
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMOF catalysts in biomass upgrading towards value-added fine chemicals†
Annika
Herbst
and
Christoph
Janiak
 *
*
Institut für Anorganische Chemie und Strukturchemie, Heinrich-Heine Universität Düsseldorf, Universitätsstraße 1, D-40225 Düsseldorf, Germany. E-mail: janiak@uni-duesseldorf.de
First published on 5th September 2016
Abstract
The development of new synthetic routes from biomass sources towards already existing molecules, which are then called bio-based molecules, or the transformation of biomass into new building blocks and materials will be of great impact. The review presents a critical comparison between metal–organic frameworks (MOFs) and other catalysts (e.g. zeolites) for biomass transformation and valorization to platform chemicals: cellulose hydrolysis to glucose, fructose or sorbitol; fructose, glucose or maltose to 5-hydroxymethylfurfural (5-HMF); sucrose to methyl lactate; furans, levulinic acid, lignin or vanillin as feedstock; triglycerides to esters and glycerol. For example, in the case of cellulose hydrolysis as well as glucose isomerization MOF-based catalysts could not compete with zeolites and sulfonated carbon which display significantly higher activity. In DMSO, MIL-101Cr-SO3H-15% and NUS-6(Hf) are among the best heterogeneous catalysts reported so far for the conversion of fructose into 5-HMF. For the glucose-to-5-HMF transformation MIL-101Cr-SO3H is only a low-to-medium activity catalyst for 5-HMF while mesoporous tantalum phosphate as well as Sn montmorillonite display significantly higher activities. On the other hand, MIL-101Cr-SO3H preferentially transformed glucose to 5-HMF over levulinic acid while the catalysts Amberlyst-15 and sulfuric acid gave mostly levulinic acid. For levulinic acid conversion to ethyl levulinate UiO-66Zr catalysts can compete with other heterogeneous catalysts for the levulinic esterification reaction. For active MOF catalysts open metal sites (coordinatively unsaturated sites) are important as the activity increases with the amount of missing linkers. The two MOFs MIL-101Cr and UiO-66 and their derivatives are used in many studies. These MOFs did not only act as catalysts themselves but also served as hosts or support to embedded catalytic species, e.g., phosphotungstic acid (PTA), ruthenium and palladium nanoparticle (Ru-NP, Pd-NP) or poly(N-bromomaleimide) catalysts. For the conversion of vanillin into 2-methoxy-4-methylphenol the selectivity of Pd@UiO-66Zr-NH2 was quantitative compared with other supported Pd catalysts (selectivity 48%). Further, MOFs were used as precursors for decomposition and carbonization due to their high porosity and uniformly distributed metal centers to yield catalytically active metal–carbonaceous materials with high thermal and chemical stability. For example, metal nanoparticles supported on nanoporous carbon (M/NC) were synthesized by carbonization and carbothermal reduction of Ru, W, V, and Ti metal precursors loaded in IRMOF-1 or IRMOF-3. Fe–Co-based MOF-derived catalysts are a highly efficient system for the conversion of 5-HMF to 2,5-diformylfuran. In water-containing reactions, the water stability of MOFs is of high importance.
1. Introduction
Valorization of waste biomass attracted tremendous research interest during the past years as it is a sustainable feedstock for a number of valuable chemicals and materials. Upgrading biomass into fuel and fine chemicals can reduce the dependence on fossil fuels. Research efforts to turn biomass into useful chemicals are summarized and evaluated in a number of excellent reviews, from which a small selection is noted here by their (abridged) title: “Redefining biorefinery” (2015),1 “Valorization of industrial waste and by-product streams” (2014),2 “Recent advancement in catalytic materials for biodiesel production” (2015),3 “Catalytic routes towards acrylic acid, adipic acid and ε-caprolactam starting from biorenewables” (2015),4 “Chemical conversion pathways for carbohydrates” (2015),5 “Cellulose and lignocellulosic biomass to chemicals and fuels, combined solvent–nanocatalysis approach” (2014),6 “Environmental performance of biomass refining into high-added value compounds” (2016),7 “Recent progress in the development of solid catalysts for biomass conversion into high value-added chemicals” (2016),8 “Catalysis for biomass and CO2 use through solar energy” (2014),9 “Targeted chemical upgrading of lignocellulosic biomass to platform molecules” (2014),10 and “Green chemistry, catalysis and valorization of waste biomass” (2016).11Importantly, it has to be the aim to use non-food biomass waste, which is, for example, generated in agricultural production, such as sugar cane bagasse, corn stover, wheat straw, rice husks and orange peel, to ensure that there is no competition with food production.11 Also waste and by-product streams from other existing industrial sectors (e.g., food industry, pulp and paper industry, biodiesel and bioethanol production) as well as marine biomass such as algae could be used as renewable resources for both biorefinery development and production of fine chemicals.2
Most of the biomass is composed of three major feedstocks: cellulose (45%), hemicelluloses (29%) and lignin (25%) (Fig. 1).
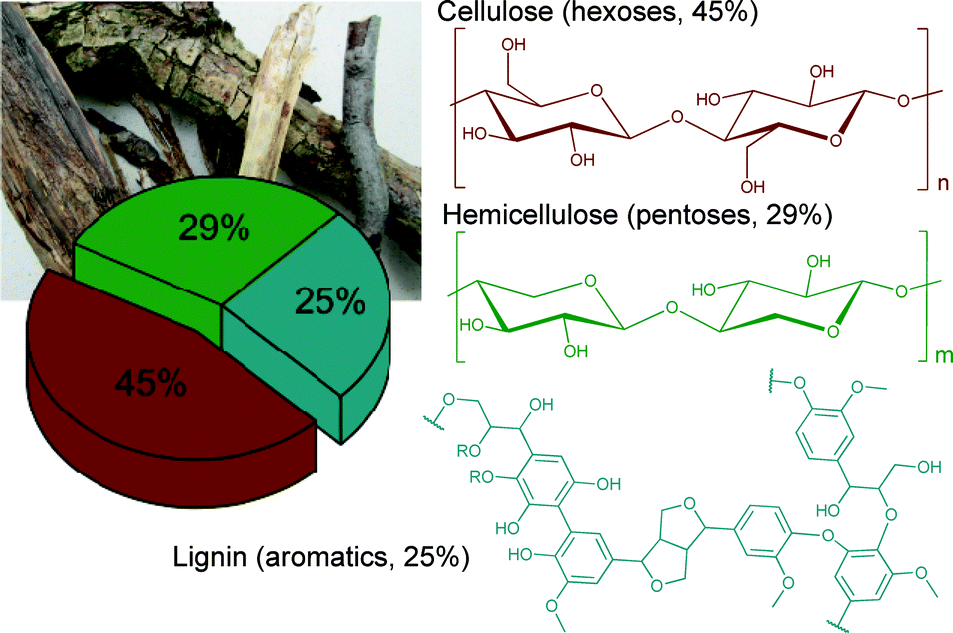 | ||
| Fig. 1 Schematic representation of woody biomass composition. Modified from ref. 1. Copyright 2015 with permission from the Royal Society of Chemistry. | ||
The hydrolysis of cellulose and hemicelluloses is the first step of their biomass valorization for an efficient production of hexoses and pentoses.12 It is also possible to convert cellulose directly into various fine-chemical products (Fig. 2). Different reactions, such as pyrolysis, hydrolysis, condensation, isomerization, deoxygenation, hydrogenation and oxidation have to be performed to obtain high value chemicals ranging from C1- to C6-based compounds (Fig. 2). A great challenge is the controlled removal of oxygen-containing functional groups without reducing the number of carbon atoms and without production of CO2.13
 | ||
| Fig. 2 Possible valuable chemicals based on carbohydrate feedstock. Figure modified from ref. 15. | ||
Especially, 5-hydroxymethylfurfural (5-HMF) and levulinic acid as well as lactic acid are discussed as so-called platform chemicals since they can be converted into important chemicals (Fig. 3). 5-HMF can be oxidized to 2,5-furandicarboxylic acid, which might replace terephthalic acid in polyethyleneterephthalate (PET) production in the future. γ-Valerolactone (Fig. 3) is a sustainable solvent and fuel additive. From both 5-HMF and γ-valerolactone, adipic acid can be obtained, which is mainly used for the production of nylon 6.6.4
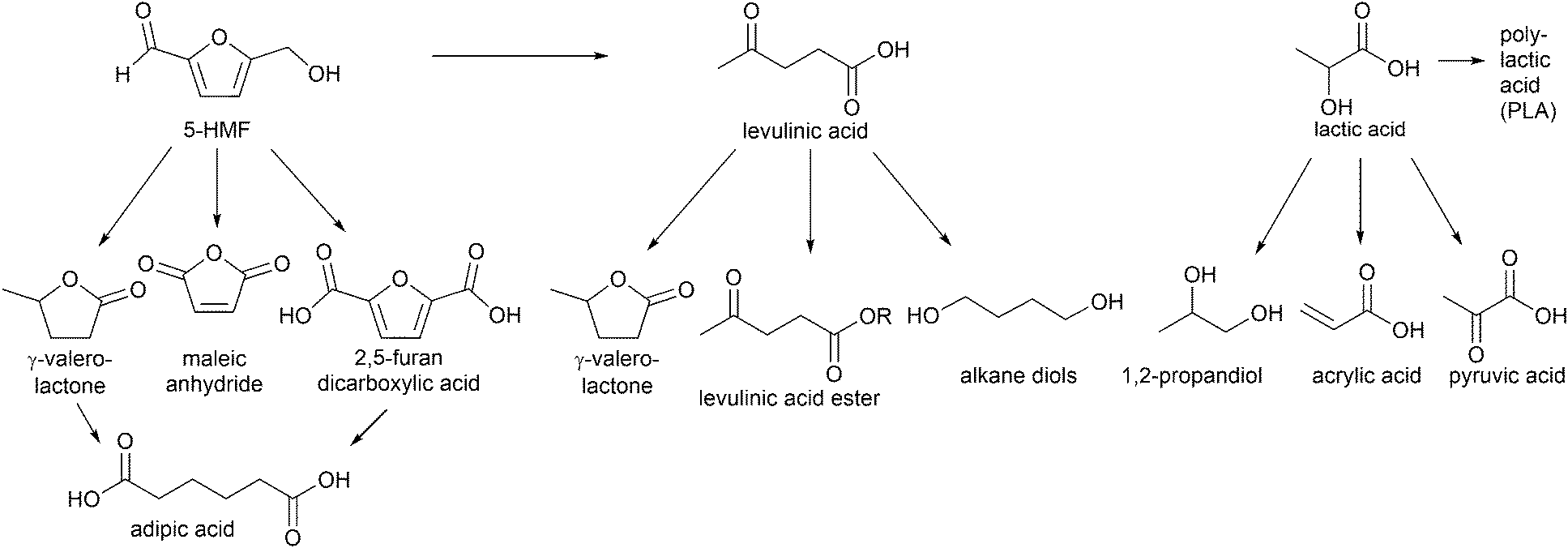 | ||
| Fig. 3 Examples of high value chemicals obtained from conversion of platform chemicals (5-HMF, levulinic acid and lactic acid). | ||
γ-Valerolactone and derivatives of levulinic acid, for instance alkyl levulinates, are suitable additives for gasoline and diesel fuels. Particular ethyl levulinate is an appropriate gasoline blendstock due to its high octane rating and lubricity, solubility and volatility. Apart from fuel additives, levulinate esters can be used as flavoring compounds and plasticizers.14
Another important platform molecule is lactic acid, which is applied not only in cosmetics and pharmaceuticals but most importantly in the polymer industry for the formation of biodegradable polylactic acid (PLA).16 Also, 1,2-propanediol and acrylic acid can be generated from lactic acid (Fig. 3). Acrylic acid derived polymers are used for superabsorbents, plastics and synthetic rubbers.4 The formation of lactic acid from carbohydrates involves multiple reactions, such as retro-aldol reaction, isomerization and 1,2-hydride shift for which Lewis acids are necessary as well as the presence of Brønsted acids for hydrolysis and dehydration. Over 90% of commercial lactic acid is biotechnologically produced by fermentation of aqueous glucose.17 This process has some drawbacks, however, such as a long reaction time because of its low reaction rates, high energy consumption and large amount of waste produced in the neutralization and purification steps.18 The platform molecules 5-HMF, levulinic acid and lactic acid will be addressed in this review.
Another major feedstock of biomass is lignin. Lignin was recognized as a potential source of aromatic compounds since it is mainly composed of aliphatic and phenolic alcohols, which are connected through aryl ether and carbon–carbon bonds. In Fig. 4 the framework of softwood lignin and common linkages found in lignin are presented.19
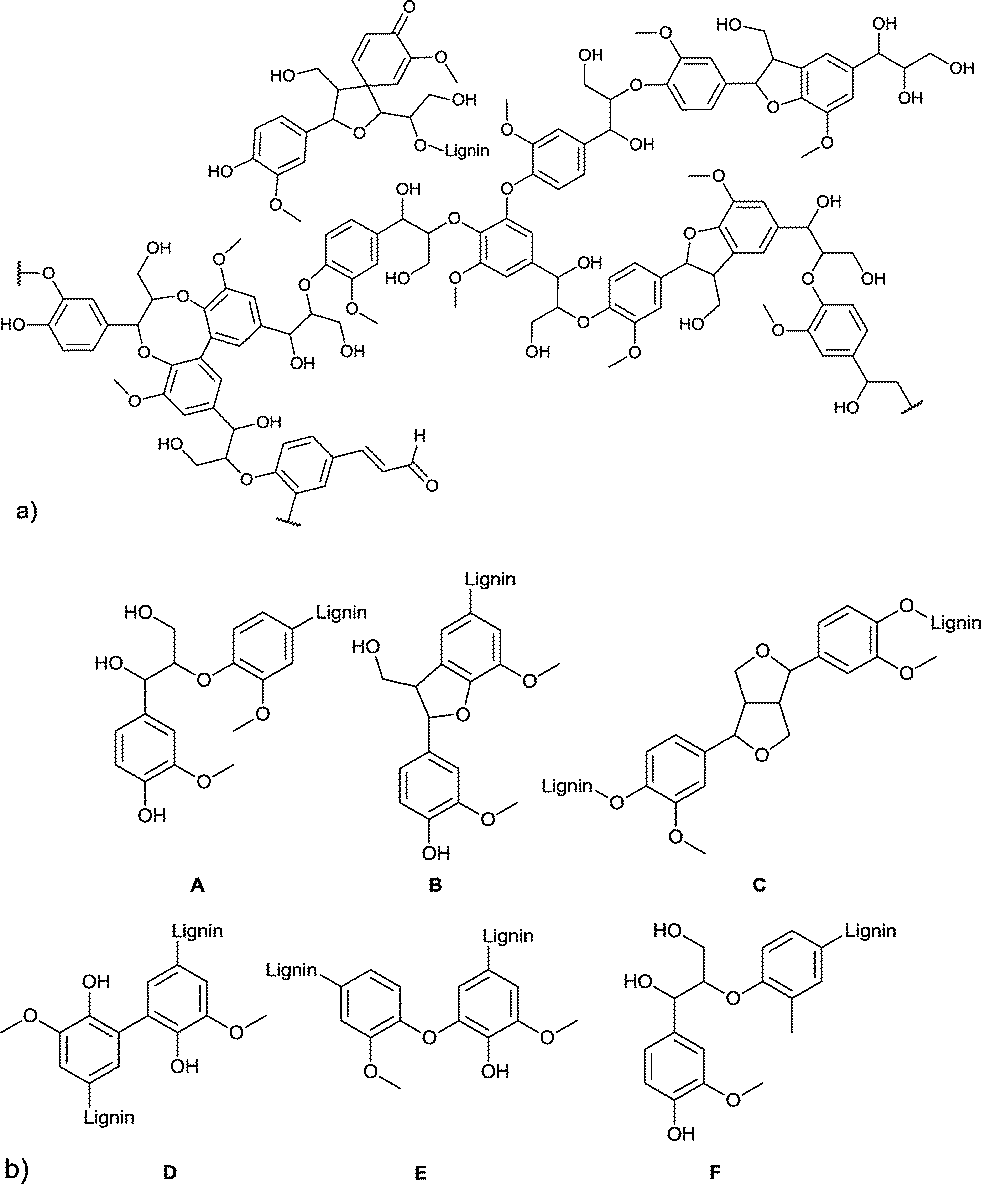 | ||
| Fig. 4 (a) Structural motifs of softwood lignin. (b) Six common linkages found in lignin: A = β-O-4, B = β-5, C = β-β′, D = 5-5′, E = 4-O-5, F = β-1′. Figures redrawn from ref. 19. | ||
However, three products are already commercially produced from lignin: vanillin, dimethyl sulfide, and dimethyl sulfoxide.20 Therefore, vanillin is often regarded as a lignin model compound for further reactions. Upton and Kasko described in their recent review possible strategies to convert lignin (regarded as a macromonomer) into polymeric materials, including among others polyurethanes, polyester and phenol-formaldehyde resins.19
One target is the development of new synthetic routes from biomass sources towards already existing molecules, which are then called bio-based molecules. An even greater impact would be to transform biomass into new building blocks and materials.11
2. Introducing the catalyst systems
2.1 Zeolite versus MOF catalysts
Versatile heterogeneous and porous catalysts currently used in industry often are zeolites. Zeolites or aluminosilicates have been first applied as catalysts in industry in 1959 as isomerization catalysts (zeolite-Y, by Union Carbide) and in 1962 as hydrocarbon cracking catalysts (zeolite-X by Mobile Oil).21 Zeolites have the empirical formula M2/nO·Al2O3·ySiO2·wH2O (y = 2–200, n = cation valence, w = water in the voids of zeolites). They consist of an infinitely extended 3D framework of AlO4 and SiO4 tetrahedra, where the Al and Si atoms are connected through oxygen bonds.21 Currently, zeolites are successfully used as heterogeneous porous catalysts in a variety of reactions and industrial processes.21,22 The most prominent reactions catalyzed by zeolites are the catalytic cracking process for oil transformation to fuels and chemicals and Friedel–Crafts reactions.22Zeolites possess strong Lewis-acid sites because of coordinatively unsaturated metal sites and can also exhibit Brønsted acidity. Zeolites are also investigated as catalysts for biomass valorization. While the focus of this review is on biomass transformation with MOF catalysts, zeolites are taken as a reference for catalytic performances. During the past 25 years a new class of porous materials called metal–organic frameworks (“organic zeolites”)23,24 emerged which may complement zeolites in future catalytic applications.25–28
Metal–organic frameworks (MOFs), also named porous coordination polymers (PCPs), consist of metal or metal-cluster nodes and bridging organic linkers which are extended in two or more typically three dimensions.25,29 By the present definition a metal–organic framework is a metal–ligand coordination network with organic ligands containing potential voids.30,31
As organic bridging units, anionic di- or tricarboxylates feature prominently.32 MOFs are crystalline materials; their molecular structure can be determined by X-ray crystallography in most cases.33 Two examples of MOFs, which are relevant to the catalytic applications in this review, are described in more detail in section 2.3.
