
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Programming membrane permeability using integrated membrane pores and blockers as molecular regulators†
Julia M.
Thomas
a,
Mark S.
Friddin
 a,
Oscar
Ces
ab and
Yuval
Elani
*ab
a,
Oscar
Ces
ab and
Yuval
Elani
*ab
aDepartment of Chemistry, Imperial College London, Exhibition Road, London, SW7 2AZ, UK. E-mail: yuval.elani10@imperial.ac.uk
bInstitute of Chemical Biology, Imperial College London, Exhibition Road, London, SW7 2AZ, UK
First published on 1st November 2017
Abstract
We report a bottom-up synthetic biology approach to engineering vesicles with programmable permeabilities. Exploiting the concentration-dependent relationship between constitutively active pores (alpha-hemolysin) and blockers allows blockers to behave as molecular regulators for tuning permeability, enabling us to systematically modulate cargo release kinetics without changing the lipid fabric of the system.
The use of biologically derived molecular components has proved to be a powerful strategy for the design and assembly of a rich and diverse range of lipid-bilayer encased microsystems. They enable a repertoire of biomimetic features to be incorporated into cell-like architectures1 including motility,2 protein synthesis,3 and enzymatic reaction cascades,4 in addition to mechanosensitive behaviours,5 active molecular transport,6 and light-activated responses.7
This repurposing of biological machinery to impart functionality into artificial systems is one of the core tenants of bottom-up synthetic biology, which has shown promise in developing a new generation of liposomal drug delivery systems,8,9 vesicle-based microreactors,4,10,11 and membrane-bound artificial cells.3,12–15 Here, we exploit this strategy to construct Giant Unilamellar Vesicles (GUVs) with user-defined permeabilities. Crucially our approach does not rely upon chemical modification or breakdown of the lipid chassis and instead exploits the interactions between embedded membrane protein pores and chemical blockers.
Fundamental to successful applications for these technologies is the ability to precisely control the exchange of materials between individual compartments and the external medium. This is critical for controlling drug release rates, modulating reaction kinetics, mediating communication between artificial cells and neighbouring biological cells,13 and regulating the interchange of nutrients and waste products.3 To date, this has been achieved by triggering the free-diffusion of materials through protein channels inserted into the membrane or by releasing the entire contents of the compartment by lysis. Alternative approaches involve generating defects in the membrane fabric, e.g. through UV irradiation,16 photocleavage,17–19 photoisomerisation,20 molecular absorbers,21,22 by taking the membrane through a phase transition,23,24 and application of shear forces,25 electric fields,11,26 osmotic gradients and changes in pH.27
These approaches rely on the application of external forces and the extent of permeability cannot be controlled systematically. They are not only binary in nature but also offer no sustainable control over bilayer permeability, especially if the compartment is destroyed in the process. Herein we address this problem by demonstrating that membrane permeability can be finely tuned by exploiting the relationship between a constitutively active membrane pore, alpha-hemolysin (α-HL), and the reversible blocker TRIMEB (heptakis(2,3,6-tri-O-methyl)-β-cyclodextrin).
α-HL is a heptameric transmembrane pore from Staphylococcus aureus that has a water-accessible channel lumen that is 1.4 nm wide at its narrowest point through which molecules can passively diffuse.28,29 It can thus be used as a molecular sieve: it is permeable to globular molecules up to 2000 Da in size30 (a category which most drugs and metabolites fall under), and longer elongated polymers,31 yet is impermeable to biological macromolecules (including DNA and proteins). This makes it suitable for use in vesicle-based enzymatic reactors and artificial cells. TRIMEB is a cyclic oligosaccharide that reversibly and non-covalently binds to the α-HL pore lumen on the cis side of the barrel near Met 113,32 restricting the passage of molecules.32,33 The extent of blockage and the proportion of time the blocker is in the bound state contributes to total leakage through the pore, a process previously quantified using optical ion flux.34 Due to the larger size of the blocker compared to the pore, it is unlikely that TRIMEB itself can fully translocate through α-HL.
