
An efficient hydrogen evolution catalyst composed of palladium phosphorous sulphide (PdP∼0.33S∼1.67) and twin nanocrystal Zn0.5Cd0.5S solid solution with both homo- and hetero-junctions†
Jingang
Song
a,
Haitao
Zhao
a,
Ranran
Sun
b,
Xiyou
Li
*ab and
Dejun
Sun
*a
aDepartment of Chemistry, Shandong University, Jinan, 250100, China. E-mail: xiyouli@sdu.edu.cn
bDepartment of Chemistry, College of Science, China University of Petroleum (East China), Qingdao, 266580, China
First published on 17th November 2016
Abstract
ZnxCd1−xS solid solutions have been successfully demonstrated as excellent visible light responsive catalysts for hydrogen evolution from water in the presence of sacrificial reagents. Especially Zn0.5Cd0.5S with a twin crystal structure is the best pristine sulphide catalyst ever reported so far due to the presence of the homo-junctions between the zinc blende (ZB) and wurtzite (WZ) segments, which improved the separation of the photon generated electron–hole pairs. However, a further improvement in the catalytic activities of the twin crystal Zn0.5Cd0.5S solid solution has never been reported. In the present work, palladium phosphorous sulphide (PdP∼0.33S∼1.67) was used for the first time as a co-catalyst and found that the photocatalytic H2 production rate and efficiency of the twin nanocrystal Zn0.5Cd0.5S solid solution in the presence of acidic or alkaline sacrificial reagents were significantly enhanced. The H2 production rates of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 under visible light illumination (λ > 420 nm) in 0.75 M ascorbic acid (pH = 2.39) and 0.7 M Na2S/0.5 M Na2SO3 (pH = 13.86) aqueous solutions are measured to be 372.12 μmol h−1 mg−1 and 246.04 μmol h−1 mg−1, respectively, which are 67 and 5.8 times higher than those of pristine twinned Zn0.5Cd0.5S solid solution. Moreover, the H2 production rate of 372.12 μmol h−1 mg−1 is the highest value ever reported so far for sulphide photocatalysts in the presence of acidic sacrificial reagents. The examination on the morphology of the Zn0.5Cd0.5S/PdP∼0.33S∼1.67 hybrid catalyst using SEM, TEM and HRTEM techniques revealed the presence of hetero-junctions between the deposited PdP∼0.33S∼1.67 and Zn0.5Cd0.5S phases, as well as homo-junctions between the ZB and WZ phases in this hybrid catalyst. The synergistic effects of homo- and hetero-junctions on the separation of the generated electron–hole pairs are responsible for the significantly promoted catalytic activities of the hybrid catalyst. This result suggests that combination of both homo- and hetero-junctions in one catalyst is a promising strategy for improving the catalytic activities of sulphide catalysts for hydrogen evolution from water.
Broader contextZnxCd1−xS solid solutions were excellent visible light responsive catalysts for hydrogen evolution from water in the presence of sacrificial reagents. Especially Zn0.5Cd0.5S with a twin crystal structure is the best pristine sulfide catalyst ever reported so far due to the presence of the homojunctions between the zinc blende (ZB) and wurtzite (WZ) segments, which improved the separation of the photon generated electron–hole pairs. However, a further improvement in the catalytic activities of the twin crystal Zn0.5Cd0.5S solid solution has never been reported. In the present work, we have successfully promoted the catalytic activity of the twin crystal Zn0.5Cd0.5S solid solution by depositing palladium phosphorous sulphide (PdP∼0.33S∼1.67) on the surface of twin crystal Zn0.5Cd0.5S solid solution and creating hetero-junctions between Zn0.5Cd0.5S and PdP∼0.33S∼1.67. Due to the presence of both the homo-junction and hetero-junction in this Zn0.5Cd0.5S/PdP∼0.33S∼1.67 nano-composite, the separation of photon induced electron–hole pairs has been improved and thus the catalytic activity was significantly improved. Combining of both homo- and hetero-junctions in one catalyst might be an efficient strategy towards a better photocatalyst for H2 evolution from water. |
1. Introduction
Due to the high energy capacity, hydrogen is considered to be an ideal fuel for a new environmentally friendly energy supply system.1,2 However, how to conveniently and cheaply get hydrogen from a sustainable and renewable source is still a critical technical challenge. Photo-catalytic splitting of water into H2 and O2, as suggested by Honda and Fujishima,3–5 is considered to be one of the most promising strategies to get hydrogen. Therefore, developing new photocatalysts for efficient hydrogen production from water under the irradiation of light attracted a lot of research interests in the past several decades.6,7 To date, several kinds of hydrogen production photocatalysts including metal and nonmetal mixed oxides,8,9 nitrides,10,11 doped perovskites,12,13 and nitrided pyrochlores,14,15 and others have been developed.16,17 In the perspective of using solar energy efficiently, photocatalysts with a visible light response are particularly important.18–20Sulphide catalysts, including CdS and ZnxCd1−xS solid solutions, are a group of promising visible-light-responsive photocatalysts due to the favourable band structure.21 The catalytic activities of these sulphide compounds are determined by the band energy levels,22,23 the morphology and the micro crystal structure.24–26 However, these pristine sulphide catalysts are subject to low catalytic activities and poor stabilities due to the fast electron–hole recombination and photocorrosion.27 Improving the separation of photoinduced hole and electron pairs is, therefore, one of the most efficient strategies to boost the activity of a sulphide catalyst. Promoting the separation of photogenerated electron–hole pairs can be achieved by creating structure junctions in a catalyst. For example, introduction of various co-catalysts into the sulphide catalyst can create hetero-junctions on the surface and thus the separation of photogenerated