MOFs entered the stage of catalysts for biomass valorization about five years ago, with the first report from Akiyama and coworkers investigating the potential of MIL-101Cr-SO3H for cellulose hydrolysis.34 A recent review of Liu and coworkers on the “catalytic transformations of organic compounds and biomass derivatives with functionalized metal–organic frameworks” touched this field and should be mentioned at this point.35
Fig. 5 compares the number of publications on zeolites and on MOFs and their respective fraction dealing with catalysis. It is obvious that catalysis is one of the main applications of zeolites, with a share of 43% of all publications on zeolites in 2015 (or about 3000 in total) (Fig. 5a). As seen in Fig. 5b a wider investigation of MOFs as catalysts started around 2008. Thus, in MOF research catalysis still contributes little; only 22% of publications in 2015 (or about 500 out of ca. 3000 altogether) addressed catalytic reactions (Fig. 5b).
 | ||
| Fig. 5 Development of the field of catalysis of (a) zeolites and (b) MOFs in comparison to the total number of publications. Source: Scifinder (search carried out in May 2016), search terms “catalysis by MOFs”, “catalysis by zeolites” compared to “MOF” and “zeolite”. For a better perspective the number of “3000” publications, corresponding to about the total number of “catalysis by zeolites” and “MOFs”, respectively, is enclosed in red rectangles. | ||
The development of MOF catalysis during the past few years was reviewed very extensively.26–28,36–38 Apart from reviews presenting a general overview about the progress, also reviews about specific fields of catalytic reactions with MOFs can be recommended, e.g., on the “introduction of chemically accessible Lewis basic sites”,39 “condensation reactions of carbonyl groups”,40 “synthesis of nitrogen-containing heterocycles”,41 “production of fine chemicals”,42 “oxidation reactions”,43 “photocatalysis”44 or “embedding of metal nanoparticles in MOFs in order to obtain cage isolated catalysts”.45 Differences in the properties of zeolites and MOFs for catalysis are depicted in Fig. 6 and summarized in Table 1.46,47
 | ||
| Fig. 6 Strengths (green) and weaknesses (red) of zeolites and MOFs, respectively. Modified from ref. 46. | ||
| Properties | Zeolites | MOFs |
|---|---|---|
| a Modified from ref. 46. | ||
| Thermal stability | Stable above 450 °C | Not stable above 300 °C |
| Chemical stability | Stable to solvents, acids, oxidizing and reducing agents | Limited chemical stability especially towards water for most cases |
| BET surface area | Around 200–500 m2 g−1 | Around 1000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 m2 g−1 000 m2 g−1 |
| Pore volume | 0.1–0.5 cm3 | Over 1 cm3 |
| Metal site density | Low | High |
| Lewis acidity | Accessible framework metal ions | Accessible framework metal ions |
| Brønsted acidity | Bridging Si(OH)/Al hydroxyl groups | Introducible through organic linker (e.g. SO3H) |
| Basicity | From framework oxygen atoms | Introducible through organic linker (e.g. NH2) |
| Active site environment | Mostly hydrophilic but can be made hydrophobic | More hydrophobic, but linker dependent |
| Additional features | Chiral functionalities, flexible and stimuli responsive behavior | |
| Reactivation | By thermal treatment | Washing procedures; thermal treatment not possible |
In general, zeolites outperform MOFs in terms of thermal and chemical stability, whereas metal–organic frameworks are superior in terms of tunability due to their composition of organic ligands and metal clusters. Therefore, liquid phase catalysis (instead of gas phase reactions) is most suitable for MOF catalysts.46 Torres-Knoop and Dubbeldam compared the pore sizes and nitrogen surface areas of ZIFs, COFs, zeolites and MOFs in the context of separation and adsorption mechanisms.48 MOFs possess a large variety of different pore sizes and geometries, up to mesopores of about 9.8 nm in diameter,49 thus overcoming diffusion limitations, which exist for microporous zeolites.47,48 Larger pores facilitate the diffusion and mass transfer of reagents and products. In comparison, zeolites display pore capacities and surface areas of one magnitude lower than MOFs.48
MOFs have the distinct advantage over zeolites that their pore environment can be tailored, for instance through postsynthetic modifications (PSMs) so that higher shape selectivity can be achieved in catalysis. By functionalizing the organic linker or metal nodes in MOFs, the chemical environment can also be controlled. The metal nodes can serve as active sites if coordinatively unsaturated sites (CUS), also termed open metal sites (OMS), are available. Due to the high crystallinity of MOFs, structure–activity relationship investigations as well as molecular simulations can be performed more profoundly in order to gain a deeper understanding.50
Regarding the above-mentioned carbohydrate- and biomass-derived platform chemicals and the products derived therefrom (Fig. 2 and 3), a valid question arises with respect to molecular dimensions and pore size for adsorption. Can MOFs adsorb, for example, glucose, 5-HMF and other molecules? This question was answered with a molecular simulation by Kruger et al. who summarized the diameter of platform molecules in comparison to catalyst pore sizes (Fig. 7).13
 | ||
| Fig. 7 Comparison of catalyst pore dimensions (blue dots) and molecular dimensions of carbohydrates (red) and biomass-derived platform chemicals (green and violet). Reprinted from ref. 13. Copyright 2012 with permission from Elsevier. | ||
Among the compared zeolite catalysts (Nu-6(2), H-ZSM5, H-BEA, H-MOR, and H-Y and the silicon–aluminum phosphate SAPO-11) H-Y displays the largest pore size with a diameter of 7.5 Å.13 The MOFs, which we cover in this review (see below), all display even larger pore diameters. For instance, MIL-101Cr has pore sizes of 29 and 34 Å with pore windows of 12 to 15 Å.51 The porosity of UiO-66 consists of octahedral cavities with diameters of 11 Å, tetrahedral cavities with a diameter of 8 Å and narrow triangular windows with a free diameter close to 6 Å (see section 2.3).52 Gupta et al. suggested in their molecular simulation study that MIL-101Cr could be suitable for glucose recovery.53 Adsorption studies of platform reagents and products, which were performed in the analyzed literature, will be mentioned.
2.2 Stability of MOFs
Biomass transformation and valorization processes involve inter alia hydrolysis, condensation, dehydration and esterification reactions where H2O is present as a starting material or generated as a product.40,42Although there are more than 20000 MOFs reported in the literature,49 only water stable MOFs would be suitable in such catalytic reactions, with the MOFs also displaying a certain degree of thermal and chemical stability. This requirement reduces the number of suitable MOF catalysts dramatically.
The hydrothermal stability of MOFs is a key and often overlooked issue for their potential applications in catalysis and other technologies when water is present or generated. Zinc-carboxylate MOFs, such as MOF-5 and the IRMOF series, have low moisture stability;54 HKUST-1 (Cu3(BTC)2) is intermediate but eventually decomposes.55,56 MIL-type compounds, including MIL-101Cr,57,58 MIL-53Al-NH2, Al-fumarate,59 CAU-10-H,60 and ZIF-8 exhibit higher water stability.61–64 Burtch et al. categorized the factors governing the water stability of MOFs into a) thermodynamic stability, including metal–ligand bond strength and lability with water and b) kinetic stability with the subcategories hydrophobicity and steric factors.65 After defining minimum testing conditions and an understanding of “water stability” the review by Burtch et al. provides a comprehensive overview of the water stability of MOFs.65
Leus and coworkers carried out a systematic study on water stability and the stability towards acids and bases as well as peroxides of selected prototypical MOFs.66 These studies confirmed and determine the following MOFs as most stable under water vapor and liquid water conditions: MIL-101Cr, MIL-53Al, MIL-53-NH2, UiO-66, UiO-66-NH2 and UiO-67.54,58,65,66
The reviews of García-García et al. and Gascon et al. are recommended for the evaluation of recent work on catalysis using MOFs in comparison with their homogeneous counterparts and with a perspective towards commercial applications. They also provided a guideline for catalytic testing practice and interpretations.50,67 Especially, MOF-catalyst stability under the applied reaction conditions has to be proven carefully, which is sometimes arguable, specifically when reactions are performed in the presence of water, since only a few MOFs have proven water stability.68
2.3 MOFs MIL-101Cr and UiO-66
The two MOFs MIL-101Cr and UiO-66 feature prominently in biomass transformations; hence their structures are briefly described here.“MIL” is the abbreviation for Materials Institute Lavoisier. Férey discovered the class of porous polycarboxylates with three-valent metal ions and named these MOFs “MILs”, such as MIL-53,69 MIL-100,70–72 MIL-88,73 and MIL-101Cr.51 MIL frameworks mostly contain the trivalent metal cations Cr3+, Fe3+, and Al3+ and benzene di- or tricarboxylate linkers.
The chromium terephthalate framework MIL-101Cr was first synthesized in 2005.51 MIL-101Cr, 3D-[Cr3(μ3-O)(bdc)3(F/OH)(H2O)2]-25H2O, is a highly porous material, exhibiting a Brunauer–Emmett–Teller (BET) surface area of up to 4000 m2 g−1.51 The secondary building unit consists of three μ3-oxido vertex-sharing chromium(III) octahedra, which are interconnected by benzene-1,4-dicarboxylate (bdc) linkers (Fig. 8a). The trinuclear units are vertices of supertetrahedra (Fig. 8b). The vertex-sharing supertetrahedra then form a three-dimensional zeotypic network (Fig. 8c) with two types of mesoporous cages (Fig. 8d). The small cage has an inner diameter of 29 Å and only pentagonal windows. The large cage has an inner diameter of 34 Å and both pentagonal and hexagonal windows (Fig. 8d). The window apertures are 12 Å or 15–16 Å, respectively (Fig. 8e).
 | ||
| Fig. 8 (a) Ligand and metal building blocks in the trinuclear {Cr3(μ3-O)(O2C-)6(F/OH)(H2O)2} building unit; (b) supertetrahedra; (c) mesoporous zeotypic network; (d) small cage with pentagonal windows and large cage with pentagonal and hexagonal windows; (e) dimensions of pentagonal and hexagonal cage window apertures. Objects in (a) to (e) are not drawn to scale. Graphics have been created from the deposited cif-file for MIL-101Cr (CSD-Refcode OCUNAK).51 | ||
MIL-101Cr has two terminal water molecules connected to the trinuclear {Cr3(μ3-O)(O2C-)6(F/OH)(H2O)2} building units with their octahedral Cr(III) ions. The aqua ligands on Cr can be removed by heating under vacuum and give coordinatively unsaturated sites (CUS, also called open metal sites, OMS). From TGA thermal stability up to 350 °C was determined.51
Originally, MIL-101Cr was synthesized hydrothermally with the addition of hydrofluoric acid as mineralizing agent but can also be obtained from HF-free synthesis. Hence, the trinuclear secondary building unit (SBU) contains either a terminal fluoride or a hydroxide ligand for charge neutrality on the third chromium atom.74 Reviews about MIL-101Cr were published by Hong in 2009 and Bhattacharjee in 2014.75,76
“UiO” stands for University of Oslo. UiO-MOFs were developed by the group of Lillerud.52 The Zr-MOF UiO-66Zr, 3D-[Zr6(μ3-O)4(μ3-OH)4(bdc)6], has a hexanuclear {Zr6(μ3-O)4(μ3-OH)4}12+ cluster with the six Zr atoms forming the vertices of an octahedron (Fig. 9, left). Together with the oxygen atoms or the carboxylate groups each Zr atom has a square antiprismatic coordination environment. Thus, the SBU is a hexanuclear cluster of six edge-sharing ZrO8 square antiprism, which is connected by benzene-1,4-dicarboxylate linkers (bdc) to 12 neighboring SBUs in a face-centered cubic (fcc) packing arrangement (Fig. 9, right). The BET surface areas for UiO-66 were reported to be in the range of 880 (ref. 77) to 1160 m2 g−1.64,78–80
 | ||
| Fig. 9 Structure of zirconium terephthalate UiO-66Zr. The SBU is a hexanuclear and octahedral cluster of six edge-sharing ZrO8 square antiprisms, which is connected to 12 neighboring SBUs in a face-centered cubic (fcc) packing arrangement. The structure was drawn from deposited cif-files under CCDC 837796 (UiO-66).81 | ||
2.4 Postsynthetic modification of MOFs
Postsynthetic modification (PSM) refers to a chemical modification of the MOF lattice after its synthesis. The structure of the MOF should be preserved upon functionalization, which is usually shown by powder X-ray diffraction (PXRD) and gas sorption experiments after the reaction.82,83 General strategies for postsynthetic modifications are depicted in Fig. 10. | ||
| Fig. 10 Postsynthetic modifications of prototypical MOFs. Modified from ref. 90. Copyright 2016 with permission from the Royal Society of Chemistry. | ||
Non-covalent PSM includes guest exchange and formation of metal nanoparticles inside the pores of a MOF, and even solvent and residue removal upon the activation procedure can be counted as non-covalent PSM.84 Covalent PSM includes all kinds of organic reactions on the linker for further functionalization.83 PSM methods under the term “building block replacement (BBR)”85 encompass solvent-assisted linker exchange (SALE),86 replacement of non-bridging ligands (ligand grafting onto metal),87 transmetalation of metal nodes (metal ion exchange) and metalation of linkers (metal ion immobilization).84,88
Decomposition of metal–organic frameworks into metal nanoparticles or porous metal oxides as well as metal supported nanoporous carbon can be considered destructive PSM using the MOF as a precursor for tailored decomposition reactions.89
2.5 Solid acid catalysts
Industrial processes for the formation of fine chemicals often use mineral acids as Brønsted-acid catalysts. Such homogenous acid catalysts are used in over-stoichiometric amounts and have to be neutralized after each batch in order to separate the products. Heterogeneous solid-acid catalysts can contribute to the case of recovery, reactivation and reuse.91 In large-scale chemical production homogenous acids were replaced by solid acids during the past years, but there is still a demand regarding such replacement in liquid phase reactions for fine chemical production.40,42,91 With respect to the production of valuable chemicals from biomass, the development of catalysts went through the same homogeneous–heterogeneous stages. Starting from homogeneous acid-catalyzed reactions, now a high potential is forecasted for heterogeneous porous catalysts.114 Since reactions often involve Lewis- and Brønsted-acid mechanisms, zeolites have already been investigated for solid-acid catalysis in biomass conversions during the past years.92MOF-based solid-acid catalysis currently attracts much attention since some MOFs can possess open metal sites, which can be active in Lewis-acid catalysis.40,93 In addition, the size and shape selectivity, which is possible through MOF pore and framework design, is interesting for acid-catalyzed reactions.50 At present MOF-based Lewis-acid catalysts are still similar or less active in direct comparison with their homogeneous or inorganic solid-acid catalysts. In a singular example where the MOF HKUST-1 was more active than H-BEA and Al-SBA-15 the higher concentration of active sites in HKUST-1 compared with H-BEA or Cu-BEA was seen as crucial together with the concerted effect of two adjacent active sites.94–96
Therefore, efforts were made to increase the number of accessible Lewis-acid sites in MOFs. De Vos and coworkers developed the concept of active site engineering.93 By adding hydrochloric acid and trifluoroacetic acid to the synthesis of UiO-66Zr, more active sites could be generated. The resulting MOF is more active in the conversion of citronellal than non-modified UiO-66Zr.97 It has been shown that both Lewis and Brønsted acidity and thereby the catalytic activity of MIL-100Fe could be enhanced through the formation of additional active sites using a postsynthetic acid treatment.93,98
When water is bound to transition metal ions as aqua ligands the pKa value of H2O (14 at 25 °C) decreases because of polarization effects. Metal ions polarize and thereby increase the acidity of their aqua ligands. For [Cr(H2O)6]3+,99,100 [CrBr(en)(H2O)3]2+,101 [Cr(en)(NH3)(H2O)3]3+,102 and some other Cr(III) aqua–amine complexes103 the first aqua-proton dissociation constant is estimated at pKa ≈4. The first acid dissociation constants for [Cr(III)(NH3)n(OH2)6−n]3+ lie between 4.4 and 5.3 depending on the number of aqua ligands and their cis, trans, fac or mer orientation.104 The acidity constant of cis-[Cr(C2O4)2(NCS)(H2O)]2− has been determined spectrophotometrically to be pKa = 7.06 ± 0.18.105
Thus, MOFs offer also Brønsted acidity through polarized aqua ligands (Fig. 11).106 A review by Jiang and Yaghi highlighted the importance of MOF-based Brønsted-acid catalysis. It is not trivial to differentiate between Lewis and Brønsted acidity in MOF-acid catalysts.107 Water on metal sites (aqua ligands) can stem from the MOF synthesis in water or can be generated during the catalyzed reaction, for instance in condensation reactions. There are also other possibilities besides aqua ligands to introduce Brønsted acidity into MOFs (Fig. 11). Acidic molecules, for example, polytungstic acid, can be encapsulated in the MOF pores during MOF synthesis, the organic linker can be functionalized with acidic moieties, such as SO3H groups, or acidic molecules other than H2O, such as trifluoroethanol, can be grafted on available metal sites.107,108
 | ||
| Fig. 11 Various Brønsted-acid sites in MOFs. Reprinted from ref. 107. Copyright 2015 with permission from the American Chemical Society. | ||
3. Scope of this review
There seems to be an unexplored potential in the field of MOF catalysis for biomass valorization. At the same time there is the question of the benefit of using MOFs for biomass catalysis with respect to other catalysts. Therefore, we aim to critically compare available studies applying MOF catalysts for the formation of platform chemicals from biomass-related starting materials (cf.Fig. 2) to other catalysts, taking strengths and weaknesses into account (cf.Fig. 6 and Table 1). Synthesis conditions, product yields and selectivity of products are given in tables in the ESI† for each chapter for MOFs and other catalysts. The focus in this review will be on acid catalysis, which is one of the most promising applications of MOFs in catalysis because (i) a high number of reactions also related to fine chemistry proceeds via acid catalysis, (ii) the catalytic activity originates from the MOF components, (iii) and it can be combined with various other mechanisms resulting in multifunctional catalysis.109 Acid-catalyzed reactions are water-containing liquid phase reactions; hence the above noted water stability of MOFs is of high importance.4. MOF catalysts in biomass transformation
4.1 Sugars as feedstock
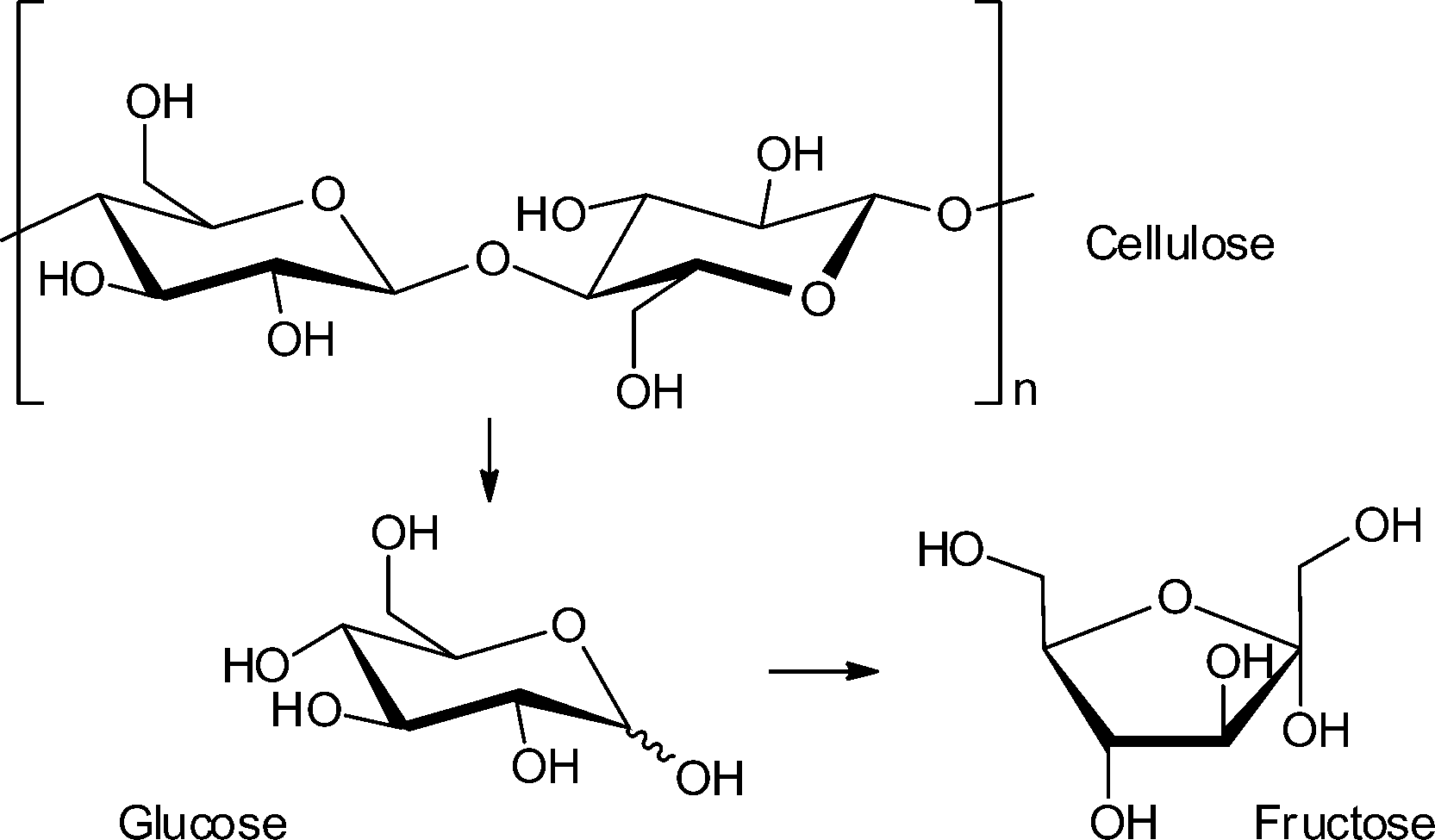 | ||
| Scheme 1 Cellulose hydrolysis to glucose and isomerization to fructose. | ||
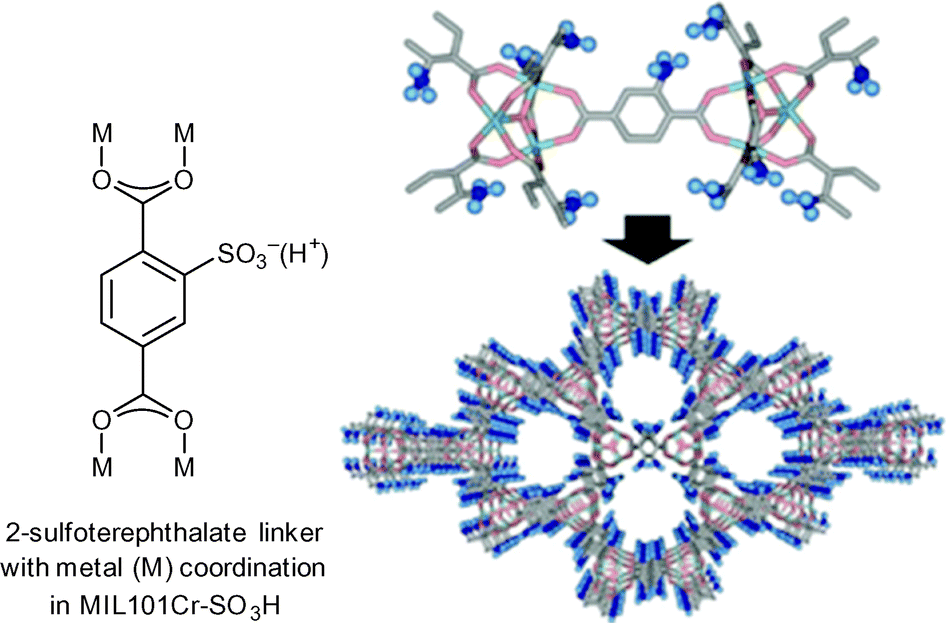 | ||
| Fig. 12 2-Sulfoterephthalate, MIL-101Cr-SO3H SBU and network. Adapted from ref. 34. Copyright 2011 with permission from John Wiley and Sons. | ||
The catalyst MIL-101Cr-SO3H gave only a very poor yield of 5.4% for mono- and disaccharides (total amount) from cellulose hydrolysis. Still, multiple run reactions over 13 cycles revealed the robustness of the catalyst. The overall low yield was explained by the poor solubility of crystalline cellulose in water.34
As a consequence, the same research group of Kitagawa investigated water soluble amylose and glucose as feedstock for isomerization to fructose and other monosaccharides. For this reaction MIL-101Cr-SO3H and other functionalized MIL-101Cr derivatives were used for the first time as catalysts.110
MIL-101Cr-SO3H gave the best glucose-to-fructose conversion with 21.6% yield (0.2 g catalyst, 25 mg glucose, water, 373 K, 24 h). NH2- and NO2-functionalized MIL-101Cr resulted in lower fructose yield (10.9% and 18.4%, respectively).110
The observed catalytic activity was ascribed to open metal sites (OMS) of the framework (Fig. 13). Simultaneous coordination by the substrate to two chromium coordination sites in the cis-position was suggested as it was reported for homogenous metal complexes.111 It is reasonable that the electron-withdrawing effect of NO2 and SO3H groups enhanced the acidity of chromium centers, which resulted in higher activity for glucose isomerization in comparison to MIL-101Cr-NH2. On the other hand, MIL-101Cr does not readily possess cis-positioned metal coordination sites (Fig. 13). To enable the hydride shift for Lewis acid-catalyzed isomerization of glucose to fructose, the bdc linkers have to partly detach from the SBU or defects have to be present in the structure leading to cis OMS. Both phenomena have been observed for MOFs97 but are not reported for MIL-101Cr. A large amount of MIL “catalyst” relative to substrate was used (200 mg catalyst vs. 25 mg glucose), so we suggest that other mechanisms than a Lewis acid-catalyzed reaction should be taken into account.
