman, Trends Pharmacol. Sci., 2016, 37, 625–640 Search PubMed.
- R. B. Woodward, F. Sondheimer, D. Taub, K. Heusler and W. McLamore, J. Am. Chem. Soc., 1952, 74, 4223–4251 CrossRef CAS.
- C. A. Roessner, J. B. Spencer, N. J. Stolowich, J. Wang, G. P. Nayar, P. J. Santander, C. Pichon, C. Min, M. T. Holderman and A. I. Scott, Chem. Biol., 1994, 1, 119–124 CrossRef CAS PubMed.
- K. C. Nicolaou, D. Vourloumis, N. Wissinger and P. S. Baran, Angew. Chem., Int. Ed., 2000, 39, 44–122 CrossRef CAS PubMed.
- J.-H. Martens, H. Barg, M. Warren and D. Jahn, Appl. Microbiol. Biotechnol., 2002, 58, 275–285 CrossRef CAS PubMed.
- D. Kadereit and H. Waldmann, Chem. Rev., 2001, 101, 3367–3396 CrossRef CAS PubMed.
- (a) W. J. Hennen, H. M. Sweers, Y. F. Wang and C. H. Wong, J. Org. Chem., 1988, 53, 4939–4945 CrossRef CAS; (b) T. Horrobin, C. H. Tran and D. Crout, J. Chem. Soc., Perkin Trans. 1, 1998, 1069–1080 RSC.
- Regioselective introduction and removal of protective groups in carbohydrate chemistry: (a) M. Filice, J. M. Guisan and J. M. Palomo, Curr. Org. Chem., 2010, 14, 516–532 CrossRef CAS; (b) G. J. Williams, R. W. Gantt and J. S. Thorson, Curr. Opin. Chem. Biol., 2008, 12, 556–564 CrossRef CAS PubMed; (c) B. La Ferla, Lipases as useful tools for the stereo- and regioselective protection and deprotection of carbohydrates, in Timely Perspectives in Carbohydrate Chemistry, ed. W. Schmid and A. Stütz, Springer-Verlag, 2002 Search PubMed.
- Recent reviews of directed evolution: (a) M. T. Reetz, Directed Evolution of Selective Enzymes: Catalysts for Organic Chemistry and Biotechnology, Wiley-VCH, Weinheim, 2016 Search PubMed; (b) A. S. Bommarius, Annu. Rev. Chem. Biomol. Eng., 2015, 6, 319–345 CrossRef CAS PubMed; (c) A. Currin, N. Swainston, P. J. Day and D. B. Kell, Chem. Soc. Rev., 2015, 44, 1172–1239 RSC; (d) C. A. Denard, H. Ren and H. Zhao, Curr. Opin. Chem. Biol., 2015, 25, 55–64 CrossRef CAS PubMed; (e) E. M. J. Gillam, J. N. Copp and D. F. Ackerley, in Methods in Molecular Biology, Vol. 1179: Directed Evolution Library Creation: Methods and Protocols, ed. E. M. J. Gillam, J. N. Copp and D. F. Ackerley, Humana, Totowa, 2nd edn, 2014 Search PubMed; (f) M. Goldsmith and D. S. Tawfik, Methods Enzymol., 2013, 523, 257–283 CAS; (g) E. M. Brustad and F. H. Arnold, Curr. Opin. Chem. Biol., 2011, 15, 201–210 CrossRef CAS PubMed; (h) C. Jäckel and D. Hilvert, Curr. Opin. Biotechnol., 2010, 21, 753–759 CrossRef PubMed; (i) N. J. Turner, Nat. Chem. Biol., 2009, 5, 567–574 CrossRef CAS PubMed; (j) S. Lutz and U. T. Bornscheuer, Protein Engineering Handbook, Wiley-VCH, Weinheim, 2009 Search PubMed.
- M. T. Reetz, A. Zonta, K. Schimossek, K. E. Jaeger and K. Liebeton, Angew. Chem., Int. Ed., 1997, 36, 2830–2832 CrossRef CAS.