electron–hole pairs can be improved and the catalytic activity of the catalysts can be promoted significantly.28 To date, various co-catalysts have been employed to combine with sulfides, including noble metals (e.g., Pt, Pd, Ru, and Rh),28 transition metal oxides (e.g., ZnO29), transition metal sulfides (e.g., MoS2,30 NiS,31 CuS,32 PdS33) and transition metal phosphides (e.g., Ni2P,34 Cu3P,35 Fe2P,36 MoP,37 Co2P,38 CoP39). These co-catalysts can either collect the electrons or scavenge the holes from the catalysts and thus the separation of photogenerated electron–hole pairs is improved. Among these numerous co-catalysts, the Pt-tipped CdSe@CdS nano composite, which consists of a CdSe seed embedded asymmetrically within a CdS nanorod and a Pt-reduction catalyst placed on the opposite end of the rod, was reported to be extremely efficient with almost 100% photon-to-hydrogen production efficiency at 455 nm.40 The efficient and steady H2 generation can be attributed to the fast hole transfer to the scavenger.41
Besides the hetero-junctions formed between the co-catalyst and the sulphide catalyst, the homo-junctions formed between the different crystal phases in a catalyst can also improve the electron–hole separation. For example, the homo-junctions formed between the zinc-blende (ZB) and wurtzite (WZ) segments in a Zn0.5Cd0.5S solid solution with a twin crystal structure can enhance the separation of photo generated electrons and holes, which makes this twin crystal Zn0.5Cd0.5S solid solution the most efficient pristine sulphide catalyst ever reported so far for hydrogen production from water without help of any co-catalyst.42,43 The improved separation of the electron–hole pairs was ascribed to the “back-to-back” potentials at the homo-junctions. However, this explanation was challenged recently by Hensen and co-workers. They confirmed the excellent catalytic activity of the twin crystal Zn0.5Cd0.5S solid solution, but found no direct correlation between the homo-junction density and the catalytic activity.44 Recently, the synchrotron X-ray diffraction and X-ray absorption studies on the ZnxCd1−xS solid solutions revealed that the photogenerated electrons and holes at the homo-junctions between ZB and WZ segments will diffuse into the CB of the WZ phase and the VB of the ZB phase, respectively, driven by the potential energy of Fermi level equilibration.45
Based on the results as mentioned above, the homo-junctions between ZB and WZ phases can facilitate the separation of electron and hole pairs. However, the further movement of electrons and holes in the bulky catalyst is limited by the boundary of the ZB and WZ segments.44,45 By combining both homo- and hetero-junctions into one catalyst, the photogenerated electron–hole pairs may achieve a more efficient separation due to their synergetic effects. With this in mind, we designed a new hybrid catalyst composed of twinned Zn0.5Cd0.5S solid solution and a tetragonal phase palladium phosphorous sulphide (PdP∼0.33S∼1.67). The PdP∼0.33S∼1.67 phase was vectorially deposited on the surface of twin crystal Zn0.5Cd0.5S solid solution by the template etching method. The resulting new catalytic composite, Zn0.5Cd0.5S/PdP∼0.33S∼1.67, presents excellent photocatalytic activities under visible light illumination (λ > 420 nm) in both 0.75 M ascorbic acid (pH = 2.39) and 0.7 M Na2S/0.5 M Na2SO3 (pH = 13.86) solutions. The H2 production rates are measured to be 372.12 μmol h−1 mg−1 (QE: 19.70%) in ascorbic acid and 246.04 μmol h−1 mg−1 (QE: 16.52%) in Na2S/Na2SO3 solution. It is worth noting that the H2 production rate of 372.12 μmol h−1 mg−1 is the highest value ever reported so far for sulfide photocatalysts in an acidic sacrificial reagent. This is also one of the few catalysts which can perform excellent catalytic activity under both acid and alkaline conditions. This result suggests that the combination of homo- and hetero-junctions in one catalyst is a promising way to improve the catalytic activity.
2. Experimental section
2.1. Preparation of twin nanocrystal Zn0.5Cd0.5S solid solution42
All chemicals were of analytical grade and were directly used without further purification. In a typical procedure, 10 mmol Zn(Ac)2·2H2O, 10 mmol Cd(Ac)2·2H2O, and 25 mmol thioacetamide were mixed with 40 mL distilled water. NaOH aqueous solution (4 M, 10 mL) was dispersed into the reaction mixture by constantly strong stirring until a homogeneous solution was formed. Then, the resulting mixture was sealed in a 100 mL Teflon-lined stainless steel autoclave and was heated to 180 °C for 24 h. The system was then cooled to ambient temperature. The final product was collected and washed with distilled water and absolute alcohol several times, and then was vacuum-dried at 80 °C for 10 h. The dried product was kept for further characterization.2.2. Preparation of Zn0.5Cd0.5S/PdP∼0.33S∼1.67
0.3 g twinned Zn0.5Cd0.5S solid solution and 0.10 g bis(triphenylphosphine) palladium(II) dichloride (Pd(PPh3)2Cl2) were mechanically mixed in a quartz boat at room temperature. The precursor was heated to 372 °C and kept for 12 h in Ar flow (30 mL min−1). The Zn0.5Cd0.5S/PdP∼0.33S∼1.67 nanoparticle was cooled to room temperature in a continuous Ar flow at a flow rate of 30 mL min−1 for 3 h. Then, the black products were collected, washed successively with benzene, absolute alcohol and distilled water, and finally dried under vacuum at 100 °C for further use.Zn0.5Cd0.5S/PdP∼0.33S∼1.67 with different molar ratios of twinned Zn0.5Cd0.5S solid solution and Pd(PPh3)2Cl2 were also prepared following the same procedures of Zn0.5Cd0.5S/PdP∼0.33S∼1.67, except that different amounts of Pd(PPh3)2Cl2 (0.02 g, 0.04 g, 0.06 g, 0.08 g and 0.12 g) were used. The products were named PdP∼0.33S∼1.67 (1, 2, 3, 4 and 6), respectively. Zn0.5Cd0.5S/PdP∼0.33S∼1.67 normally represents the product prepared with 0.10 g Pd(PPh3)2Cl2, i.e. PdP∼0.33S∼1.67 (5).
