
The mechanism of H2 and H2O desorption from bridging hydroxyls of a TiO2(110) surface†
Ruimin
Wang
ab and
Hongjun
Fan
*ab
aState Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, Liaoning Province, P. R. China. E-mail: fanhj@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, 19 A Yuquan Road, Shijingshan District, Beijing 100049, P.R. China
First published on 2nd December 2016
Abstract
The photocatalytic H2 production from H2O over TiO2 has attracted tremendous attention in recent years, and great progress has been achieved in light adsorption and water dissociation. As a comparison, the H2 and O2 production over a pure TiO2 surface has not been successful yet. Recently, the desorption of bridging hydroxyls (BBOHs) to form H2 has been found to be possible on a TiO2(110) surface. However, the yield of H2 is low, and the majority of BBOHs desorb as H2O. Here, for the first time, we have systematically studied the mechanism of H2 desorption and the competition of H2 and H2O desorption on a TiO2(110) surface with DFT methods. We found that the generally believed pathway, the direct coupling of BBOHs, is not the most facile pathway. We propose that the reaction undergoes a Ti–H key intermediate, which then forms an O–Hδ+⋯Hδ−–Ti type dehydrogenation transition state. On a stoichiometric surface, the barriers for H2 desorption and H2O desorption are 2.20 eV and 1.09 eV, respectively. However, with an increase in BBO vacancies (Ovs, created by H2O desorption), the barrier for H2 desorption decreases, while the barrier for H2O desorption increases. Specifically, H2 desorption was found to be easier than H2O desorption with our two Ovs TiO2 model. Therefore, our results predict that H2O desorption is dominant at the beginning, then H2 desorption becomes more and more competitive. This prediction perfectly fits with the experimental observations. Furthermore, the stability of the key Ti–H intermediates and the possible ways for changing the conditions of H2 desorption have also been discussed.
Introduction
Titania (TiO2) has emerged as one of the most important metal oxides in a large number of technological areas, including heterogeneous catalysis, electronic devices, photocatalysis, biomaterials, and so on.1–7 TiO2-based photocatalysis has grown into a fascinating research field since Fujishima and Honda's first reports on water splitting on TiO2 and its potential application in clean hydrogen production.8 The key problems in water splitting include light adsorption, water dissociation, H2 production and O2 production etc. In the past decades, great progress has been achieved in the fields of light adsorption and water dissociation. However, the H2 production over a TiO2 surface has not been successful until very recently14–18 by heating the photocatalyzed methanol/TiO2(110) surface.15 As shown in the literature and our previous work (ref. 33), the dissociation of MeOH on TiO2 affords aldehyde and bridging hydroxyls (BBOHs). The observation of H2 from MeOH shows that H2 can be formed by BBOHs, which is not clear before. The dissociation of H2O also affords BBOHs, together with the hydroxyl group on a five-coordinated titanium (Ti5c) site. Therefore, the observation of H2 from MeOH is crucial; it tells us that in principle H2 can also be formed by the dissociation of H2O over a TiO2 surface. However, currently the temperature for H2 desorption is >500 K, and H2O is already desorbed at this temperature (in the case of MeOH, the production of aldehyde leaves BBOH on the surface which cannot desorb as MeOH). If we can reduce the H2 desorption temperature to which the H2O can adsorb on the surface, the water splitting for hydrogen production could be available (although not catalytically unless the hydroxyl group on Ti5c can also be removed). Understanding the mechanism is one of the promising ways for designing new systems with better reactivity, and we expected that the mechanistic study of H2 desorption from a BBOH-covered TiO2(110) surface could be helpful in increasing the yield of H2 for the methanol/TiO2(110) system, as well as in providing valuable ideas to realize the H2 from the H2O/TiO2(110) system.The most commonly observed and investigated hydrogen adatoms on the rutile TiO2(110) surface are BBOHs that can be achieved by various methods.9–15 The largest challenge for the H2 desorption over TiO2(110) is the competition of H2O desorption. Besides that, BBOHs can also migrate into the TiO2 bulk. DFT calculations have shown that on a stoichiometric surface the barrier for H2O desorption is 0.9–1.1 eV,14,18 and the barrier for BBOH migration to bulk is also about 1.1 eV.12,17,18 However, the direct coupling of two BBOHs to form H2 is much difficult, with a barrier of 1.6–1.85 eV from nudged elastic band (NEB) calculations.14,18 These theoretical studies support the findings that it is indeed much easier for the BBOHs to diffuse into the bulk or produce H2O compared to the formation of H2 on the surface. On the experimental side, Michael A. Henderson found that BBOHs desorb as H2O at temperatures between 500 and 550 K.16 However, Yin et al. used high BBOH coverage samples and no corresponding H2O (or H2) temperature-programmed desorption (TPD) signal has been observed; instead, they supported that the BBOHs migrated into the TiO2 bulk.12 Afterwards, Y. Du et al. found a H2O TPD peak at 500 K, and no H2 product was detected from the highly hydroxylated TiO2(110) surface.14
Surprisingly, very recently our colleagues' TPD experiments have shown the desorption of H2O at about 520 K and the desorption of H2 at ∼50 K higher.15 Although the ratio of H2 is low, it is the first time that H2 production has been observed on a TiO2(110) gas–solid interface. The H2O yield increased very fast at first and then reached a plateau, and after that, the H2 yield was increasing faster than that of H2O.15 Later on, H2 desorption on an anatase(101) surface has also been observed by our colleagues.19 Wu et al. found a H2 peak at 207 K using thermal desorption spectroscopy (TDS) spectra, and they proposed that H–Ti species could be the photoactive species.21
These important observations put a big challenge to the mechanistic studies. Previous theoretical calculations have shown that for the 1 ML (1 ML = 5.2 × 1014 molecules cm−2) BBOHs coverage model, using the NEB method with 12 images, the direct coupling of two BBOHs to form H2 has a barrier of 1.85 eV (ref. 18) which is much higher than the barrier of H2O desorption. It is known that H2 desorption is facilitated by the reducing ability of the surface since it requires two electrons to be transferred from the surface to two H+ (BBOH) to form H2, while H2O desorption does not involve electron transfer between the surface and H2O, thus is less sensitive to the reducing ability of the surface. With a decrease in BBOH concentration, the surface becomes less reducing, and the H2 desorption barrier is expected to be higher, while in ref. 14, using the NEB method with 7 images, the barrier for the 1/2 ML BBOHs coverage model is found to be only 1.60 eV. We know that the barrier evaluated by the NEB method is highly sensitive to the number of images used in the calculation. Therefore, we have re-evaluated the direct coupling mechanism using the newly developed climbing-image nudged elastic band (CI-NEB) method38,39 which is designed to locate the transition state directly. We found that for the 1 ML BBOHs coverage model, the H2 desorption barrier is 1.80 eV, which is almost the same as that in ref. 18. While for the 1/2 ML BBOHs coverage model, the barrier is 2.00 eV. To further evaluate this mechanism, we have calculated the barrier using the 1/4 ML BBOHs coverage model (which is more close to the experimental conditions), and we obtained an even higher barrier of 2.66 eV (shown in Fig. 1). Based on these results, we think that possibly the H2 is not formed by the direct coupling of two BBOHs as the communities have thought before, and it undergoes a new mechanism which has not been addressed yet.