 | ||
| Fig. 13 (a) MIL-101Cr SBU, X = F, OH or solvent; (b) homogeneous metal catalyzed hydride shift in glucose-to-fructose isomerization. The figure was redrawn from ref. 111. | ||
For the conversion of amylose it was shown that fructose can only be obtained when hydrochloric acid served as co-catalyst for the initial conversion of amylose to glucose. MIL-101Cr-SO3H was described as stable in acidic environment, but no sorption or PXRD data was given to prove that the catalyst was unchanged after the reaction. After long reaction times (48–120 h) 5-HMF and levulinic acid were also detected but conversion rates from glucose were not reported.110
 | ||
| Scheme 2 Cellulose and cellobiose into sorbitol. | ||
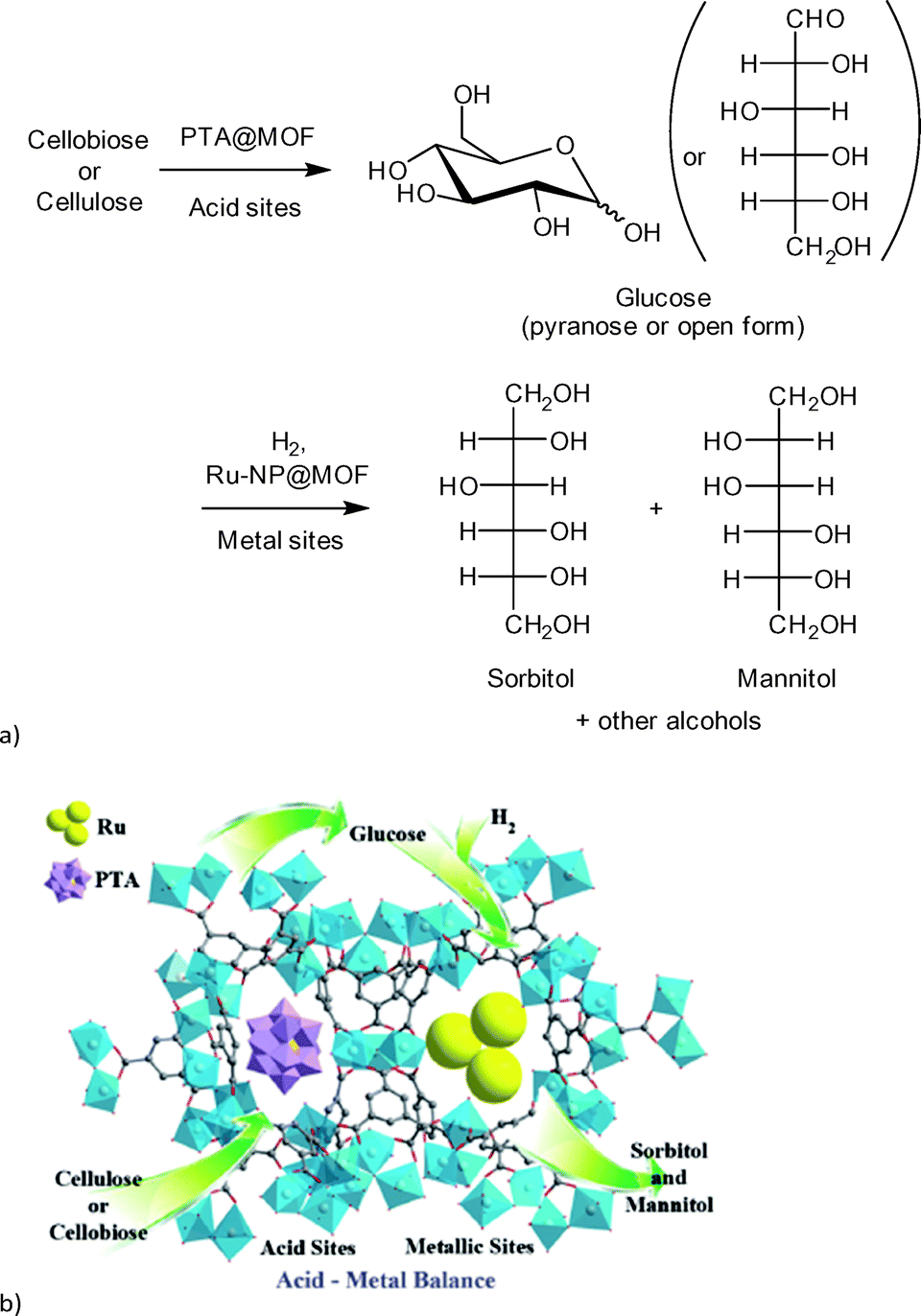 | ||
| Fig. 14 (a) Reaction scheme of cellulose conversion to sorbitol with Ru(nanoparticle)/PTA@MIL-100Cr; (b) proposed function of MOF composite catalyst. Redrawn and reprinted from ref. 112. Copyright 2013 with permission from John Wiley and Sons. | ||
A MOF was chosen as support because the phosphotungstic acid (PTA = H3PW12O40) loading could be controlled effectively by using the “ship in a bottle” approach. A sorbitol yield of 57.9% was achieved by adjusting the ratio of acid (PTA) and ruthenium nanoparticle metal catalyst. Importantly, the MOF alone showed no significant catalytic activity. Only very low amounts of glucose (0.5%), ethylene glycol and glycerol (4.1% combined) were measured. For cellobiose an even higher sorbitol yield of 95.1% was reached using Ru/PTA@MIL-100Cr.112 Reference experiments using only MIL-100Cr or PTA@MIL-100Cr gave low amounts of glucose and ethylene glycol/glycerol (13.5%, 7.2% and 11.3%, 5.6%, respectively). Ru@MIL-100Cr resulted in a sorbitol yield of 56%, which illustrates the need for careful balance of acidity.112 This example shows the advantages of MOFs as host matrix. It would be very interesting to investigate this reaction using a strong Brønsted acid MOF such as MIL-101Cr-SO3H. The selectivity of products can be significantly changed towards the production of ethylene glycol (44.6%) using basically the same catalysts by adjusting the amount of PTA and ruthenium as well as temperature parameters. As MOF support, HKUST-1 instead of MIL-100Cr was used.113
Competitive heterogeneous catalysts applied in the conversion of biomass derived compounds are sulfonated resins, such as Amberlyst, EBD resins and Nafion, nanoporous metal oxides (sulfated protonated metal oxides such as SO42−/ZrO2), hydrotalcites, activated carbon and ordered mesoporous carbon as well as silica (MCM, SBA) and zeolites.114
In the case of cellulose hydrolysis as well as glucose isomerization MOF-based catalysts cannot compete with other heterogeneous catalysts. Zeolites and sulfonated carbon display significantly higher activity (Table S2 in the ESI†).115,116
Only the PTA@MIL-100Cr catalyst can match the activity of other heterogeneous catalysts for the conversion of cellulose into sorbitol and exhibited higher selectivity than 1% Rh–5% Ni/mesoporous carbon and H4SiW12O40–Ru/C.117,118
 | ||
| Scheme 3 Conversion of fructose to 5-HMF. | ||
Bromberg and coworkers investigated the conversion of fructose into 5-HMF using MIL-101Cr embedded in a cross-linked, catalytically active poly(N-bromomaleimide) (PMAi-Br) polymer (Fig. 15).122
 | ||
| Fig. 15 Schematic representation of MIL-101Cr@polymer composite. Reprinted with permission from ref. 122. Copyright 2014, American Chemical Society. | ||
The amorphous polymer network filled the MOF pores to some extent, but the BET surface areas of the composite were still between 1100 and 1600 m2 g−1, which showed a certain accessibility of the pores. PMAi-Br is a polymeric, water- and solvent-insoluble analogue of N-bromosuccinimide (NBS). Homogeneous NBS is an active catalyst in the dehydration of fructose and other monosaccharides since it is a source of electrophilic bromine. Fig. 16 illustrates the proposed mechanism of fructose dehydration to 5-HMF involving a nucleophilic attack of a Br− ion.
 | ||
| Fig. 16 Coordination of the N(–Br) atom of PMAi-Br as a polymer ligand to the Cr OMS in MIL-101Cr releases a bromide anion for a nucleophilic attack on the fructofuranosyl oxocarbenium ion acccording to the suggested mechanism for the fructose to 5-HMF dehydration.122 Redrawn with permission from ref. 122. | ||
PMAi-Br alone already gave a 5-HMF yield of 50% from fructose after 1 h, but the MIL-101Cr-PMAi-Br composite achieved a significantly higher 5-HMF yield of 86% after 1 h (Fig. 17). The authors explained the increased activity with the higher surface area of MOF-PMAi-Br in comparison to the non-porous polymer and also with a synergism in which the polymer ligand PMAi-Br is coordinated to the unsaturated sites of MIL-101Cr and thereby releases Br− ions (Fig. 16).122
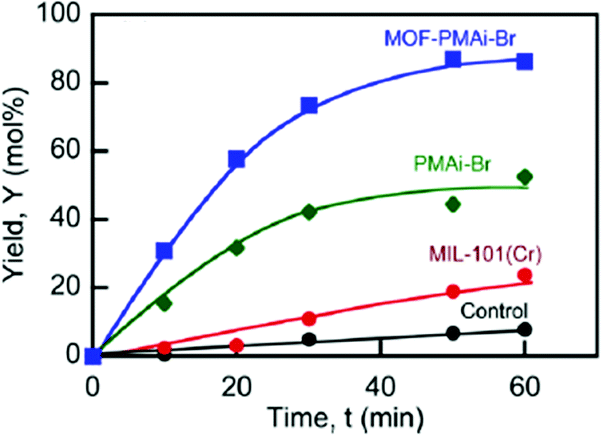 | ||
| Fig. 17 5-HMF yield (%) from fructose at 100 °C. Conditions: DMSO, 5 mL; D-fructose concentration (Cfo), 0.555 mol L−1; catalyst loading, 50 mg mL−1. Control: DMSO solvent with no catalysts added. Reprinted with permission from ref. 122. Copyright 2014, American Chemical Society. | ||
5-HMF formation was also observed without catalyst in DMSO as a solvent (“Control” in Fig. 17), which is in line with the previous study of Zhang et al.119 MIL-101Cr in DMSO gave a 5-HMF yield of 24% after 1 h (Fig. 17).
For the dehydration of glucose with MIL-101Cr-PMAi-Br a 5-HMF yield of 7% and 16% after 2 and 6 h, respectively, was reported and was used as an argument against the leaching of chromium species from MIL-101Cr since soluble chromium salts are known to catalyze the isomerization of glucose to fructose (see also section 4.1.4).122
Chen et al. reported the conversion of fructose into 5-hydroxymethylfurfural by different sulfonic acid functionalized MOFs using DMSO as solvent at 120 °C for 1 h.123 The supposedly water stable MOFs MIL-101Cr, UiO-66 and MIL-53Al had been functionalized by postsynthetic reaction with chlorosulfonic acid.123 All three SO3H-functionalized MOFs were active in fructose dehydration. For MIL-101Cr-SO3H different degrees of sulfonic acid functionalization from 3 mol% to 15 mol% revealed an increase in 5-HMF yield with the SO3H content. At 120 °C, after 1 h 5-HMF was obtained in 90% yield with MIL-101Cr-SO3H-15% (Fig. 18). Adsorption experiments indicated that MIL-101Cr-SO3H-15% showed the highest adsorption of fructose (0.006 mmol g−1), whereas MIL-53Al-SO3H-8.2% adsorbed the most 5-HMF (0.09 mmol g−1) (changes were measured with HPLC). For the SO3H-functionalized MOFs the 5-HMF adsorption is significantly higher than the fructose uptake.123
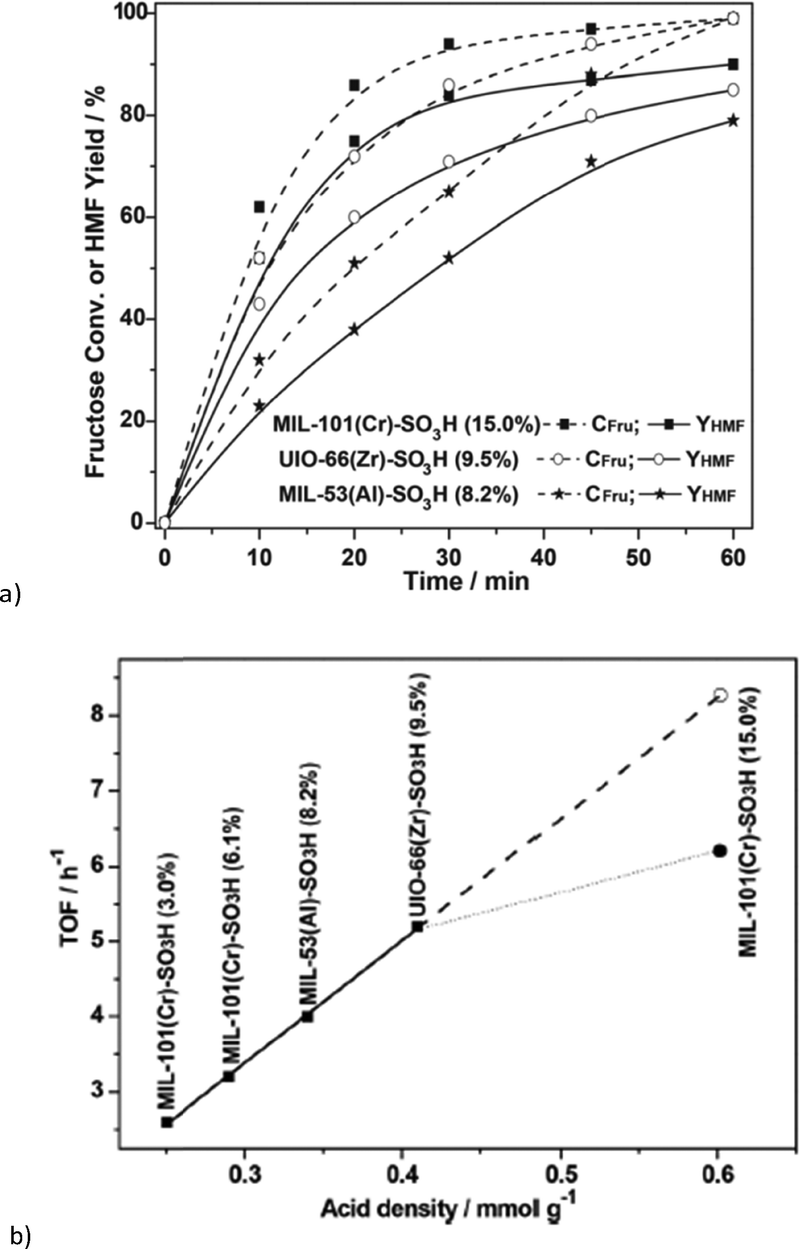 | ||
| Fig. 18 (a) Comparison of catalytic performance of different MOF-SO3H on fructose to 5-HMF transformation. Fructose conversion (dotted lines) also includes the formation of by-products (polymeric humins) to 5-HMF. Conditions: fructose (500 mg), MOF-SO3H (300 mg), DMSO (5 mL), 120 °C; (b) TOFs versus molar sulfonic acid-site density of MOF-SO3H. (TOFs for the fructose-to-HMF transformation were measured at t = 10 min, given as amount of consumed fructose per amount of MOF-SO3H per hour. Conditions: fructose (500 mg), MOF-SO3H (300 mg), DMSO (5 mL), 120 °C, 10 min). Reprinted from ref. 123. Copyright 2014 with permission from the Royal Society of Chemistry. | ||
The turnover frequency (TOF) of fructose transformation showed a linear relation with the sulfonic acid content of MOF-SO3H from 3% to 9.5% SO3H derivatization, irrespective of the actual MOF (MIL-101Cr, MIL-53Al or UiO-66Zr). Hence, the catalytic activity of MOF-SO3H was ascribed to the Brønsted-acidic SO3H groups, independently from the parent MOF. Only for MIL-101Cr-SO3H-15% is the TOF lower than expected from extrapolation (6.2 h−1 instead of 8.3 h−1) (Fig. 18, bottom). For MIL-101Cr-SO3H-15% the authors ascribed this discrepancy to the resulting lower accessible pore volume.123 It could also be suggested, however, that the TOF levelling off with SO3H content stems from diffusion control of the reaction rate. The pore volume of MIL-53Al-8.2% is already lower than that of MIL-101Cr-6.2%. Above a certain number of active sites a further increase will not lead to an increase in reaction rate if the diffusion rate limit has already been reached or is being approached.