2.3. Characterization
Scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDS) were performed using a JSM 6700F microscope at an accelerating voltage of 5 kV and linked with an Oxford Instruments X-ray analysis system. For SEM imaging, C (1–2 nm) was sputtered onto the grids to prevent charging effects and to improve the image clarity. High-resolution transmission electron microscopy (TEM, STEM and HRTEM) analyses were conducted using a JEM 2100 microscope at an accelerating voltage of 200 kV. For TEM imaging, a small amount of product was dispersed in ethanol by ultrasonic treatment for 30 min, and then, one drop of the resulting solution was placed onto a carbon-coated copper grid and dried at room temperature. The element contents of samples were also determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis using an IRIS Intrepid II XRP instrument.Thermogravimetric (TG) and differential thermal analysis (DTA) were performed on a TA Instruments 2950 thermogravimetric analyzer equipped with an EGA furnace. The Pd(PPh3)2Cl2 sample (5 mg) was placed in a platinum pan and heated at a rate of 10 °C min−1 to 800 °C in an atmosphere of argon flow (180 mL min−1).
Fourier transform infrared spectra (FTIR) were recorded for the samples dispersed in KBr pellets with 2 cm−1 resolution using an α ALPHA-T spectrometer.
XRD patterns were obtained on an X-ray diffractometer (type HZG41B-PC) using Cu Kα radiation at a scan rate of 0.05° 2θ s−1.
XPS measurements were performed in an ultrahigh vacuum VG ESCALAB 210 electron spectrometer equipped with a multi-channel detector. The spectra were excited with Mg Kα (1253.6 eV) radiation (operated at 200 W) of a twin anode in the constant analyzer energy mode with a pass energy of 30 eV. All the binding energies were referenced to the C 1s peak at 284.8 eV of the surface adventitious carbon.
The Brunauer–Emmett–Teller (BET) specific surface area (SBET) of the powders was measured by nitrogen adsorption using a Micromeritics ASAP 2020 nitrogen adsorption apparatus. The distribution of the pore size was calculated from desorption isotherms by the Barret–Joyner–Halender (BJH) method, assuming a cylindrical pore model.
Fluorescence emission spectra were recorded by using an Edinburgh FLS920 phosphorimeter equipped with a continuous Xe-900 xenon lamp. The emission quantum yields were measured at room temperature using a calibrated integrating sphere as a sample chamber, and Specpure BaSO4 as a reflecting standard.
UV-vis diffused reflectance spectra of the samples were obtained for the dry-pressed disk samples using a UV-vis spectrometer (UV2550, Shimadzu, Japan). BaSO4 was used as a reflectance standard.
2.4. Photocatalytic hydrogen production
Photocatalytic reactions of hydrogen production from water were conducted in a gas-closed system with a top irradiation Pyrex cell. A 300 W xenon lamp (18 A) equipped with a cut-off filter (λ > 420 nm) was used to provide incident visible light and the area of the surface irradiated in the cylinder reactor was about 15.9 cm2. In a typical photocatalytic experiment, 1 mg of the catalyst was dispersed with a constant stirring in 100 mL sacrificial reagent aqueous solution (20% lactic acid (pH = 1.64), or 0.75 M ascorbic acid (pH = 2.39), or 20% TEOA (pH = 12.59), or 0.7 M Na2S and 0.5 M Na2SO3 (pH = 13.86)). Before irradiation, the system was bubbled with nitrogen for 30 min to remove the air inside and to ensure that the reaction system is under anaerobic conditions. The amount of hydrogen generated was determined with online gas chromatography (GC7900, TCD, nitrogen as a carrier gas and a 5 Å molecular sieve column). All glassware was carefully rinsed with distilled water prior to use.The apparent quantum efficiency (QE) was measured under similar photocatalytic reaction conditions. 1 mg of the catalyst was dispersed with a constant stirring in 100 mL sacrificial reagent aqueous solution. The apparent quantum efficiency was measured using a 420 nm band pass filter and an irradiatometer. The QE was finally calculated according to the equation:43

The power of the light was 0.153 J s−1, corresponding to the number of incident photons (3.23 × 1017 photons per second), which was measured using an irradiatometer (FZ-A, Beijing Normal University Optical Instrument).