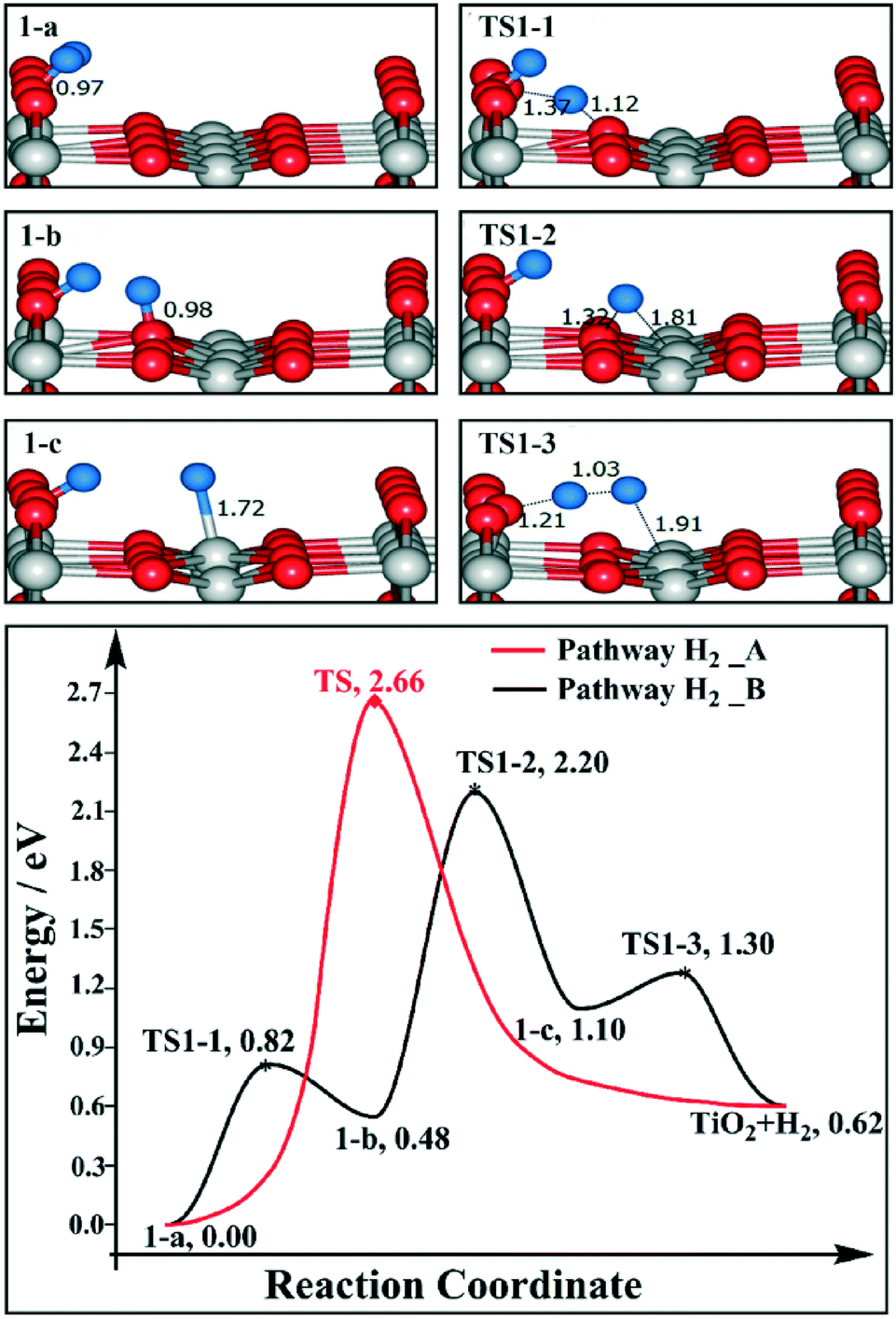 | ||
| Fig. 1 Reaction profiles for H2 desorption on 1/4 ML BBOHs on a stoichiometric TiO2(110) surface. The side views of selected intermediates and transition states are shown in the figure with simplified schematic diagrams. | ||
Indeed, it is not surprising that the direct coupling of BBOHs has a very high barrier, since both BBOHs bear strong positive charges. There are many well studied dehydrogenation/hydrogenation systems which happen under mild conditions. In general, these systems bear less polarized M–H bonds or have both Hδ+ and Hδ− coexisting in the system (shown in Scheme 1).22–30 The dehydrogenation is therefore facilitated by the less electrostatic repulsion between two H atoms. On a TiO2(110) surface, such mechanisms are also available. Two types of Ti–H species can be candidates for key intermediates of dehydrogenation: the end on Ti5c–H and the bridging Ti–H–Ti at the BBO vacancies (Ov) where H occupied the place of the missing BBO (Ti5c-Ov−H).
 | ||
| Scheme 1 Typical mechanisms for dehydrogenation under mild conditions. | ||
In this work, we have systematically investigated the mechanism of H2 desorption, as well as the competition between H2 and H2O desorption on a TiO2(110) surface. We propose that the reaction undergoes a Ti–H key intermediate, which then forms an O–Hδ+⋯Hδ−–Ti type transition state. The Ov plays an important role in both H2 desorption and H2O desorption. Our mechanism successfully explains the experimental observations, especially the competition between H2 desorption and H2O desorption. Furthermore, we have also discussed the possible ways to increase the yield of H2 desorption on the TiO2(110) surface.
Methods
As shown in our previous studies,32,33 all of our calculations were performed using the Vienna ab initio simulation package code34,35 and plane augmented wave potential.36 The wave function was expanded by the plane wave, with a kinetic cut-off of 400 eV and a density cut-off of 650 eV. The generalized gradient approximation with the spin-polarized Perdew–Burke–Ernzerhof functional37 was used to determine the optimized molecular structures of TiO2(110).Unless specified, an efficient force reversed method31 was used to locate the transition state (TS). The CI-NEB methods (constrained minimization and climbing-image nudged elastic band methods)38,39 were used for TSs which are challenging using the force reversed method, and in this case the minimum energy pathway for each elementary reaction was discretized by a total of ten images between the initial and final states.
Our surface model was cut out of a six-layer slab TiO2 crystal to expose the (110) surface32,33,40–44 and fixed on the bottom two layers of the atoms. All Ti5c sites on the bottom layer were saturated with water molecules to maintain the bulk coordination environment.32,33 The periodically repeated slabs on the surface were decoupled by 15 Å vacuum gaps. A Monkhorst–Pack grid45 of (2 × 1 × 2) k-points was used for the 4 × 2 surface unit cell. Isolated gas-phase molecules were optimized in a (15 × 15 × 15) unit cell with a single k-point.
The reaction energy (ΔE), the H2O desorption energy (ΔEH2O_desorption) and the H2 desorption energy (ΔEH2_desorption) were defined as follows:
| ΔE = Efin state − Einit state | (1) |
| ΔEH2O_desorption = EH2O + Eslab+Ov+(n−2)BBOHs − Eslab+n×BBOHs | (2) |
| ΔEH2_desorption = EH2 + Eslab+(n−2)×BBOHs − Eslab+n×BBOHs | (3) |
Results
The mechanism of H2 and H2O recombinative desorption from a stoichiometric TiO2(110) surface
As mentioned above, the direct coupling of the two BBOHs to produce H2 (pathway H2_A) is rather hard, and we will search for alternative mechanisms. On a stoichiometric TiO2(110) surface, only Ti5c is unsaturated and able to form a Ti5c–H intermediate. We then consider a mechanism that starts with BBOH migration to Ti5c to obtain Ti5c–H. Then this negatively charged hydrogen atom couples with the positively charged BBOH to form H2. In this mechanism, the key transition state for H2 desorption is BBO–Hδ+⋯Hδ−–Ti5c. We denote this pathway as pathway H2_B. Since 1 ML BBOHs are not available experimentally, we have investigated the H2 desorption on 1/4 and 1/2 ML BBOHs models using a 4 × 2 unit cell of TiO2(110).The reaction coordinates of pathway H2_B for the 1/4 ML BBOHs model are shown in Fig. 1. The H transfers from the BBO site (1-a) to the sub-surface oxygen (Os) site (1-b) are endothermic by 0.48 eV, with a barrier of 0.82 eV (TS1-1). The H transfers from the Os site (1-b) to the Ti5c site (1-c) are highly endothermic (0.62 eV), and the barrier is also much higher (1.72 eV, TS1-2). The barrier for the coupling of BBO–H and Ti5c–H to produce H2 (TS1-3) is 0.20 eV. The H2 desorption energy is 0.62 eV, and the rate-determining step is the H transfers from Os to Ti5c with an overall barrier of 2.20 eV. BBOH can also transfer to Ti5c directly (pathway H2_C). We have tried very hard to locate its transition state using the force reversed method, but we are not successful. Therefore, we calculated the barrier using the CI-NEB method, and the barrier we obtained is 2.19 eV (see Fig. S1†) which is almost the same as pathway H2_B.
We have also considered the pathway where H2 has been formed by the coupling of sub-surface hydroxyls (Os–H) and Ti5c–H (pathway H2_D). However, the intermediate ready for dehydrogenation is already higher in energy than TS1-3 by 0.45 eV (see Fig. S2†), and therefore this is not the preferred pathway.
Based on these results, we propose that pathway H2_B is the most facile pathway for dehydrogenation of BBOHs to form H2 on a stoichiometric TiO2(110) surface. Its overall barrier is lower than that of pathway H2_A by more than 0.4 eV.
The optimized structures are also shown in Fig. 1. The optimized bond lengths for BBO–H, Os–H, and Ti5c–H are 0.97, 0.98 and 1.72 Å, respectively. In Ts1-1, r(BBO–H) is 1.37 Å, and r(H–Os) is 1.12 Å. In TS1-2, r(Os–H) is 1.32 Å and r(H–Ti5c) is 1.81 Å. In Ts1-3, r(BBO–H), r(H–H), and r(H–Ti5c) are 1.21 Å, 1.03 Å and 1.91 Å, respectively.