Kinetic studies of MIL-101Cr-SO3H-15% (kobs 2.01 h−1, activation energy 55 kJ mol−1) and MIL-101Cr-PMAi-Br (kobs 2.97 h−1, activation energy 53 kJ mol−1) revealed a slightly better performance of MIL-101Cr-PMAi-Br.122,123
The carbohydrate feedstocks inulin, sucrose and cellobiose gave moderate 5-HMF yields of 46%, 44%, and 24%, respectively, with MIL-101Cr-SO3H-15% as catalyst. Glucose conversion using MIL-101Cr-SO3H-15% in DMSO or in the ionic liquid [BMIM]Cl resulted in very low 5-HMF yields of 7–8%.123
Hu and coworkers reported the superior performance of a hafnium based MOF named NUS-6(Hf) for the dehydration of fructose to 5-HMF.124 NUS-6(Hf) and NUS-6(Zr) were hydrothermally synthesized at (only) 80 °C in water with acetic acid as modulator and are largely isostructural to UiO-66, albeit with 2-sulfoterephthalate instead of the terephthalate linker and with partially missing linkers. Defect formation led to the formation of additional mesopores around 4 nm (Fig. 19) according to the Barrett–Joyner–Halenda (BJH) calculated pore size distribution from nitrogen sorption isotherms. NUS-6(Hf) and NUS-6(Zr) exhibited BET surface areas of 550 and 530 m2 g−1, respectively. The linker 2-sulfotherephthalate was previously reported to give unstable UiO-type frameworks. Here the use of a modified synthesis procedure for NUS-6(Zr) and NUS-6(Hf) allowed them to be activated under retention of the structure, which was important for their application in catalysis.124
 | ||
| Fig. 19 1 × 2 × 2 super unit cell of NUS-6(Hf) with mesopores indicated by yellow spheres. Reprinted with permission from ref. 124. Copyright 2016, American Chemical Society. | ||
The first attempts of fructose dehydration with NUS-6 were performed in water as solvent but resulted only in negligible amounts of 5% 5-HMF; hence the authors adjusted their solvent to DMSO following the work of Chen et al.123 Subsequently, NUS-6(Zr) (3.5 mol% catalyst) gave quantitative conversion of fructose (50 mg) with a yield of 5-HMF of 84% after 1 h. Fructose conversion (solid lines and symbols in Fig. 20) also includes the formation of by-products (e.g. polymeric humins). NUS-6(Hf) outperformed all presently known other MOF or heterogeneous catalysts114 with a 5-HMF yield of 98% after 1 h using the same reaction conditions (Fig. 20).
 | ||
| Fig. 20 Kinetic study of fructose dehydration to 5-HMF with NUS-6 and DMST catalysts (DMST = dimethyl-2-sulfoterephthalate). Conditions: fructose (50 mg), 3.5 mol% catalyst (2.7 mg for DMST, 35 mg for NUS-6(Zr), 50 mg for NUS-6(Hf)), DMSO (1 mL), 100 °C, 1 h. Reprinted with permission from ref. 124. Copyright 2016, American Chemical Society. | ||
Catalysis with the homogeneous linker dimethyl-2-sulfoterephthalate (DMST) resulted in faster conversion of fructose than with heterogeneous MOF NUS-6 catalysts but gave a lower yield (and selectivity) of 5-HMF (Fig. 20).
As seen from the above studies, Brønsted-acidic MOF catalysts can compete with other heterogeneous catalysts, such as Amberlyst125,126 and Nafion127 in the dehydration of fructose to 5-HMF. However, in water or biphasic (aqueous–organic) solvent mixtures, MOFs have not been very successful yet for fructose conversion although numerous other heterogeneous catalysts were shown to be active in the presence of water.128 For comparison of MOFs with heterogeneous catalysts only fructose-to-5-HMF reactions in DMSO have thus been considered. DMSO is apparently inhibiting humin formation to a large extent. Humins are insoluble polymers, complex carbonaceous materials which usually form as undesired by-products in acid-catalyzed conversion of biomass.129
In DMSO MIL-101Cr-SO3H-15% and NUS-6(Hf) are among the best heterogeneous catalysts reported so far for the conversion of fructose into 5-HMF (Table S3†).128 Although high 5-HMF yields are reported from fructose dehydration in DMSO, product separation is problematic due to the formation of toxic sulfur compounds during high temperature DMSO distillation. Therefore, low boiling solvents or water would be required if a technical process is envisioned.130
More sustainable than using fructose as feedstock would be the so-called “second-generation biomass feedstocks” from non-food sources of crops (stems, leaves and husks) or agricultural and forestry waste. Therefore, processes must be optimized to obtain 5-HMF from glucose or even cellulose (Fig. 2).11
 | ||
| Scheme 4 Conversion of glucose to 5-HMF. | ||
For the conversion of glucose involving MOF catalysts the highest reported 5-HMF yield was low, 16% after 6 h using MIL-101Cr-PMAi-Br, and the activity was mainly ascribed to PMAi-Br (release of Br− nucleophiles) instead of the MOF MIL-101Cr (cf.Fig. 17).122
Herbst et al. compared the activity of MIL-101Cr, its nitro, sulfonic acid and mixed nitro/sulfonic acid derivatives, MIL-101Cr, MIL-101Cr-NO2, MIL-101Cr-NO2/SO3H, MIL-101Cr-SO3H(33%) and MIL-101Cr-SO3H(100%), for glucose conversion into 5-HMF.134 For the first time a MOF was successfully applied in a biphasic water-based solvent mixture, namely THF:H2O (v:v 39:1) for the glucose-to-5-HMF conversion. MIL-101Cr-SO3H was found to achieve 29% conversion of glucose to 5-HMF after 24 h (Fig. 21).134
 | ||
| Fig. 21 (a) 5-HMF yield from glucose with different MIL-101Cr catalysts. (b) Time-dependent conversion for MIL-101Cr-SO3H (glucose 223 mg, 1.24 mmol, catalyst 5.22 × 10−5 mol, 5 mL THF/H2O v:v 39:1, 130 °C, 24 h). Reprinted from ref. 134. Copyright 2016 with permission from the Royal Society of Chemistry. | ||
When the reaction with MIL catalysts was carried out in pure THF no product was formed, revealing the indispensability of water for the glucose-to-5-HMF conversion. MIL-101Cr-SO3H preferentially led to 5-HMF over levulinic acid (molar ratio 1:0.3), while the catalysts Amberlyst-15 and sulfuric acid form mostly levulinic acid with 5-HMF to levulinic acid ratios of 1:3 and 1:10, respectively (Fig. 22).134 At the same time, MIL-101Cr-NO2 is most selective, yielding only 5-HMF and showing no formation of levulinic acid. Using 5-HMF as substrate did not result in any conversion to levulinic acid with MIL-101Cr-SO3H, thereby ruling out the catalytic formation of levulinic acid from 5-HMF.134
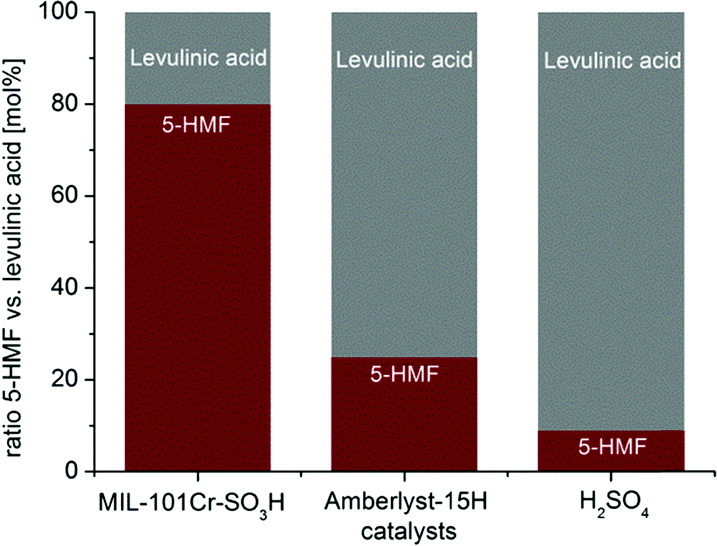 | ||
| Fig. 22 Comparison between MIL-101Cr-SO3H, Amberlyst-15H and H2SO4 for the ratio between 5-HMF and levulinic acid from glucose conversion by 1H NMR. Conditions: catalyst MIL-101Cr-SO3H 5.22 × 10−5 mol, glucose 223 mg (1.24 mmol) in 5 mL THF:H2O v:v 39:1, 130 °C. For Amberlyst-15 and H2SO4 the reaction was conducted with 1.57 × 10−4 mol of the catalyst under the same conditions. Reprinted from ref. 134. Copyright 2016 with permission from the Royal Society of Chemistry. | ||
Conversion of maltose resulted in 50% 5-HMF yield (saccharide solutions were 5 wt%) with MIL-101Cr-SO3H catalyst (Fig. 23).134
 | ||
| Fig. 23 Conversion of maltose to 5-HMF. | ||
Catalyst recycling experiments showed that MIL-101Cr-SO3H stays porous and crystalline but becomes deactivated apparently through fouling by humin formation. With the use of ethanol as an alternative reaction medium the formation of insoluble humins could be prevented, but the yield of 5-HMF and 5-ethyl-HMF decreased to 5% and 11%, respectively, after 24 h (Fig. 24).134
 | ||
| Fig. 24 Ethanol as alternative reaction medium for the formation of 5-ethyl-HMF. | ||
Su and coworkers also investigated the activity of MIL-101Cr-SO3H in the glucose-to-5-HMF conversion.135 They used a solvent mixture of γ-valerolactone (GVL) and H2O (9:1) at 150 °C. The glucose-to-catalyst molar ratio can be calculated as 5:1 or 2:1 (three Cr ions per catalyst formula unit), thereby taking into account that only 60% of the linkers had a SO3H group. In the study of Herbst et al., the glucose-to-MIL-SO3H ratio was 24:1. Su et al. synthesized MIL-101Cr-SO3H from monosodium sulfoterephthalic acid, HF and Cr(NO3)3 instead of CrO3. The average particle size of MIL-101Cr-SO3H was 400 nm and the BET surface area was 1700 m2 g−1.135 The acidity was calculated from back-titration experiments with saturated NaCl to be 1.61 mmol g−1. Unfortunately, no titration curves were given. Su et al. performed their experiments using 30 mg of glucose, 30 mg of MIL-101Cr-SO3H, 1 mL of solvent (GVL/H2O 9:1) and different temperatures from 110 °C to 150 °C. Increasing the temperature to 150 °C resulted in a maximum yield of 45% 5-HMF. The reported 5-HMF selectivity of 46% went together with the formation of humins. Also, the lower yield of 5-HMF in pure water (12%) is in line with the results of Herbst et al. Using pure GVL gave a 5-HMF yield of 26% after 2 h. Probably due to shorter reaction times and lower substrate-to-catalyst ratio recycle experiments lasting for 2 h each showed no loss in activity for at least 5 runs.135
In terms of the mechanism, Su et al. referred to the 1,2-hydride shift, proven for Lewis-acidic zeolites. Through HPLC experiments small amounts of fructose could be identified, although the possibility of alternative intermediates is not discussed. They suggested second-order kinetics for the reaction. In fixed bed reactions, a 5-HMF yield between 35% and 45% over 56 h was obtained.135
It is generally more difficult to obtain 5-HMF from glucose with high selectively and yield using heterogeneous catalysts.128
In contrast to fructose conversion, for the glucose-to-5-HMF transformation MIL-101Cr-SO3H is only a low-to-medium activity catalyst with moderate yields of below 30% for 5-HMF.136 Mesoporous tantalum phosphate as well as Sn montmorillonite (Sn-Mont) display significantly higher 5-HMF yields in a shorter time under similar conditions.137,138 MIL-101Cr-SO3H displays similar activity to zirconium phosphate (ZrPO, 24% yield in H2O/methyl isobutyl ketone (MIBK), 165 °C, 6 h).139
 | ||
| Scheme 5 Conversion of sucrose to methyl lactate. | ||
ZIF-8 and ZIF-67 are both composed of 2-methylimidazolate linkers and display the same sodalite type structure.140,141 They differ in the metal, which is Zn for ZIF-8 and Co for ZIF-67. ZIF-8 achieved a methyl lactate yield of 35% (24 h, 160 °C, 225 mg = 0.65 mmol sucrose, 160 mg = 0.70 mmol “catalyst”, methanol) (Fig. 25a). Low amounts (<2%) of pyruvaldehyde dimethyl acetal (PADA) and 1,1,2,2-tetramethoxypropane (TMP) as well as (<4%) non-identified products were also observed. By variation of the already stoichiometric “catalyst” amount from 160 mg to 500 mg (substrate:catalyst molar ratio 1:1 and 1:3, respectively) a slight increase in yield to 42% was measured. In direct comparison with Zn(NO3)2, ZIF-8 outperformed the metal salt since the Zn nitrate gave only 22% yield of methyl lactate. In contrast, ZIF-67 is less active than Co(NO3)2, with 19% versus 26% yield. The higher activity of ZIF-8 over ZIF-67 is ascribed to the smaller particle size (150 nm vs. 1 μm) and stronger Lewis-acidic Zn over Co sites.18