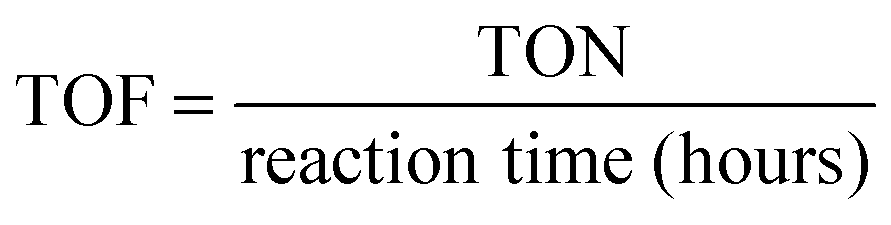
The turnover number (TON) and turnover frequency (TOF) were calculated by using the following equations:34
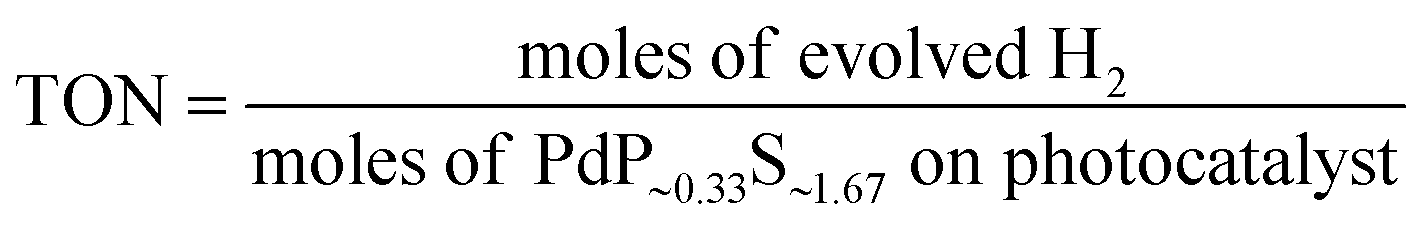
2.5. Photoelectrochemical measurements
Photocurrents were measured by using the CHI760D electrochemical work station (Chenhua Instrument, Shanghai, China) with a standard three-electrode system. The working electrode was a photocatalyst coated ITO with an active area of ca. 1.5 cm2, the counter electrode was a Pt sheet, and the reference electrode was Hg/Hg2+ (saturated KCl). A sacrificial reagent aqueous solution (0.75 M ascorbic acid (pH = 2.39) (A) or 0.7 M Na2S and 0.5 M Na2SO3 (pH = 13.86)) was used as an electrolyte. The working electrode was illuminated using a solar simulator with a UV cut-off filter (λ > 420 nm) (100 mW cm−2) during photocurrent measurements. The working electrode was prepared following the procedures as listed below: 0.01 g of the photocatalyst (twinned Zn0.5Cd0.5S or Zn0.5Cd0.5S/PdP∼0.33S∼1.67) was ground with 0.5 mL water to make a slurry. The slurry was then coated onto a 3 cm × 1 cm ITO glass electrode with a doctor blade (film thickness: ca. 300 nm). The working electrodes were dried at 100 °C overnight to evaporate all of the water. Then, the photoresponsive signals of the samples were measured under chopped light at 0.0 V.3. Results and discussion
3.1 Preparation and characterization of twinned Zn0.5Cd0.5S solid solution
The twinned Zn0.5Cd0.5S solid solution was prepared following the literature.42 The morphology of the prepared twinned Zn0.5Cd0.5S solid solution was examined by SEM, TEM, SAED and the results are shown in Fig. S1 (ESI†). Black and white strips can be identified in the TEM images of the prepared nanoparticles of Zn0.5Cd0.5S solid solution, suggesting the presence of a strip-like structure on the surface of the particles. In the high-resolution TEM (HRTEM) images, boundaries of the twin crystal structure can be identified.42 The twin crystal structure was further supported by the fast Fourier transform (FFT) pattern as shown in Fig. S1C (ESI†).43 All the results suggested that the prepared Zn0.5Cd0.5S solid solution has a twin crystal structure. Because the Zn/Cd ratio in the solid solution is very important for photocatalytic activity, the elementary composition of the twinned Zn0.5Cd0.5S was also determined by the inductively coupled plasma atomic emission spectrometry (ICP-AES) and the electron dispersive spectrum (EDS). The results are shown in Tables S1 and S2 (ESI†). The results revealed that the Zn/Cd ratio in the solid solution is approximately 0.5/0.5, which is in accordance with the initial feeding ratio between Zn and Cd in the reaction mixture.3.2 Preparation and structure characterization of Zn0.5Cd0.5S/PdP∼0.33S∼1.67
Thermogravimetric (TG) and differential thermal analysis (DTA) in an argon atmosphere revealed that Pd(PPh3)2Cl2 molecules begin to decompose at 364 °C, which is different from the pyrolysis process in air (Fig. S2, ESI†).46 Tri(m-tolyl)phosphorus, a similar compound with triphenylphosphorus, has been used as a phosphorus source for the preparation of various metal phosphides.47 The pyrolysis temperature was 370 °C. Therefore, our reaction temperature was set at 372 °C for the purpose of speeding up the decomposition of Pd(PPh3)2Cl2 molecules. At this temperature, following the template etching method,48–52 we have successfully deposited a nanocomposite with a composition of PdP∼0.33S∼1.67 onto the surface of the twin crystal Zn0.5Cd0.5S solid solution with Pd(PPh3)2Cl2 as a precursor. The complete decomposition of Pd(PPh3)2Cl2 was further supported by the IR spectra of the nanocomposite, the mixture of Pd(PPh3)2Cl2 and twinned Zn0.5Cd0.5S solid solution, and the pristine twin crystal Zn0.5Cd0.5S solid solution (Fig. S3, ESI†). There is no IR signal of Pd(PPh3)2Cl2 remaining in the spectrum of the product, indicating the complete decomposition of Pd(PPh3)2Cl2.As revealed by the images of SEM and TEM (Fig. 1A and B), nanoparticles with a diameter in the range of 30–120 nm were formed. In the TEM images, strip-like structures can be identified, which can be assigned to the structure of twinned Zn0.5Cd0.5S solid solution.42,43 Some smaller nanoparticles are distributed on the surface of Zn0.5Cd0.5S solid solution, which are the PdP∼0.33S∼1.67 nanocomposite. The HRTEM image shows that part of the Zn0.5Cd0.5S particles are in close contact with the deposited PdP∼0.33S∼1.67 (Fig. 1C), indicating the formation of heterojunctions. The SAED patterns as shown in the insets of Fig. 1C revealed a twin crystal structure for the Zn0.5Cd0.5S part and a tetragonal single crystal structure for the deposited PdP∼0.33S∼1.67 segments. The lattice spacing of ca. 0.325 nm (Fig. 1D) in the Zn0.5Cd0.5S part belongs to the (111) diffraction plane of Zn0.5Cd0.5S.42,43 The similar lattice spacing (0.325 nm) was found for the PdP∼0.33S∼1.67 particles, which can be assigned to the (002) diffraction plane of a tetragonal PdP∼0.33S∼1.67 crystal.53
