
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemoenzymatic synthesis of polypeptides containing the unnatural amino acid 2-aminoisobutyric acid†
Kousuke
Tsuchiya
* and
Keiji
Numata
*
Enzyme Research Team, Biomass Engineering Research Division, RIKEN Center for Sustainable Resource Science, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan. E-mail: kosuke.tsuchiya@riken.jp; keiji.numata@riken.jp
First published on 5th May 2017
Abstract
Polypeptides containing 2-aminoisobutyric acid (Aib) units as an unnatural amino acid residue were synthesized by papain-catalyzed chemoenzymatic polymerization of a tripeptide ethyl ester L-Ala-Aib-L-Ala-OEt in an aqueous medium. The Aib-containing polypeptide adopted an α-helix conformation in both the solid and solution phases, which was induced by the periodic Aib residue.
Polypeptides are bio-based polymeric materials with various primary structures composed of 20 natural L-amino acids. The structural diversity in polypeptides offers a wide range of physical properties, which makes synthetic polypeptides attractive materials. Integrating unnatural amino acids into the polypeptide backbones enables the fabrication of novel functionalities by altering their structures;1,2 similarly, natural living systems incorporate rare amino acids such as N-methylglycine and selenocysteine into proteins for specific functions.3,4 The introduction of α,α-disubstituted amino acids into polypeptide architectures is a powerful way to control their secondary structures.5,6 2-Aminoisobutyric acid (Aib), one of the unnatural amino acids, is known to be a strong helical promoter when it is incorporated into a polypeptide sequence.7 Oligopeptides containing Aib residues exhibit a strong tendency to form 310- or α-helical secondary structures.8–10
Conventional synthetic methods based on solid phase peptide synthesis can be used to construct sophisticated sequences containing unnatural amino acid residues, although the costly and tedious multistep synthesis limits the attainable chain length and sequence.11 Among the solution syntheses of polypeptides, chemoenzymatic polymerization of amino acid ester derivatives catalyzed by proteases has advantages that include a facile, green synthesis in aqueous media and a more atom-economical process than conventional solution methods that use condensing agents.12 Various amino acid derivatives can be chemoenzymatically converted not only into linear polypeptides13–19 but also into polypeptide architectures with unique shapes such as star-shaped and telechelic-type polypeptides.20–22 Furthermore, some proteases, in particular papain, exhibit a relatively broad substrate specificity, which allows unnatural amino acid derivatives to be involved in chemoenzymatic polymerization. For example, in our previous study, ω-aminoalkanoates, monomer units for synthetic polyamides such as nylon, were successfully copolymerized with diethyl L-glutamate by papain-catalyzed chemoenzymatic polymerization.23 However, the resulting polymer showed quite low nylon contents even using an excess amount of ω-aminoalkanoates in feed, and contaminated with the homopolymer of glutamate monomer. In the present work, we examined the feasibility of polymerizing the non-natural amino acid Aib via chemoenzymatic polymerization using papain. The Aib units were successfully introduced into the polypeptide backbone through the use of an Aib-containing tripeptide ethyl ester as a monomer (Scheme 1).