- M. T. Reetz, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5716–5722 CrossRef CAS PubMed.
- S. Lutz and W. M. Patrick, Curr. Opin. Biotechnol., 2004, 15, 291–297 CrossRef CAS PubMed.
- (a) M. R. Parikh and I. Matsumura, J. Mol. Biol., 2005, 352, 621–628 CrossRef CAS PubMed; (b) M. T. Reetz, S. Prasad, J. D. Carballeira, Y. Gumulya and M. Bocola, J. Am. Chem. Soc., 2010, 132, 9144–9152 CrossRef CAS PubMed.
- (a) M. T. Reetz, Angew. Chem., Int. Ed., 2011, 50, 138–174 CrossRef CAS PubMed; (b) C. G. Acevedo-Rocha, S. Hoebenreich and M. T. Reetz, Directed Evolution Library Creation: Methods and Protocols, 2014, pp. 103–128 Search PubMed.
- M. T. Reetz, S. Wilensek, D. Zha and K. E. Jaeger, Angew. Chem., Int. Ed., 2001, 40, 3589–3591 CrossRef CAS PubMed.
- M. T. Reetz, M. Bocola, J. D. Carballeira, D. Zha and A. Vogel, Angew. Chem., Int. Ed., 2005, 44, 4192–4196 CrossRef CAS PubMed.
- W. M. Patrick and A. E. Firth, Biomol. Eng., 2005, 22, 105–112 CrossRef CAS PubMed.
- (a) J.-L. Reymond, Enzyme Assays, John Wiley & Sons, 2006 Search PubMed; (b) C. G. Acevedo-Rocha, R. Agudo and M. T. Reetz, J. Biotechnol., 2014, 191, 3–10 CrossRef CAS PubMed.
- ISM as a new concept for engineering enzyme catalysis: (a) M. T. Reetz, Max-Planck-Society Yearbook, 2005, pp. 327–331 Search PubMed; (b) M. T. Reetz, L. W. Wang and M. Bocola, Angew. Chem., Int. Ed., 2006, 45, 1236–1241 CrossRef CAS PubMed; (c) M. T. Reetz and J. D. Carballeira, Nat. Protoc., 2007, 2, 891–903 CrossRef CAS PubMed; (d) Enhancing the affinity of ligands to a protein (hormone receptor) by using the best mutant of a saturation mutagenesis library as the template for randomization at another site: K. Chockalingam, Z. Chen, J. A. Katzenellenborgen and H. Zhao, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5691–5696 CrossRef CAS PubMed.
- (a) Z. Sun, R. Lonsdale, L. Wu, G. Li, A. Li, J. Wang, J. Zhou and M. T. Reetz, ACS Catal., 2016, 6, 1590–1597 CrossRef CAS; (b) Z. Sun, R. Lonsdale, A. Ilie, G. Li, J. Zhou and M. T. Reetz, ACS Catal., 2016, 6, 1598–1605 CrossRef CAS.
- Reviews of directed evolution of CYPs:10f (a) S. Kumar and J. R. Halpert, Biochem. Biophys. Res. Commun., 2005, 338, 456–464 CrossRef CAS PubMed; (b) G. Grogan, Curr. Opin. Chem. Biol., 2011, 15, 241–248 CrossRef CAS PubMed; (c) G.-D. Roiban and M. T. Reetz, Chem. Commun., 2015, 51, 2208–2224 RSC.