Supported by single-channel electrical measurements in droplet interface bilayers (DIBs), we show that this relationship can be leveraged to carefully control the rate of release of a fluorescent dye contained within α-HL functionalised GUVs. We achieve this by regulating the concentration of TRIMEB co-encapsulated inside the GUVs, which acts as an adapter molecule, enabling the permeability of the GUV to be precisely pre-determined (Fig. 1A). Our approach does not rely upon chemical modification to the vesicles or the application of externally applied forces, and does not lead to the destruction of the membrane chassis.
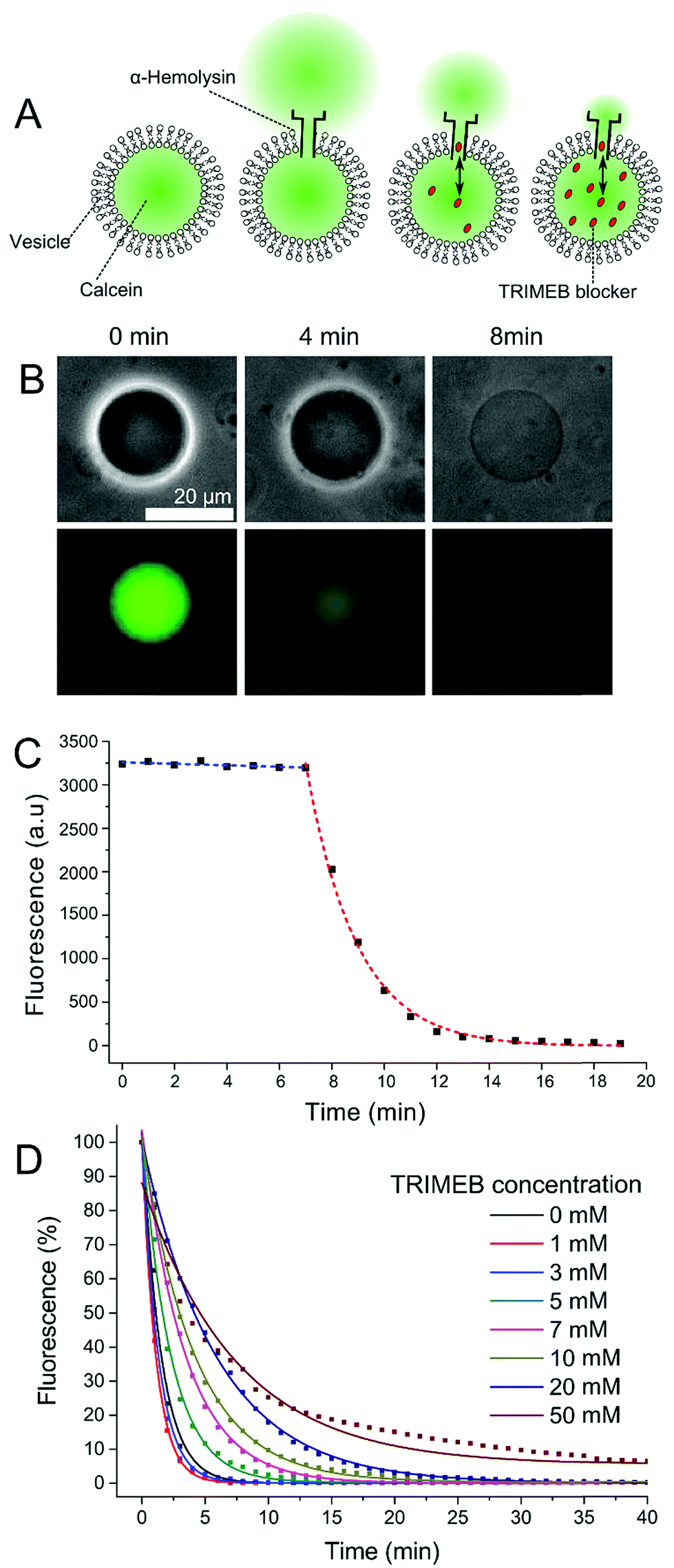 | ||
| Fig. 1 Vesicle permeability fluorescence assay. (A) Schematic of the vesicle/α-HL/TRIMEB system. The presence of blocker reduces calcein flux through the pore. (B) Phase contrast and fluorescence microscopy images after a pore insertion event. As the internal and external content of the vesicle equilibrates the contrast of the membrane contour reduces. (C) Typical trace of vesicle fluorescence over time. Following a lag phase (blue) an exponential decay is seen (red) due to the formation of a pore and efflux of calcein. (D) Average vesicle fluorescence levels over time with different TRIMEB concentrations (lag phase removed). The larger the concentration of blocker the slower the decay. | ||
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) GUVs in TRIS buffer (25 mM Tris–HCl; 500 mM KCl; pH 8.0) were prepared by the emulsion phase transfer technique.35 The effect of blocker concentration was studied by encapsulating 1 mM calcein with TRIMEB concentrations ranging from 1–50 mM. An inverted fluorescence microscope (FITC filter; 200 ms exposure) was used to quantify dye leakage, with vesicle contours and mean fluorescence extracted using a thresholding function of an image analysis software (Image J). Data from