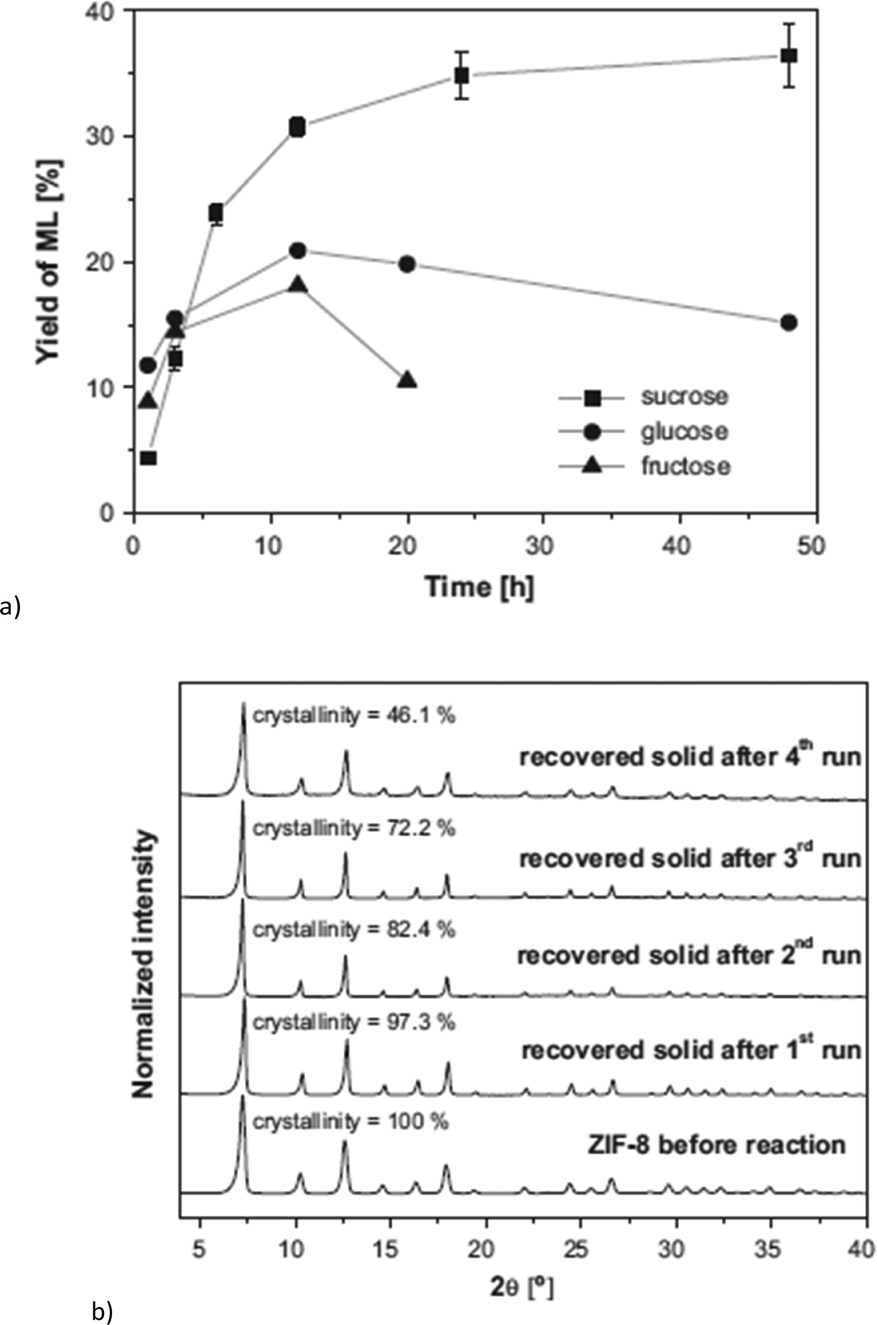 | ||
| Fig. 25 (a) Yield of methyl lactate (ML) obtained over ZIF-8 using different sugars as reactants. Error bars in the case of sucrose derived from at least 4 different experiments (reaction conditions: 160 °C, 160 mg of catalyst and 225 mg of sugar, methanol); (b) XRD patterns of ZIF-8 before the reaction (fresh catalyst) and recovered solids after the various catalytic cycles, including percentage of crystallinity, referred to catalyst before reaction. Reprinted from ref. 18. Copyright 2016 with permission from Elsevier. | ||
After reuse of the ZIF-8 catalyst over four cycles the methyl lactate yield decreased from 35% in the first cycle to 27% in the fourth cycle. PXRD after each cycle shows that the bulk ZIF-8 material stayed crystalline and its structure was retained (Fig. 25b). However, the BET surface area decreased from 1391 m2 g−1 to 757 m2 g−1 indicating deactivation of the catalyst through fouling processes. It is known that in carbohydrate conversion various products including insoluble humins can be formed.129
Different activation processes for ZIF-8, such as methanol washing, calcination at different temperatures and vacuum drying were investigated, with the latter method giving the best results. The discrepancy of sugar conversion (for ZIF-8 around 98%) and total identified product yield of (only) 12–40% depending on reagent (sucrose or glucose) showed the significance of unwanted and unidentified side reactions. The higher yield for sucrose in comparison with glucose and fructose in the conversion to methyl lactate (Fig. 25a) was ascribed to the slower hydrolysis of sucrose in water to 1 eq. glucose and 1 eq. fructose, resulting in fewer side reactions. Since sucrose is a disaccharide and too large to diffuse into the ZIF-8 pores, which are 11.6 Å in diameter (with pore windows of 3.4 Å),142 hydrolysis occurs at the pore mouth. The molecular diameter of the monosaccharides glucose and fructose was given as 8.5 Å which is still larger than the pore window. Even though adsorption of carbohydrates was stated as possible by referencing to the described gate effect of ZIF-8 (ref. 143) and adsorption of caffeine (6.1 × 7.6 Å),144 it remains questionable if catalysis really occurs inside the MOF pores. The group of Taarning et al. investigated the activity of the tin-containing heterogeneous catalysts Sn-Beta, Sn-MCM-41 and Sn-MFI for the conversion of sucrose using similar conditions. The performance of ZIF-8 (35% methyl lactate yield) is better than that of Sn-MCM-41 (28%) and Sn-MFI (24%) but Sn-Beta gave a higher yield of 57% methyl lactate (Table S3†).145,146
4.2 Furans as feedstock
 | ||
| Scheme 6 (a) Conversion of 5-HMF to 2,5-dihydroxymethyl-tetrahydrofuran (DHMTHF) and (b) conversion of furfural to furfuryl alcohol. | ||
Alternatively, Chen et al. described the presence of amine moieties in different MOFs (MIL-101Cr, MIL-53Al, MIL-101Al-NH2, and MIL-53Al-NH2) as a key feature not only for the formation of uniform and well-dispersed Pd-NPs but also for the preferential adsorption of 2,5-dihydroxymethylfuran (DHMF) over 5-HMF. In turn this is supposed to enable further hydrogenation to 2,5-dihydroxymethyl-tetrahydrofuran (DHMTHF). The higher adsorption of DHMF is ascribed to its more hydrophilic nature as well as the improved hydrogen bonding interactions of DHMF compared to 5-HMF. The stronger H-bonding interactions through two hydroxyl groups of DHMF may hamper the dissociation of DHMF from coordinating metal sites and can lead to further hydrogenation to DHMTHF.147
The procedure for Pd-NP encapsulation has to be regarded critically since the reaction involves the treatment of activated MIL-101Al-NH2 in H2O with hydrochloric acid followed by reduction of the Pd precursor H2PdCl4 with NaBH4 at 0 °C.147 The detailed study of Leus et al. showed that MIL-101Al-NH2 is not stable under aqueous and acidic conditions.66 In addition, the surface areas of the parent MOFs were unusually low compared with the literature values. Nevertheless, the supposed material Pd@MIL-101Al-NH2 was probed in a 5-recycle run experiment in the hydrogenation of 5-HMF to DHMTHF (Scheme 6). After the 5th cycle, product selectivity decreased from 96% (1st cycle) to 80%. Pd leaching was determined to only 0.03% by ICP-AES analysis. The other tested catalysts (Pd@MIL-101Cr, Pd@MIL-53Al and Pd@MIL-53Al-NH2) showed lower activity than Pd@MIL-101Al-NH2 but still moderate to good selectivity for DHMTHF. Generally, the amino-modified MILs displayed a better product selectivity than the comparable non-amino supports, which underlines the above stated role of amine groups.147
 | ||
| Scheme 7 Conversion of 5-HMF to 2,5-diformylfuran (DFF). | ||
The work of Fang et al. presented the first application of MOF-derived metal–carbonaceous materials for biomass transformation.149 As the parent MOF material the mixed-metal MOF MIL-45b formed by CoCl2·4H2O, fine Fe powder and trimesic acid in H2O at 180 °C was used. The characterized MOF was pyrolized by heating under an argon atmosphere at different temperatures from 500 °C to 800 °C.149
The resulting FeCo/C catalysts were characterized by XRD, TEM, XPS and EDX. EDX analysis revealed the homogeneous distribution of an Fe and Co nanoparticle phase within the carbon particle. Different metal and metal oxide species were detected by XRD and XPS. The real composition of the catalyst remained inconclusive but was described by Fang et al. as an Fe/Co alloy. The lowest calcination temperature of 500 °C seemed to yield hollow nanoparticles of approximately 60 nm (Fig. 26). FeCo/C(500) exhibited a BET surface area of 243 m2 g−1 which decreased with higher calcination temperature along with the catalytic 5-HMF conversion.149
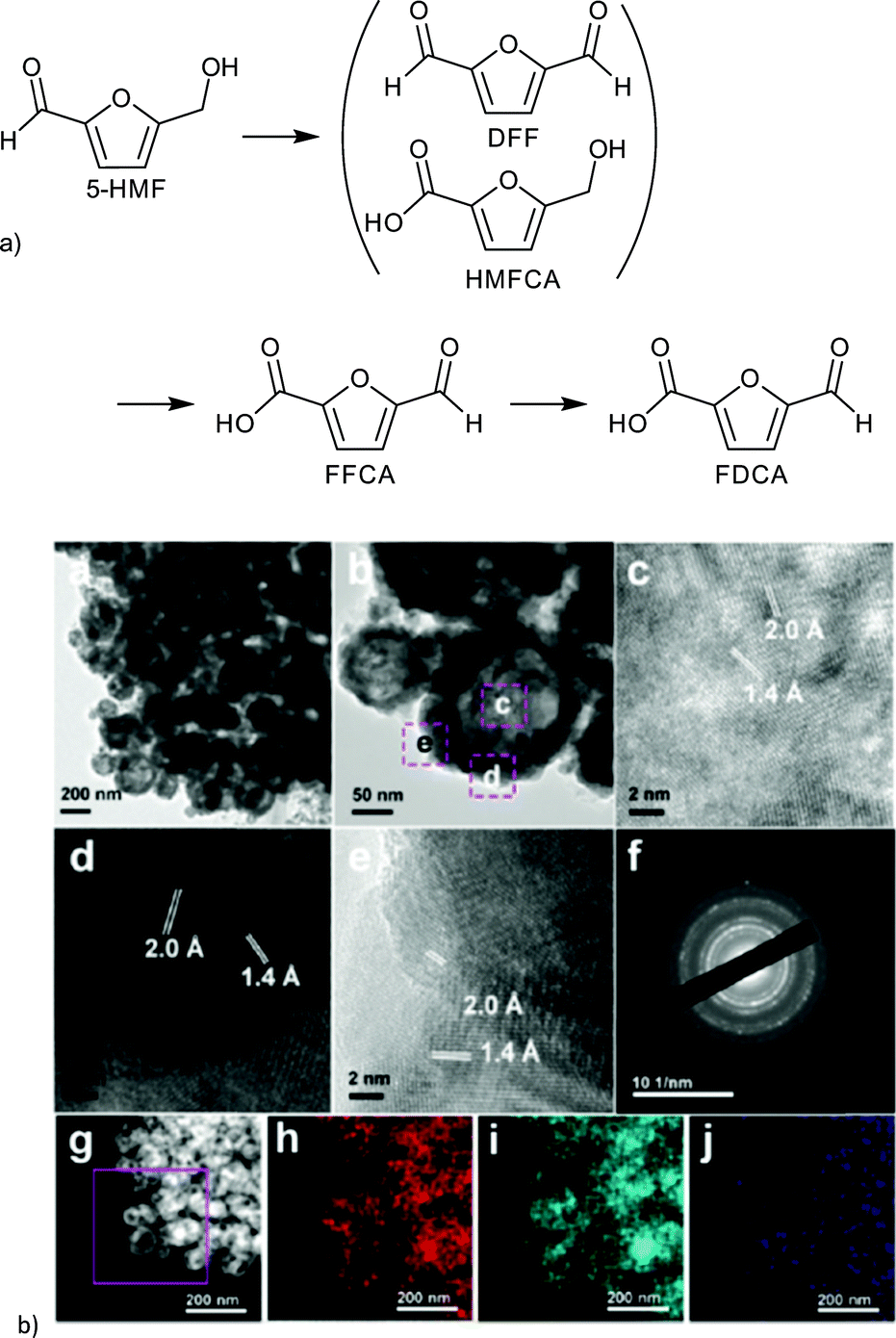 | ||
| Fig. 26 (a) Products of HMF oxidation. (b) (a–e) HRTEM images of FeCo/C(500); (f) SAED pattern; (g) HAADF-STEM images of FeCo/C(500) and the corresponding elemental mappings of (h) Fe, (i) Co and (j) C. Redrawn and reprinted from ref. 149. Copyright 2016, with permission from the Royal Society of Chemistry. | ||
The change in several parameters such as temperature, O2 pressure, solvent and calcination temperature of the catalyst on the 5-HMF oxidation was investigated. After optimizing the reactions conditions (to 100 °C, 1 MPa O2, 6 h, toluene, Na2CO3) a yield of >99% DFF was obtained. FeCo/C(500) could be easily recovered by magnetic separation and reused up to six runs without any significant loss in reactivity.149
Using the non-noble Fe–Co-based MOF-derived catalysts the authors presented a sustainable, cost-effective, and highly efficient system for the conversion of 5-HMF to DFF.149 The activity of FeCo/C(500) was compared to that of several noble metal catalysts and was found to have a comparable high activity under similar or even milder reaction conditions (Table S4†).150,151
4.3 Levulinic acid as feedstock
 | ||
| Scheme 8 Conversion of levulinic acid to γ-valerolactone. | ||
The resulting composites consisted of metal nanoparticles supported on nanoporous carbon. The materials exhibited BET surface areas of 900 m2 g−1 up to 2300 m2 g−1 and mesoporosity. The embedded nanoparticles had sizes between 3–8 nm (M/NC3 derived from M(Ru,W,V,Ti)@IRMOF-3) and 10–18 nm (M/NC1 derived from M(Ru,W,V,Ti)@IRMOF-1). The smaller nanoparticle size in M/CN3 was traced to the ability of IRMOF-3 to coordinate metal precursors via amino groups, leading to site isolation and minimization of aggregation of metal nanoparticles during CCR. Metal loadings differed significantly from 1 up to 10 wt%, depending on the kind of incorporated metal. The Zn ions from the MOF framework (IRMOF-1 and -3) were completely removed, as proven by X-ray fluorescence analysis.152
The synthesis procedure of Kim et al.152 and Fang et al.149 differed in the calcination temperature (500–800 °C vs. 1000 °C) and an additional passivation in O2 flow (Kim et al.). Nevertheless, the most significant difference was the MOF material, since for MIL-45b no experimental surface area was stated, whereas the IRMOF materials exhibited BET surface areas between 1500 and 320 m2 g−1 (IRMOF-3, before and after metal impregnation) and 800–330 m2 g−1 (IRMOF-1, before and after metal impregnation).152
M/NC3 materials were shown to be highly active catalysts for liquid phase conversion of model compounds and derivatives of lignocellulosic biomass.152
Benzyl alcohol as a model compound for aromatic groups in lignin was oxidized into benzaldehyde. Levulinic acid as a raw material obtainable from lignocellulosic biomass was hydrogenated into γ-valerolactone and esterified to methyl levulinate using different M/NC materials. In all cases M/NC3 materials derived from amino-terephthalate IRMOF3 showed better performance than M/NC1 from terephthalate IRMOF-1 which correlates with the observed smaller sized M-NPs (M = Ru, W, V, Ti).152 Best results were obtained for the hydrogenation of levulinic acid using Ru/NC3. γ-Valerolactone was obtained with 97% yield and 100% selectivity under the following conditions: levulinic acid (1.36 mmol), H2O (20 mL), 2 MPa N2, 130 °C, 6 h, Ru/NC3 (Ru 8.64 μmol), catalyst:reactant 1:160.152
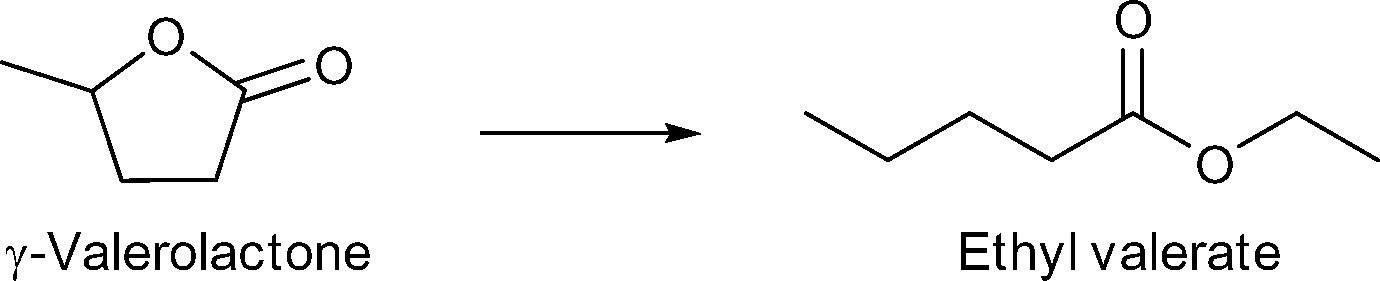 | ||
| Scheme 9 Conversion of γ-valerolactone to ethyl valerate. | ||
MIL-101Cr-SO3H(25–100%) not only acted as support for Pd NPs but also had an influence on the selectivity. Ring-opening, hydrodeoxygenation and hydrogenation starting from γ-valerolactone can yield products 1–4 in Fig. 27, with ethyl valerate 4 having been the desired product in the work of Zhang et al.
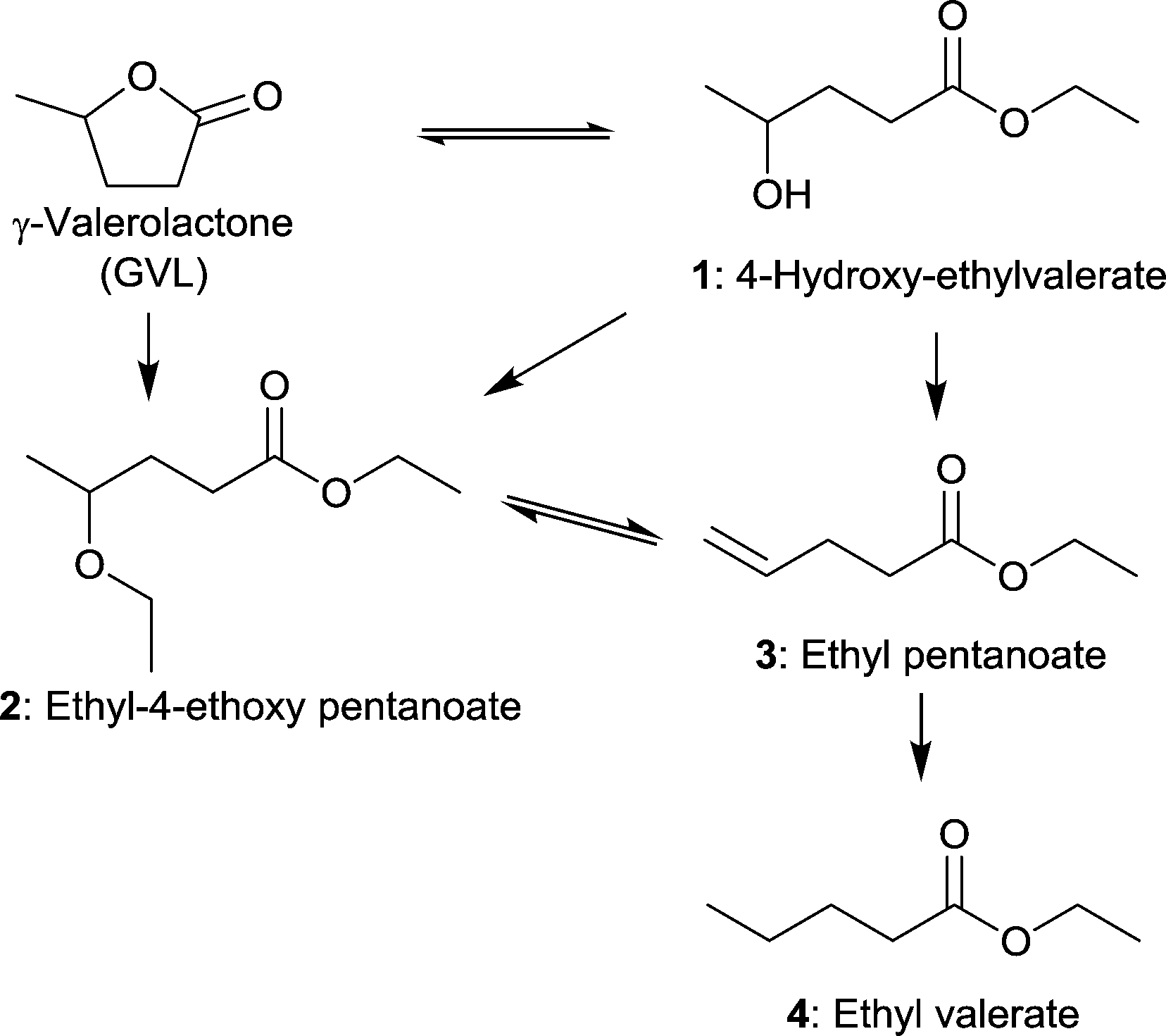 | ||
| Fig. 27 Reaction pathways of GVL conversion in ethanol; obtainable products 1–4 with MIL-101Cr-SO3H or Pd@MIL-101Cr-SO3H are depicted. Additional by-products were omitted for clarity. Modified from ref. 153. | ||
The authors carefully compared the activity and selectivity of MOF catalysts MIL-101Cr-SO3H with the Pd-NP supported Pd@MIL-101Cr-SO3H catalysts.