- Examples of rational design of CYP mutants:36 (a) P. J. Loida and S. G. Sligar, Biochemistry, 1993, 32, 11530–11538 CrossRef CAS PubMed; (b) J. Reinen, G. Vredenburg, K. Klaering, N. P. Vermeulen, J. N. Commandeur, M. Honing and J. C. Vos, J. Mol. Catal. B: Enzym., 2015, 121, 64–74 CrossRef CAS; (c) I. K. Jóźwik, F. M. Kiss, Ł. Gricman, A. Abdulmughni, E. Brill, J. Zapp, J. Pleiss, R. Bernhardt and A. M. W. Thunnissen, FEBS J., 2016, 283, 4128–4148 CrossRef PubMed; (d) E. Roduner, W. Kaim, B. Sarkar, V. B. Urlacher, J. Pleiss, R. Gläser, W. D. Einicke, G. A. Sprenger, U. Beifuß and E. Klemm, ChemCatChem, 2013, 5, 82–112 CrossRef CAS; (e) M. Dietrich, T. A. Do, R. D. Schmid, J. Pleiss and V. B. Urlacher, J. Biotechnol., 2009, 139, 115–117 CrossRef CAS PubMed; (f) X. Liu, Z.-B. Wang, Y.-N. Wang and J.-Q. Kong, Molecules, 2016, 21, 760 CrossRef PubMed; (g) A. Eichler, L. Gricman, S. Herter, P. P. Kelly, N. J. Turner, J. Pleiss and S. L. Flitsch, ChemBioChem, 2016, 17, 426–432 CrossRef CAS PubMed.
- P. J. Loida and S. G. Sligar, Protein Eng., 1993, 6, 207–212 CrossRef CAS PubMed.
- S. G. Bell, X. Chen, R. J. Sowden, F. Xu, J. N. Williams, L.-L. Wong and Z. Rao, J. Am. Chem. Soc., 2003, 125, 705–714 CrossRef CAS PubMed.
- I. Schlichting, J. Berendzen, K. Chu, A. M. Stock, S. A. Maves, D. E. Benson, R. M. Sweet, D. Ringe, G. A. Petsko and S. G. Sligar, Science, 2000, 287, 1615–1622 CrossRef CAS PubMed.
- (a) A. Glieder, E. T. Farinas and F. H. Arnold, Nat. Biotechnol., 2002, 20, 1135–1139 CrossRef CAS PubMed; (b) M. W. Peters, P. Meinhold, A. Glieder and F. H. Arnold, J. Am. Chem. Soc., 2003, 125, 13442–13450 CrossRef CAS PubMed.
- (a) P. Meinhold, M. W. Peters, A. Hartwick, A. R. Hernandez and F. H. Arnold, Adv. Synth. Catal., 2006, 348, 763–772 CrossRef CAS; (b) B. M. van Vugt-Lussenburg, M. C. Damsten, D. M. Maasdijk, N. P. Vermeulen and J. N. Commandeur, Biochem. Biophys. Res. Commun., 2006, 346, 810–818 CrossRef CAS PubMed.
- M. Landwehr, L. Hochrein, C. R. Otey, A. Kasrayan, J.-E. Bäckvall and F. H. Arnold, J. Am. Chem. Soc., 2006, 128, 6058–6059 CrossRef CAS PubMed.
- C. J. Whitehouse, S. G. Bell, H. G. Tufton, R. J. Kenny, L. C. Ogilvie and L.-L. Wong, Chem. Commun., 2008, 966–968 RSC.
- W. L. Tang, Z. Li and H. Zhao, Chem. Commun., 2010, 46, 5461–5463 RSC.
- S. Q. Pham, G. Pompidor, J. Liu, X.-D. Li and Z. Li, Chem. Commun., 2012, 48, 4618–4620 RSC.
- R. Agudo, G. D. Roiban and M. T. Reetz, ChemBioChem, 2012, 13, 1465–1473 CrossRef CAS PubMed.
- (a) F. P. Guengerich, Drug Metab. Rev., 2004, 36, 159–197 CrossRef CAS PubMed; (b) F. P. Guengerich, Nat. Rev., 2002, 1, 359–366 CAS.
- (a) S. Kille, F. E. Zilly, J. P. Acevedo and M. T. Reetz, Nat. Chem., 2011, 3, 738–743 CrossRef CAS PubMed; (b) C. Ritter, N. Nett, C. G. Acevedo-Rocha, R. Lonsdale, K. Kräling, F. Dempwolff, S. Höbenreich, P. L. Graumann, M. T. Reetz and E. Meggers, Angew. Chem., Int. Ed., 2015, 54, 13440–13443 CrossRef CAS PubMed; (c) K. T. Nguyen, C. Virus, N. Günnewich, F. Hannemann and R. Bernhardt, ChemBioChem, 2012, 13, 1161–1166 CrossRef CAS PubMed; (d) M. Geier, A. Braun, P. Fladischer, P. Stepniak, F. Rudroff, C. Hametner, M. D. Mihovilovic and A. Glieder, FEBS J., 2013, 280, 3094–3108 CrossRef CAS PubMed.