 | ||
| Fig. 1 SEM (A), TEM (B), HRTEM (C and D) and SAED (the inset of C) images of Zn0.5Cd0.5S/PdP∼0.33S∼1.67. | ||
The STEM image and the corresponding EDX elemental mapping of the resulted nanocomposite as shown in Fig. S4 A–F (ESI†) clearly indicate the presence of Zn, Cd, S, Pd and P elements in the sample. Zn and Cd distribute uniformly in the same area, suggesting the formation of solid solution between ZnS and CdS. The distribution of Pd, S, and P covers the same area, suggesting the presence of PdPxSy. Because the diffraction patterns as shown in Fig. 1C have proved that the deposited particles on the surface of Zn0.5Cd0.5S are single crystals, these particles should not be a simple mixture of PdPx and PdSy, but a pure compound composed of Pd, P, and S elements instead. Because there is no extra S atom was added in the reaction mixture, the S atoms in this compound must be grabbed from Zn0.5Cd0.5S via ionic exchange reactions.32,49
To determine the composition of the prepared hybrid catalyst, both ICP-AES and EDS experiments were performed and the results are summarized in Tables S1 and S2 (ESI†). The atomic ratio between Zn and Cd is still 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, suggesting no changes in the composition of Zn0.5Cd0.5S solid solution during the reaction. But a little increase in the percentage of S relative to that of Zn and Cd is observed, which may be ascribed to the conversion of a small amount of Zn0.5Cd0.5S into PdPxSy during the etching reaction. When Zn0.5Cd0.5S changed into PdPxSy, the S atoms were captured by PdPxSy, and the excess Zn2+ and Cd2+ were washed away in the following purification procedures. Therefore, the number of S atoms captured by PdPxSy should be equal to the sum of the numbers of Zn2+ and Cd2+ lost during the reaction. In the pristine twin crystal Zn0.5Cd0.5S, the total atomic percentage of Zn and Cd is 50.52%. This atomic percentage decreased to 48.90% in Zn0.5Cd0.5S/PdPxSy. The total loss of Zn and Cd during the reaction is about 1.62%, so the total atomic percentage of the S atom captured by PdPxSy should be about 1.62%. A similar result (1.64%) was calculated from the ICP results. The atomic percentage of Pd and P in the final product is measured to be around 0.94% and 0.31%, respectively, so the Pd/P ratio in PdPxSy particles should be close to 3/1, and a Pd/S ratio close to 1/1.6. Of course the dilution effect caused by the introduction of PdPxSy into Zn0.5Cd0.5S will also lead to a decrease in the atomic percentage of Zn and Cd. But the atomic percentage of Pd in the nanocomposite is only less than 1%, therefore, this dilution effect on the percentage of Zn and Cd should be neglectable.
:1, suggesting no changes in the composition of Zn0.5Cd0.5S solid solution during the reaction. But a little increase in the percentage of S relative to that of Zn and Cd is observed, which may be ascribed to the conversion of a small amount of Zn0.5Cd0.5S into PdPxSy during the etching reaction. When Zn0.5Cd0.5S changed into PdPxSy, the S atoms were captured by PdPxSy, and the excess Zn2+ and Cd2+ were washed away in the following purification procedures. Therefore, the number of S atoms captured by PdPxSy should be equal to the sum of the numbers of Zn2+ and Cd2+ lost during the reaction. In the pristine twin crystal Zn0.5Cd0.5S, the total atomic percentage of Zn and Cd is 50.52%. This atomic percentage decreased to 48.90% in Zn0.5Cd0.5S/PdPxSy. The total loss of Zn and Cd during the reaction is about 1.62%, so the total atomic percentage of the S atom captured by PdPxSy should be about 1.62%. A similar result (1.64%) was calculated from the ICP results. The atomic percentage of Pd and P in the final product is measured to be around 0.94% and 0.31%, respectively, so the Pd/P ratio in PdPxSy particles should be close to 3/1, and a Pd/S ratio close to 1/1.6. Of course the dilution effect caused by the introduction of PdPxSy into Zn0.5Cd0.5S will also lead to a decrease in the atomic percentage of Zn and Cd. But the atomic percentage of Pd in the nanocomposite is only less than 1%, therefore, this dilution effect on the percentage of Zn and Cd should be neglectable.