a minimum of 10 GUVs were taken for each experimental condition (full experimental details are available in the ESI†). α-HL was added to the external solution as water soluble monomers (final concentration 50 ng μL−1) which proceeded to bind to the membrane and assemble into the pore complex.
After a variable lag phase of between 5 and 60 minutes (Fig. 1B and C), GUV fluorescence was found to decay exponentially following the addition of the protein. We attribute this lag to the time taken for α-HL monomers to bind to the membrane and to oligomerize into a functional pore. Depending on blocker concentration, the GUV interior was indistinguishable from background after 5–80 minutes. The contrast of the GUV under phase contrast microscopy also faded over time as the composition of the interior approached that of the exterior (as observed elsewhere).36 The smaller the concentration of blocker, the quicker the fluorescence decay (Fig. 1D).
The permeability of our system (Peff) can be defined by the equation below:
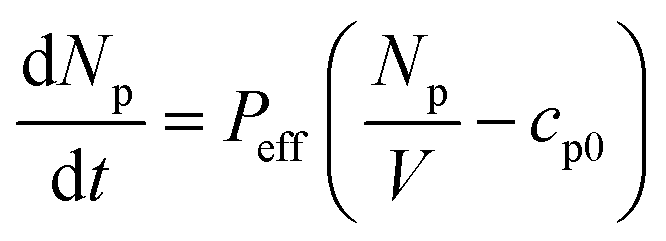 | (1) |
Here, Np is the number of encapsulated fluorescent particles, V is vesicle volume, and cp0 is the concentration of calcein outside the vesicle. As external calcein is removed during GUV preparation, and as the volume of the GUV is negligible compared to that of the external solution we can assume that cp0 = 0 at all times.
Only GUVs where r > 5 μm were analysed as these could easily be resolved using our setup. By fitting total GUV fluorescence per unit volume (which is proportional to dye concentration)37 to the equation above we extract Peff for GUVs containing embedded pores and variable amounts of blocker (Fig. 2). We note that in this case, it is not the intrinsic membrane permeability that is being measured, but the permeability of the entire vesicle/α-HL/TRIMEB assembly, eliminating the dependence on membrane area.