To better compare the H2 and H2O desorption reactions, we have re-evaluated the H2O desorption pathway with our theoretical model. The results are shown in Fig. 2. The H transfers from one BBO to another BBO to produce H2O is endothermic by 0.40 eV, with a barrier of 0.82 eV (TS2-1). The H2O desorption energy is 1.09 eV. We denote this pathway as pathway H2O_A, and the results are pretty similar to those in ref. 14 and 18. In TS2-1, r(BBO–H) and r(H–BBOH) are 1.21 Å and 1.27 Å, respectively.
 | ||
| Fig. 2 Reaction profiles for H2O desorption on 1/4 ML BBOHs on a stoichiometric TiO2(110) surface. The side views of selected intermediates and transition states are shown in the figure with simplified schematic diagrams. | ||
We have also investigated another pathway, denoted as pathway H2O_B, for H2O desorption. Firstly, H transfers from BBO to Os to form Os–H and then transfers from Os to another BBO to form H2O. The first transition state is TS1-1 in pathway H2_B, and the second transition state is TS2-2 as shown in Fig. 2. In TS2-2, r(Os–H) is 1.16 Å, and r(H–BBOH) is 1.30 Å. The overall barrier is 1.09 eV.
Furthermore, we have considered other relative positions of two BBOHs. If the two BBOHs are on the same BBO row (but not adjacent), they can easily migrate to adjacent sites (1-a) firstly and then follow the H2 and H2O desorption pathways as discussed above. The calculated barrier of H migration between BBO sites is 0.93 eV, which is pretty similar to that in ref. 20. The energy differences of the isomers with different relative BBOH positions are less than 0.15 eV. It suggests that no matter where the BBOH starting positions were, for H2 desorption, the rate-determining step is always the H transfers from Os to Ti5c (TS1-2); for H2O desorption, the rate-determining step is always the H2O desorbed from the stoichiometric surface.
Meanwhile, when two H atoms are adsorbed on different BBO rows, one H atom should transfer to the Ti5c for H2 desorption, and two H atoms should transfer to the same row for H2O desorption. In both cases, the rate-determining steps are the H transfers from Os to Ti5c, and the barriers are comparable to that for pathway H2_B. As shown above, the barrier is pretty high; therefore in the following studies, we didn't consider the cases where two H atoms are adsorbed on different BBO rows.
We have also calculated the reaction profiles of H2 and H2O desorption for the 1/2 ML BBOHs model (as shown in Fig. 3 and 4). For pathway H2_B, the H transfers from the BBO site (3-a) to the Os site (3-b) are endothermic by 0.56 eV, with a barrier of 0.76 eV (TS3-1). The H transfers from the Os site (3-b) to the Ti5c site (3-c) are also endothermic (0.16 eV), and the barrier is much higher (1.28 eV, TS3-2). The barrier for the coupling of BBO–H and Ti5c–H to produce H2 (TS3-3) is 0.09 eV. The H2 desorption energy is 0.08 eV, and the rate-determining step is the H transfers from Os to Ti5c with an overall barrier of 1.84 eV. The desorption energy of H2O is 1.00 eV. For pathway H2O_A, the coupling of two BBOHs to produce H2O is endothermic by 0.49 eV, with a barrier of 0.90 eV (TS4-1). By pathway H2O_B, the coupling of Os–H and BBO–H to produce H2O has a barrier of 0.23 eV (TS4-2). Therefore, with the 1/2 ML BBOHs model, the H2 desorption barrier (1.84 eV) is also much higher than the H2O desorption barrier (1.00 eV), which is the same as in the case of the 1/4 ML BBOHs model (see Fig. 1 and 2 and 3 and 4, respectively). In addition, with an increase in BBOHs, the H2 desorption barrier decreases, and the H2O desorption barrier does not change obviously. Therefore, the barrier difference between H2 and H2O desorption becomes smaller.
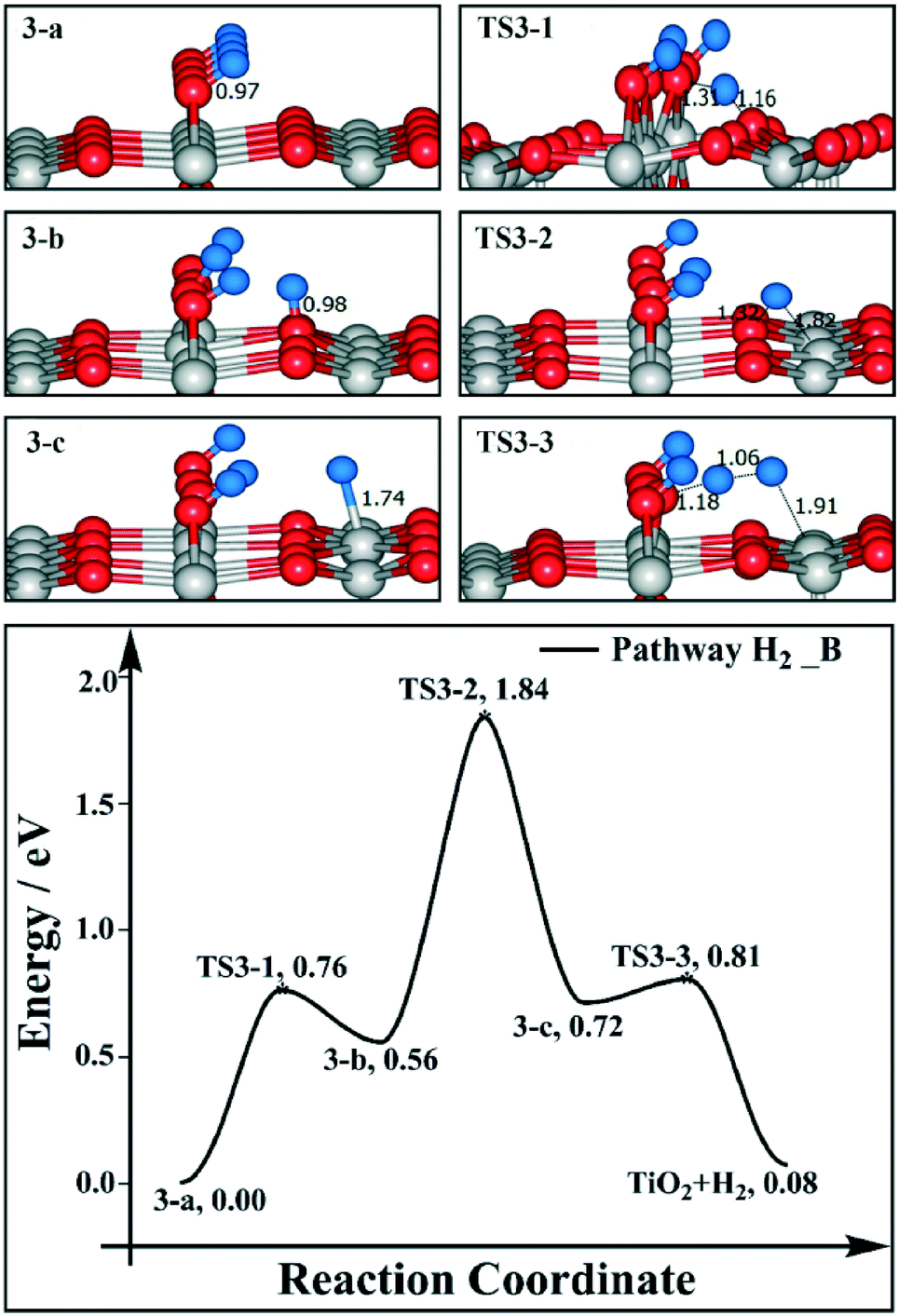 | ||
| Fig. 3 Reaction profiles for H2 desorption on 1/2 ML BBOHs on a stoichiometric TiO2(110) surface. The side views of selected intermediates and transition states are shown in the figure with simplified schematic diagrams. | ||
 | ||
| Fig. 4 Reaction profiles for H2O desorption on 1/2 ML BBOHs on a stoichiometric TiO2(110) surface. The side views of selected intermediates and transition states are shown in the figure with simplified schematic diagrams. | ||
Therefore, although we proposed new mechanisms, we also predict that the H2 desorption is much harder than the H2O desorption on the stoichiometric TiO2(110) surface. Thus, the H2O desorption is dominant at the beginning of desorption, which is in good agreement with the experimental observations.15 Since H2O desorption leaves Ovs on the surface, later on we will investigate how do these Ovs affect the H2 and H2O desorption.