Previously, three pure phases of PdSxPy, namely PdPS, PdS1.33P0.67, and PdS1.67P0.33, have been successively prepared by the reactions of Pd, S and P at extremely high temperature and pressure.52 All of them have essentially a 2:1 ratio of anions to cations. Their formation depends mainly on the feeding atomic ratio of Pd:P:S in the starting materials. During the charge ratio of Pd:P:S = 1:1:>3, a metal phase PdP0.33S1.67 was formed. Based on the facts that the calculated atomic percentage of Pd, P and S in the final hybrid catalyst is close to PdP0.33S1.67, and the tetragonal structure revealed by the SAED patterns shown in Fig. 1C corresponds well with the tetragonal cell for PdS1.67P0.33 in the literature,53 and more importantly, the 0.325 nm lattice spacing deduced from the HRTEM image in Fig. 1D corresponds well with the (002) facet spacing of tetragonal PdP0.33S1.67,53 we speculated that the resulted hybrid catalyst should have a composition close to PdP∼0.33S∼1.67. This speculation was further supported by the XRD diffraction pattern of the hybrid catalyst as shown in Fig. 2, which will be discussed later in the next part. Therefore, we use Zn0.5Cd0.5S/PdP∼0.33S∼1.67 to stand for the hybrid catalyst in the following text.
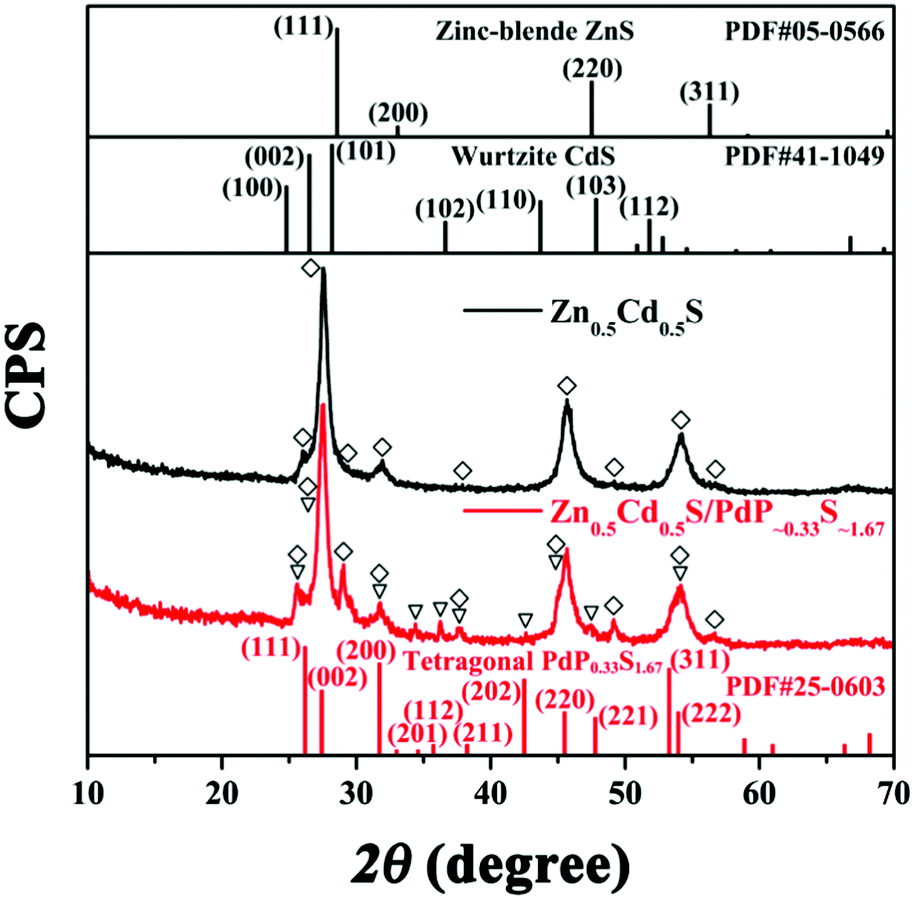 | ||
| Fig. 2 The X-ray diffraction patterns of twinned Zn0.5Cd0.5S (black) and Zn0.5Cd0.5S/PdS∼1.67P∼0.33 (red). The vertical bars show the cubic phase ZnS (JCPDS card no. 05-0566), hexagonal phase CdS (JCPDS Card No. 41-1049), and the tetragonal phase PdP0.33S1.67 (JCPDS card no. 25-0603) powder diffraction patterns from the Joint Committee on Powder Diffraction Standards (JCPDS) card. | ||
We have also noted that the tetragonal phase PdP∼0.33S∼1.67 segments were mostly deposited on the surface of the ZB phase of twinned Zn0.5Cd0.5S solid solution, which might be attributed to the template effects of the ZB phase of twinned Zn0.5Cd0.5S solid solution on the crystal growth of PdP∼0.33S∼1.67 caused by the precise matching between their lattice spaces (Fig. S5, ESI†).54 These kinds of intimate heterojunctions between Zn0.5Cd0.5S and PdP∼0.33S∼1.67 components should be very favourable for vectorial transfer of the charge carriers within the hybrids and thus improve the separation of electron–hole pairs.55
3.3 X-ray diffraction (XRD) experiments of twinned Zn0.5Cd0.5S solid solution and Zn0.5Cd0.5S/PdP∼0.33S∼1.67
The X-ray diffraction (XRD) patterns of twinned Zn0.5Cd0.5S, Zn0.5Cd0.5S/PdP∼0.33S∼1.67, together with the standard diffraction patterns of the cubic phase ZnS (JCPDS card no. 05-0566), hexagonal phase CdS (JCPDS card no. 41-1049), and tetragonal phase PdP0.33S1.67 (JCPDS card no. 25-0603) are shown in Fig. 2.56 It can be seen that the diffraction pattern of Zn0.5Cd0.5S exhibits multiphase characteristics in comparison with that of the cubic phase and the hexagonal phase. This result indicated the formation of the twinned Zn0.5Cd0.5S solid solution.42,43 Besides the diffraction peaks of twinned Zn0.5Cd0.5S solid solution, the diffraction peaks of (111), (002), (200), (201), (112), (211), (202), (220), (221), (311), and (222) of tetragonal phase PdP0.33S1.67 can also be identified in the diffraction pattern of the hybrid catalyst, which suggests again that the cocatalyst is tetragonal phase PdP∼0.33S∼1.67. This corresponds well with the results of the HRTEM, ICP and EDX experiments as discussed above. The XRD patterns of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 and twinned Zn0.5Cd0.5S solid solution after calcined at 372 °C as shown in Fig. S6 (ESI†) show no diffraction peaks of impurities,54 indicating the good stability of the Zn0.5Cd0.5S/PdP∼0.33S∼1.67 composite.3.4 X-ray photoelectron spectroscopy (XPS) of twinned Zn0.5Cd0.5S solid solution and Zn0.5Cd0.5S/PdP∼0.33S∼1.67