- (a) Y. Yang, J. Liu and Z. Li, Angew. Chem., Int. Ed., 2014, 53, 3120–3124 CrossRef CAS PubMed; (b) K. Zhang, B. M. Shafer, M. D. Demars, H. A. Stern and R. Fasan, J. Am. Chem. Soc., 2012, 134, 18695–18704 CrossRef CAS PubMed.
- I. Kaluzna, A. De Vries and D. Mink, Chimi. Oggi. Chem. Today, 2015, 33, 2 Search PubMed.
- (a) R. Lonsdale, J. N. Harvey and A. J. Mulholland, J. Phys. Chem. Lett., 2010, 1, 3232–3237 CrossRef CAS; (b) R. Lonsdale, J. N. Harvey and A. J. Mulholland, J. Phys. Chem. B, 2010, 114, 1156–1162 CrossRef CAS PubMed.
- (a) E. W. Svastits, J. D. Dawson, R. Breslow and S. H. Gellman, J. Am. Chem. Soc., 1985, 107, 6427–6428 CrossRef CAS; (b) J. A. McIntosh, P. S. Coello, C. C. Farwell, Z. J. Wang, J. C. Lewis, T. R. Brown and F. H. Arnold, Angew. Chem., Int. Ed., 2013, 52, 9309–9312 CrossRef CAS PubMed; (c) R. Singh, M. Bordeaux and R. Fasan, ACS Catal., 2014, 4, 546–552 CrossRef CAS PubMed; (d) R. Singh, J. N. Kolev, R. A. Sutera and R. Fasan, ACS Catal., 2015, 5, 1685–1691 CrossRef CAS PubMed.
- Reviews of halogenases: (a) D. G. Fujimori and C. T. Walsh, Curr. Opin. Chem. Biol., 2007, 11, 553–560 CrossRef CAS PubMed; (b) X. Chen and V. Pée, Acta Biochim. Biophys. Sin., 2008, 40, 183–193 CrossRef CAS PubMed; (c) L. C. Blasiak and C. L. Drennan, Acc. Chem. Res., 2009, 42, 147–155 CrossRef CAS PubMed; (d) D. R. Smith, S. Grüschow and R. J. Goss, Curr. Chem. Biol., 2013, 17, 276–283 CrossRef CAS PubMed; (e) M. Ayala, L. Segovia and E. Torres, Mini-Rev. Med. Chem., 2016, 16, 1100–1111 CrossRef CAS PubMed; (f) V. Agarwal, Z. D. Miles, J. M. Winter, A. S. Eustáquio, A. A. El Gamal and B. S. Moore, Chem. Rev., 2017 DOI:10.1021/acs.chemrev.6b00571.
- J. Payne, M. Andorfer and J. C. Lewis, Methods Enzymol., 2016, 575, 93–126 CAS.
- (a) C. Dong, F. Huang, H. Deng, C. Schaffrath, J. B. Spencer, D. O'Hagan and J. H. Naismith, Nature, 2004, 427, 561–565 CrossRef CAS PubMed; (b) H. Deng, S. L. Cobb, A. R. McEwan, R. P. McGlinchey, J. H. Naismith, D. O'Hagan, D. A. Robinson and J. B. Spencer, Angew. Chem., Int. Ed., 2006, 45, 759–762 CrossRef CAS PubMed.
- X. Zhu, W. De Laurentis, K. Leang, J. Herrmann, K. Ihlefeld, K.-H. Pee and J. H. Naismith, J. Mol. Biol., 2009, 391, 74–85 CrossRef CAS PubMed.