The mechanism of H2 and H2O recombinative desorption from the TiO2(110) model of an Ov
The H2 desorption barrier is higher than the H2O desorption barrier by 0.84–1.11 eV on a stoichiometric TiO2(110) surface, and the desorption of H2O leaves Ovs on the TiO2(110) surface. Each Ov results in two adjacent Ti5c-Ovs (the five coordinated Ti at the Ov) which can be used to form bridging Ti5c-Ov−H−Ti5c-Ov species. These titanium hydride species can also couple with BBOH to form new mechanisms for H2 desorption.With these ideas, we have investigated the mechanism of H2O and H2 desorption on the 1/4 ML BBOHs model with an Ov. We have considered the following three cases: 1) an Ov and two BBOHs are on different BBO rows; 2) two BBOHs are on different sides of an Ov; 3) two BBOHs are on the same side of an Ov.
For the first case where an Ov and two BBOHs are on different BBO rows, H2 can be formed via pathway H2_B (as shown in Fig. 5). The H transfers from the BBO site (5-a) to the Os site (5-b) are endothermic by 0.56 eV, with a barrier of 0.77 eV (TS5-1). The H transfers from the Os site (5-b) to the Ti5c site (5-c) is also endothermic (0.44 eV), and the barrier is much higher (1.56 eV, TS5-2). The barrier for the coupling of BBO–H and Ti5c–H to produce H2 (TS5-3) is 0.19 eV. Finally, the H2 desorption energy is 0.51 eV. The H2O desorption energy is 1.10 eV. For pathway H2O_A, the coupling of two BBO–Hs to produce H2O is endothermic by 0.42 eV, with a barrier of 0.82 eV (TS6-1). For pathway H2O_B, the coupling of Os–H and BBO–H to produce H2O has a barrier of 0.25 eV (TS6-2). The H2 desorption barrier (2.12 eV) and H2O desorption barrier (1.10 eV) were found to be almost the same as the 1/4 ML BBOHs model on a stoichiometric surface (see Fig. 1 and 2). Therefore, we came into a conclusion that if the Ov is far from BBOHs, it will have minor effects on H2 desorption and H2O desorption.
 | ||
| Fig. 5 Reaction profiles for H2 desorption of the model where an Ov and two BBOHs are on different BBO rows. The side views of selected intermediates and transition states are shown in the figure with simplified schematic diagrams. | ||
In the case of two BBOHs on different sides of an Ov, the Ti5c-Ov may be involved in the reaction. We have considered three pathways for H2 desorption. The first one is pathway H2_B as discussed above. In the second pathway (denoted as pathway H2_E), one H migrates from BBO to Os and then migrates to Ti5c-Ov. The H2 is therefore formed through a transition state featured by BBO–Hδ+⋯Hδ−–Ti5c-Ov. The third pathway (denoted as pathway H2_F) is pretty similar to pathway H2_E, except that H migrates from BBO to Ti5c-Ov directly but not through Os.
For pathway H2_B (as shown in Fig. 7), the migration of H from the BBO site (7-Ba) to the Os site (7-Bb) is endothermic by 0.47 eV, with a barrier of 0.68 eV (TS7-B1). The H transfers from the Os site (7-Bb) to the Ti5c site (7-Bc) are also endothermic (0.38 eV), and the barrier is much higher (1.57 eV, TS7-B2). Then, the migration of H from the Ti5c site (7-Bc) to the Os site (7-Bd) is exothermic by −0.16 eV, with a barrier of 1.15 eV (TS7-B3). The H transfers from the Os site (7-Bd) to the Ti5c site (7-Be) are endothermic by 0.06 eV, with a barrier of 1.16 eV (TS7-B4). The barrier for BBO–H and Ti5c–H to produce H2 (TS7-B5) is 0.15 eV. Finally, the H2 desorption energy is 0.20 eV. For pathway H2_E, the migration of H from the BBO site to the Os site is the same as that in pathway H2_B; the migration of H from the Os site (7-Bb) to the Ov site (7-Ec) is exothermic by −0.22 eV, with a barrier of 1.43 eV (TS7-E2). The barrier for the coupling of BBO–H and Ti5c-Ov–H to produce H2 (TS7-E3) is 0.76 eV. For pathway H2_F, the migration of H directly from the BBO site (7-Ba) to the Ov site (7-Ec) is endothermic by 0.25 eV, with a barrier of 1.90 eV (TS7-F1). Then, the H2 is formed as that in pathway H2_E. The overall barriers for the three pathways are 2.04 eV, 1.90 eV and 1.90 eV, respectively, and it has the trend B > E ≈ F. They are all lower than those for the 1/4 ML BBOHs model on a stoichiometric surface (see Fig. 1), as well as for the case of an Ov and two BBOHs on different BBO rows (see Fig. 5), but a little higher than that for the 1/2 ML BBOHs model on a stoichiometric surface (see Fig. 3).
The optimized structures are also shown in Fig. 7. In Ts7-E2, r(Os–H) is 1.29 Å, and r(H–Ti5c-Ov) is 1.84 Å. In Ts7-E3, r(BBO–H), r(H–H), and r(H–Ti5c-Ov) are 1.21 Å, 1.04 Å and 1.91 Å, respectively. In TS7-F1, r(BBO–H) is 1.40 Å and r(H–Ti5c-Ov) is 1.75 Å.
We have calculated the H2O desorption profile for the model where two BBOHs are on different sides of an Ov. For the H2O desorption, H could be initially transferred to the Ov site, and then H2O is produced by the BBO–H and Ti5c-Ov–H. The H2O desorption energy is 1.47 eV. The rate-determining step is the H transfers from Os to Ti5c-Ov (TS7-E2) with an overall barrier of 1.90 eV, which is the same as H2 desorption. But the H2O desorption energy is much higher than the barrier of BBO–H and Ti5c–H to produce H2 (TS7-B5, 0.15 eV). Therefore, under these conditions, H2 desorption is more competitive than H2O desorption.
In the case of the BBOHs that are on the same side of an Ov (8-a), we have also considered three pathways for H2 desorption. For pathway H2_B, the rate-determining step is the H transfers from Os to Ti5c with an overall barrier of 1.89 eV. For pathway H2_E, the rate-determining step is the H transfers from Os to Ti5c-Ov with an overall barrier of 1.87 eV. For pathway H2_F, the rate-determining step is the H transfers from BBO to Ti5c-Ov with an overall barrier of 1.87 eV. These results are similar to the case of two BBOHs that on different sides of an Ov (see Fig. 7), which suggests that the relative positions of BBOHs and Ovs have mirror effects on H2 desorption.
We have also calculated the H2O desorption profile for the model where two BBOHs are on the same side of an Ov. For pathway H2O_A, the H transfers from the BBO site (8-a) to the adjacent BBO site (8-b) costs 0.15 eV, with a barrier of 1.26 eV (TS8-1). The coupling of two BBOHs to produce H2O is endothermic by 0.44 eV, with a barrier of 0.96 eV (TS8-2). For pathway H2O_B, the H transfers from the BBO site (8-a) to the Os site (8-d) are endothermic by 0.48 eV, with a barrier of 0.89 eV (TS8-3); the H transfers from the Os site (8-d) to the BBO site (8-b) are exothermic by −0.33 eV, with a barrier of 0.45 eV (TS8-4); then the H transfers from the BBO site (8-b) to the Os site (8-e) are endothermic by 0.32 eV, with a barrier of 0.72 eV (TS8-5); the coupling of Os–H and BBO–H to produce H2O has a barrier of 0.43 eV (TS8-6). Finally, the H2O desorption energy is 1.47 eV (as shown in Fig. 8). Therefore in this case, the H2O desorption barrier (1.47 eV) is higher than those on 1/4 or 1/2 ML BBOHs on a stoichiometric surface (see Fig. 2 and 4, respectively) or on the model with an Ov and two BBOHs that are on different BBO rows (see Fig. 6), but it is less than the model with two BBOHs that are on different sides of an Ov (TS7-E2). We propose that this is because in this model the H2O desorption will create two adjacent Ov sites, exposing a highly unsaturated Ti4c (four-coordinated titanium) site, and the location of BBOHs and Ovs has played a decisive role in the H2O desorption pathway with an Ov model.
 | ||
| Fig. 6 Reaction profiles for H2O desorption of the model where an Ov and two BBOHs are on different BBO rows. The side views of selected intermediates and transition states are shown in the figure with simplified schematic diagrams. | ||
We have only considered limited relative positions of the Ov sites and the BBOHs. However, given the fact that the H migration between BBO sites is facile (with a barrier of 1.26 eV) under the conditions of H2 desorption, our proposed pathways are expected to be also valid for other relative positions of Ov sites and the BBOHs if they are in the same BBO row.