Significant GVL conversions of 39% to 44% were obtained for MIL-101Cr-SO3H(50%) and for MIL-101Cr-SO3H(100%), respectively (Table 2). The reaction temperature was about 200 °C. When the temperature was raised to 250 °C (entry 7) a conversion of 86% for GVL to three different products was achieved. Concerning product formation a trend could be observed: the higher the SO3H content, the more ethyl pentanoate (3) was obtained, whereas the main product is ethyl-4-ethoxy pentanoate (2) with 66% to 70% selectivity for MIL-101Cr-SO3H(50,80100%).153
| Selectivity (%)b | ||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Sulfur/Crc | Conv. (%) | 1 | 2 | 3 |
| a Table taken from ref. 153. Copyright 2014 with permission from the Royal Society of Chemistry. b 1: 4-Hydroxy-ethylvalerate, 2: ethyl-4-ethoxy pentanoate, 3: ethyl pentanoate; see Fig. 27. c Catalyst 100 mg, GVL 10 mmol, ethanol 5.8 mL, 200 °C, 10 h, 3 MPa N2. d 250 °C | ||||||
| 1 | Blank | — | 0 | 0 | 0 | 0 |
| 2 | MIL-101Cr | 0 | 3 | 100 | 0 | 0 |
| 3 | MIL-101Cr-SO3H(25) | 0.20 | 8 | 94 | 6 | 0 |
| 4 | MIL-101Cr-SO3H(50) | 0.40 | 39 | 20 | 66 | 14 |
| 5 | MIL-101Cr-SO3H(80) | 0.45 | 45 | 11 | 69 | 20 |
| 6 | MIL-101Cr-SO3H(100) | 0.80 | 44 | 14 | 70 | 16 |
| 7 | MIL-101Cr-SO3H(100)d | — | 86 | 0 | 3 | 90 |
When raising the temperature to 250 °C, 90% selectivity for ethyl pentanoate 3 could be achieved, which was traced to its endothermic, only entropically favored formation. Ethyl pentanoate is a promising candidate as a bio-based solvent with a vapor pressure higher than that of GVL but lower than those of common octane boosters; hence ethyl pentanoate is also a valuable product obtainable with MIL-101Cr-SO3H.153
Comparing the conversions and product selectivity of MIL-101Cr-SO3H to Pd@MIL-101Cr-SO3H(X) substantial differences were observed. First, Pd@MIL-101Cr-SO3H(50) gave the desired product ethyl valerate 4 with 26% selectivity (35% conversion), whereas product 3 was absent. Pd@MIL-101Cr-SO3H(100) gave the highest conversion at 200 °C with 51% and product selectivity of 66% towards ethyl-4-ethoxy pentanoate 2 and 26% to 4 (Fig. 28). A physical mixture of MIL-101Cr-SO3H(100) and Pd@C gave a higher selectivity towards ethyl valerate (44%) but lower conversion (42%), from which the authors concluded that Pd nanoparticles in close proximity to SO3H groups show lower C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO bond hydrogenation activity. To increase the yield of the desired ethyl valerate 4 using Pd@MIL-101Cr-SO3H(100) the temperature was raised to 250 °C and a selectivity of 83% to 4 at a conversion of 98% were achieved. Reusability experiments of Pd@MIL-101Cr-SO3H(80) revealed a dramatic change in product selectivity after the first run from ethyl valerate 4 to ethyl pentanoate 3 (Fig. 28).153
C and CO bond hydrogenation activity. To increase the yield of the desired ethyl valerate 4 using Pd@MIL-101Cr-SO3H(100) the temperature was raised to 250 °C and a selectivity of 83% to 4 at a conversion of 98% were achieved. Reusability experiments of Pd@MIL-101Cr-SO3H(80) revealed a dramatic change in product selectivity after the first run from ethyl valerate 4 to ethyl pentanoate 3 (Fig. 28).153
 | ||
| Fig. 28 Reusability and product distribution of Pd@MIL-101-SO3H(80) catalyst in GVL ring opening reactions. Reaction conditions: 10 mmol of GVL, 5820 mL of ethanol, 250 °C, 10 h, 3 MPa H2. Reprinted from ref. 153. Copyright 2014 with permission from the Royal Society of Chemistry. | ||
The authors explain this change in selectivity by an aggregation of the Pd-NPs resulting in deactivation and framework collapse. Leaching of Pd species was excluded by a hot filtration test and ICP-AES analysis. However, initial TEM analysis before the catalytic reaction already showed larger Pd-NPs (20 nm) for MIL-101Cr-SO3H(80 and 100). No data were given which confirmed the integrity of the MIL-101Cr-SO3H framework after being reacted under the quite harsh condition of 250 °C.153 Therefore, although the results are promising for the valorization of biomass, the final catalyst stability remained inconclusive.
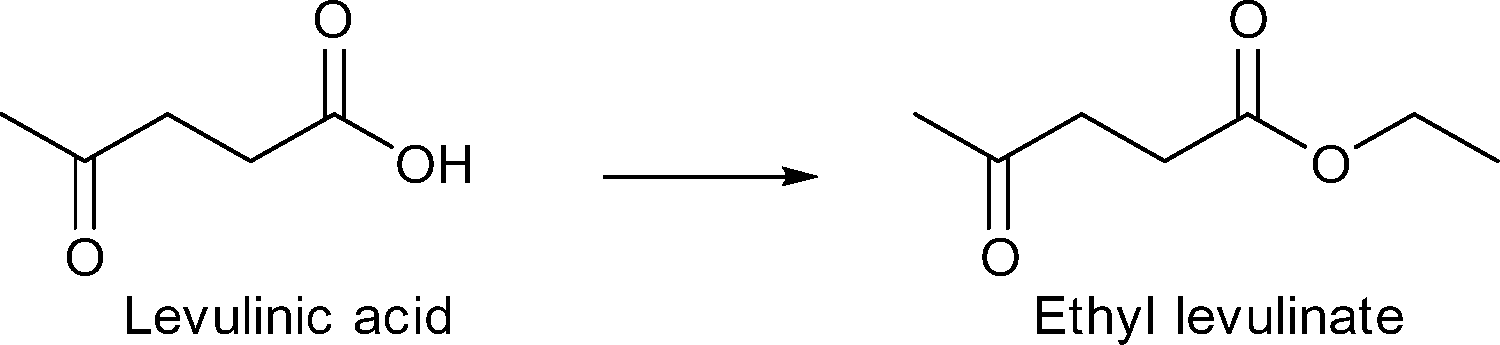 | ||
| Scheme 10 Esterification of levulinic acid to ethyl levulinate. | ||
Recently, Cirujano and coworkers investigated the activity of UiO-66Zr and UiO-66Zr-NH2 for the esterification of levulinic acid with ethanol, butanol and long chain alcohols (C12, C16, C18).155
Under the applied conditions (levulinic acid, LA 1 mmol, molar ratio EtOH:LA = 15:1, 8 h, 78 °C, catalyst 1.8 mol% Zr with respect to LA), both UiO-66Zr and UiO-66Zr-NH2 converted levulinic acid quantitatively into ethyl levulinate after 8 h. Both MOFs displayed very similar activities of 94% and 95% yield, respectively. The ratio of ethanol:LA was reduced to 5:1 for comparison with literature results (T = 78 °C). UiO-66Zr gave activities of 49% (4 h/78 °C) and 73% (4 h/100 °C). In comparison, Amberlyst-15 (ref. 156) (5 h/70 °C) gave 55% conversion and sulfated TiO2 (ref. 156) gave 40% (5 h/70 °C). UiO-66Zr displayed higher conversions than sulfated ZrO2, the zeolite H-ZSM-5 and mesoporous silica H-MCM-22. On the other hand, desilicated zeolite loaded with dodecatungstophosphoric acid (DTPA/DHZSM-5) (82% at 4 h/78 °C)157 and sulfated TiO2 nanorods (83% at 4 h/105 °C)158 performed better than UiO-66Zr even when increasing the temperature to 100 °C because of lower alcohol excess.155 Nevertheless, the above stated comparison points out that UiO-66Zr catalysts can compete with current state-of-the-art catalysts for the levulinic esterification reaction to ethyl levulinate.
The authors went deeper into materials characterization and investigated the influence of linker deficiency, which is inherent to UiO materials,97 on the catalytic activity. Different batches of the same material were found to have strongly different activities leading to a 9-fold increase from the less active to the most active material (Fig. 29).155
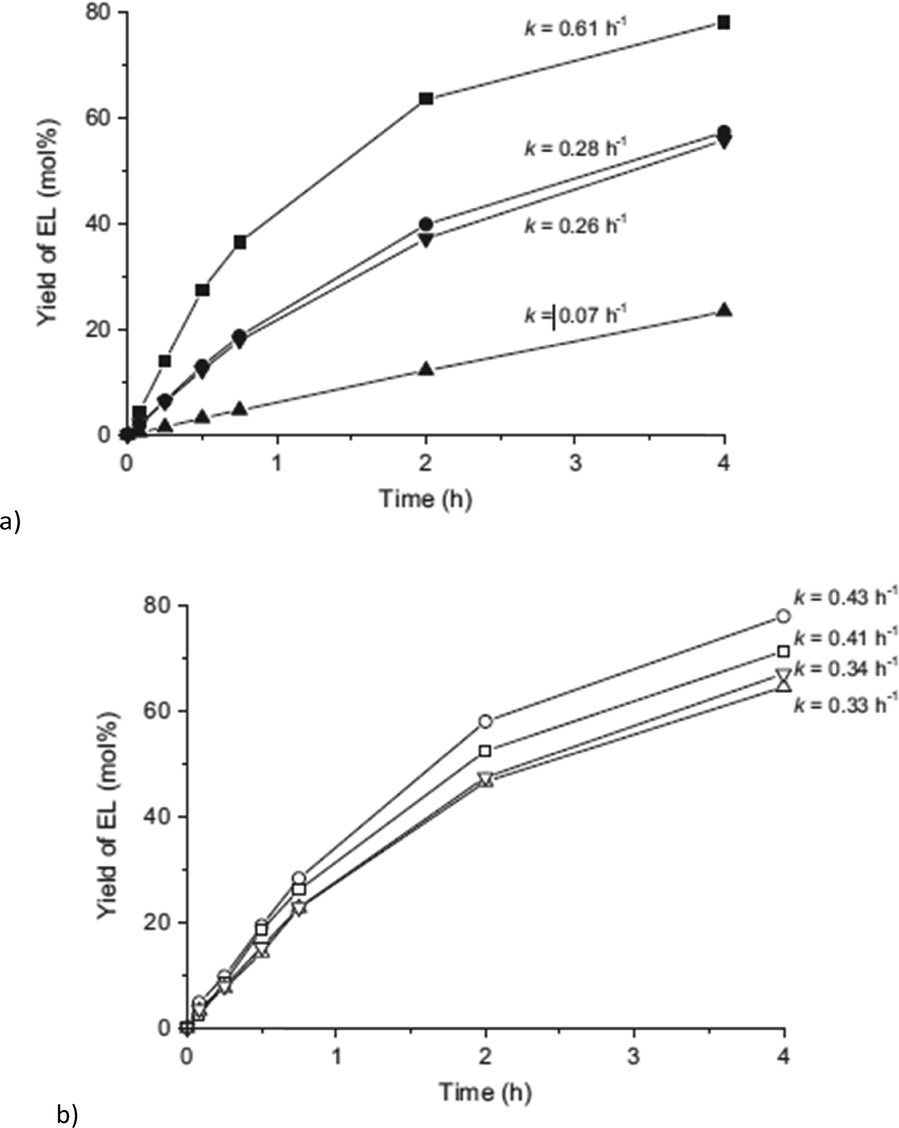 | ||
| Fig. 29 Yield of ethyl levulinate (EL) obtained over (a) UiO-66, batches B1 to B4 (from top to bottom), (b) UiO-66-NH2, batches A1 to A4 (from top to bottom). For each curve, the calculated pseudo-first-order kinetic rate constant is indicated. Reprinted from ref. 155. Copyright 2015 with permission from Elsevier. | ||
Linker deficiencies were calculated by the method of Valenzano et al.,159 where thermogravimetric analysis was used to determine the relative weight loss of the organic ligand (above 300 °C) with respect to the amount expected from the stoichiometry of the ideal structure (solid residue calculated as ZrO2). The derived linker deficiencies (%) correlated with the corresponding rate constants (k, h−1) (Fig. 29 and Table 3). The linker deficiencies have been calculated from the linker to cluster ratios (coordination number, c.n., Table 3), revealing in all cases a lower ratio than the expected value of 12 for the ideal UiO-66 structure. The higher the amount of missing linkers, the higher the activity of the MOF catalyst, which demonstrates the importance of OMS for the reaction.
| Material | Sample batch | k (h−1) | %def | Av c.n. | d c (nm) |
|---|---|---|---|---|---|
| a Taken from ref. 155. Copyright 2015 with the permission from Elsevier. | |||||
| UiO-66 | B1 | 0.61 | 13.2 | 10.42 | 120 ± 5 |
| B2 | 0.28 | 7.2 | 11.14 | 65 ± 5 | |
| B3 | 0.26 | 4.8 | 11.42 | 200 ± 10 | |
| B4 | 0.07 | 2.5 | 11.70 | 95 ± 5 | |
| UiO-66-NH2 | A1 | 0.43 | 6.3 | 11.24 | 45 ± 10 |
| A2 | 0.41 | 8 | 11.04 | 65 ± 5 | |
| A3 | 0.34 | 2.5 | 11.70 | 110 ± 5 | |
| A4 | 0.33 | 2.1 | 11.75 | 103 ± 9 | |
| UiO-66-NO2 | N1 | 0.07 | 0.4 | 11.95 | 95 ± 5 |
UiO-66Zr-NO2 was obtained with a linker-to-cluster ratio very close to 12 (Fig. 29) and as expected the catalytic activity was low. By comparing UiO-66Zr with UiO-66Zr-NH2, it is evident that not only defect formation but also the amino groups influenced the catalyst performance. Batches B4 of Ui-66Zr and A3 of UiO-66Zr-NH2 displayed the same degree of linker deficiency (2.5%) and have similar particle sizes, but the rate constant of A3 is almost 5-times higher than that of B4 (Table 3). This indicates a supporting effect of the amino group of UiO-66Zr-NH2. A possible reaction pathway proposed by the authors is depicted in Fig. 30.155
 | ||
| Fig. 30 (a) Defect formation through a missing linker. Two Zr atoms have a vacancy and two Zr ions from different {Zr6} clusters are coordinated by hydroxido or chlorido anions in order to retain framework neutrality. (b) Proposed reactivation pathway over UiO-66-NH2. Redrawn from ref. 155. Copyright 2015 with permission from Elsevier. | ||
The authors attributed the different grades of linker deficiency to small differences in nucleation and crystal growth rates as well as stochastic variations in temperature, time or reagent concentrations. Recalculation of activities based on the experimentally determined Zr content gave higher values comparable with that of homogeneous p-toluenesulfonic acid.155
Esterification products using longer chain alcohols (C12–OH to C18–OH) are much larger in size; therefore it can be expected that conversion takes place at the MOF surface instead of inside the pores. This correlates with a much slower reaction rate (for C16–OH, 66%/8 h, 82%/20 h).155
4.4 Triglycerides to esters and glycerol (Scheme 11)
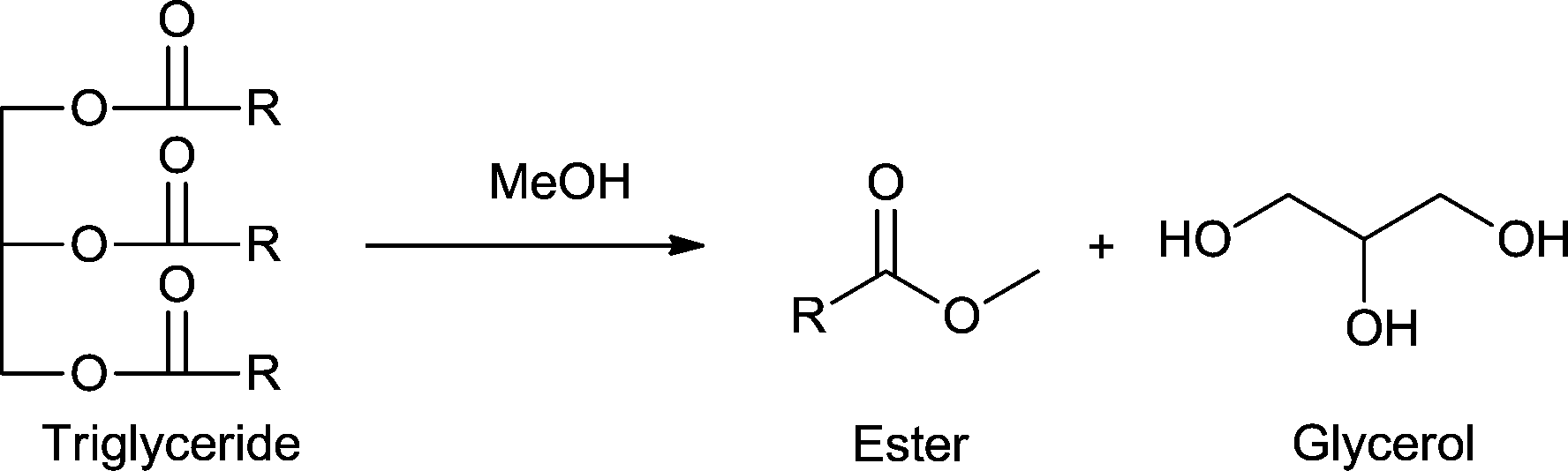 | ||
| Scheme 11 Transesterification of triglycerides to the corresponding ester and glycerol. | ||
The transesterification of vegetable oil by a metal–organic framework was investigated first by Chizallet et al. in 2010 using unfunctionalized ZIF-8.163 After detailed experimental and theoretical studies they concluded that the acido-basic sites were located at the external surface of the material or at defects but not in the microporosity of ZIF-8. Surface N-moieties and OH groups as basic sites were shown to be particularly interesting for the combined activation of alcohols and esters and were proposed as the active sites.163
Chen and coworkers reported in 2014 the transesterification of triglycerides catalyzed by an amine-functionalized MOF.164 MOF-5, IRMOF-10 and MIL-53Al-NH2 were selected as pre-catalysts and modified with amines using i) dative modification (grafting) of ethylenediamine (ED) or 4-dimethylaminopyridine (DMAP) to unsaturated metal sites located at the secondary building units of MOFs and (ii) covalent modification of the amine-tagged organic linkers within MIL-53Al-NH2 by alkylation with 2-dimethylaminoethyl chloride.
The ethylenediamine (ED)-grafted MOF-5-ED and IRMOF-10-ED catalysts exhibited high conversions in the liquid phase transesterification with methanol of glyceryl triacetate (R = Me in Scheme 11; both >99.9% after 3 h) and glyceryl tributyrate (R = butyl in Scheme 11; both >99.9% after 6 h). After upscaling the reaction (starting from 302 mg of glyceryl tributyrate) to 2.1 g of glyceryl tributyrate, conversions were slightly lower with 89% and 99% for MOF-5-ED and IRMOF-10-ED, respectively, after 10 h.164
MIL-53Al-NH2 showed only a very low yield (4%) in the conversion of glyceryl triacetate compared with 2-dimethylaminoethyl functionalized MIL-53Al-NH2 (99.9% yield). For the conversion of glyceryl tributyrate the catalytic activity (TOF) towards transesterification and the basic site density of amine-grafted MOFs revealed a linear correspondence (Fig. 31a and b).164 The basic site density was determined by acid–base titration with aqueous HCl as titrant in aqueous KCl solution.164 MOF-5 and other Zn-carboxylate based IRMOFs are, however, unstable in water,65 so it is not clear how reliable the obtained values were.