- M. L. Hillwig, Q. Zhu, K. Ittiamornkul and X. Liu, Angew. Chem., Int. Ed., 2016, 55, 5780–5784 CrossRef CAS PubMed.
- A. Lang, S. Polnick, T. Nicke, P. William, E. P. Patallo, J. H. Naismith and K. H. van Pée, Angew. Chem., Int. Ed., 2011, 50, 2951–2953 CrossRef CAS PubMed.
- S. Brown and S. E. O'Connor, ChemBioChem, 2015, 16, 2129–2135 CrossRef CAS PubMed.
- (a) S. A. Shepherd, C. Karthikeyan, J. Latham, A.-W. Struck, M. L. Thompson, B. R. Menon, M. Q. Styles, C. Levy, D. Leys and J. Micklefield, Chem. Sci., 2015, 6, 3454–3460 RSC; (b) S. A. Shepherd, B. R. Menon, H. Fisk, A. W. Struck, C. Levy, D. Leys and J. Micklefield, ChemBioChem, 2016, 17, 821–824 CrossRef CAS PubMed; (c) J. Latham, J.-M. Henry, H. H. Sharif, B. R. K. Menon, S. A. Sheperd, M. F. Greaney and J. Micklefield, Nat. Chem., 2016, 7, 11873–11880 Search PubMed.
- C. B. Poor, M. C. Andorfer and J. C. Lewis, ChemBioChem, 2014, 15, 1286–1289 CrossRef CAS PubMed.
- J. T. Payne, C. B. Poor and J. C. Lewis, Angew. Chem., Int. Ed., 2015, 54, 4226–4230 CrossRef CAS PubMed.
- M. C. Andorfer, H. J. Park, J. Vergara-Coll and J. C. Lewis, Chem. Sci., 2016, 7, 3720–3729 RSC.
- Reviews of Baeyer–Villiger reactions: (a) G. R. Krow, Org. React., 1993, 43, 251–798 CAS; (b) M. Renz and B. Meunier, Eur. J. Org. Chem., 1999, 737–750 CrossRef CAS; (c) G.-J. ten Brink, I. W. C. E. Arends and R. A. Sheldon, Chem. Rev., 2004, 104, 4105–4123 CrossRef PubMed; (d) C. Jiménez-Sanchidrián and J. R. Ruiz, Tetrahedron, 2008, 64, 2011–2026 CrossRef.
- (a) F. Grein, A. C. Chen, D. Edwards and C. M. Crudden, J. Org. Chem., 2006, 71, 861–872 CrossRef CAS PubMed; (b) L. Reyes, J. R. Alvarez-Idaboy and N. Mora-Diez, J. Phys. Org. Chem., 2009, 22, 643–649 CrossRef CAS; (c) Y. Itoh, M. Yamanaka and K. Mikami, J. Org. Chem., 2013, 78, 146–153 CrossRef CAS PubMed.
- Examples of asymmetric Baeyer–Villiger reactions using chiral synthetic catalysts: (a) C. Bolm, G. Schlingloff and K. Weickhardt, Angew. Chem., Int. Ed., 1994, 33, 1848–1849 CrossRef; (b) G. Strukul, Angew. Chem., Int. Ed., 1998, 37, 1198–1209 CrossRef; (c) A. V. Malkov, F. Friscourt, M. Bell, M. E. Swarbrick and P. Kočovský, J. Org. Chem., 2008, 73, 3996–4003 CrossRef CAS PubMed; (d) Y. Imada, T. Iida, S.-I. Murahashi and T. Naota, Angew. Chem., Int. Ed., 2005, 44, 1704–1706 CrossRef CAS PubMed; (e) G. Peris and S. J. Miller, Org. Lett., 2008, 10, 3049–3052 CrossRef CAS PubMed; (f) S. Xu, Z. Wang, X. Zhang and K. Ding, Angew. Chem., Int. Ed., 2008, 47, 2840–2843 CrossRef CAS PubMed; (g) L. Zhou, X. Liu, J. Ji, Y. Zhang, X. Hu, L. Liu and X. Feng, J. Am. Chem. Soc., 2012, 134, 17023–18026 CrossRef CAS PubMed; (h) L. Zhou, X. Liu, J. Ji, Y. Zhang, W. Wu, Y. Liu, L. Lin and X. Feng, Org. Lett., 2014, 16, 3938–3941 CrossRef CAS PubMed; (i) N. C. Abascal and S. J. Miller, Org. Lett., 2016, 18, 4646–4649 CrossRef CAS PubMed.