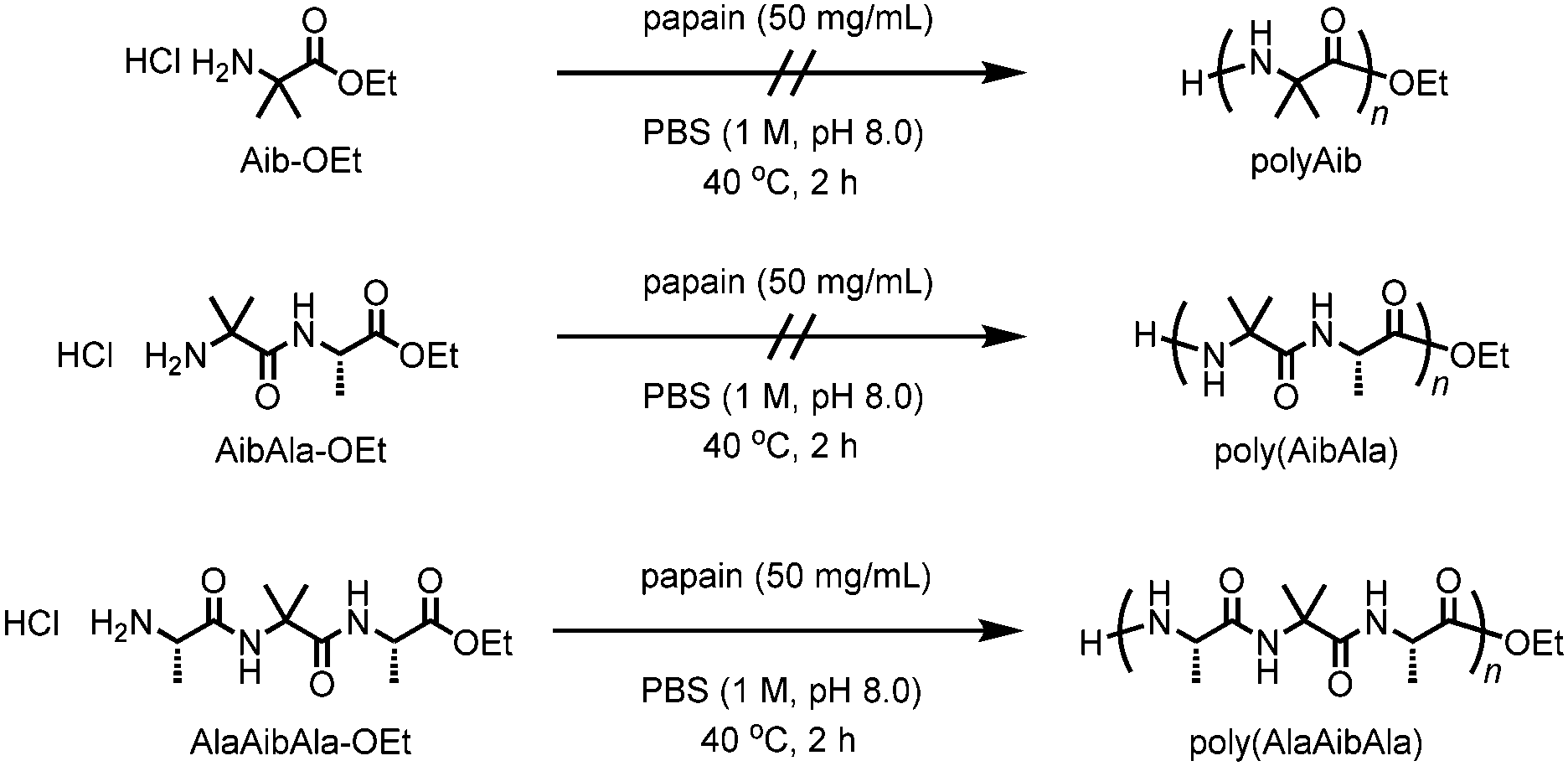 | ||
| Scheme 1 Chemoenzymatic polymerization of Aib-containing monomers using papain. | ||
According to previous studies, papain was selected as a reasonable candidate for the catalysis of Aib polymerization because of its relatively broad substrate selectivity.12 Papain-catalyzed polymerization can produce the polypeptides that not only contain various natural amino acids but also contain unnatural amino acids such as ω-aminoalkanoates.23 The chemoenzymatic polymerization of Aib-OEt was performed using papain in phosphate buffer (1 M, pH 8.0) at 40 °C; the results are listed in Table 1. However, unlike the polymerization of hydrophobic amino acids such as alanine or leucine,15,16 no precipitation occurred after 2 h (run 1). Neither changing the feed concentration of Aib-OEt (0.1 to 2.0 M) nor copolymerization with L-alanine ethyl ester (Ala-OEt) improved the polymerization, resulting in no precipitation. We assumed that the inactivity of Aib-OEt in papain-catalyzed polymerization was due to the lack of an α-proton, which is essential for the recognition of DL isomers.24 Indeed, the polymerization of D-alanine ethyl ester (D-Ala-OEt) did not proceed in the presence of papain. Therefore, we designed dipeptide and tripeptide ethyl esters containing an Aib unit: Aib-L-Ala ethyl ester (AibAla-OEt) and L-Ala-Aib-L-Ala ethyl ester (AlaAibAla-OEt), respectively. Dipeptide esters are suitable substrates in protease-catalyzed polymerization to afford alternating copolypeptides.25–27 AibAla-OEt and AlaAibAla-OEt were synthesized as the HCl salts by the conventional solution method.8 No polymerization of AibAla-OEt proceeded (run 2), whereas the copolymerization of AibAla-OEt with Ala-OEt afforded a precipitate in low yield (run 4). In contrast, the polymerization of AlaAibAla-OEt afforded a precipitate in moderate yields (run 5 to 8), which was maximized at a feed concentration of 0.25 M (run 6). Time course study of the chemoenzymatic polymerization of AlaAibAla-OEt was also performed to investigate the kinetics as shown in Fig. S4 (ESI†). The monomer conversion increased up to 44% within 1 h, showing a tendency similar to the chemoenzymatic polymerization of natural amino acid monomers.19,21 Among the monomers used, only AlaAibAla-OEt could be homopolymerized by papain to afford poly(AlaAibAla), indicating that the L-Ala unit is favorable for recognition by papain in the chemoenzymatic polymerization. High concentrations of AlaAibAla-OEt deteriorated the polymerization efficiency, and no polypeptide was obtained at feed concentration of 1.0 M or greater (run 9).