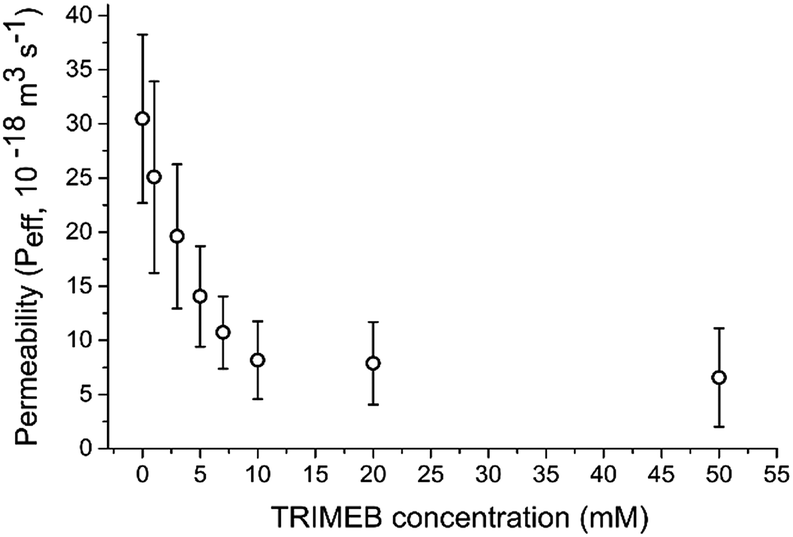 | ||
| Fig. 2 Permeability values associated with the vesicle/α-HL/TRIMEB system with different concentrations of TRIMEB. Error bars = 1 standard deviation; n > 10. | ||
An increase in blocker concentration led to slower dye leakage kinetics, with Peff ranging from 6.6 × 10−18 m3 s−1 to 30.5 × 10−18 m3 s−1 in the conditions tested. Between 0 mM and 10 mM TRIMEB a linear relationship between blocker concentration and Peff was observed (Pearson's r value = 0.96; Radj2 = 0.91), with Peff decreasing 2.2 × 10−18 m3 s−1 with every 1 mM increase in TRIMEB. A highly significant difference in Peff was observed between eight conditions tested (ANOVA, p < 0.0001). These results show that blocker concentration can be used to tune membrane permeability in a quantitative manner.
As the blocker sterically prohibits passage of material through the pore, it stands to reason that changing the type of blocker will change Peff. Indeed, using an analogous blocker, γ-cyclodextrin (γ-CD) with –OH as opposed to –OMe constituents on the sugar rings revealed a significantly larger Peff for 20 mM of blocker (7.87 × 10−18 m3 s−1 for TRIMEB and 1.58 × 10−17 m3 s−1 for γ-CD; unpaired t-test, n = 10, p < 0.001). Blocker type is therefore a further parameter that can be varied in achieving defined permeabilities.
It is likely that in our system we measure single protein insertion events. We do not see a deviation from the original exponential decay curve mid-way through (which would indicate a second pore inserting). Furthermore, a probabilistic interpretation of our system makes multiple pore insertion events unlikely. Typically, only 7–10% of the GUVs leak during the course of the experiments. Assuming this is the baseline rate of single-pore insertion, and neglecting any cooperative pore insertion processes, the probability of two or more proteins inserting is between 0.5 and 1%. We also note that the permeability of the system as a whole is being measured, and it is clear that TRIMEB can be used to effectively modulate vesicle permeabilities at a given α-HL concentration, regardless of whether there is one or several pores inserting.
To further probe our system at a single channel level we performed electrical measurements of droplet interface bilayers (DIBs)38 (Fig. 3A). DIBs are planar bilayers formed when two monolayer-coated water-in-oil droplets are brought into contact. Inserting agar-coated Ag/AgCl electrodes into the droplets allows electrical interrogation. We formed DIBs from 1.5 μL water droplets containing 100 nm vesicles (formed by extrusion) made from 1,2-diphytanoyl-sn-glycero-3-phosphocholine (DPhPC), with one droplet containing α-HL (ground electrode), and the other TRIMEB. A holding potential of 100 mV was applied in voltage-clamp mode, and the bilayer current was measured (full experimental details available in the ESI†).