X-ray photoelectron spectra (XPS) of the hybrid catalyst have been recorded to determine the chemical composition and valence states of the elements in the sample. The binding energy is calibrated by taking the C 1s peak at 284.6 eV as reference. The spectra of the overall survey scan of twinned Zn0.5Cd0.5S and Zn0.5Cd0.5S/PdP∼0.33S∼1.67 indicate the presence of Zn, Cd, S, Pd and P (Fig. S7, ESI†). Fig. 3A and B compares the high resolution XPS spectra of Zn 2p and Cd 3d of the samples. The sharp peaks located at 1023.9 and 1046.9 eV as shown in Fig. 3A can be assigned to the Zn 2p3/2 and Zn 2p1/2 levels, respectively.57,58 The peaks of Cd 3d5/2 and Cd 3d3/2 are found at 404.8 and 411.6 eV in Fig. 3B.57 The peaks of Zn 2p3/2, Zn 2p1/2, Cd 3d5/2 and Cd 3d3/2 are all sharp and symmetrical, demonstrating that the valences of Zn and Cd in the samples are both +2.57,58 | ||
| Fig. 3 The high resolution XPS spectrum of Zn 2p (A) and Cd 3d (B) of twinned Zn0.5Cd0.5S (black) and Zn0.5Cd0.5S/PdP∼0.33S∼1.67 (red) photocatalysts, S 2p of the twinned Zn0.5Cd0.5S photocatalyst (C), Pd 3d of the Zn0.5Cd0.5S/PdP∼0.33S∼1.67 photocatalyst (D), S 2p of the Zn0.5Cd0.5S/PdP∼0.33S∼1.67 photocatalyst (E) and P 2p of the Zn0.5Cd0.5S/PdP∼0.33S∼1.67 photocatalyst (F). | ||
In addition, the peaks observed at 335.6 and 336.2 eV belong to Pd 3d5/2 (Fig. 3D), while the peaks at 340.9 and 341.6 eV can be assigned to Pd 3d3/2.34,59,60 The difference (5.4 eV) between Pd 3d5/2 and Pd 3d3/2 can be attributed to the spin orbit coupling.59 The splitting of the Pd 3d5/2 and Pd 3d3/2 peaks is because the electronegativity value of S is higher than that of P, which is expected to result in significant changes in the binding energy positions of the core level of Pd. These binding energy values are very close to those observed for Pd in PdPS (335.7 eV and 336.2 eV for Pd 3d5/2; 340.9 eV and 341.5 eV for Pd 3d3/2),61 but different from those of palladium sulphide (336.6 eV for Pd 3d5/2 and 341.8 eV for Pd 3d3/2)59 and pure Pd clusters (335.3 eV for 3d5/2 and 340.0 eV for 3d3/2).61 Therefore, the valence of Pd in PdP∼0.33S∼1.67 should be +2, not 0.34,59,60
In the S 2p region, the band can be deconvoluted into two peaks (161.2–161.3 eV and 162.3–162.4 eV), which can be attributed to the S 2p3/2 and S 2p1/2 levels, respectively (Fig. 3C and E).62 The difference between S 2p3/2 and S 2p1/2 levels is also due to the spin orbit coupling.61,62 The binding energies of S 2p3/2 and S 2p1/2 of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 are relatively smaller than that of pure S (2p3/2, 164 eV), which is because of the interactions of S with metal cations as has been reported previously.30,59,63 It is also obvious that the binding energy of S 2p of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 shifts toward the lower energy in comparison with that of the pristine twinned Zn0.5Cd0.5S solid solution (2p3/2, 161.7 eV), which can be attributed to the presence of PdP∼0.33S∼1.67 in which S interacted with Pd.48 The P 2p peaks (Fig. 3F) are observed at 128.5 eV and 129.3 eV, which are similar to that observed for P 2p in PdPS (128.6 eV and 129.2 eV)61 and, therefore, can be assigned to P 2p3/2 and P 2p1/2 levels of PdP∼0.33S∼1.67, respectively. The peak at about 133.0 eV can be assigned to phosphate species formed during synthesis.34
Based on the results as mentioned above, we can conclude that we have successfully prepared a hybrid catalyst based on a twinned Zn0.5Cd0.5S solid solution with PdP∼0.33S∼1.67 as a co-catalyst. The resulted catalyst contains about 1.68% Pd and 0.17% P in weight. It must be noted that we have actually prepared a series of hybrid catalysts with different contents of Pd and P. Their catalytic activities for H2 evolution from water were tested and the results are summarized in Fig. S8 (ESI†). The hybrid catalyst with 1.68% Pd and 0.17% P (wt%) showed the best catalytic activity among these series of hybrid catalysts, so we show here only the characterization of this material.