| Run | Monomer | Conc. (M) | Yieldb (%) | Aib contentc (mol%) |
|---|---|---|---|---|
| a Polymerization was carried out using monomer (HCl salt) and papain (50 mg mL−1) in phosphate buffer (1 M, pH 8.0) at 40 °C for 2 h. All the reactions were triplicated. b Precipitate was collected by centrifugation, washed with water, and lyophilized. c The Aib content in the polymer was estimated by 1H NMR. d Molar ratio was 1/1. e Molar ratio was 1/2. | ||||
| 1 | Aib-OEt | 1.0 | 0 | — |
| 2 | AibAla-OEt | 0.50 | 0 | — |
| 3 | AibAla-OEt/Ala-OEtd | 1.0 | 0 | — |
| 4 | AibAla-OEt/Ala-OEte | 1.5 | 3.5 ± 1.9 | 4.9 ± 0.16 |
| 5 | AlaAibAla-OEt | 0.17 | 15 ± 7.3 | 31 ± 0.87 |
| 6 | AlaAibAla-OEt | 0.25 | 30 ± 4.2 | 28 ± 0.55 |
| 7 | AlaAibAla-OEt | 0.33 | 12 ± 5.0 | 25 ± 1.2 |
| 8 | AlaAibAla-OEt | 0.50 | 4.9 ± 3.6 | 23 ± 1.2 |
| 9 | AlaAibAla-OEt | 1.0 | 0 | — |
The structures of the obtained polypeptides were characterized by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry and 1H nuclear magnetic resonance (NMR) spectroscopy. The copolypeptide from AibAla-OEt and Ala-OEt, poly(AibAla-r-Ala) (run 4), showed two series of peaks derived from polyAla and poly(AibAla-r-Ala) with ethyl ester and carboxylic acid terminal groups (Fig. S1, ESI†). In addition to the formation of polyAla, only one unit of Aib was incorporated in poly(AibAla-r-Ala), indicating a low affinity of AibAla-OEt for papain. The 1H NMR spectrum of poly(AibAla-r-Ala) exhibited a new signal compared to the spectrum of polyAla; this new signal was assigned to the methyl group of the Aib unit at 1.53 ppm (Fig. S2b, ESI†), and the proportion of Aib was estimated to be 4.9 mol%. By contrast, distinctive peaks derived from the tetramer and pentamer of the AlaAibAla unit appeared in the MALDI-TOF mass spectra of poly(AlaAibAla) (Fig. 1a), indicating successful polymerization of AlaAibAla-OEt using papain. The molecular weight of poly(AlaAibAla) was in the range from 900 to 1800, which is similar to the results of other hydrophobic polypeptide synthesis by papain.16,19 This is due to the limited solubility of the resulting polypeptides in the buffer solution, causing the precipitation during the reaction. The molecular weight dispersity was assumed to be narrow from MALDI-TOF mass spectra, although the exact value could not be estimated because of poor solubility of poly(AlaAibAla) in common eluents for size exclusion chromatography. In addition to the peaks corresponding to the tetramer and pentamer, a series of peaks with an interval of 71 m/z corresponding to an Ala unit were also observed. This insertion of additional Ala units into poly(AlaAibAla) can occur via transamidation during polymerization.25,28 Hydrolysis of the amide bonds in the poly(AlaAibAla) backbone may also cause the introduction of Ala units. However, no peak assignable to the polypeptide with a hydrolyzed C-terminal (i.e., carboxylic acid terminal) was detected in the MALDI-TOF mass spectra. Thus, transamidation is assumed to have been the major pathway for Ala insertion. The peaks corresponding to the Ala-inserted poly(AlaAibAla) became more intense when the feed concentration of AlaAibAla-OEt was increased (Fig. S3, ESI†), indicating that transamidation likely occurred at a higher concentration of substrate.29 In the 1H NMR spectrum of poly(AlaAibAla), the peak derived from the methyl protons of the Ala units shifted and overlapped with the peak of the Aib methyl groups at 1.45 ppm (Fig. S2c, ESI†). This substantial change in the chemical shift of the Ala units is assumed to reflect a structural transition of poly(AlaAibAla) compared to polyAla. The Aib contents in the poly(AlaAibAla)s were also estimated from 1H NMR spectra as listed in Table 1. When the polymerization was carried out at the low concentration (0.17 M), the Aib content was 31 mol%, which corresponded to the theoretical value of an authentic AlaAibAla homopolymer (33 mol%). The Aib content decreased with an increase in feed concentration of AlaAibAla-OEt, because of the Ala insertion, namely, transamidation, revealed by MALDI-TOF mass spectra.