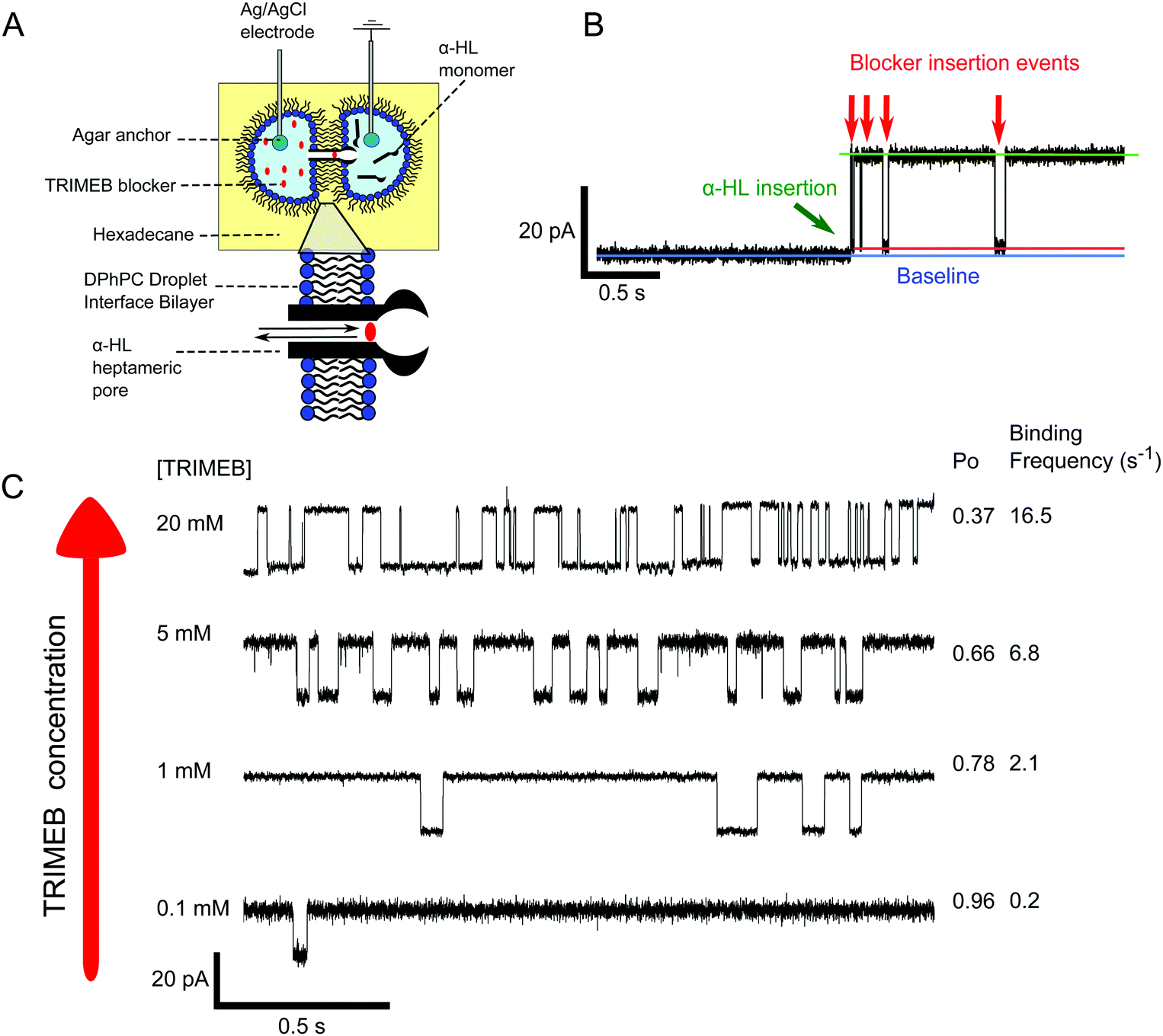 | ||
| Fig. 3 Electrical interrogation of the system using DIBs. (A) Schematic of the experimental setup. (B) Trace of a typical protein insertion event followed by TRIMEB blocking events. By comparing the baseline (blue) to the fully open (green) and blocked (red) traces ca. 92% of the pore was estimated to be blocked. (C) Increasing TRIMEB concentration leads to more frequent blocking events and lower open probabilities. | ||
Single pore opening events were observed as ca. 19 pA jumps in the baseline current (in accordance with previous findings),39 and subsequent pore blockage events by TRIMEB detected as blockades in the current (Fig. 3B). Analysis of the trace revealed that 92% of the pore was sterically blocked by the TRIMEB, which is in line with previous reports.33 As expected, increasing TRIMEB concentration led to more frequent blocking events and lower open probabilities (Po) (Fig. 3C), which reinforces our fluorescence microscopy results.