The mechanism of H2 and H2O recombinative desorption from the TiO2(110) model of two Ovs
Our calculations show that the H2 desorption barrier is much higher than the H2O desorption barrier on stoichiometric TiO2(110) models. However, the barriers become closer when an Ov is introduced on the surface. Thus it is very interesting to know how the competition changes if we introduce more Ovs. Is it possible that at the end the H2 desorption can be more facile than the H2O desorption? Although our 4 × 2 slab with an Ov reflects 1/8 ML of Ovs, which is already not small in reality, we know that the Ovs are not perfectly distributed equally on the TiO2(110) surface, and there are Ovs which are more closer to each other, which is in line with a local region with a higher Ov concentration.Thus we have investigated the mechanism H2 desorption and H2O desorption on 1/4 ML BBOHs TiO2(110) surface model with two Ovs. We have considered four cases concerning the relative positions of the two Ovs: two BBOHs are on different sides of two adjacent Ovs; two BBOHs are on the same side of two adjacent Ovs; two alternate Ovs are near the BBOHs; one Ov is near the BBOHs, and the other Ov is on the different BBO row of the BBOH case.
Firstly, in the case of two BBOHs that are on different sides of two adjacent Ovs, we have evaluated four possible pathways for H2 desorption. The first three pathways are pathway H2_B, pathway H2_E and pathway H2_F as shown above. In the fourth pathway, each BBOH migrates to an Ov site, and then the two Ti5c-Ov–H couple with each other to give H2 (pathway H2_G).
For pathway H2_B, the H transfers from the BBO site (9-Ba) to the Os site (9-Bb) are endothermic by 0.72 eV, with a barrier of 0.87 eV (TS9-B1). The H transfers from the Os site (9-Bb) to the Ti5c site (9-Bc) are also endothermic (0.03 eV), and its barrier is also much higher (1.20 eV, TS9-B2). The barrier for the coupling of BBO–H and Ti5c–H to produce H2 (TS9-B3) is 0.03 eV. The H2 desorption energy is −0.01 eV. For pathway H2_E, the H transfers from the BBO site (9-Ba) to the Os site (9-Eb) are endothermic by 0.65, with a barrier of 0.84 eV (TS9-E1); the H transfers from the Os site (9-Eb) to the Ov site (9-Ec) are exothermic by −0.96 eV, with a barrier of 0.92 eV (TS9-E2); the H transfers from the Ov site (9-Ec) to the adjacent Ov site (9-Ed) are endothermic by 0.17 eV, with a barrier of 0.53 eV (TS9-E3). The barrier for BBO–H and Ti5c-Ov–H to produce H2 (TS9-E4) is 0.99 eV. Meanwhile, we have also studied the formation of H2 from Ti5c-Ov–H and Os–H (pathway H2_H, see Fig. S3†), and we found that it is similar to those from BBO–H and Ti5c-Ov–H in pathway H2_E. For pathway H2_F, the migration of H directly from the BBO site (9-Ba) to the Ov site (9-Ec) is exothermic, with a barrier of 1.66 eV (TS9-F1). The coupling of BBO–H and Ti5c-Ov–H to produce H2 is the same as that in pathway H2_E. For pathway H2_G, the migration of other H from the BBO site (9-Ec) to the Ov site (9-Ge) is endothermic by 0.26 eV, with a barrier of 1.97 eV (TS9-G1). The coupling of the two adjacent Ti4c-Ov–Hs to produce H2 has a barrier of 0.43 eV (TS9-G2). The overall barriers of the four pathways are 1.92 eV, 1.57 eV, 1.66 eV and 1.97 eV, respectively, and it has the trend G ≈ B > E ≈ F. The barriers of pathways H2_E and H2_F are all smaller than those on the stoichiometric surface models (see Fig. 1 and 3) and the models with an Ov (see Fig. 5 and 7). Because the barrier of H2_B and H2_G are much higher than those of other pathways, we will not calculate it anymore for other surface models with two Ovs. Besides, if the BBOHs are on the same side of two adjacent Ovs, the rate-determining step is also the H transfers from Os to the Ov site (TS9-E2). It is similar to the case where two BBOHs are on different sides of two adjacent Ovs, and we didn't show the details any more.
 | ||
| Fig. 7 Reaction profiles for H2 desorption of the model where two BBOHs are on different sides of an Ov. The side views of selected intermediates and transition states are shown in the figure with simplified schematic diagrams. | ||
As mentioned above, the locations of BBOHs and Ovs are both crucial for the H2O desorption. Thus, we have studied the mechanism of H2O desorption on two BBOHs that are on the same side of two adjacent Ovs, firstly. For pathway H2O_A, the coupling of two BBO–Hs to produce H2O is endothermic by 0.91 eV, with a barrier of 1.25 eV (TS10-1); for pathway H2O_B, the Os–H and BBO–H produce H2O with a barrier of 0.43 eV (TS10-2). The H2O desorption energy in this model is found to be 1.81 eV (as shown in Fig. 10), which is much higher than that on the stoichiometric model and the models with an Ov. We think that this is because, with this two Ovs model, the H2O desorption will create three adjacent Ov sites, exposing two highly unsaturated Ti4c sites. If two BBOHs are on different sides of two adjacent Ovs, the H atom could initially be transferred to the Ov site (9-Ec), and then H2O is produced by the BBO–H and Ti5c-Ov–H. The H2O desorption energy is also 1.81 eV which is much higher than the barrier of H2 formation (TS9-E4, 0.99 eV). Therefore, if two BBOHs are on different sides of Ovs, H2 desorption is more competitive than H2O desorption.
We are curious if the results discussed above highly depend on the relative position of the two Ovs. Therefore we also calculated the reaction profile of H2 desorption and H2O desorption on the 1/4 ML BBOHs TiO2(110) surface model with two alternative Ovs. For H2 desorption, we have considered pathways H2_E and H2_F. For pathway H2_E, the migration of H from the BBO site (11-Ea) to the Os site (11-Eb) is endothermic by 0.58 eV, with a barrier of 0.72 eV (TS11-E1); the H migration from the Os site (11-Eb) to the Ov site (11-Ec) is highly exothermic by −1.19 eV, with a barrier of 0.69 eV (TS11-E2). The barrier for BBO–H and Ti5c-Ov–H to produce H2 (TS11-E3) is 1.23 eV. Finally, the H2 desorption energy is 0.25 eV. By pathway H2_F, the barrier of H directly migrating from the BBO site to the Ov site is 1.41 eV (TS11-F1). For the H2O desorption pathway, we considered that the H would be initially transferred to the Ov site, and then H2O is produced by the BBO–H and Ti5c-Ov–H. The formation of H2O is endothermic by 1.27 eV, with a barrier of 1.96 eV (TS12-1), and the H2O desorption energy is 1.94 eV (as shown in Fig. 12). The overall barrier is 2.55 eV. Therefore, for this model, we also found that H2 desorption is easier than H2O desorption. The barrier for H2 desorption is even lower than that in the model with two adjacent Ovs (see Fig. 9 and 11), which suggest that the relative positions of the Ovs are important for H2 desorption. The rate-determining barriers of the two alternative Ov models, 1.27–1.41 eV, fit with the H2 desorption temperature in the TPD experiments.15
In order to further understand the role of the relative position of Ovs in the H2 desorption, we calculated the case where one Ov is near the BBOHs, while the other Ov is on the different BBO row of BBOHs. In this case, for pathway H2_E, the migration of H from the BBO site (13-Ea) to the Os site (13-Eb) is endothermic by 0.44 eV, with a barrier of 0.63 eV (TS13-E1); the H transfers from the Os site (13-Eb) to the Ov site (13-Ec) are exothermic by 0.26 eV, with a barrier of 1.37 eV (TS13-E2). The barrier for the coupling of BBO–H and Ti5c-Ov–H to produce H2 (TS13-E3) is 0.75 eV. Finally, the H2 desorption energy is 0.12 eV. By pathway H2_F, the barrier of the H that directly transfers from the BBO site (13-Ea) to the Ov site (13-Ec) is 1.84 eV (TS13-F1, as shown in Fig. 13). The results are similar to those for the case of two BBOHs that are on different sides of an Ov (see Fig. 7). These results suggest that if the Ov is not on the same row of BBO–H, it has a minor effect on H2 desorption.
Furthermore, the barrier of H migration between BBO sites is 1.25 eV, which is pretty similar to those for the stoichiometric surface models and one Ov models. Thus our proposed pathways are expected to be also valid for other relative positions of Ovs sites and the BBOHs.