 | ||
| Fig. 1 (a) MALDI-TOF mass spectrum of poly(AlaAibAla) prepared by papain-catalyzed polymerization (run 5 in Table 1) and (b) schematic for the substrate specificity of papain for Aib-containing monomers for the (i) acylation and (ii) aminolysis steps of chemoenzymatic polymerization. | ||
The mechanism of protease-catalyzed polymerization is known to proceed through several steps, including substrate activation by a catalytic triad in proteases and aminolysis to the activated acyl intermediate. Subsites are located next to a catalytic Cys residue in the active site of papain that recognize suitable substrates for both the activation and aminolysis reactions.24 Aib-OEt and AibAla-OEt clearly possess a low affinity for papain based on the results of the polymerization (Fig. 1b). Aib-OEt appears to be mismatched with the subsite next to the active site and therefore neither acylation by a thiol group nor aminolysis takes place. AibAla-OEt can function as an electrophile in the activation step, as indicated by the slight copolymerization with Ala-OEt. However, the amine group of AibAla-OEt is located near the α-carbon of the Aib unit, resulting in a mismatch during the aminolysis step. By contrast, the Aib units of AlaAibAla-OEt can occupy a subsites one unit farther from the active site that is more tolerant of D-substrates.24 Thus, AlaAibAla-OEt can participate as an electrophile and a nucleophile in the activation and the aminolysis steps, respectively, to propagate the polypeptide chain.
Infrared (IR) spectra of the polypeptides were collected to investigate the effect of the Aib residue on the secondary structures (Fig. 2a). The amide I region corresponding to the stretching vibration mode of carbonyl groups (1700–1600 cm−1) exhibits a different profile depending on the secondary structure of the polypeptide.30,31 A strong peak assignable to a β-sheet conformation was recorded at 1630 cm−1 in the IR spectrum of polyAla. A shoulder from 1700 to 1650 cm−1 appeared in the IR spectrum of poly(AibAla-r-Ala); however, the peak for the β-sheet structure was still detected because of the low Aib content. By contrast, the spectrum of poly(AlaAibAla) showed a distinctively different peak at 1660 cm−1, which is assignable to an α-helical conformation. This dramatic change in the secondary structure from β-sheet to α-helix was induced by the introduction of Aib units into the polyAla backbone. In particular, polypeptides consisting of an XaaXaaAib triad tend to assemble into a helical conformation because of the steric hindrance of the methyl substituent at the α-carbon.7,10
 | ||
| Fig. 2 (a) IR spectra of Aib-containing polypeptides and (b) CD spectra of the polypeptides in 2,2,2-trifluoroethanol solution (100 mM). Poly(AlaAib-r-Ala): run 4; poly(AlaAibAla): run 5 in Table 1. | ||
The secondary structures of the polypeptides in the solution phase were analyzed by circular dichroism (CD) spectroscopy. The CD spectra of the obtained polypeptides in 2,2,2-trifluoroethanol (100 μM) are displayed in Fig. 2b. The CD spectrum of polyAla exhibited a negative peak at 218 nm and a positive peak at 193 nm, indicating the formation of a β-sheet structure.32 However, the peak intensity was quite low because of the poor solubility of polyAla. Poly(AibAla-r-Ala) exhibited a CD profile similar to that of polyAla, with a slight deviation at approximately 190 nm. By contrast, the CD spectrum of poly(AlaAibAla) was typical of α-helix structures in which two negative peaks at 218 and 208 nm and a positive peak at 191 nm were detected.33 The negative Cotton effect clearly indicates that the α-helix structure of poly(AlaAibAla) has a right-handed screw sense that originates from the L-Ala residues.