This system could be further optimised in future by using blockers with other channels that are not constitutively active, and by engineering α-HL mutants which have longer blocker residency times.40 Other issues that need to be addressed for delivery applications are precise control over initiation of content release, and methods to prevent the spontaneous release of contents which limits long term storage.
In conclusion, we have developed a new method to pre-program the permeability of vesicles with fine control using a strategy that offers significant advantages over existing methods, especially as no chemical modification or vesicle lysis is required. In this approach, the structural fabric of the system remains the same with only the formulation (i.e. concentration of an encapsulated blocker) changing. The process of vesicle manufacture can therefore be decoupled from modulation of permeability. In principle, our method could also be applied to other channels and blockers in order to develop more elaborate synthetic bio-systems with further degrees of control, and coupled, for example, to stimuli-responsive channel opening processes. This ability to finely attenuate vesicle permeability could have a significant impact on the next generation of drug delivery agents, soft-matter micro/nanoreactors, and artificial cells.
This work was supported by the EPSRC via grant EP/J017566/1 and by EPSRC Fellowship EP/N016998/1 awarded to YE.
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. Trantidou, M. Friddin, Y. Elani, N. J. Brooks, R. V. Law, J. M. Seddon and O. Ces, ACS Nano, 2017, 11, 6549–6565 CrossRef CAS PubMed.
- M. Guix, C. C. Mayorga-Martinez and A. Merkoçi, Chem. Rev., 2014, 114, 6285–6322 CrossRef CAS PubMed.
- V. Noireaux and A. Libchaber, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17669–17674 CrossRef CAS PubMed.
- Y. Elani, R. V. Law and O. Ces, Nat. Commun., 2014, 5, 5305 CrossRef CAS PubMed.
- K. Charalambous, P. J. Booth, R. Woscholski, J. M. Seddon, R. H. Templer, R. V. Law, L. M. Barter and O. Ces, J. Am. Chem. Soc., 2012, 134, 5746–5749 CrossRef CAS PubMed.
- H. E. Findlay, N. J. Harris and P. J. Booth, Sci. Rep., 2016, 6, 39349 CrossRef CAS PubMed.
- V. R. Schild, M. J. Booth, S. J. Box, S. N. Olof, K. R. Mahendran and H. Bayley, Sci. Rep., 2017, 7, 46585 CrossRef PubMed.
- A. Samad, Y. Sultana and M. Aqil, Curr. Drug Delivery, 2007, 4, 297–305 CrossRef CAS.
- Y. Elani, R. V. Law and O. Ces, Ther. Delivery, 2015, 6, 541–543 CrossRef CAS PubMed.
- P. Walde, A. Goto, P.-A. Monnard, M. Wessicken and P. L. Luisi, J. Am. Chem. Soc., 1994, 116, 7541–7547 CrossRef CAS.
- A. Fischer, A. Franco and T. Oberholzer, ChemBioChem, 2002, 3, 409–417 CrossRef CAS PubMed.
- P. L. Luisi, F. Ferri and P. Stano, Naturwissenschaften, 2006, 93, 1–13 CrossRef CAS PubMed.
- R. Lentini, N. l. Y. Martín, M. Forlin, L. Belmonte, J. Fontana, M. Cornella, L. Martini, S. Tamburini, W. E. Bentley and O. Jousson, ACS Cent. Sci., 2017, 3, 117–123 CrossRef CAS PubMed.
- Y. Elani, Biochem. Soc. Trans., 2016, 44, 723–730 CrossRef CAS PubMed.
- A. Salehi-Reyhani, O. Ces and Y. Elani, Exp. Biol. Med., 2017, 242, 1309–1317 CrossRef PubMed.
- A. Yavlovich, A. Singh, S. Tarasov, J. Capala, R. Blumenthal and A. Puri, J. Therm. Anal. Calorim., 2009, 98, 97 CrossRef CAS PubMed.
- Y. Wan, J. K. Angleson and A. G. Kutateladze, J. Am. Chem. Soc., 2002, 124, 5610–5611 CrossRef CAS PubMed.