- Reviews of BVMOs:64 (a) M. M. Kayser, Tetrahedron, 2009, 65, 947–974 CrossRef CAS; (b) J. D. Stewart, Curr. Org. Chem., 1998, 2, 195–216 CAS; (c) M. D. Mihovilovic, Curr. Org. Chem., 2006, 10, 1265–1287 CrossRef CAS; (d) D. E. Torres PazmiÇo, H. M. Dudek and M. W. Fraaije, Curr. Opin. Chem. Biol., 2010, 14, 138–144 CrossRef PubMed; (e) H. Leisch, K. Morley and P. Lau, Chem. Rev., 2011, 111, 4165–4222 CrossRef CAS PubMed; (f) F. Hollmann, I. W. Arends, K. Buehler, A. Schallmey and B. Bühler, Green Chem., 2011, 13, 226–265 RSC; (g) M. Bucko, P. Gemeiner, A. Schenkmayerova, T. Krajcovic, F. Rudroff and M. D. Mihovilovic, Appl. Microbiol. Biotechnol., 2016, 100, 6585–6599 CrossRef CAS PubMed.
- (a) M. T. Reetz, B. Brunner, T. Schneider, F. Schulz, C. M. Clouthier and M. M. Kayser, Angew. Chem., Int. Ed., 2004, 43, 4075–4078 CrossRef CAS PubMed; (b) A. Kirschner and U. T. Bornscheuer, Appl. Microbiol. Biotechnol., 2008, 81, 465–472 CrossRef CAS PubMed.
- Review of directed evolution of BVMOs: Z. G. Zhang, L. P. Parra and M. T. Reetz, Chem. – Eur. J., 2012, 18, 10160–10172 CrossRef CAS PubMed.
- (a) I. Polyak, M. T. Reetz and W. Thiel, J. Am. Chem. Soc., 2012, 134, 2732–2741 CrossRef CAS PubMed; (b) I. Polyak, M. T. Reetz and W. Thiel, J. Phys. Chem. B, 2013, 117, 4993–5001 CrossRef CAS PubMed.
- M. D. Mihovilovic, F. Rudroff, A. Winninger, T. Schneider, F. Schulz and M. T. Reetz, Org. Lett., 2006, 8, 1221–1224 CrossRef CAS PubMed.
- C. M. Clouthier, M. M. Kayser and M. T. Reetz, J. Org. Chem., 2006, 71, 8431–8437 CrossRef CAS PubMed.
- (a) E. Malito, A. Alfieri, M. W. Fraaije and A. Mattevi, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13157–13162 CrossRef CAS PubMed; (b) C. Rodríguez, G. de Gonzalo, D. E. T. Pazmino, M. W. Fraaije and V. Gotor, Tetrahedron: Asymmetry, 2008, 19, 197–203 CrossRef.
- (a) M. T. Reetz and S. Wu, Chem. Commun., 2008, 5499–5501 RSC; (b) M. T. Reetz and S. Wu, J. Am. Chem. Soc., 2009, 131, 15424–15432 CrossRef CAS PubMed; (c) H. M. Dudek, G. de Gonzalo, D. E. T. Pazmiño, P. Stępniak, L. S. Wyrwicz, L. Rychlewski and M. W. Fraaije, Appl. Environ. Microbiol., 2011, 77, 5730–5738 CrossRef CAS PubMed.
- Z. G. Zhang, G. D. Roiban, J. P. Acevedo, I. Polyak and M. T. Reetz, Adv. Synth. Catal., 2013, 355, 99–106 CrossRef CAS.