In general, polyAla most commonly forms β-sheet structures,34,35 even in solvents that stabilize α-helix conformations, such as 2,2,2-trifluoroethanol.36 The introduction of Aib units, which is known to be a strong helical inducer, into the polyAla backbone caused the distinctive transition of the secondary structure from β-sheet to an α-helix conformation. The poly(AlaAibAla)s prepared from different feed concentrations of AlaAibAla-OEt showed almost identical CD profiles regardless of the structural variations (i.e., the difference in the amount of Ala insertion) as depicted in Fig. 1a. This indicates that introducing the AlaAibAla triad, or perhaps only the Aib unit to a certain extent, into the polyAla backbone can trigger an enormous conformational change without the design of elaborate sequences, resulting in the formation of a definite α-helix structure.
In summary, we demonstrated the successful chemoenzymatic polymerization of an Aib-containing tripeptide ester, AlaAibAla-OEt, by papain in an aqueous buffer solution, resulting in the formation of poly(AlaAibAla). The obtained poly(AlaAibAla) was found to adopt an α-helical conformation in both the solid and solution phases by IR and CD spectroscopic analyses. The polymerization results of three types of Aib-containing monomers suggest that appropriate control of the affinity for papain is essential for achieving the papain-catalyzed polymerization of unnatural amino acids. That is, the chemoenzymatic polymerization of unnatural amino acids, even with little or no substrate specificity in proteases, can reasonably proceed when Ala units (or potentially any natural amino acids) are present on both sides which is essential for recognition at the active site in proteases. This methodology can broadly extend the versatility of chemoenzymatic synthesis for the precise construction of polypeptide-based architectures. This technique for synthesizing polypeptides consisting of unnatural amino acid residues can be further applied to the fabrication of various artificial polypeptide materials with unique structures.
This work was financially supported by Impulsing Paradigm Change through Disruptive Technologies Program (ImPACT). This work was supported by JST ERATO Grant Number JPMJER1602, Japan.
Notes and references
- Y. Goto and H. Suga, J. Am. Chem. Soc., 2009, 131, 5040–5041 CrossRef CAS PubMed.
- T. Passioura and H. Suga, Chem. Commun., 2017, 53, 1931–1940 RSC.
- L. Moroder, J. Pept. Sci., 2005, 11, 187–214 CrossRef CAS PubMed.
- S. Agrawal, A. Adholeya and S. K. Deshmukh, Front. Pharmacol., 2016, 7, 333 Search PubMed.
- C. Toniolo, G. M. Bonora, A. Bavoso, E. Benedetti, B. di Blasio, V. Pavone and C. Pedone, Biopolymers, 1983, 22, 205–215 CrossRef CAS.
- M. Oba, M. Tanaka, M. Kurihara and H. Suemune, Helv. Chim. Acta, 2002, 85, 3197–3218 CrossRef CAS.
- J. Venkatraman, S. C. Shankaramma and P. Balaram, Chem. Rev., 2001, 101, 3131–3152 CrossRef CAS PubMed.
- E. Galoppini and M. A. Fox, J. Am. Chem. Soc., 1996, 118, 2299–2300 CrossRef CAS.
- J. Solà, M. Helliwell and J. Clayden, J. Am. Chem. Soc., 2010, 132, 4548–4549 CrossRef PubMed.
- Y. Demizu, M. Doi, Y. Sato, M. Tanaka, H. Okuda and M. Kurihara, Chem. – Eur. J., 2011, 17, 11107–11109 CrossRef CAS PubMed.