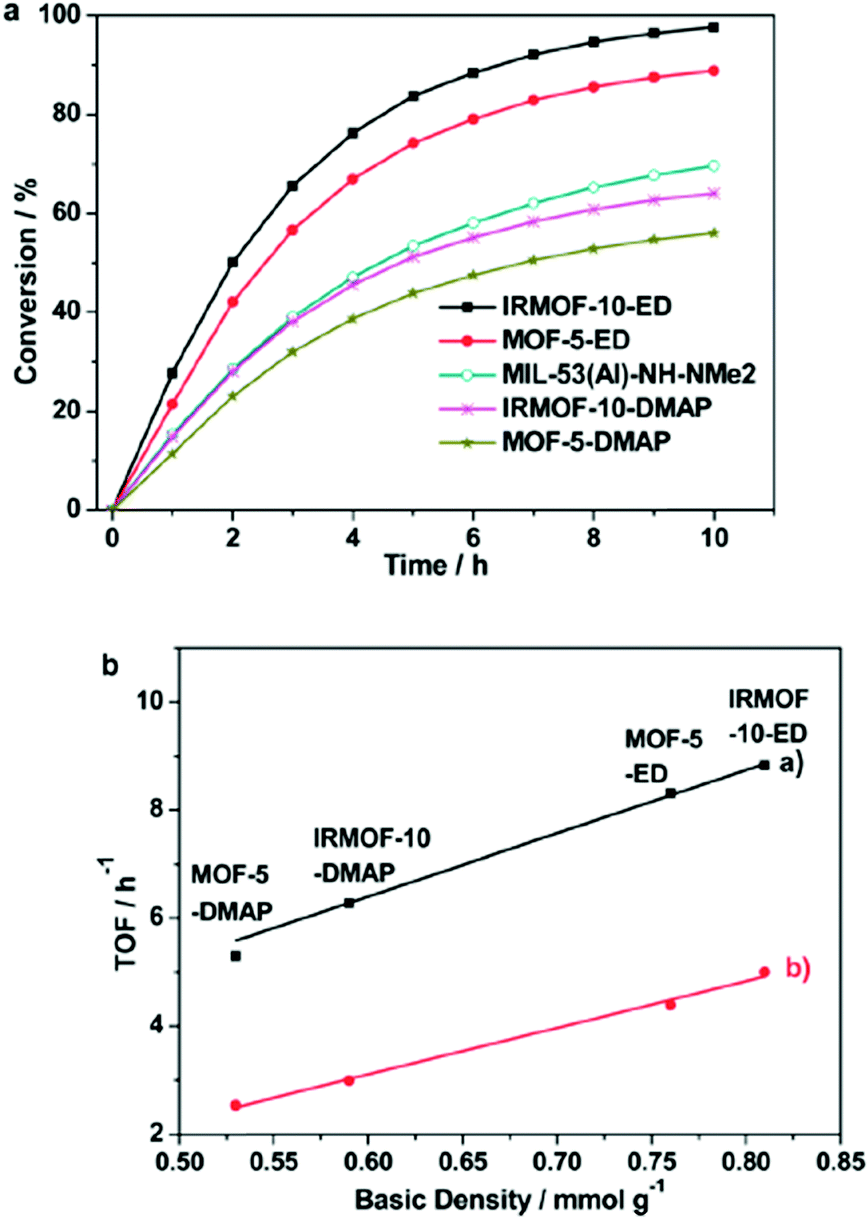 | ||
| Fig. 31 (a) Comparison of the catalytic activities of amine-functionalized MOFs for the transesterification of glyceryl tributyrate with methanol (reaction conditions: glyceryl tributyrate 2.1 g; methanol 8.0 mL; catalyst 80 mg, temperature 60 °C). (b) Linear correlations between the basic site density of amine-functionalized MOFs with their catalytic activity in the transesterification of (a) glyceryl triacetate (black line) and (b) glyceryl tributyrate (red line) with methanol. Reprinted from ref. 164. Copyright 2014 with permission from the Royal Society of Chemistry. | ||
The powder X-ray diffractogram after the reaction is shown only for IRMOF-10-ED, which largely agreed with the diffractogram of the parent IRMOF-10, although cycling experiments revealed a loss in activity of about 37% from the first to the fourth run. The authors explained this decrease with leaching of ethylenediamine.164
Often algae oil, sunflower oil or other natural feedstocks are used for transesterification. These natural products are not comparable to glyceryl triacetate as the model compound which makes a fair comparison difficult. Zieba et al. described the activity of a heterogeneous Zn catalyst (zinc hydroxy nitrate, Zn5(OH)8(NO3)2·2H2O) at similar conditions (5 wt% catalyst, glyceryl triacetate:methanol 1:29 molar ratio, 3 h, 50 °C) for the reaction of glyceryl triacetate with methanol.165 This Zn catalyst gave only 52% methyl acetate after 3 h which is lower than that for grafted MOF-5, IRMOF-10 and for MIL-53Al-NH-NMe2.
4.5 Feedstock lignin
 | ||
| Fig. 32 Reactions catalyzed by IRMOF-74(I, II). Redrawn with permission from ref. 166. Copyright 2016, American Chemical Society. | ||
IRMOF-74(I) and IRMOF-74(II) were selected because of i) sufficient thermal and chemical stability, ii) large enough channels for substrate diffusion, iii) high density of open metal sites (OMS) for C–O bond activation. IRMOF-74(I) is synthesized from Mg2+ and 2,5-dihydroxyterephthalate with a mixture of DMF/ethanol and H2O as solvent from a solvothermal reaction. IRMOF-74(II) contains Mg2+ and 3,3′-dihydroxy-[1,1′-biphenyl]-4,4′-dicarboxylate as linker. Fig. 33 shows the large channels of IRMOF-74(I) and (II) with diameters of 1.4 and 2.2 nm, respectively. The longest intramolecular distances in all three substrates range between 9 Å and 14 Å (straight line between the blue-highlighted atoms in Fig. 33). Therefore, the pores were large enough to accommodate all substrates.166
 | ||
| Fig. 33 (a) Pore diameters of IRMOF-74(I) and IRMOF-74(II) and largest intramolecular distances in the aromatic ether compounds, corresponding to a straight line between the blue-highlighted atoms in the optimized geometries. (b) Conversion efficiencies of the substrates into the corresponding hydrocarbon and phenol at 120 °C under 10 bar hydrogen. Reprinted with permission from ref. 166. Copyright 2016, American Chemical Society. | ||
Both IRMOFs were proven to be stable under the applied conditions (45 mg catalyst, 5.0 mL p-xylene, 120 °C, 16 h, 10 bar H2) by solvent stability tests, TGA, PXRD and analysis of the supernatant reaction solution by elemental analysis. It is noted that the authors reported partial dissolution of IRMOF-74(I) in H2O. The authors did not provide information about substrate amounts; as a consequence, no statement about substrate-to-catalyst ratio can be made.
IRMOF-74(I) and (II) revealed catalytic activity for the hydrogenolysis of ethers generating phenol and the corresponding aromatic hydrocarbon (Fig. 32) for three substrates (Fig. 33). Higher conversions were obtained with IRMOF-74(II) with, for example, 39% conversion of PPE versus 12% with IRMOF-74(I) (Fig. 33). Product selectivity was slightly higher for IRMOF-74(I) from all three substrates. For example product selectivity from PPE for 1 and 4 was 87% and 91% for IRMOF-74(I) versus 83% and 87% for IRMOF-74(II).166 Based on the reaction stoichiometry (cf.Fig. 32) equal amounts of the two products would be expected. Selectivity distributions showed lower amounts of the alkyl benzenes 1, 2 and 3 with respect to phenol which was explained by the volatility of the arene compounds and was experimentally verified by residual gas analysis (RGA). Although conversions of ethers with MOF catalysts were low to moderate, without catalyst no reaction occurred, supporting the role of MOF as a catalyst. The difference in surface area between the two MOFs was small, 1627 m2 g−1 for IRMOF-74(I) and 1736 m2 g−1 for IRMOF-74(II). According to the authors the higher conversions with IRMOF-74(II) point to reactions occurring within the MOF pores. Therefore, DFT calculations were performed which indicated that the OMS in MOF pores bind and orient the substrate. Furthermore the OMS are proposed to play a role in activating or orienting H2. All findings conclude that substrate confinement may play a key role in MOF catalysis, but has to be investigated in more detail.
In addition to the pure IRMOFs also TiClx and Ni NPs have been encapsulated into the IRMOF-74 pores, since they are known to catalyze aryl ether hydrogenolysis.166 By this modification, substrate conversion was significantly enhanced to up to 82% for Ni@IRMOF-74(II) in the case of PPE (see Fig. 33) with high selectivities towards 1 of 96% and 4 of 98%. Conversions with pure Raney Ni as catalyst were similar (76%) to that for Ni@IRMOF-74(II), but selectivity was lower, giving 81% of 1 and 75% of 4. Besides, substantial amounts of cyclohexanol were found which was not observed using the M@IRMOF-74-catalysts. The activity order was Ni@IRMOF-74 > Ti@IRMOF-74 > IRMOF-74, regardless of substrate. Ni@IRMOF-74 could be reused up to 5 times without loss in activity and retained its structure as confirmed by PXRD.166
 | ||
| Scheme 12 Hydrodeoxygenation of vanillin to 2-methoxy-4-methyl phenol. | ||
The conditions for vanillin conversion were 2 mmol of vanillin, 50 mg of catalyst, 20 mL of H2O, 0.5 MPa of H2 at 100 °C for Pd@MIL-101Cr-SO3H and 90 °C for Pd@UiO-66-NH2.167,168 Vanillin was first hydrogenated to vanillin alcohol and then converted to 2-methoxy-4-methylphenol (Scheme 12), as proven for both catalysts by GC. The catalytic activities of Pd@MIL-101Cr-SO3H and Pd@UiO-66Zr-NH2 were very similar, the latter one converted vanillin quantitatively to 2-methoxy-2-methylphenol within 1 h and Pd@MIL-101Cr-SO3H with a selectivity of 91% (96% conversion). A reference experiment with Pd@MIL-101Cr gave a lower conversion (67%) and product selectivity (58% 2-methoxy-4-methylphenol).167
Moreover, the UiO-66Zr-NH2 support showed a higher adsorption of the substrate and the intermediate than UiO-66Zr, although the amino-modification possessed a lower BET surface area and pore volume. It was suggested that the adsorption of vanillin or vanillin alcohol by amine-functionalized MOFs is enhanced through the hydrophilic nature of the substrate molecule as well as hydrogen-bonding interactions between the substrate molecule and the MOF.
Both catalysts could be re-used seven times without any loss in activity and selectivity. The presence of free amine moieties in the frameworks of Pd@UiO-66Zr-NH2 was assumed to play a key role in the formation of uniform, well-dispersed and leaching-resistant palladium nanoparticles on the support. For direct catalysis the amine moieties were proposed to interact with the substrate molecule but this suggestion was not proven.168 Compared with other supported Pd catalysts such as Pd@CM170 (CM = carbonaceous microspheres, conversion >99%, 2-methoxy-4-methylphenol selectivity 48%) selectivity of Pd@UiO-66Zr-NH2 (conversion 100%) towards 4-methoxy-4-methyl phenol (100%) was higher (Table S5†).169
5. Conclusion and perspective
From the presented studies it can be concluded that MOFs can display a similar or better activity in selected reactions than heterogeneous catalysts such as zeolites or metal oxides. This is the case, for example, in fructose dehydration in DMSO or in reactions where moderate acidity leads to the best results. In glucose dehydration and in cellulose hydrolysis MOFs cannot (yet) compete with other state-of-the-art catalysts. Interestingly, in metal@MOF catalyzed reactions, such as hydrogenation, MOFs not only seem to serve as a support for the catalytically active metal but also seem to have a significant influence on product selectivity. This was, for example, the case in the hydro-deoxygenation of vanillin to 2-methoxy-4-methylphenol with Pd@MIL-101Cr-SO3H or Pd@UiO-66Zr-NH2.167,168 The following features especially distinguish MOFs from zeolites and metal oxides and are worthy of further development to enable new catalytic pathways to sustainable fine chemicals:• Tunable Lewis and Brønsted acidity through the type and degree of functionalization and defect engineering;
• Uniform nature of open metal sites (OMS) and other catalytically active sites within the MOF;
• Hydrogen bonding interactions through linkers and thereby anchoring and orienting substrates and intermediates;
• Modulation of hydrophilicity/hydrophobicity and acidity/basicity in terms of micro-environment in the pores, thereby controlling the adsorption of substrates/intermediates.
In order to develop efficient MOF catalysts for biomass catalysis a better understanding has to be gained regarding:
• Interplay between organic linker modifications and metal incorporation in MOF supports;
• Pore size versus pore confinement;
• Hierarchical meso/macroporous MOFs for faster mass transport;
• Better water stability of MOFs;
• Humin formation and solutions for prevention;170
• Improving MOF characterization methods regarding their catalytic properties.
Method development is fundamental, aiming at a deeper understanding for instance in structure–property relations. There is the need for a reliable determination of the acidity and basicity of MOFs. Currently, liquid acid or base titration is commonly used, but this involves usually proton exchange with sodium chloride or other salts which does not lead to satisfactory results. Recently, Klet submitted the first detailed report about reliable acid–base titration for MOFs.171 For acidity/basicity measurements, most of the time MOF stability data (after titration) and experimental data (e.g. titration curves) are not reported, which makes it difficult to verify if the MOF remained stable under the applied conditions.
Methods such as temperature programmed desorption (usually performed with NH3) have to be further developed to be applicable to temperatures below 300 °C, with less basic probe molecules and new methods for acid–base characterization of MOFs have to be found.
Another arguable point is about free and accessible pore space. The usual characterization of a MOF pre-catalyst material involves PXRD for crystallinity and MOF identification, nitrogen volumetric sorption at 77 K for (BET) surface area measurement and pore volume determination and NMR digestion for the degree of linker functionalization. All three measurements give only an average value. The pore volume determined by N2 sorption is seen as equally accessible also for larger substrate molecules. What about the distribution of functional groups within the MOF particle? For example, (postsynthetic) functionalization could occur primarily in the outer sphere of the MOF particle or within the pore mouths. Small dinitrogen molecules will diffuse through the entire MOF particle, but do larger molecules still enter the smaller functionalized pores? How many of the functional sites are really available for catalysis? How deep do the different molecules diffuse into the MOF particle? This has a significant influence on catalyst activity, as TON and TOF values are usually calculated by assuming the number of active sites from the molecular formula. The diffusion path length would have consequences for particle size engineering which could reduce the amount of MOF material needed. Importantly, methods which offer spatial resolution within one MOF particle are necessary to gain a better understanding of MOF catalysis.
In addition, a reliable method to quantify hydrophilicity or hydrophobicity of the pore size environment to the best of our knowledge has not been reported yet. This could help to optimize substrate uptake by MOFs as well as to investigate MOF substrate/intermediate interactions.
Taking the numerous possible applications of MOFs into account, there is an urgent need for MOFs which are stable in acidic or basic aqueous solution. Intensive research is going on, aiming at a better understanding of structure–property relations to improve hydrothermal stability in order to advance MOFs to real applications.68,172,173
Finally, we want to encourage the MOF community to address work on MOF catalysis in the context of biomass-based fine chemical production.
References
- D. Esposito and M. Antonietti, Chem. Soc. Rev., 2015, 44, 5821–5853 RSC.
- A. A. Koutinas, A. Vlysidis, D. Pleissner, N. Kopsahelis, I. Lopez Garcia, I. K. Kookos, S. Papanikolaou, T. H. Kwan and C. S. K. Lin, Chem. Soc. Rev., 2014, 43, 2587–2627 RSC.
- M. R. Avhad and J. M. Marchetti, Renewable Sustainable Energy Rev., 2015, 50, 696–718 CrossRef CAS.
- R. Beerthuis, G. Rothenberg and R. N. Shiju, Green Chem., 2015, 17, 1341–1361 RSC.
- C. Chatterjee, F. Pong and A. Sen, Green Chem., 2015, 17, 40–71 RSC.
- S. Dutta and S. Pal, Biomass Bioenergy, 2014, 62, 182–197 CrossRef CAS.
- S. González-García, B. Gullón, S. Rivas, G. Feijoo and M. T. Moreira, J. Cleaner Prod., 2016, 120, 170–180 CrossRef.
- M. Hara, K. Nakajima and K. Kamata, Sci. Technol. Adv. Mater., 2016, 16, 34903–34925 CrossRef PubMed.
- P. Lanzafame, G. Centi and S. Perathoner, Chem. Soc. Rev., 2014, 43, 7562–7580 RSC.
- J. S. Luterbacher, D. Martin Alonso and J. A. Dumesic, Green Chem., 2014, 16, 4816–4838 RSC.
- R. A. Sheldon, J. Mol. Catal. A: Chem., 2016, 422, 3–12 CrossRef CAS.
- R. Rinaldi and F. Schüth, ChemSusChem, 2009, 2, 1096–1107 CrossRef CAS PubMed.
- J. S. Kruger, V. Nikolakis and D. G. Vlachos, Curr. Opin. Chem. Eng., 2012, 1, 312–320 CrossRef CAS.
- D. J. Hayes, Catal. Today, 2009, 145, 138–151 CrossRef CAS.
- C.-H. Zhou, X. Xia, C.-X. Lin, D.-S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- S. Inkinen, M. Hakkarainen, A.-C. Albertsson and A. Södergård, Biomacromolecules, 2011, 12, 523–532 CrossRef CAS PubMed.
- J. Becker, A. Lange, J. Fabarius and C. Wittmann, Curr. Opin. Biotechnol., 2015, 36, 168–175 CrossRef CAS PubMed.
- B. Murillo, B. Zornoza, O. de La Iglesia, C. Téllez and J. Coronas, J. Catal., 2016, 334, 60–67 CrossRef CAS.
- B. M. Upton and A. M. Kasko, Chem. Rev., 2016, 116, 2275–2306 CrossRef CAS PubMed.
- J. E. Holladay, J. F. White, J. J. Bozell and D. Johnson, Pacific Northwest National Laboratory, US Department of Energy, 2007 Search PubMed.
- Zeolites in industrial separation and catalysis, ed. S. Kulprathipanja, Wiley-VCH, Weinheim, 2010 Search PubMed.
- I. Fechete, Y. Wang and J. C. Védrine, Catal. Today, 2012, 189, 2–27 CrossRef CAS.
- C. Janiak, Angew. Chem., 1997, 109, 1499–1502 CrossRef.
- C. Janiak, Angew. Chem., Int. Ed. Engl., 1997, 36, 1431–1434 CrossRef CAS.
- C. Janiak and J. K. Vieth, New J. Chem., 2010, 34, 2366–2388 RSC.
- A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804–6849 RSC.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- C. Janiak, Dalton Trans., 2003, 2781–2804 RSC.
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O'Keeffe, M. P. Suh and J. Reedijk, CrystEngComm, 2012, 14, 3001–3004 RSC.
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O'Keeffe, M. P. Suh and J. Reedijk, Pure Appl. Chem., 2013, 85, 1715–1724 CrossRef CAS.
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle, M. Bosch and H.-C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- J.-P. Zhang, P.-Q. Liao, H.-L. Zhou, R.-B. Lin and X.-M. Chen, Chem. Soc. Rev., 2014, 43, 5789–5814 RSC.
- G. Akiyama, R. Matsuda, H. Sato, M. Takata and S. Kitagawa, Adv. Mater., 2011, 23, 3294–3297 CrossRef CAS PubMed.
- X. Liu, H. Zhang, F. Chang, S. Huang, K. Yang and S. Yang, Curr. Org. Chem., 2016, 20, 761–776 CrossRef CAS.
- D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502–7513 CrossRef CAS PubMed.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Commun., 2012, 48, 11275–11288 RSC.
- Y. Kim and S. Huh, CrystEngComm, 2016, 18, 3524–3550 RSC.
- A. Dhakshinamoorthy, M. Opanasenko, J. Cejka and H. Garcia, Adv. Synth. Catal., 2013, 355, 247–268 CAS.
- A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2014, 43, 5750–5765 RSC.
- A. Dhakshinamoorthy, M. Opanasenko, J. Čejka and H. Garcia, Catal. Sci. Technol., 2013, 3, 2509–2540 CAS.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Catal. Sci. Technol., 2011, 1, 856–867 CAS.
- J.-L. Wang, C. Wang and W. Lin, ACS Catal., 2012, 2, 2630–2640 CrossRef CAS.
- A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2012, 41, 5262–5284 RSC.
- A. Dhakshinamoorthy, M. Alvaro, A. Corma and H. Garcia, Dalton Trans., 2011, 40, 6344–6360 RSC.