- I. A. Mirza, B. J. Yachnin, S. Wang, S. Grosse, H. Bergeron, A. Imura, H. Iwaki, Y. Hasegawa, P. C. Lau and A. M. Berghuis, J. Am. Chem. Soc., 2009, 131, 8848–8854 CrossRef CAS PubMed.
- Y. Itoh, M. Yamanaka and K. Mikami, Org. Lett., 2003, 5, 4803–4806 CrossRef CAS PubMed.
- J. Rehdorf, M. D. Mihovilovic and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2010, 49, 4506–4508 CrossRef CAS PubMed.
- H. L. van Beek, R. T. Winter, G. R. Eastham and M. W. Fraaije, Chem. Commun., 2014, 50, 13034–13036 RSC.
- K. Balke, S. Schmidt, M. Genz and U. T. Bornscheuer, ACS Chem. Biol., 2015, 11, 38–43 CrossRef PubMed.
- P. Černuchová and M. D. Mihovilovic, Org. Biomol. Chem., 2007, 5, 1715–1719 Search PubMed.
- A. Corma, L. T. Nemeth, M. Renz and S. Valencia, Nature, 2001, 412, 423–425 CrossRef CAS PubMed.
- B. J. Yachnin, M. B. McEvoy, R. J. MacCuish, K. L. Morley, P. C. Lau and A. M. Berghuis, ACS Chem. Biol., 2014, 9, 2843–2851 CrossRef CAS PubMed.
- C. C. Ryerson, D. P. Ballou and C. Walsh, Biochemistry, 1982, 21, 2644–2655 CrossRef CAS PubMed.
- J. P. Acevedo, M. T. Reetz, J. A. Asenjo and L. P. Parra, Enzyme Microb. Technol., 2017, 100, 60–70 CrossRef CAS PubMed.
- (a) D. H. Kwan, I. Constantinescu, R. Chapanian, M. A. Higgins, M. P. Kötzler, E. Samain, A. B. Boraston, J. N. Kizhakkedathu and S. G. Withers, J. Am. Chem. Soc., 2015, 137, 5695–5705 CrossRef CAS PubMed; (b) J. Ihssen, J. Haas, M. Kowarik, L. Wiesli, M. Wacker, T. Schwede and L. Thöny-Meyer, Open Biol., 2015, 5, 140227 CrossRef PubMed; (c) R. Han, L. Liu, H.-D. Shin, R. R. Chen, J. Li, G. Du and J. Chen, Appl. Environ. Microbiol., 2013, 79, 7562–7568 CrossRef CAS PubMed; (d) J. van der Borght, C. Chen, L. Hoflack, L. Van Renterghem, T. Desmet and W. Soetart, Appl. Environ. Microbiol., 2011, 77, 6939–6944 CrossRef CAS PubMed; (e) N. Wagner, A. Bosshart, J. Failmezger, M. Bechtold and S. Panke, Angew. Chem., Int. Ed., 2015, 54, 4182–4186 CrossRef CAS PubMed; (f) K. Schmölzer, T. Czabany, C. Luley-Goedl, T. Pavkov-Keller, D. Ribitsch, H. Schab, K. Gruber, H. Weber and B. Nidetzky, Chem. Commun., 2015, 51, 3083–3086 RSC; (g) S. C. Dodani, G. Kiss, J. K. B. Cahn, Y. Su, V. S. Pande and F. H. Arnold, Nat. Chem., 2016, 8, 419–425 CrossRef CAS PubMed; (h) J. Bai, J. Yang, Y. Zhou and Q. Yang, Process Biochem., 2015, 50, 1760–1766 CrossRef CAS; (i) T. Vanhercke, P. Shrestha, A. G. Green and S. P. Singh, J. Biol. Chem., 2011, 286, 12860–12869 CrossRef CAS PubMed.
- (a) X. Feng, J. Sanchis, M. T. Reetz and H. Rabitz, Chem. – Eur. J., 2012, 18, 5646–5654 CrossRef CAS PubMed; (b) D. Quaglia, M. C. C. J. C. Ebert, P. F. Mugford and J. Pelletier, PLoS One, 2017, 12, e0171741 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |