Synthesis of sinter-resistant Au@silica catalysts derived from Au25 clusters†
Received
25th August 2016
, Accepted 6th December 2016
First published on 6th December 2016
Abstract
Gold clusters exhibit remarkable catalytic activity for many reactions such as carbon monoxide oxidation, alcohol, alkene, and hydrocarbon oxidations, and reduction reactions at low temperatures. However, several previous studies show that Au clusters undergo problematic sintering at temperatures above 250 °C, which makes them unsuitable catalysts for high-temperature oxidation reactions. Here we report the coating of Au25(11-MUA)18 clusters (where 11-MUA = mercaptoundecanoic acid) by silica to produce sinter-resistant Au@SiO2 catalysts. The structure of the resulting materials before and after calcination at temperatures up to 650 °C was followed by TEM and extended X-ray absorption fine structure spectroscopy (EXAFS) analyses, which showed that the Au@SiO2 catalysts created were much more stable to sintering compared to control materials; with average particles sizes of 2.2 nm after calcination at 250 °C and just over 3 nm after calcination at 650 °C. In addition, we explored the activity of the resulting materials for the 4-nitrophenol reduction and styrene epoxidation reactions; results clearly showed that the Au surfaces are accessible for reactants and that the kinetics of 4-nitrophenol reduction was directly related to the dispersion of the Au particles, as measured via the first shell Au–Au coordination numbers by EXAFS. Styrene epoxidation results show that the Au@SiO2 materials have excellent activity and recyclability.
Introduction
Gold catalysts, especially below 2 nm in size, have attracted much attention in catalysis owing to their well-understood structures and broad catalytic activity.1–5 Several theoretical studies have shown that small clusters (Au14, Au25, Au28, Au38, etc.) can dissociate molecular oxygen by transferring electron charge to 2π* of dioxygen,6,7 which is a crucial step in oxidation reactions. For example, ceria supported Au25 clusters have shown moderate catalytic activity for carbon monoxide oxidation at room temperature.8 A number groups have shown that Au25(SR)18 (in the form of free clusters or supported clusters) are effective catalysts for oxidation and reduction reactions at moderate temperatures.9,10 However, activation of pre-synthesized clusters without destroying their integrity remains a significant challenge in the field. Many groups have shown that Au catalysts showed sintering and corresponding drops in activity for supported-Au catalysts after heating at moderate temperatures.11,12 For example, Haruta et al. observed that the Au supported on TiO2 catalyst calcined at 500 °C showed lesser catalytic activity for CO oxidation than the catalyst calcined at 200 °C, due to sintering of Au clusters at higher calcination temperatures.13 The melting point of metals decreases with particle size,14 and most metals show significant surface mobility and sintering when approaching their Tammann temperature (half the melting point of a metal, in K).15 Thermal stability of catalysts is crucial because many industrially important reactions, such as hydrocracking,16 partial oxidation,17 and complete combustion18 are carried out at higher temperatures of 300 °C to 600 °C. Thus due to their thermal instability, application of Au catalysts in industry is restricted.
Previously our group studied the thermal stability of Au25(SC8H9)18 clusters on carbon supports by EXAFS and TEM analysis which showed that at temperatures above 200 °C, thiol stabilizers start to decompose and Au clusters begin to sinter.19 The size of the Au clusters increased up to 1.9 nm and 2.1 nm after heating at 250 °C and 350 °C for 1.5 h, respectively. Other groups have shown similar results on a variety of supports, particularly at high loadings.20,21 Zhu et al. carried out thermogravimetric analysis (TGA) studies which showed that Aun(SR)m begins to lose its stabilizing ligands at 200 °C and decomposes completely at 250 °C.20 Sintering in nanoparticle catalysts is mainly due to the agglomeration of small particles owing to their mobility on supports at a higher temperature. The agglomeration of Au (and other metal) nanoparticles can be prevented by a number of strategies. One popular strategy is to encapsulate the nanoparticles with a metal oxide shell (either core@shell or yolk@shell structures),22,23 overcoating with metal oxides via atomic vapour deposition,24 or spatially isolating them in mesoporous silica25 or MOF frameworks.26
Nearly all attempts to make core@shell or yolk@shell clusters have involved much larger nanoparticles, which have lower metal surface areas and thus are less practical for catalysis. Two previous attempts have been made to encapsulate Au clusters with a silica shell.27,28 Pradeep et al. previously synthesized glutathione-protected Au25 clusters encapsulated with silica shell, with many clusters embedded in a silica matrix, but did not study the thermal stability of the resulting materials.27 Devi et al. synthesized silica colloids containing many ca. 1.5 nm Au clusters stabilized by mercaptoalkylammonium chloride stabilizers.28 However, while the resulting particles were catalytically active for H2O2 oxidation of a peroxidase substrate, the Au clusters were found to undergo significant sintering at 250 °C thermal treatments, with 90% of the particles above 3 nm in size.
In this paper, we synthesized Au25(11-MUA)18 clusters and then encapsulated the clusters with silica shells. The thermal stability of the resulting materials was probed by TEM and EXAFS analysis. TEM images of Au@SiO2 catalysts revealed that majority of the particles are still less than 2 nm in size after calcination at 650 °C, although there is some minor sintering of particles to 2–4 nm in size which is due to agglomeration of multiple Au25(11-MUA)18 clusters in single silica particles. In order to show that the Au clusters are much more sinter-resistant in the silica matrix, we coated Au25(11-MUA)18 clusters onto silica spheres and calcined at two different temperatures (250 °C and 650 °C) and compared the samples and their catalytic activity with Au25@SiO2 catalysts. There was dramatic differences in the catalytic activity of silica-encapsulated vs. the non-encapsulated clusters, which was due to the tremendous sintering seen for non-encapsulated clusters. For encapsulated clusters, only a small reduction of the catalytic activity for 4-nitrophenol reduction reaction over Au25@SiO2 catalysts was seen upon increased calcination temperature, which correlates well with the slight increase in EXAFS first shell Au–Au coordination numbers. Styrene epoxidation reactions using Au25@SiO2 particles show that materials calcined at 250 °C retain excellent activity and strong recyclability, while materials calcined at 650 °C significantly outperform their non-encapsulated counterparts
Experimental section
Materials
Hydrogen tetrachloroaurate(III) trihydrate (HAuCl4·3H2O, 99.9% on metal basis, Aldrich), 11-mercaptoundecanoic acid (11-MUA, 95%, Sigma-Aldrich), tetraoctylammonium bromide (TOAB, 98%, Aldrich), sodium borohydride (NaBH4, 98%, EMD), tetraethylorthosilicate (TEOS, 98%, Aldrich), 4-nitrophenol (PNP, 99%, Alfa Aesar), tert-butyl hydroperoxide (TBHP, 70 wt% in H2O, Sigma-Aldrich), acetonitrile (CH3CN, 99.9% Fischer Scientific), and styrene (99%, Sigma-Aldrich) were used as received. Ammonia (30%), tetrahydrofuran (THF, high purity) and ethanol (100%) were purchased from EMD, Fischer Scientific and Commercial Alcohol respectively. Milli-Q water was used for synthesis.
Synthesis of Au25(11-MUA)18 clusters
The synthetic procedure for Au25(11-MUA)18 clusters has been documented previously by our group.29 In a typical synthesis, TOAB (333.5 mg, 1.2 eq.) was added to HAuCl4·3H2O solution (200.0 mg) in 20 ml THF in a round bottom flask. The solution was stirred until it turned from yellow to orange-red. After that 11-MUA (545.9 mg, 5 eq. in 5 mL THF) was added to the flask and the solution was stirred until it became colourless. The solution was then cooled using an ice bath followed by the addition of ice-cold NaBH4 (75.66 mg, 4 eq. in 2 mL water) dropwise and the solution was monitored using UV-vis spectroscopy. The addition of NaBH4 was continued until the characteristic absorption peaks of Au25(11-MUA)18 clusters were seen by UV-vis spectroscopy. The solution was then centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 2 min, and the precipitate of larger particles was discarded. The remaining Au25(11-MUA)18 cluster solution in THF was then cooled in an ice bath and additional NaBH4 (28.38 mg, 1.5 eq. in 2 mL water) was added dropwise until a brown precipitate was obtained. The residue was centrifuged and washed with THF twice, followed by re-dissolution in water. Au25(11-MUA)18 clusters were precipitated out with a few drops of dilute acetic acid with an approximate pH of 3. The precipitate was washed with water twice and redissolved in THF. One drop of NaBH4 (1.5 eq. in 2 mL water) was added to the THF solution, which re-precipitated out Au25(11-MUA)18 clusters. The precipitate was then collected using centrifugation and washed with THF. 10 mg of dried Au25(11-MUA)18 clusters were dispersed in Millipore water (6 mL) and diluted with 25 mL ethanol.
000 rpm for 2 min, and the precipitate of larger particles was discarded. The remaining Au25(11-MUA)18 cluster solution in THF was then cooled in an ice bath and additional NaBH4 (28.38 mg, 1.5 eq. in 2 mL water) was added dropwise until a brown precipitate was obtained. The residue was centrifuged and washed with THF twice, followed by re-dissolution in water. Au25(11-MUA)18 clusters were precipitated out with a few drops of dilute acetic acid with an approximate pH of 3. The precipitate was washed with water twice and redissolved in THF. One drop of NaBH4 (1.5 eq. in 2 mL water) was added to the THF solution, which re-precipitated out Au25(11-MUA)18 clusters. The precipitate was then collected using centrifugation and washed with THF. 10 mg of dried Au25(11-MUA)18 clusters were dispersed in Millipore water (6 mL) and diluted with 25 mL ethanol.
Synthesis of silica encapsulated Au25(11-MUA)18 clusters
The silica encapsulation strategy is depicted in Scheme 1. In order to minimise the formation of multiple clusters in a single silica shell, 1.0 ml of the above Au25(11-MUA)18 cluster solution was diluted with 10 mL ethanol. Then the diluted Au25(11-MUA)18 clusters and ammonia (1.0 mL) were added to a solution of TEOS (10 μL) in ethanol (10 mL) dropwise under moderate stirring. After 12 h of stirring the material was collected by centrifugation and washed several times with ethanol and water. The silica encapsulated clusters are labelled as Au25@SiO2 in this manuscript for brevity. The Au25@SiO2 materials were dried at 100 °C and then calcined at different temperatures 250 °C, 350 °C, 450 °C, 550 °C and 650 °C for 3 h in a stream of air, to give the resulting Au@SiO2 materials.
 |
| | Scheme 1 General scheme for the synthesis of silica encapsulated Au25(11-MUA)18 clusters. | |
Synthesis of Au25(MUA)18 clusters on silica spheres (control samples)
Synthesis of silica spheres: to synthesize silica spheres, we followed the same procedure as above, without Au25(11-MUA)18 clusters. In a typical synthesis, 6 mL water is diluted with 25 mL ethanol, and then 1 mL of a water/ethanol mixture is added into the vial and the whole mixture was diluted with 10 mL ethanol. Then this very diluted water/ethanol mixture and ammonia (1 mL) was added to the TEOS (10 μL) and ethanol (10 mL) solution dropwise under moderate stirring. The solution was then kept stirring for 12 h, then collected by centrifugation and washed several times with ethanol and water.
Deposition of Au25(11-MUA)18 clusters on silica spheres: 1.0 ml of the aqueous Au25(11-MUA)18 cluster solution was added to the dried silica sphere sample and stirred. After 2 h stirring, the solvent was evaporated using a rotary evaporator followed by drying on a Schlenk line apparatus. The metal loading was maintained as 2.0 wt% Au. Au25(11-MUA)18 clusters supported on silica spheres were dried at 100 °C and then calcined at two different temperatures 250 °C and 650 °C for 3 h in a stream of air. The non-encapsulated catalysts have been labelled as Au25/SiO2.
Characterization
UV-vis absorption spectra of the Au25(11-MUA)18 clusters were analysed using a Varian Cary 50 Bio UV-vis spectrometer. The morphology of the Au25@SiO2 materials was analysed by a HT7700 TEM operating at 100 kV. The size of 200 particles was measured for each sample using ImageJ software to calculate average particle sizes and standard deviations and create size distribution histograms.30 Extended X-ray absorption fine structure (EXAFS) spectroscopic analysis was performed at HXMA beamline 061D-1 (energy range 5–30 keV, resolution 1 × 10−4 ΔE/E) at the Canadian Light Source. The storage ring electron energy and ring current were 2.9 GeV and 250 mA, respectively. All data was collected in transmission mode. The energy for the Au-L3 edge (11919 eV) was selected by using a Si(111) double crystal monochromator with Rh-coated 100 nm long KB mirror. Higher harmonics were removed by detuning the double crystal monochromator. Data fitting was carried out using the Demeter software package.31 In order to fit the data, the amplitude reduction factor was fixed at 0.9 for all the data, which was the value obtained from fitting of the Au foil data. Metal loadings of Au25@SiO2 and Au25/SiO2 were analysed by a Varian Spectra AA 55 Atomic Absorption Spectroscope. Nitrogen adsorption/desorption isotherms were collected by using a Micromeritics ASAP2020 system (Norcross GA). The BET model was used to calculate the specific surface area of catalysts.
Catalytic activity for 4-nitrophenol reduction
2.0 mL of 0.10 mM 4-nitrophenol and 1.0 mg Au25@SiO2 were mixed in a quartz cuvette and the UV-vis spectra were collected. After adding ice cold 0.5 mL 0.1 M NaBH4 in water to the cuvette, the spectrum was analysed immediately, to get the initial concentration of 4-nitrophenolate, and the progress of the reaction was monitored at 2 min time intervals. Catalytic results were reproduced several times for each sample, with errors <5%.
Styrene epoxidation
Catalytic tests were carried out in a 100 mL round bottom flask fitted with a reflux condenser. 20 mg catalyst, 920 μL styrene, and 2.0 mL of the TBHP solution were added to 5.0 mL acetonitrile and then kept for stirring for 24 h at 82 °C.32 The substrate to Au mole ratio is 3920:1. Products were analysed by a gas chromatograph (7890A, Agilent Technologies) equipped with a HP-5 column using a flame ionization detector. For recyclability tests, the catalyst was removed from the catalytic mixture by centrifugation, washed with acetone, and dried at 50 °C before reuse.
Results and discussion
UV-vis absorption studies of the as-synthesised Au25(11-MUA)18 clusters in water/ethanol mixtures show three peaks at 680 nm, 446 nm, and 398 nm which are attributed to HOMO–LUMO transitions as shown in Fig. 1.33 The UV-vis spectrum gives clear evidence for the formation of Au25 clusters. Previously we have also used MALDI-MS analysis to further strengthen this assignment.29 The clusters were copiously purified by taking advantage of the poor solubility of the carboxylate-terminated clusters in THF in basic conditions as well as poor solubility in water under acidic conditions. TEM images (Fig. S1, ESI†) of the Au25(11-MUA)18 clusters show cluster cores with a size of ca. 1 nm in agreement with previous results.34
 |
| | Fig. 1 UV-vis spectrum of Au25(11-MUA)18 clusters in water/ethanol mixture. | |
The Au25(11-MUA)18 clusters were then coated with silica via sol–gel synthesis using NH3 as a catalyst. Au25 cluster solutions were significantly diluted before silica coating to minimize the number of clusters per silica sphere in the final materials and control the final metal loading. Fig. 2 shows TEM images of the as-synthesized silica coated Au25(11-MUA)18 clusters. The TEM images suggest that the clusters are encapsulated in the silica matrix, stronger evidence for encapsulation comes from comparisons of calcinations of encapsulated clusters vs. control samples shown below. It should be noted that many silica spheres contain multiple clusters, although some particles seem to have only a single Au25(11-MUA)18 cluster. From TEM analysis, the average silica sphere diameter is 40 nm and clusters are ca. 1.1 ± 0.3 nm in size (the slightly larger sizes are likely an artefact of the inability to focus on each individual particle in the three-dimensional silica spheres, causing some particles to appear larger as they are out of focus). The Au loading in silica matrix was maintained as 2.0 wt% which was confirmed by atomic absorption spectroscopy (AAS). Control samples were also made which consisted of Au25(11-MUA)18 clusters decorated on the surface of silica spheres, as shown in Fig. 3a.
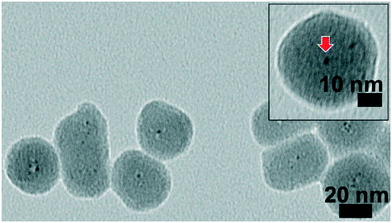 |
| | Fig. 2 TEM image of as-synthesized Au25(11-MUA)18@SiO2 (inset; enlarged image). | |
 |
| | Fig. 3 TEM images of Au25/SiO2 control samples a) as synthesized, b) calcined at 250 °C, c) calcined at 650 °C. | |
A major issue with many Au systems is the propensity for Au clusters or nanoparticles to sinter at moderate calcination temperatures; previous studies have shown that temperatures of at least 250 °C are needed to remove thiol stabilizers from the Au cluster surfaces, with even higher temperatures needed to completely oxidize the disulphide biproducts.19 To investigate the sinter-resistance of the Au25@SiO2 catalysts, they were calcined at temperatures up to 650 °C followed by TEM analysis. Fig. 4 shows TEM images of the samples upon sintering at temperatures between 250 °C and 650 °C (2.2 ± 1.0 nm at 250 °C and 3.2 ± 2.0 nm at 650 °C); very little growth in the average particle size was seen. Particle size histograms showed a high population of <2 nm sized particles even after calcination at higher temperatures (Fig. 5). As shown in Fig. 3b and c, control samples consisting of Au25(11-MUA)18 clusters on the surface of silica spheres showed a much greater degree of sintering, with average particle sizes of 3.2 ± 1.7 nm at 250 °C and 15.5 ± 10.0 nm at 650 °C. The particle size histogram for the non-encapsulated control samples is shown in Fig. S2 (ESI†), and is quite distinct from the Au@SiO2 histogram in Fig. 5, with a number of particles >6 nm in size even after calcination at 250 °C, and nearly all particles >5 nm in size after calcination at 650 °C. Thus, it is quite evident that the encapsulation of the Au25 clusters in silica greatly promotes their stability to sintering at higher temperatures.
 |
| | Fig. 4 TEM images of Au25@SiO2 calcined at a) 250 °C, b) 350 °C, c) 450 °C, d) 550 °C, e) and f) 650 °C (inset; enlarged image). | |
 |
| | Fig. 5 Histogram of Au25@SiO2 catalysts calcined at different temperatures. | |
The Au25@SiO2 materials were also examined by nitrogen adsorption/desorption isotherms, which are shown in Fig. S3,† along with the corresponding BJH pore-size distributions. The BET surface area of the catalyst increased from 23 m2 g−1 to 70 m2 g−1 after calcination at 250 °C. Calculated BJH pore-size distributions show a broad distribution of mesopores centred at 9 nm after calcination at 250 °C that are not present in the initial as-synthesized sample. Pore volumes of micropores increased from 0.0012 cm3 g−1 to 0.0092 cm3 g−1 after calcination at 250 °C, while mesopore pore volumes increased from 0.051 cm3 g−1 to 0.111 cm3 g−1. We believe the resulting porosity was caused by removal of the MUA as well as residual solvent from the silica spheres. Importantly, the silica spheres have some porosity, which may allow for catalytic accessibility of the Au clusters.
To further understand changes to the Au clusters before and after calcination, Au L3-edge EXAFS analysis was carried out in transmission mode on the hard X-ray microanalysis beamline (HXMA) at the Canadian Light Source (CLS). Fig. 6 shows the Au L3 edge EXAFS k-space and phase-corrected R-space spectra of the as-synthesized Au25(11-MUA)18@SiO2 clusters prior to calcination. The black line represents the experimental Fourier transformed EXAFS spectra, and the red line represents the simulated EXAFS fit for Au25(11-MUA)18 clusters. Others have previously collected X-ray crystallographic data of Au25(SR)18 clusters which have shown that they have a core–shell morphology, in which the core is composed of an Au13 icosahedron, in which the central atom is surrounded by 12 Au atoms while the shell consists of six S–Au–S–Au–S staple motifs.35 Twelve out of the twenty faces of the icosahedron are surrounded by six staple motifs, with sulfur atoms directly attached to 12 Au atoms of the icosahedron core. A multishell fitting approach which has previously been documented by Zhang's group was used to fit the Au–S and three Au–Au contributions of the clusters.36 In order to fit the data, we first fit the Au–S contribution by using Au–S model data obtained from the standard Au25(SR)18 structure.36,37 After fitting the parameters for Au–S contribution, we fixed those values followed by fitting all first shell Au–Au coordination modes (there are three Au–Au first shell interactions: the first two involve Au atoms in core and the third involves Au staple atoms). Coordination number (CN) values for all Au–Au contributions were fixed based on the crystal structure of the clusters. The final EXAFS fitting parameters are shown in Table 1. The Au–S bond length was found to be 2.31(1) Å which matches well with crystallographic data of Au25L18 clusters in which L = phenylethanethiol (see Table S1, ESI†).38 For the Au first shell fit the Au–Au(core) R value of 2.76(2) Å is attributed to the distance between the central Au atom and the surface Au atom of the icosahedron core and 6 of the Au surface atom pairs, whereas 2.91(6) Å is the bond distance between the other surface Au atom pairs of the icosahedron (Au–Au(surf)). The last Au–Au bond length, 3.3(1) Å is attributed to the distance between the surface Au atoms and the staple Au atoms. These values agree well with those seen for other Au clusters,19 and thus the EXAFS data fitting clearly shows that the basic core structure of Au25(11-MUA)18 clusters are similar to that of Au25 clusters using other thiolate ligands.
 |
| | Fig. 6 Au L3 edge EXAFS fitting in a) k space and b) phase-corrected R space of as-synthesized Au25(11-MUA)18@SiO2. | |
Table 1 EXAFS fitting parameters of as-synthesized Au25(11-MUA)18@SiO2
| Type |
CN |
R/Å |
σ
2/Å2 |
E
o shift (eV) |
| Au–S |
1.3 |
2.31(1) |
0.001 |
0.73 |
| Au–Au(core) |
1.44 |
2.76(2) |
0.002(2) |
2.5(3) |
| Au–Au(surf) |
1.92 |
2.91(6) |
0.01(1) |
2.5(3) |
| Au–Au(staple) |
2.88 |
3.3(1) |
0.10(3) |
2.5(3) |
The Au25@SiO2 materials calcined at different temperatures were also analyzed by EXAFS spectroscopy at the Au L3 edge. Phase-corrected R-space EXAFS spectra are shown in Fig. 7, while fitted data are found in Table 2. Individual k and phase-corrected R space spectra of Au25@SiO2 calcined at different temperatures are shown in Fig. S4 and S5 (ESI†) respectively. Au–S contributions are seen around 2.3 Å, while Au–Au contributions are present between 2.8 Å and 3.2 Å. An fcc Au model was used for fitting the single shell Au–Au fit, as some growth in cluster sizes was seen which prevented the use of the previous Au25 model. After calcination at 250 °C, an Au–S contribution was not observed in the data, which suggests that thiol stabilizers are completely removed from the gold surface, which is consistent with previous work using other thiolate ligands.19 After calcination at 250 °C, the first shell coordination number for the Au–Au (NAu–Au) contribution is 9.6(7) which suggests that the average number of atoms per particle is ∼300, which works out to a ca. 2.1 nm average particle size.39 As many of the silica spheres contain multiple Au clusters, this is consistent with some sintering of clusters within silica spheres, and is also consistent with TEM analysis above. EXAFS data suggest that on average ca. 12 clusters sinter together by 250 °C. As the calcination temperature increases, the average NAu–Au slightly increases and the NAu–Au of catalysts calcined at 350 °C, 450 °C, 550 °C, and 650 °C are 9.8(7), 10.0(7), 10.2(7), and 10.5(6), respectively which suggest that the average number of atoms in the Au particles grows slowly at higher calcination temperatures. Thus, EXAFS results suggest that there is some sintering of particles, albeit from TEM analyses there are still a large number of <2 nm particles at 650 °C.
 |
| | Fig. 7 Au L3-edge EXAFS data in phase-corrected R space of Au@SiO2 catalysts calcined at different temperatures. | |
Table 2 EXAFS fitting parameters of as-synthesized Au25@SiO2 catalysts calcined at different temperatures
| Catalyst |
CN (Au–Au) |
R/Å (Au–Au) |
σ
2/Å2 |
E
o shift (eV) (Au–Au) |
| Au25@SiO2 calcined at 250 °C |
9.6(7) |
2.84(1) |
0.010(2) |
6.1(8) |
| Au25@SiO2 calcined at 350 °C |
9.8(7) |
2.843(8) |
0.010(1) |
5.7(7) |
| Au25@SiO2 calcined at 450 °C |
10.0(7) |
2.851(7) |
0.009(1) |
5.6(7) |
| Au25@SiO2 calcined at 550 °C |
10.2(7) |
2.860(8) |
0.009(1) |
6.7(7) |
| Au25@SiO2 calcined at 650 °C |
10.5(6) |
2.856(6) |
0.009(1) |
6.5(5) |
Catalytic activity
4-Nitrophenol reduction reaction.
One of the major concerns upon coating clusters or nanoparticles with silica overlayers is whether or not the metal surfaces are still catalytically accessible. To probe whether the Au clusters could be accessed prior to and after sintering the Au25@SiO2 materials, the materials were probed as catalysts for the reduction of 4-nitrophenol. The catalytic activity for the 4-nitrophenol reduction reaction is an excellent gauge of the catalytically accessible surface area of metallic catalysts.40 A typical UV-vis spectra reaction profile is shown in Fig. S6;† the original solution has a peak at 317 nm which corresponds to 4-nitrophenol.41 After adding NaBH4 to the solution, the peak shifted to 400 nm which indicates the deprotonation of 4-nitrophenol to form nitrophenolate. The intensity of peak due to 4-nitrophenolate decreased as a function of time and a new peak appeared at 300 nm, which has been previously attributed to the formation of 4-aminophenol.42 In the absence of Au25@SiO2 catalysts, no activity for nitrophenol reduction was seen. Table 3 shows the rate constants for 4-nitrophenolate reduction reactions, which are calculated using pseudo-first-order kinetics.43,44 All materials showed a linear relationship between ln[Ct]/[Co] and reaction time (min), as shown in Fig. S7.† As the calcination temperature increases, the rate constants for the 4-nitrophenol reduction reaction was slightly reduced from 1.1 × 10−1 min−1 at 250 °C to 1.78 × 10−2 min−1 at 650 °C, which is likely due to the small increase in Au particle size, and thus decrease in catalytically available surface area. Fig. 8 shows the correlation of reaction rate constant with the coordination number of Au as determined by EXAFS at each calcination temperature; a strong relationship was seen between NAu–Au and the rate constant for 4-nitrophenol reduction. There is a drop in activity beyond calcination temperatures of 350 °C that cannot be explained by coordination number changes alone; this may be due to partial condensation of the silica matrix which leads to mass-transfer issues.
Table 3 Rate constant k (min−1) for 4-nitrophenol reduction reaction
| Catalyst |
k (min−1) |
| Au25@SiO2 calcined at 250 °C |
1.1 × 10−1 |
| Au25@SiO2 calcined at 350 °C |
1.0 × 10−1 |
| Au25@SiO2 calcined at 450 °C |
3.75 × 10−2 |
| Au25@SiO2 calcined at 550 °C |
2.80 × 10−2 |
| Au25@SiO2 calcined at 650 °C |
1.78 × 10−2 |
| Au25/SiO2 calcined at 250 °C |
1.75 × 10−1 |
| Au25/SiO2 calcined at 650 °C |
4.82 × 10−4 |
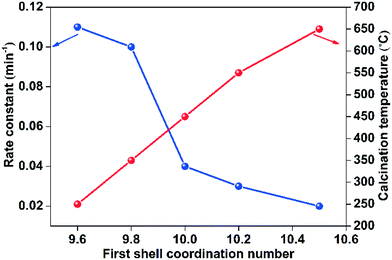 |
| | Fig. 8 Plot of first shell coordination number of Au@SiO2 catalysts vs. rate constant for 4-nitrophenol reduction reaction and calcination temperature. | |
Control samples in which silica spheres were decorated with Au25(11-MUA)18 clusters on the surface, followed by calcination at 250 °C and 650 °C were also examined (Fig. S8†). The encapsulated clusters (e.g. Au25@SiO2) showed a slightly lower activity than the Au25/SiO2 control samples upon calcination at 250 °C; this is likely due to mass transfer issues caused by the silica shell.45 The rate constant for the control samples for 4-nitrophenol reduction dropped from 1.75 × 10−1 min−1 for samples treated at 250 °C to 4.82 × 10−4 min−1 for samples treated at 650 °C, which was due to the tremendous sintering of gold clusters on the surface of the silica support as shown earlier by TEM. The Au25@SiO2 sample calcined at 650 °C had a rate constant that was over 35 times that of the control samples treated at the same temperature. Thus, this cluster encapsulation strategy allows for the formation of well-dispersed, supported-Au catalysts that can withstand high temperature operating and/or calcination conditions. We do note that for mild catalytic reactions such as CO oxidation, Au25 clusters can be activated under mild thermal conditions without significant adverse sintering of the Au cluster catalysts.46
Styrene epoxidation.
Results for styrene epoxidation reactions over Au25@SiO2 and Au25/SiO2 catalysts calcined at two different temperatures (250 °C and 650 °C) are shown in Table 4. The Au25@SiO2 catalyst calcined at 250 °C showed 70.0% conversion for this reaction, with a selectivity towards styrene oxide (SO) of 92.3% (the other product is benzaldehyde (BA)). This is higher than selectivities seen for larger Au particles,32 but in line with that reported for Au25 clusters supported on hydroxyapatite (HAP) by others.47 After calcination at 650 °C, conversion was slightly reduced to 62.3% with similar selectivity for SO, which is consistent with the drop in catalytic activity seen in the 4-nitrophenol reduction reactions above due to the slightly lower surface area of catalysts calcined at higher temperatures. Recyclability studies for Au25@SiO2 catalysts calcined at 250 °C are shown in Table S2;† after five cycles the catalyst showed the same activity for the reaction. This is significant, as others have shown that Au clusters supported on HAP or silica showed slight deactivation over multiple cycles.20 Au25@SiO2 catalysts calcined at 250 °C and 650 °C had average turnover frequencies (TOFs) of 116 ± 9 h−1 and 103 ± 10 h−1, respectively. These catalysts also show excellent catalytic activity for the styrene epoxidation reaction, in line with the best results in the literature.32,47–49 For example, Tsukuda et al. reported that Au25/HAP had an average TOF of 114 h−1.47 Wu et al. demonstrated that silica supported Au nanoparticles with an average particle size of 6.4 nm had an average TOF of 99.4 h−1.32 Non-encapsulated catalysts (Au25/SiO2) calcined at 250 °C had slightly higher conversion of styrene, with very slightly higher selectivity for styrene oxide (>93%); however, conversions dropped to 15.1% upon calcination of the control samples at 650 °C. These results are also consistent with 4-nitrophenol reduction results, such that non-encapsulated catalyst show slightly higher activities at moderate calcination temperatures due to the absence of mass-transfer issues, while higher calcination temperatures lead to tremendous sintering and a corresponding loss of catalyst surface area and activity. In order to compare TONs of the catalysts, we also took total surface Au atoms into account. When accounting for the relative surface areas; samples calcined at 650 °C showed higher adjusted TONs compared to Au25@SiO2 calcined at 250 °C. This may be due the removal of disulphide by-products from the catalyst at higher temperatures.
Table 4 Conversion (%) and TONs for styrene epoxidation reactions over 24 h
| Catalyst |
Conversion (%) |
Selectivity |
TONa |
Adj. TONb |
| SO (%) |
BA (%) |
|
TON = moles of product/moles of Au.
TON = moles of product/moles of surface Au.
|
| Au25@SiO2 calcined at 250 °C |
70.0 |
92.3 |
7.6 |
2800 |
5100 |
| Au25@SiO2 calcined at 650 °C |
62.3 |
91.8 |
8.3 |
2500 |
6500 |
| Au25/SiO2 calcined at 250 °C |
75.6 |
93.7 |
6.3 |
3000 |
8000 |
| Au25/SiO2 calcined at 650 °C |
15.1 |
93.4 |
6.6 |
600 |
7500 |
Conclusions
Herein we have synthesized silica encapsulated Au25(11-MUA)18 clusters, followed by calcination to give active catalysts which have minimal sintering of the Au clusters. To the best of our knowledge, sinter-resistant Au25(11-MUA)18 clusters encapsulated with silica shells have not been documented elsewhere. Even after calcination at 650 °C, the majority of the particles are below 2 nm. Catalytic studies for 4-nitrophenolate reduction reaction and styrene epoxidation reactions over the catalysts show that the active sites on the gold surface are accessible to the reactants. As the calcination temperature increases, the rate constant of the reaction slightly decreased, which is likely due to the increased coordination number of gold atoms, as individual clusters within the silica spheres sinter to form slightly larger particles. Finally, Au25@SiO2 catalysts calcined at 250 °C showed remarkable catalytic activity, selectivity and recyclability for styrene epoxidation reactions.
Acknowledgements
We would like to acknowledge the National Sciences and Engineering Research Council of Canada (NSERC) for financial support. We thank Ning Chen at the Canadian Light source (CLS) for assistance with XAS measurements. Hiwa Salimi and Leila Dehabadi are acknowledged for AAS and N2 adsorption analysis respectively. EXAFS experiments described in this paper were performed at the Canadian Light Source, which is supported by the Natural Sciences and Engineering Research Council of Canada, the National Research Council Canada, the Canadian Institutes of Health Research, the Province of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan.
Notes and references
- Y. Zhu, H. Qian, B. A. Drake and R. Jin, Angew. Chem., Int. Ed., 2010, 49, 1295–1298 CrossRef CAS PubMed.
- S. Sreedhala, V. Sudheeshkumar and C. P. Vinod, Catal. Today, 2015, 244, 177–183 CrossRef CAS.
- O. Lopez-Acevedo, K. A. Kacprzak, J. Akola and H. Häkkinen, Nat. Chem., 2010, 2, 329–334 CrossRef CAS PubMed.
- M. Haruta and M. Daté, Appl. Catal., A, 2001, 222, 427–437 CrossRef CAS.
- Y. Zhao, A. Mpela, D. I. Enache, S. H. Taylor, D. Hildebrandt, D. Glasser, G. J. Hutchings, M. P. Atkins and M. S. Scurrell, Stud. Surf. Sci. Catal., 2007, 163, 141–151 CrossRef CAS.
- B. Yoon, H. Häkkinen and U. Landman, J. Phys. Chem. A, 2003, 107, 4066–4071 CrossRef CAS.
- A. Sanchez, S. Abbet, U. Heiz, W. D. Schneider, H. Häkkinen, R. N. Barnett and U. Landman, J. Phys. Chem. A, 1999, 103, 9573–9578 CrossRef CAS.
- X. Nie, H. Qian, Q. Ge, H. Xu and R. Jin, ACS Nano, 2012, 6, 6014–6022 CrossRef CAS PubMed.
- Y. Zhu, H. Qian, M. Zhu and R. Jin, Adv. Mater., 2010, 22, 1915–1920 CrossRef CAS PubMed.
- A. Shivhare, S. J. Ambrose, H. Zhang, R. W. Purves and R. W. J. Scott, Chem. Commun., 2013, 49, 276–278 RSC.
- C. M. Yang, M. Kalwei, F. Schüth and K. J. Chao, Appl. Catal., A, 2003, 254, 289–296 CrossRef CAS.
- G. M. Veith, A. R. Lupini, S. Rashkeev, S. J. Pennycook, D. R. Mullins, V. Schwartz, C. A. Bridges and N. J. Dudney, J. Catal., 2009, 262, 92–101 CrossRef CAS.
- F. Boccuzzi, A. Chiorino, M. Manzoli, P. Lu, T. Akita, S. Ichikawa and M. Haruta, J. Catal., 2001, 202, 256–267 CrossRef CAS.
- P. Buffat and J. P. Borel, Phys. Rev. A, 1976, 13, 2287–2298 CrossRef CAS.
- J. Lu, B. Fu, M. C. Kung, G. Xiao, J. W. Elam, H. H. Kung and P. C. Stair, Science, 2012, 335, 1205–1208 CrossRef CAS PubMed.
- K. C. Park and S. K. Ihm, Appl. Catal., A, 2000, 203, 201–209 CrossRef CAS.
- S. S. Bharadwaj and L. D. Schmidt, Fuel Process. Technol., 1995, 42, 109–127 CrossRef CAS.
- O. Deutschmann, L. I. Maier, U. Riedel, A. H. Stroemman and R. W. Dibble, Catal. Today, 2000, 59, 141–150 CrossRef CAS.
- A. Shivhare, D. M. Chevrier, R. W. Purves and R. W. J. Scott, J. Phys. Chem. C, 2013, 117, 20007–20016 CAS.
- Y. Zhu, H. Qian and R. Jin, Chem. – Eur. J., 2010, 16, 11455–11462 CrossRef CAS PubMed.
- J. E. Martin, J. Odinek, J. P. Wilcoxon, R. A. Anderson and P. Provencio, J. Phys. Chem. B, 2003, 107, 430–434 CrossRef CAS.
- S. H. Joo, J. Y. Park, C.-K. Tsung, Y. Yamada, P. Yang and G. A. Somorjai, Nat. Mater., 2009, 8, 126–131 CrossRef CAS PubMed.
- S. Wang, M. Zhang and W. Zhang, ACS Catal., 2011, 1, 207–211 CrossRef CAS.
- A. Cao, R. Lu and G. Veser, Phys. Chem. Chem. Phys., 2010, 12, 13499–13510 RSC.
- H. Song, R. M. Rioux, J. D. Hoefelmeyer, R. Komor, K. Niesz, M. Grass, P. Yang and G. A. Somorjai, J. Am. Chem. Soc., 2006, 128, 3027–3037 CrossRef CAS PubMed.
- G. Lu, S. Li, Z. Guo, O. K. Farha, B. G. Hauser, X. Qi, Y. Wang, X. Wang, S. Han, X. Liu, J. S. DuChene, H. Zhang, Q. Zhang, X. Chen, J. Ma, S. C. J. Loo, W. D. Wei, Y. Yang, J. T. Hupp and F. Huo, Nat. Chem., 2012, 4, 310–316 CrossRef CAS PubMed.
- M. A. Habeeb Muhammed and T. Pradeep, Small, 2011, 7, 204–208 CrossRef CAS PubMed.
- A. Samanta, B. B. Dhar and R. N. Devi, J. Phys. Chem. C, 2012, 116, 1748–1754 CAS.
- A. Shivhare, L. Wang and R. W. J. Scott, Langmuir, 2015, 31, 1835–1841 CrossRef CAS PubMed.
- X. Liu, M. Atwater, J. Wang and Q. Huo, Colloids Surf., B, 2007, 58, 3–7 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- J. Liu, F. Wang, S. Qi, Z. Gu and G. Wu, New J. Chem., 2013, 37, 769–774 RSC.
- Y. Lu and W. Chen, Chem. Soc. Rev., 2012, 41, 3594–3623 RSC.
- Z. Wu, J. Suhan and R. Jin, J. Mater. Chem., 2009, 19, 622–626 RSC.
- J. Akola, M. Walter, R. L. Whetten, H. Häkkinen and H. Grönbeck, J. Am. Chem. Soc., 2008, 130, 3756–3757 CrossRef CAS PubMed.
- M. A. Macdonald, D. M. Chevrier, P. Zhang, H. Qian and R. Jin, J. Phys. Chem. C, 2011, 115, 15282–15287 CAS.
- G. A. Simms, J. D. Padmos and P. Zhang, J. Chem. Phys., 2009, 131, 214703 CrossRef PubMed.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS PubMed.
- H. G. Fritsche and R. E. Benfield, Z. Phys. D: At., Mol. Clusters, 1993, 26, 15–17 CrossRef CAS.
- S. Wunder, F. Polzer, Y. Lu, Y. Mei and M. Ballauff, J. Phys. Chem. C, 2010, 114, 8814–8820 CAS.
- F. Dong, W. Guo, S. K. Park and C. S. Ha, Chem. Commun., 2012, 48, 1108–1110 RSC.
- K. Kuroda, T. Ishida and M. Haruta, J. Mol. Catal. A: Chem., 2009, 298, 7–11 CrossRef CAS.
- M. Schrinner, F. Polzer, Y. Mei, Y. Lu, B. Haupt, M. Ballauff, A. Göldel, M. Drechsler, J. Preussner and U. Glatzel, Macromol. Chem.
Phys., 2007, 208, 1542–1547 CrossRef CAS.
- B. Baruah, G. J. Gabriel, M. J. Akbashev and M. E. Booher, Langmuir, 2013, 29, 4225–4234 CrossRef CAS PubMed.
- J. Lee, J. C. Park and H. Song, Adv. Mater., 2008, 20, 1523–1528 CrossRef CAS.
- Z. Wu, D. E. Jiang, A. K. P. Mann, D. R. Mullins, Z. A. Qiao, L. F. Allard, C. Zeng, R. Jin and S. H. Overbury, J. Am. Chem. Soc., 2014, 136, 6111–6122 CrossRef CAS PubMed.
- Y. Liu, H. Tsunoyama, T. Akita and T. Tsukuda, Chem. Commun., 2010, 46, 550–552 RSC.
- N. S. Patil, R. Jha, B. S. Uphade, S. K. Bhargava and V. R. Choudhary, Appl. Catal., A, 2004, 275, 87–93 CrossRef CAS.
- Y. Jin, P. Wang, D. Yin, J. Liu, H. Qiu and N. Yu, Microporous Mesoporous Mater., 2008, 111, 569–576 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TEM images of Au25(SR)18 clusters, particle size histograms, gas adsorption data, and EXAFS fitting plots for calcined samples and nitrophenolate reduction and styrene oxidation catalytic results. See DOI: 10.1039/c6cy01822j |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*