- B. Merrifield, Science, 1986, 232, 341–347 CAS.
- K. Numata, Polym. J., 2015, 47, 537–545 CrossRef CAS.
- H. Uyama, T. Fukuoka, I. Komatsu, T. Watanabe and S. Kobayashi, Biomacromolecules, 2002, 3, 318–323 CrossRef CAS PubMed.
- G. Li, A. Vaidya, K. Viswanathan, J. Cui, W. Xie, W. Gao and R. A. Gross, Macromolecules, 2006, 39, 7915–7921 CrossRef CAS.
- G. Li, V. K. Raman, W. Xie and R. A. Gross, Macromolecules, 2008, 41, 7003–7012 CrossRef CAS.
- P. J. Baker and K. Numata, Biomacromolecules, 2012, 13, 947–951 CrossRef CAS PubMed.
- K. Numata and P. J. Baker, Biomacromolecules, 2014, 15, 3206–3212 CrossRef CAS PubMed.
- Y. Ma, R. Sato, Z. Li and K. Numata, Macromol. Biosci., 2016, 16, 151–159 CrossRef CAS PubMed.
- J. M. Ageitos, K. Yazawa, A. Tateishi, K. Tsuchiya and K. Numata, Biomacromolecules, 2016, 17, 314–323 CrossRef CAS PubMed.
- J. M. Ageitos, P. J. Baker, M. Sugahara and K. Numata, Biomacromolecules, 2013, 14, 3635–3642 CrossRef CAS PubMed.
- J. Fagerland, A. Finne-Wistrand and K. Numata, Biomacromolecules, 2014, 15, 735–743 CrossRef CAS PubMed.
- K. Tsuchiya and K. Numata, Macromol. Biosci., 2016, 16, 1001–1008 CrossRef CAS PubMed.
- K. Yazawa and K. Numata, Polymers, 2016, 8, 194 CrossRef.
- I. Schechter and A. Berger, Biochem. Biophys. Res. Commun., 1967, 27, 157–162 CrossRef CAS PubMed.
- X. Qin, A. C. Khuong, Z. Yu, W. Du, J. Decatur and R. A. Gross, Chem. Commun., 2013, 49, 385–387 RSC.
- X. Qin, W. Xie, S. Tian, J. Cai, H. Yuan, Z. Yu, G. L. Butterfoss, A. C. Khuong and R. A. Gross, Chem. Commun., 2013, 49, 4839–4841 RSC.
- G. Li, J. Wu, X. Qin, J. Zhu, K. Viswanathan, H. Dong, P. Somasundaran and R. A. Gross, Langmuir, 2014, 30, 6889–6896 CrossRef CAS PubMed.
- D. Tessaro, L. Cerioli, S. Servi, F. Viani and P. D'Arrigo, Adv. Synth. Catal., 2011, 353, 2333–2338 CrossRef CAS.
- D. Combes and P. Lozano, Ann. N. Y. Acad. Sci., 1993, 672, 409–414 CrossRef.
- W. Huang, S. Krishnaji, O. R. Tokareva, D. Kaplan and P. Cebe, Macromolecules, 2014, 47, 8107–8114 CrossRef CAS.
- O. S. Rabotyagova, P. Cebe and D. L. Kaplan, Macromol. Biosci., 2010, 10, 49–59 CrossRef CAS PubMed.
- E. Iizuka and J. T. Yang, Proc. Natl. Acad. Sci. U. S. A., 1966, 55, 1175–1182 CrossRef CAS.
- P. Argos and J. K. MohanaRao, Methods Enzymology, Academic Press, 1986, vol. 130, pp. 185–207 Search PubMed.
- L. M. Shinchuk, D. Sharma, S. E. Blondelle, N. Reixach, H. Inouye and D. A. Kirschner, Proteins, 2005, 61, 579–589 CrossRef CAS PubMed.
- G. Xu, L. Gong, Z. Yang and X. Y. Liu, Soft Matter, 2014, 10, 2116–2123 RSC.
- F. D. Sönnichsen, J. E. Van Eyk, R. S. Hodges and B. D. Sykes, Biochemistry, 1992, 31, 8790–8798 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and experimental procedures, 1H NMR spectra, and MALDI-TOF mass spectra. See DOI: 10.1039/c7cc03095a |
| This journal is © The Royal Society of Chemistry 2017 |