- N. J. Wymer, O. V. Gerasimov and D. H. Thompson, Bioconjugate Chem., 1998, 9, 305–308 CrossRef CAS PubMed.
- V. C. Anderson and D. H. Thompson, Biochim. Biophys. Acta, Biomembr., 1992, 1109, 33–42 CrossRef CAS.
- K. Kano, Y. Tanaka, T. Ogawa, M. Shimomura and T. Kunitake, Photochem. Photobiol., 1981, 34, 323–329 CrossRef CAS.
- B. Khoobehi, C. A. Char, G. A. Peyman and K. M. Schuele, Lasers Surg. Med., 1990, 10, 303–309 CrossRef CAS PubMed.
- K. A. Dendramis, P. B. Allen, P. J. Reid and D. T. Chiu, Chem. Commun., 2008, 4795–4797 RSC.
- A. Blicher, K. Wodzinska, M. Fidorra, M. Winterhalter and T. Heimburg, Biophys. J., 2009, 96, 4581–4591 CrossRef CAS PubMed.
- D. Papahadjopoulos, K. Jacobson, S. Nir and I. Isac, Biochim. Biophys. Acta, Biomembr., 1973, 311, 330–348 CrossRef CAS.
- A.-L. Bernard, M.-A. Guedeau-Boudeville, V. Marchi-Artzner, T. Gulik-Krzywicki, J.-M. di Meglio and L. Jullien, J. Colloid Interface Sci., 2005, 287, 298–306 CrossRef CAS PubMed.
- R. Dimova, N. Bezlyepkina, M. D. Jordö, R. L. Knorr, K. A. Riske, M. Staykova, P. M. Vlahovska, T. Yamamoto, P. Yang and R. Lipowsky, Soft Matter, 2009, 5, 3201–3212 RSC.
- J. Du and S. P. Armes, J. Am. Chem. Soc., 2005, 127, 12800–12801 CrossRef CAS PubMed.
- L. Song, M. R. Hobaugh, C. Shustak, S. Cheley, H. Bayley and J. E. Gouaux, Science, 1996, 274, 1859–1865 CrossRef CAS PubMed.
- E. Gouaux, J. Struct. Biol., 1998, 121, 110–122 CrossRef CAS PubMed.
- R. Fussle, S. Bhakdi, A. Sziegoleit, J. Tranum-Jensen, T. Kranz and H.-J. Wellensiek, J. Cell Biol., 1981, 91, 83–94 CrossRef CAS PubMed.
- J. J. Kasianowicz, E. Brandin, D. Branton and D. W. Deamer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13770–13773 CrossRef CAS.
- L.-Q. Gu, O. Braha, S. Conlan, S. Cheley and H. Bayley, Nature, 1999, 398, 686–690 CrossRef CAS PubMed.
- A. J. Heron, J. R. Thompson, B. Cronin, H. Bayley and M. I. Wallace, J. Am. Chem. Soc., 2009, 131, 1652–1653 CrossRef CAS PubMed.
- O. K. Castell, J. Berridge and M. I. Wallace, Angew. Chem., Int. Ed., 2012, 51, 3134–3138 CrossRef CAS PubMed.
- S. Pautot, B. J. Frisken and D. Weitz, Langmuir, 2003, 19, 2870–2879 CrossRef CAS.
- P. Peterlin, G. Jaklič and T. Pisanski, Meas. Sci. Technol., 2009, 20, 055801 CrossRef.
- M. Kaltenbach, S. R. Devenish and F. Hollfelder, Lab Chip, 2012, 12, 4185–4192 RSC.
- H. Bayley, B. Cronin, A. Heron, M. A. Holden, W. L. Hwang, R. Syeda, J. Thompson and M. Wallace, Mol. BioSyst., 2008, 4, 1191–1208 RSC.
- G. Villar, A. J. Heron and H. Bayley, Nat. Nanotechnol., 2011, 6, 803–808 CrossRef CAS PubMed.
- L.-Q. Gu, S. Cheley and H. Bayley, J. Gen. Physiol., 2001, 118, 481–494 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/c7cc05423h |
| This journal is © The Royal Society of Chemistry 2017 |