Discussion
The effect of two BBOHs versus an Ov in H2 and H2O desorption
From the above discussion, we know that the concentration of Ovs play an important role in the H2 and H2O desorption. It has been suggested that the two adjacent BBOHs and an Ov made equal contributions to the band-gap states.46–48 Based on our results, we are able to discuss whether BBOHs and an Ov also have similar effects on chemical reactions such as H2 and H2O desorption. We have made comparisons for two different models, which have similar electronic properties. The first is 1/2 ML BBOHs on a stoichiometric surface versus 1/4 ML BBOHs on the same side of an Ov. We found that the H2 desorption is slightly preferred for the first case (rate-determining barrier, 1.84 eV versus 1.87 eV), while the H2O desorption is highly preferred for the first case (rate-determining barrier, 1.00 eV versus 1.47 eV, see Fig. 4versusFig. 8, respectively). Concerning the different pathways for H2 desorption, we found that pathway H2_B which involves Ti5c–H is more sensitive to the concentration of BBOHs, while H2_E or H2_F which involves Ti5c-Ov is more sensitive to the concentration of Ovs. In the second comparison, we start with an Ov near the BBOHs, and then we compare the effect of an Ov or two BBOHs that are on different BBO row of BBOHs. The calculated H2 desorption barriers for the two cases are 1.81 eV (as discussed above, see Fig. 13) and 1.82 eV (as shown in Fig. 14), respectively. | ||
| Fig. 8 Reaction profiles for H2O desorption of the model where two BBOHs are on the same side of an Ov. The side views of selected intermediates and transition states are shown in the figure with simplified schematic diagrams. | ||
 | ||
| Fig. 9 Reaction profiles for H2 desorption of the model where two BBOHs are on different sides of two adjacent Ovs. The side views of selected intermediates and transition states are shown in the figure with simplified schematic diagrams. | ||
 | ||
| Fig. 10 Reaction profiles for H2O desorption of the model where two BBOHs are on the same side of two adjacent Ovs. The side views of selected intermediates and transition states are shown in the figure with simplified schematic diagrams. | ||
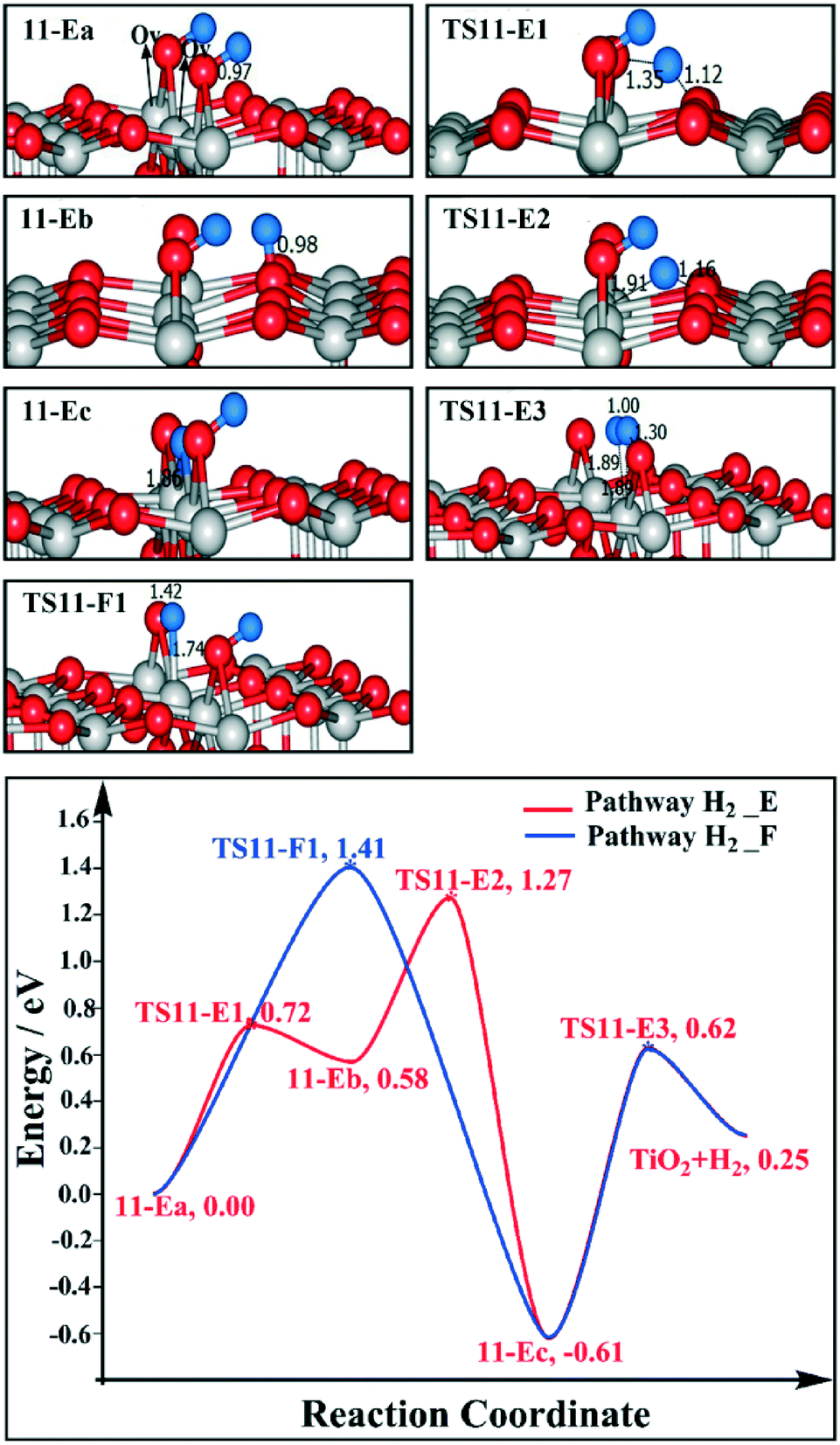 | ||
| Fig. 11 Reaction profiles for H2 desorption of the model where two alternative Ovs are near the BBOHs. The side views of selected intermediates and transition states are shown in the figure with simplified schematic diagrams. | ||
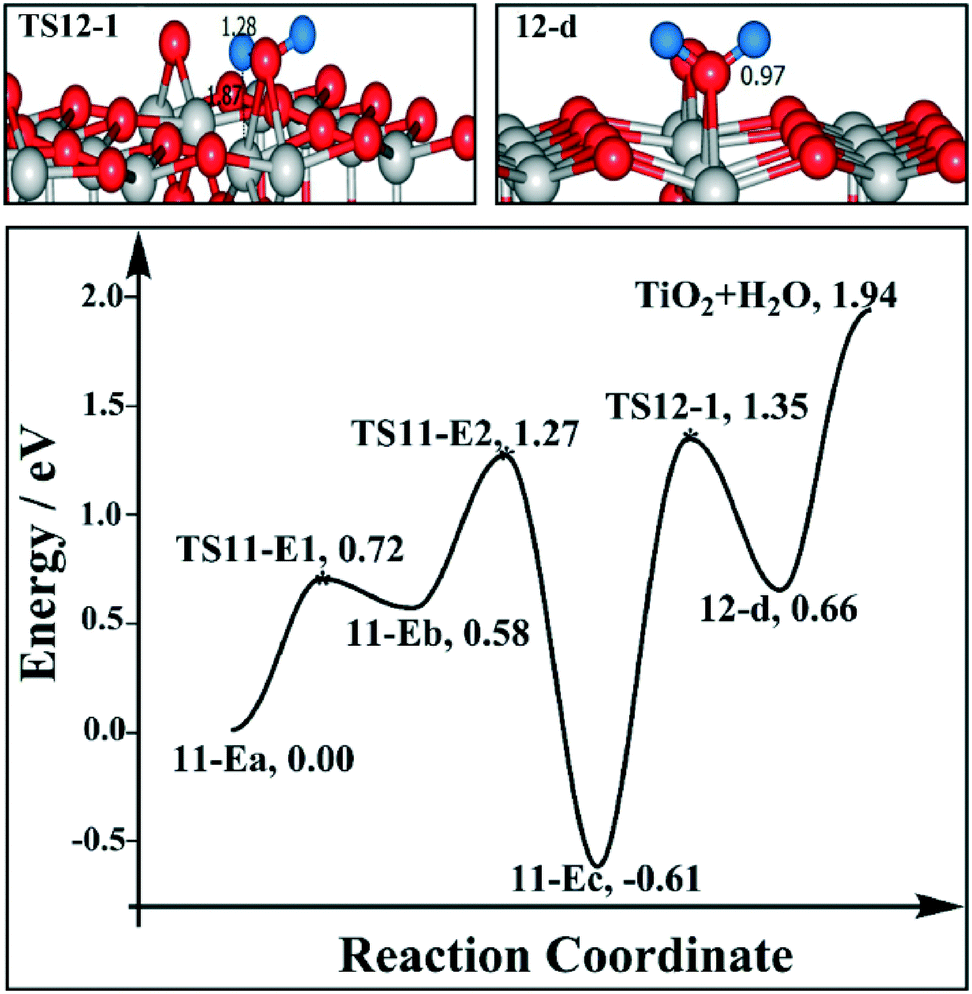 | ||
| Fig. 12 Reaction profiles for H2O desorption of the model where two alternate Ovs are near the BBOHs. The side views of selected intermediates and transition states are shown in the figure with simplified schematic diagrams. | ||
 | ||
| Fig. 13 Reaction profiles for H2 desorption of the model where one Ov is near the BBOHs, while the other Ov is on the different BBO row of BBOHs. The side views of selected intermediates and transition states are shown in the figure with simplified schematic diagrams. | ||
 | ||
| Fig. 14 Reaction profiles for H2 desorption of the model where an Ov is near BBOHs, while two BBOHs are on the different BBO row of BBOHs. The side views of selected intermediates and transition states are shown in the figure with simplified schematic diagrams. | ||
Therefore, we predict that if two BBOHs or an Ov is far from BBOHs, they have similar but very small effects for H2 desorption. Otherwise, if they are near the BBOHs, they have very different effects for H2O desorption, and mirror effects for H2 desorption. H2O desorption is more influenced by the surface structure, and desorption near the Ov is much more difficult, while H2 desorption is more controlled by the electronic properties on the surface where BBOHs and Ovs contributed similarly. This is reasonable since in the H2 desorption the surface is oxidized, while in the H2O desorption it is not.