- M. Opanasenko, Catal. Today, 2014, 243, 2–9 CrossRef.
- A. Torres-Knoop and D. Dubbeldam, ChemPhysChem, 2015, 16, 2046–2067 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- P. García-García, M. Müller and A. Corma, Chem. Sci., 2014, 5, 2979–3007 RSC.
- G. Férey, Science, 2005, 309, 2040–2042 CrossRef PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- K. M. Gupta, K. Zhang and J. Jiang, Sci. Rep., 2015, 5, 12821 CrossRef CAS PubMed , (1–9).
- J. J. Low, A. I. Benin, P. Jakubczak, J. F. Abrahamian, S. A. Faheem and R. R. Willis, J. Am. Chem. Soc., 2009, 131, 15834–15842 CrossRef CAS PubMed.
- F. Jeremias, D. Fröhlich, C. Janiak and S. K. Henninger, New J. Chem., 2014, 38, 1846–1852 RSC.
- C. Janiak and S. K. Henninger, Chimia, 2013, 67, 419–424 CrossRef CAS PubMed.
- J. Ehrenmann, S. K. Henninger and C. Janiak, Eur. J. Inorg. Chem., 2011, 4, 471–474 CrossRef.
- A. Khutia, H. U. Rammelberg, T. Schmidt, S. Henninger and C. Janiak, Chem. Mater., 2013, 25, 790–798 CrossRef CAS.
- F. Jeremias, D. Fröhlich, C. Janiak and S. K. Henninger, RSC Adv., 2014, 4, 24073–24082 RSC.
- D. Fröhlich, S. K. Henninger and C. Janiak, Dalton Trans, 2014, 43, 15300–15304 RSC.
- P. Küsgens, M. Rose, I. Senkovska, H. Fröde, A. Henschel, S. Siegle and S. Kaskel, Microporous Mesoporous Mater., 2009, 120, 325–330 CrossRef.
- T. Wu, L. Shen, M. Luebbers, C. Hu, Q. Chen, Z. Ni and R. I. Masel, Chem. Commun., 2010, 6120–6122 RSC.
- F. Jeremias, A. Khutia, S. K. Henninger and C. Janiak, J. Mater. Chem., 2012, 22, 10148–10151 RSC.
- F. Jeremias, V. Lozan, S. K. Henninger and C. Janiak, Dalton Trans, 2013, 42, 15967–15973 RSC.
- N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS PubMed.
- K. Leus, T. Bogaerts, J. de Decker, H. Depauw, K. Hendrick, H. Vrielinck, V. van Speybroeck and P. van der Voort, Microporous Mesoporous Mater., 2016, 226, 110–116 CrossRef CAS.
- J. Gascon, A. Corma, F. Kapteijn and F. X. Llabrés i Xamena, ACS Catal., 2014, 4, 361–378 CrossRef CAS.
- N. Qadir, S. A. Said and H. M. Bahaidarah, Microporous Mesoporous Mater., 2015, 201, 61–90 CrossRef CAS.
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Férey, Chem. – Eur. J., 2004, 10, 1373–1382 CrossRef CAS PubMed.
- G. Férey, C. Serre, C. Mellot-Draznieks, F. Millange, S. Surble, J. Dutour and I. Margiolaki, Angew. Chem., Int. Ed., 2004, 43, 6296–6301 CrossRef PubMed.
- P. Horcajada, S. Surblé, C. Serre, D.-Y. Hong, Y.-K. Seo, J.-S. Chang, J.-M. Grenèche, I. Margiolaki and G. Férey, Chem. Commun., 2007, 2820–2822 RSC.
- C. Volkringer, D. Popov, T. Loiseau, G. Férey, M. Burghammer, C. Riekel, M. Haouas and F. Taulelle, Chem. Mater., 2009, 21, 5695–5697 CrossRef CAS.
- C. Serre, F. Millange, S. Surblé and G. Ferey, Angew. Chem., Int. Ed., 2004, 43, 6285–6289 CrossRef PubMed.
- T. Zhao, F. Jeremias, I. Boldog, B. Nguyen, S. K. Henninger and C. Janiak, Dalton Trans., 2015, 44, 16791–16801 RSC.
- D.-Y. Hong, Y. K. Hwang, C. Serre, G. Férey and J.-S. Chang, Adv. Funct. Mater., 2009, 19, 1537–1552 CrossRef CAS.
- S. Bhattacharjee, C. Chen and W.-S. Ahn, RSC Adv., 2014, 4, 52500–52525 RSC.
- C. Zlotea, D. Phanon, M. Mazaj, D. Heurtaux, V. Guillerm, C. Serre, P. Horcajada, T. Devic, E. Magnier, F. Cuevas, G. Férey, P. L. Llewellyn and M. Latroche, Dalton Trans, 2011, 40, 4879–4881 RSC.
- G. E. Cmarik, M. Kim, S. M. Cohen and K. S. Walton, Langmuir, 2012, 28, 15606 CrossRef CAS PubMed.
- P. M. Schoenecker, C. G. Carson, H. Jasuja, C. J. J. Flemming and K. S. Walton, Ind. Eng. Chem. Res., 2012, 51, 6513–6519 CrossRef CAS.
- H. Jasuja and K. S. Walton, J. Phys. Chem. C, 2013, 117, 7062–7068 CAS.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- K. K. Tanabe and S. M. Cohen, Chem. Soc. Rev., 2011, 40, 498–519 RSC.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed.
- J. D. Evans, C. J. Sumby and C. J. Doonan, Chem. Soc. Rev., 2014, 43, 5933–5951 RSC.
- P. Deria, J. E. Mondloch, O. Karagiaridi, W. Bury, J. T. Hupp and O. K. Farha, Chem. Soc. Rev., 2014, 43, 5896–5912 RSC.
- O. Karagiaridi, W. Bury, J. E. Mondloch, J. T. Hupp and O. K. Farha, Angew. Chem., 2014, 126, 4618–4628 CrossRef.
- P. Deria, W. Bury, I. Hod, C.-W. Kung, O. Karagiaridi, J. T. Hupp and O. K. Farha, Inorg. Chem., 2015, 54, 2185–2192 CrossRef CAS PubMed.
- C. K. Brozek and M. Dincă, Chem. Soc. Rev., 2014, 43, 5456–5467 RSC.
- Y. Song, X. Li, L. Sun and L. Wang, RSC Adv., 2015, 5, 7267–7279 RSC.
- Q. Wang, J. Bai, Z. Lu, Y. Pan and X. You, Chem. Commun., 2016, 52, 443–452 RSC.
- A. Corma and H. García, Chem. Rev., 2003, 103, 4307–4366 CrossRef CAS PubMed.
- M. Moliner, Dalton Trans., 2014, 43, 4197–4208 RSC.
- P. Valvekens, F. Vermoortele and D. de Vos, Catal. Sci. Technol., 2013, 3, 1435–1445 CAS.
- E. Pérez-Mayoral and J. Čejka, ChemCatChem, 2011, 3, 157–159 CrossRef.
- E. Pérez-Mayoral, Z. Musilova, B. Gil, B. Marszalek, M. Položij, P. Nachtigall and J. Čejka, Dalton Trans, 2012, 41, 4036–4044 RSC.
- M. Položij, E. Pérez-Mayoral, J. Čejka, J. Hermann and P. Nachtigall, Catal. Today, 2013, 204, 101–107 CrossRef.
- F. Vermoortele, B. Bueken, G. Le Bars, B. van de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier, V. van Speybroeck, C. Kirschhock and D. E. de Vos, J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed.
- F. Vermoortele, R. Ameloot, L. Alaerts, R. Matthessen, B. Carlier, E. V. R. Fernandez, J. Gascon, F. Kapteijn and D. E. de Vos, J. Mater. Chem., 2012, 22, 10313–10321 RSC.
- F. Xu, H. R. Krouse and T. W. Swaddle, Inorg. Chem., 1985, 267–270 CrossRef CAS.
- K. Emerson and W. M. Graven, J. Inorg. Nucl. Chem., 1959, 11, 309–313 CrossRef CAS.
- R. G. Hughes and C. S. Garner, Inorg. Chem., 1968, 7, 1988–1993 CrossRef CAS.
- T. J. Williams and C. S. Garner, Inorg. Chem., 1969, 8, 1639–1645 CrossRef CAS.
- Mechanisms of Inorganic Reactions, ed. F. Basolo and R. G. Pearson, John Wiley & Sons, Inc, New York, 1967 Search PubMed.
- P. Andersen, A. Dossing, J. Gerup and M. Rude, Acta Chem. Scand., 1990, 44, 346–352 CrossRef CAS.
- K. R. Ashley and S. Kulprathipanja, Inorg. Chem., 1972, 11, 444–447 CrossRef CAS.
- A. Herbst, A. Khutia and C. Janiak, Inorg. Chem., 2014, 53, 7319–7333 CrossRef CAS PubMed.
- J. Jiang and O. M. Yaghi, Chem. Rev., 2015, 115, 6966–6997 CrossRef CAS PubMed.
- A. Vimont, H. Leclerc, F. Maugé, M. Daturi, J. Lavalley, S. Surblé, C. Serre and G. Férey, J. Phys. Chem. C, 2007, 111, 383–388 CAS.
- A. Dhakshinamoorthy and H. Garcia, ChemSusChem, 2014, 7, 2392–2410 CrossRef CAS PubMed.
- G. Akiyama, R. Matsuda, H. Sato and S. Kitagawa, Chem. – Asian J., 2014, 9, 2772–2777 CrossRef CAS PubMed.
- J. Tang, X. Guo, L. Zhu and C. Hu, ACS Catal., 2015, 5, 5097–5103 CrossRef CAS.
- J. Chen, S. Wang, J. Huang, L. Chen, L. Ma and X. Huang, ChemSusChem, 2013, 6, 1545–1555 CrossRef CAS PubMed.
- S. Wang, J. Chen and L. Chen, Catal. Lett., 2014, 144, 1728–1734 CrossRef CAS.
- L. Wang and F.-S. Xiao, Green Chem., 2015, 17, 24–39 RSC.
- M. Moliner, Y. Roman-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164–6168 CrossRef CAS PubMed.
- A. Onda, T. Ochi and K. Yanagisawa, Green Chem., 2008, 10, 1033–1037 RSC.
- J. Pang, A. Wang, M. Zheng, Y. Zhang, Y. Huang, X. Chen and T. Zhang, Green Chem., 2012, 14, 614–617 RSC.
- J. Geboers, S. van de Vyver, K. Carpentier, K. de Blochouse, P. Jacobs and B. Sels, Chem. Commun., 2010, 46, 3577–3579 RSC.
- Y. Zhang, V. Degirmenci, C. Li and E. J. M. Hensen, ChemSusChem, 2011, 4, 59–64 CrossRef CAS PubMed.
- H. B. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597–1600 CrossRef CAS PubMed.
- M. M. Ozer, Y. Jia, Z. Zhang, J. R. Thompson and H. H. Weitering, Science, 2007, 316, 1594–1597 CrossRef PubMed.
- L. Bromberg, X. Su and T. A. Hatton, Chem. Mater., 2014, 26, 6257–6264 CrossRef CAS.
- J. Chen, K. Li, L. Chen, R. Liu, X. Huang and D. Ye, Green Chem., 2014, 16, 2490–2499 RSC.
- Z. Hu, Y. Peng, Y. Gao, Y. Qian, S. Ying, D. Yuan, S. Horike, N. Ogiwara, R. Babarao, Y. Wang, N. Yan and D. Zhao, Chem. Mater., 2016, 28, 2659–2667 CrossRef CAS.
- K.-I. Shimizu, R. Uozumi and A. Satsuma, Catal. Commun., 2009, 10, 1849–1853 CrossRef CAS.
- C. Lansalot-Matras and C. Moreau, Catal. Commun., 2003, 4, 517–520 CrossRef CAS.
- Z. Huang, W. Pan, H. Zhou, F. Qin, H. Xu and W. Shen, ChemSusChem, 2013, 6, 1063–1069 CrossRef CAS PubMed.
- T. Wang, M. W. Nolte and B. H. Shanks, Green Chem., 2014, 16, 548–572 RSC.
- G. Tsilomelekis, M. Orella, Z. Lin, Z. Cheng, W. Zheng, V. Nikolakis and D. Vlachos, Green Chem., 2016, 18, 1983–1993 RSC.
- A. Osatiashtiani, A. F. Lee, D. R. Brown, J. A. Melero, G. Morales and K. Wilson, Catal. Sci. Technol., 2014, 4, 333–342 CAS.
- L. Yang, G. Tsilomelekis, S. Caratzoulas and D. G. Vlachos, ChemSusChem, 2015, 8, 1334–1341 CrossRef CAS PubMed.
- R. Weingarten, A. Rodriguez-Beuerman, F. Cao, J. S. Luterbacher, D. M. Alonso, J. A. Dumesic and G. W. Huber, ChemCatChem, 2014, 6, 2229–2234 CrossRef CAS.
- R. Bermejo-Deval, M. Orazov, R. Gounder, S.-J. Hwang and M. E. Davis, ACS Catal., 2014, 4, 2288–2297 CrossRef CAS.
- A. Herbst and C. Janiak, New J. Chem., 2016, 40, 7958–7967 RSC.
- Y. Su, G. Chang, Z. Zhang, H. Xing, B. Su, Q. Yang, Q. Ren, Y. Yang and Z. Bao, AIChE J., 2016 DOI:10.1002/aic.15356 , (manuscript online June 24, 2016).
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- I. Jiménez-Morales, A. Teckchandani-Ortiz, J. Santamaría-González, P. Maireles-Torres and A. Jiménez-López, Appl. Catal., B, 2014, 144, 22–28 CrossRef.
- J. Wang, J. Ren, X. Liu, J. Xi, Q. Xia, Y. Zu, G. Lu and Y. Wang, Green Chem., 2012, 14, 2506–2512 RSC.
- V. V. Ordomsky, J. van der Schaaf, J. C. Schouten and T. A. Nijhuis, ChemSusChem, 2013, 6, 1697–1707 CrossRef CAS PubMed.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed.
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O′Keeffe and O. M. Yaghi, Science, 2008, 319, 939–943 CrossRef CAS PubMed.
- S. B. Novaković, G. A. Bogdanović, C. Heering, G. Makhloufi, D. Francuski and C. Janiak, Inorg. Chem., 2015, 54, 2660–2670 CrossRef PubMed.
- S. A. Moggach, T. D. Bennett and A. K. Cheetham, Angew. Chem., Int. Ed., 2009, 48, 7087–7089 CrossRef CAS PubMed.
- N. Liédana, A. Galve, C. Rubio, C. Téllez and J. Coronas, ACS Appl. Mater. Interfaces, 2012, 4, 5016–5021 Search PubMed.
- M. S. Holm, Y. J. Pagán-Torres, S. Saravanamurugan, A. Riisager, J. A. Dumesic and E. Taarning, Green Chem., 2012, 14, 702–706 RSC.
- C. M. Osmundsen, M. S. Holm, S. Dahl and E. Taarning, Proc. R. Soc. A, 2012, 468, 2000–2016 CrossRef CAS.
- J. Chen, R. Liu, Y. Guo, L. Chen and H. Gao, ACS Catal., 2014, 5, 722–733 CrossRef.
- Q. Yuan, D. Zhang, L. van Haandel, F. Ye, T. Xue, E. J. Hensen and Y. Guan, J. Mol. Catal. A: Chem., 2015, 406, 58–64 CrossRef CAS.
- R. Fang, R. Luque and Y. Li, Green Chem., 2016, 18, 3152–3157 RSC.
- G. D. Yadav and R. V. Sharma, Appl. Catal., B, 2014, 147, 293–301 CrossRef CAS.
- A. Takagaki, M. Takahashi, S. Nishimura and K. Ebitani, ACS Catal., 2011, 1, 1562–1565 CrossRef CAS.
- J. Kim, G. T. Neumann, N. D. McNamara and J. C. Hicks, J. Mater. Chem. A, 2014, 2, 14014–14027 CAS.
- D. Zhang, F. Ye, Y. Guan, Y. Wang and E. J. M. Hensen, RSC Adv., 2014, 4, 39558–39564 RSC.
- H. J. Bart, J. Reidetschlager, K. Schatka and A. Lehmann, Ind. Eng. Chem. Res., 1994, 33, 21–25 CrossRef CAS.
- F. Cirujano, A. Corma and F. Llabrés i Xamena, Chem. Eng. Sci., 2015, 124, 52–60 CrossRef CAS.
- D. R. Fernandes, A. S. Rocha, E. F. Mai, C. J. Mota and V. Teixeira da Silva, Appl. Catal., A, 2012, 425, 199–204 CrossRef.
- K. Y. Nandiwale, S. K. Sonar, P. S. Niphadkar, P. N. Joshi, S. S. Deshpande, V. S. Patil and V. V. Bokade, Appl. Catal., A, 2013, 460, 90–98 CrossRef.
- Z. Li, R. Wnetrzak, W. Kwapinski and J. J. Leahy, ACS Appl. Mater. Interfaces, 2012, 4, 4499–4505 CAS.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- A. Sivasamy, K. Y. Cheah, P. Fornasiero, F. Kemausuor, S. Zinoviev and S. Miertus, ChemSusChem, 2009, 2, 278–300 CrossRef CAS PubMed.
- M. Koberg and A. Gedanken, Energy Environ. Sci., 2012, 5, 7460–7469 CAS.
- D. Sun, Y. Yamada, S. Sato and W. Ueda, Appl. Catal., B, 2016, 193, 75–92 CrossRef CAS.
- C. Chizallet, S. Lazare, D. Bazer-Bachi, F. Bonnier, V. Lecocq, E. Soyer, A. Quoineaud and N. Bats, J. Am. Chem. Soc., 2010, 35, 12365–12377 CrossRef PubMed.
- J. Chen, R. Liu, H. Gao, L. Chen and D. Ye, J. Mater. Chem. A, 2014, 2, 7205–7213 CAS.
- A. Zięba, A. Pacuła and A. Drelinkiewicz, Energy Fuels, 2010, 24, 634–645 CrossRef.
- V. Stavila, R. Parthasarathi, R. W. Davis, F. El Gabaly, K. L. Sale, B. A. Simmons, S. Singh and M. D. Allendorf, ACS Catal., 2016, 6, 55–59 CrossRef CAS.
- F. Zhang, Y. Jin, Y. Fu, Y. Zhong, W. Zhu, A. A. Ibrahim and M. S. El-Shall, J. Mater. Chem. A, 2015, 3, 17008–17015 CAS.
- F. Zhang, S. Zheng, Q. Xiao, Y. Zhong, W. Zhu, A. Lin and M. Samy El-Shall, Green Chem., 2016, 18, 2900–2908 RSC.
- Z. Zhu, H. Tan, J. Wang, S. Yu and K. Zhou, Green Chem., 2014, 16, 2636 RSC.
- J.-P. Lange, Angew. Chem., 2015, 127, 13382–13394 CrossRef.
- R. C. Klet, Y. Liu, T. C. Wang, J. T. Hupp and O. K. Farha, J. Mater. Chem. A, 2016, 4, 1479–1485 CAS.
- J. Canivet, J. Bonnefoy, C. Daniel, A. Legrand, B. Coasne and D. Farrusseng, New J. Chem., 2014, 38, 3102–3111 RSC.
- K. Tan, N. Nijem, Y. Gao, S. Zuluaga, J. Li, T. Thonhauser and Y. J. Chabal, CrystEngComm, 2015, 17, 247–260 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Tables with catalytic conditions, product and by-product yields for MOF and non-MOF catalysts in biomass conversion. See DOI: 10.1039/c6ce01782g |
| This journal is © The Royal Society of Chemistry 2017 |