The competition of H2 and H2O desorption on a TiO2(110) surface
As shown by the experiments, H2 desorption and H2O desorption are competing reactions on a TiO2(110) surface.15 The H2O yield increases pretty faster at the beginning and then reaches a plateau; after that, the H2 yield was increasing faster than that of H2O. Based on our theoretical results, we can also compare the competition of H2 and H2O desorption. At the beginning of the reaction, there are BBOHs on the surface, while Ovs are rare. According to our calculations on the stoichiometric model, the H2O desorption barrier is 1.09 eV, which is much lower that the H2 desorption barrier; therefore, H2O desorption is dominated at this stage. However, H2O desorption creates Ovs, which means that there are more and more Ovs on the surface along with the H2O desorption. As shown in Fig. 15, the calculated H2O desorption barriers increases from 1.00 to 1.09 eV for stoichiometric models to 1.1–1.90 eV for the Ov models and to 1.81–2.55 eV for two Ovs models. It suggests that although the H2O desorption is the dominating reaction at the beginning, however, as the reaction goes, the H2O desorption becomes harder and harder. Certainly, because in reality the surface is not extremely reduced, there are always local regions which have similar structures as the Ov models and are better for H2O desorption than the two Ovs structures. Therefore even at the late stage of the reaction, the actual barrier for H2O desorption is estimated to be close to that in the Ov models but not the two Ovs models. | ||
| Fig. 15 The comparison of H2 and H2O desorption on models with different Ovs coverage. | ||
As a comparison, the H2 desorption is pretty hard for a stoichiometric surface. However, as shown in Fig. 15, the calculated H2 desorption barrier decreases slightly from 1.84 to 2.20 eV for stoichiometric models to 1.82–2.12 eV for the Ov models, and then decreases dramatically to 1.27–1.81 eV for two Ovs models. It suggests that the H2 desorption is pretty hard at the beginning; however, as the H2O desorption goes, the H2 desorption becomes easier and easier. For the two Ovs models, we actually predicted that H2 desorption becomes easier than H2O desorption. These predictions perfectly fit with the experimental observations in ref. 15.
We have discussed above that the H2 desorption is mostly controlled by the electronic effect, more specifically, the reducing power of the TiO2 surface. Then another question is how to understand that H2O desorption makes the H2 desorption easier since H2O desorption does not provide additional electrons to the surface. We think that there are two reasons. Firstly, H2O desorption creates Ovs which enable pathways H2_E and H2_F. These two pathways are the most preferred pathways for H2 desorption, especially when the concentration of Ov on the surface is large. Secondly, as shown in ref. 48, the Ti 3d electrons induced by BBOH and Ov are distributed in a local region, rather than on the whole surface. Under the H2 desorption conditions, BBOH can diffuse easily along the BBO row, while Ov cannot. Therefore, there are chances that desorption already creates a significant amount of Ovs. Then we have two Ovs close to each other, which result in local regions with more Ti 3d electrons and stronger reducing power. As suggested by our two Ovs models, H2 is ready to be formed on such local regions.
Besides, M. A. Henderson and coworkers have found that BBOHs can diffuse to the rutile bulk.49 Y. Du et al. have shown that the diffusion of BBOH to the bulk has a barrier of about 1.1 eV,14 which is lower than the H2 desorption barrier. The energy changes for the H diffusion are quite small, suggesting that the H diffusion between the surface and bulk is reversible. H2 and H2O desorption happens at a relatively high temperature, and under these conditions the desorption has a significant entropic drive. Therefore, we expect the BBOHs in the bulk can also diffuse to the surface, followed by desorption as H2O or H2.
Beside the Ovs, as shown above, BBOHs also have strong effects on the H2 desorption. On the stoichiometric surface model, the H2 desorption barriers of our proposed mechanism for 1/4 ML and 1/2 ML BBOHs are 2.20 eV and 1.84 eV, respectively, while the H2 desorption barriers of the direct coupling of two BBOHs for 1/4 ML and 1/2 ML BBOHs are 2.66 eV, 2.00 eV, respectively. In each case, our mechanism has a smaller barrier. As a comparison, the H2O desorption barrier is similar for 1/4 ML and 1/2 ML BBOHs. As discussed above, this is because when there are more BBOHs or Ovs on the TiO2(110) surface, the surface has stronger reducibility.
The locations of BBOHs and Ovs are also relevant. When the BBOHs are on the same side of the Ov, for H2 desorption, the rate-determining steps are always the H transfers from the Os site to the Ov site; for H2O desorption, the rate-determining steps are always the H2O desorbed from the surface. In both cases, the desorption barrier does not change obviously with the locations of BBOHs and Ovs. When the BBOHs are on different sides of the Ov, for H2 desorption, the rate-determining step is still the H transfers from the Os site to the Ov site, but for H2O desorption, the H would be initially transferred to the Ov site, and then H2O is produced by the BBO–H and Ti5c-Ov–H. In these cases, H2 desorption is more competitive than H2O desorption.
Although we did not exhaust all possible relative positions of BBOHs and Ovs, the general picture for H2O desorption and H2 desorption should be valid since BBOHs can easily diffuse along the BBO rows. When the initial coverage of BBOHs is small, there is much less chance for the surface to build up local regions with high Ov concentration, and H2O desorption will be the only available process. Perhaps this is the reason why for a long time H2 desorption on a TiO2(110) surface has not been observed.
The Ti–H species
We propose that the Ti–H species is the key intermediate for H2 desorption, and its stability is crucial for the rate-determining barrier. There are two types of Ti–H species, the Ti5c–H and Ti5c-Ov–H. The Ti5c–H has also been assumed to respond to H2 desorption under other conditions.14,21 Here for the first time we have proposed the complete reaction profiles involving Ti–H species for H2 desorption on a TiO2(110) surface. The Ti5c-Ov–H is more stable than the Ti5c–H species. However, for all stoichiometric and Ov models, the formation of Ti–H species is endothermic, suggesting that the Ti–H species are pretty unstable. The stability of the Ti–H species increases with an increase in the Ov, which is the key reason for the decrease in the H2 desorption barrier. In our two Ovs models, the formation of Ti5c-Ov–H could even be exothermic.Nevertheless, to our knowledge, Ti–H species on a TiO2(110) surface has never been observed experimentally. Can we actually observe it? Our answer is that it will be pretty hard in the current H2 desorption experiments. Although we predict that the formation of Ti5c-Ov–H species can be exothermic for the local regions with high Ov concentration, in each case the formation of Ti5c-Ov–H is the rate-determining step, and the reaction between Ti5c-Ov–H and BBOH to form H2 is very fast. We think that it is possible to observe these Ti5c-Ov–H species by sputtering H atoms to a surface with a very high Ov concentration.
Conclusions
In summary, we have investigated the mechanism of H2 and H2O desorption from BBOH of a TiO2(110) surface. Our results suggest that it is unlikely for two BBOHs to couple directly to form H2, and we propose that the reaction undergoes a Ti–H key intermediate, which then forms an O–Hδ+⋯Hδ−–Ti type dehydrogenation transition state. The H2 desorption is pretty hard on a stoichiometric surface. However, it becomes easier with an increase in Ovs which is generated by H2O desorption. As a comparison, the H2O desorption is pretty easy on a stoichiometric surface but becomes harder and harder. In the case of close Ovs that are generated by H2O desorption, H2 desorption could be easier than H2O desorption on these local regions. Therefore, our theoretical results suggest that H2O desorption is the major product at the beginning of the reaction. If the initial BBOH concentration is high enough, at the late stage of the reaction the H2 desorption becomes a competitive reaction. These results fit with the experimental observations very well.The H2 production is a key process for photocatalytic H2O splitting. Currently, the H2 yield is pretty low and the desorption temperature is pretty high on the TiO2(110) surface. Based on our results, possible ways to improve the current reaction system include the following: a) use the surface with a high Ovs concentration; b) specifically design close Ovs on the surface; c) other surface modifications to increase the reducing power of the surface; d) introduce other metals which have more stable M–H species. With these strategies, if the H2 desorption temperature can be reduced to lower than that for H2O desorption at Ti5c, we expect that the H2 desorption from the H2O/TiO2 system could be available.
Acknowledgements
This work was supported by the National Science Foundation of China (21210004, 21673224) and the Strategic pilot science and technology project of the Chinese Academy of Sciences (XDB17010200).References
- M. R. Hoffmann, S. T. Martin, W. Y. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- T. L. Thompson and J. T. Yates, Chem. Rev., 2006, 106, 4428–4453 CrossRef CAS PubMed.
- M. A. Henderson, S. Otero-Tapia and M. E. Castro, Faraday Discuss., 1999, 114, 313–319 RSC.
- X. Q. Gong, A. Selloni, O. Dulub, P. Jacobson and U. Diebold, J. Am. Chem. Soc., 2008, 130, 370–381 CrossRef CAS PubMed.
- M. A. Henderson, Surf. Sci. Rep., 2011, 66, 185–297 CrossRef CAS.
- C. L. Pang, R. Lindsay and G. Thornton, Chem. Soc. Rev., 2008, 37, 2328–2353 RSC.
- A. Fujishima, X. Zhang and D. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- W. S. Epling, C. H. F. Peden, M. A. Henderson and U. Diebold, Surf. Sci., 1998, 412, 333–343 CrossRef.
- S. Suzuki, K. Fukui, H. Onishi and Y. Iwasawa, Phys. Rev. Lett., 2000, 84, 2156–2159 CrossRef CAS PubMed.
- M. Kunat, U. Burghaus and C. Wöll, Phys. Chem. Chem. Phys., 2004, 6, 4203–4207 RSC.
- X. L. Yin, M. Calatayud, H. Qiu, Y. Wang, A. Birkner, C. Minot and C. Wöll, ChemPhysChem, 2008, 9, 253–256 CrossRef CAS PubMed.
- N. G. Petrik and G. A. Kimmel, J. Phys. Chem. C, 2009, 113, 4451–4460 CAS.
- Y. Du, N. G. Petrik, N. A. Deskins, Z. Wang, M. A. Henderson, G. A. Kimmel and I. Lyubinetsky, Phys. Chem. Chem. Phys., 2012, 14, 3066–3074 RSC.
- C. B. Xu, W. S. Yang, Q. Guo, D. X. Dai, M. D. Chen and X. M. Yang, J. Am. Chem. Soc., 2013, 135, 10206–10209 CrossRef CAS PubMed.
- M. A. Henderson, Surf. Sci., 1996, 355, 151–166 CrossRef CAS.
- G. H. Enevoldsen, H. P. Pinto, A. S. Foster, M. C. R. Jensen, W. A. Hofer, B. Hammer, J. V. Lauritsen and F. Besenbacher, Phys. Rev. Lett., 2009, 102, 136103 CrossRef PubMed.
- P. M. Kowalski, B. Meyer and D. Marx, Phys. Rev. B, 2009, 79, 115410 CrossRef.
- C. B. Xu, W. S. Yang, Q. Guo, D. X. Dai, M. D. Chen and X. M. Yang, J. Am. Chem. Soc., 2014, 136, 602–605 CrossRef CAS PubMed.
- S. C. Li, Z. Zhang, D. Sheppard, B. D. Kay, J. M. White, Y. Du, I. Lyubinetsky, G. Henkelman and Z. Dohnalek, J. Am. Chem. Soc., 2008, 130, 9080–9088 CrossRef CAS PubMed.
- Z. F. Wu, W. H. Zhang, F. Xiong, Q. Yuan, Y. K. Jin, J. L. Yang and W. X. Huang, Phys. Chem. Chem. Phys., 2014, 16, 7051–7057 RSC.
- P. Chen, Z. T. Xiong, J. Z. Luo, J. Y. Lin and K. L. Tan, J. Phys. Chem. B, 2003, 107, 10967–10970 CrossRef CAS.
- Z. T. Xiong, G. T. Wu, H. J. Hu and P. Chen, Adv. Mater., 2004, 16, 1522–1525 CrossRef CAS.
- W. Luo, J. Alloys Compd., 2004, 381, 284–287 CrossRef CAS.
- J. Lu and Z. G. Z. Fang, J. Phys. Chem. B, 2005, 109, 20830–20834 CrossRef CAS PubMed.
- W. L. Xu, G. T. Wu, W. Yao, H. J. Fan, J. S. Wu and P. Chen, Chem. – Eur. J., 2012, 18, 13885–13892 CrossRef CAS PubMed.
- T. A. Rokob, A. Hamza, A. Stirling, T. Soos and I. Papai, Angew. Chem., Int. Ed., 2008, 47, 2435–2438 CrossRef CAS PubMed.
- A. Comas-Vives and G. Ujaque, J. Am. Chem. Soc., 2013, 135, 1295–1305 CrossRef CAS PubMed.
- Y. Duan, L. Li, M. W. Chen, C. B. Yu, H. J. Fan and Y. G. Zhou, J. Am. Chem. Soc., 2014, 136, 7688–7700 CrossRef CAS PubMed.
- A. Paul and C. B. Musgrave, Angew. Chem., Int. Ed., 2007, 46, 8153–8156 CrossRef CAS PubMed.
- K. J. Sun, Y. H. Zhao, H. Y. Su and W. X. Li, Theor. Chem. Acc., 2012, 131, 1–10 CAS.
- R. M. Wang and H. J. Fan, Sci. China., Chem., 2015, 58, 614–619 CrossRef CAS.
- Q. Guo, C. B. Xu, Z. F. Ren, W. S. Yang, Z. B. Ma, D. X. Dai, H. J. Fan, T. K. Minton and X. M. Yang, J. Am. Chem. Soc., 2012, 134, 13366–13373 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B, 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B, 1996, 54, 11169–11186 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B, 1994, 50, 17953–17979 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jonsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- G. Henkelman and H. Jonsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- J. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- J. Zhao, J. Yang and H. Petek, Phys. Rev. B, 2009, 80, 235416 CrossRef.
- J. Paier, R. Hirsch, M. Marsman and G. Kresse, J. Chem. Phys., 2005, 122, 234102 CrossRef PubMed.
- R. Sanchez de Armas, J. Oviedo, M. A. San Miguel and J. F. Sanz, J. Phys. Chem. C, 2007, 111, 10023–10028 CAS.
- E. V. Stefanovich and T. N. Truong, Chem. Phys. Lett., 1999, 299, 623–629 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188–5192 CrossRef.
- R. L. Kurtz, R. Stockbauer, T. E. Madey, E. Roman and J. L. Desegovia, Surf. Sci., 1989, 218, 178–200 CrossRef CAS.
- M. A. Henderson, W. S. Epling, C. H. F. Peden and C. L. Perkins, J. Phys. Chem. B, 2003, 107, 534–545 CrossRef CAS.
- C. Di Valentin, G. Pacchioni and A. Selloni, Phys. Rev. Lett., 2006, 97, 166803–166806 CrossRef PubMed.
- M. I. Nandasiri, V. Shutthanandan, S. Manandhar, A. M. Schwarz, L. Oxenford, J. V. Kennedy, S. Thevuthasan and M. A. Henderson, J. Phys. Chem. Lett., 2015, 6, 4627–4632 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Reaction profiles for pathways H2_C, H2_D and H2_H are shown. The optimized individual structures of the Cartesian coordinates from theory. See DOI: 10.1039/c6cy02007k |
| This journal is © The Royal Society of Chemistry 2017 |