3.5 Visible light-driven H2 evolution of twinned Zn0.5Cd0.5S solid solution and Zn0.5Cd0.5S/PdP∼0.33S∼1.67
The prepared Zn0.5Cd0.5S/PdP∼0.33S∼1.67 sample, together with the twinned Zn0.5Cd0.5S solid solution, was tested as a catalyst for H2 evolution with different sacrificial reagents. The results are summarized in Fig. 4A and supported by Movie S1 (ESI†). With 0.75 M ascorbic acid aqueous solution (pH 2.39) as a sacrificial reagent, Zn0.5Cd0.5S/PdP∼0.33S∼1.67 gave a H2 evolution rate of 372.12 μmol h−1 mg−1, which is 67 times higher than that of the pristine twinned Zn0.5Cd0.5S solid solution (5.55 μmol h−1 mg−1) measured under the same conditions. The H2 production rate of 372.12 μmol h−1 mg−1 is obviously larger than those reported previously in the literature,64–67 and is the highest value achieved so far for sulfide photocatalysts in the presence of acidic sacrificial reagents. The photocatalytic activities of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 in other sacrificial reagents (20% lactic acid (pH = 1.64), 20% triethanol amine (TEOA, pH = 12.59), 0.7 M Na2S/0.5 M Na2SO3 (pH = 13.86)) were also investigated (Fig. 4A). The photocatalytic H2 evolution rate of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 increased 25 times in 20% lactic acid aqueous solution, 21 times in 20% TEOA aqueous solution, in comparison with those of twinned Zn0.5Cd0.5S solid solution tested under the same conditions. Although twinned Zn0.5Cd0.5S solid solution showed a considerable photocatalytic hydrogen evolution rate with Na2S/Na2SO3 as a sacrificial reagent, the photocatalytic hydrogen evolution rate of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 is still 5.28 times higher than that of twinned Zn0.5Cd0.5S solid solution (Table S3, ESI†). In order to verify the accuracy of these H2 evolution rates as mentioned above, the photocatalytic experiments were also conducted with a 10 mg catalyst under the same conditions. The results (Fig. S9A and Table S4, ESI†) are similar to those measured under the same conditions using a 1 mg catalyst. All these results suggest that the H2 evolution rates we measured are reliable. These remarkable H2 evolution rates indicate that Zn0.5Cd0.5S/PdP∼0.33S∼1.67 performs high photocatalytic H2 evolution activities under both acidic and alkaline conditions. As previously reported, most of the transition metal phosphide cocatalysts show high catalytic activity in acidic sacrificial reagent solution, but may not be active or even unstable in basic sacrificial reagent solutions.35–39 On the other hand, most of the transition metal sulphide cocatalysts can only perform high catalytic activity under alkaline conditions.31–33 A sulphide H2 evolution catalyst with excellent catalytic activity under both acidic and alkaline conditions is rarely reported in the literature.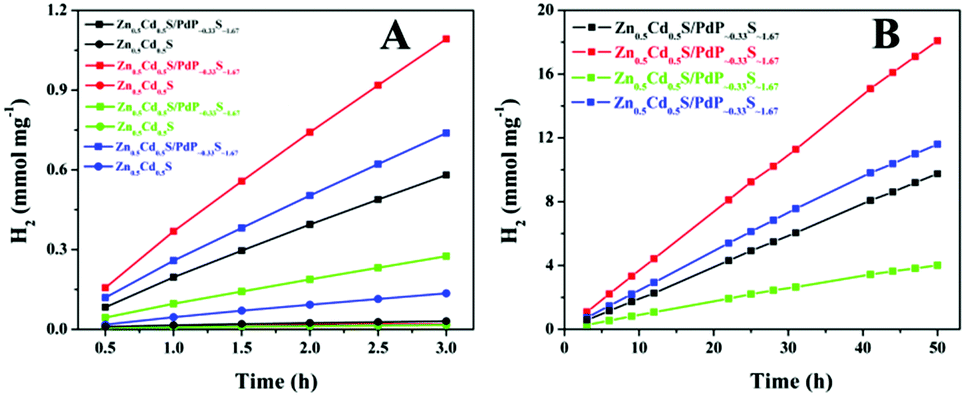 | ||
| Fig. 4 The rate of H2 evolution (A) and long-term H2 evolution (B) of twinned Zn0.5Cd0.5S (solid circle) and Zn0.5Cd0.5S/PdP∼0.33S∼1.67 (solid square) photocatalysts in 20% lactic acid (pH = 1.64) (black), 0.75 M ascorbic acid (pH = 2.39) (red), 20% TEOA (pH = 12.59) (green), 0.7 M Na2S and 0.5 M Na2SO3 (pH = 13.86) (blue) sacrificial reagent aqueous solution. | ||
The apparent quantum yields (QEs) of the Zn0.5Cd0.5S/PdP∼0.33S∼1.67 photocatalyst for H2 evolution were measured for a three-hour reaction under the illumination of a monochromatic light at 420 nm (Fig. S10 and Table S5, ESI†). The measured QEs for Zn0.5Cd0.5S/PdP∼0.33S∼1.67 in lactic acid, ascorbic acid, TEOA and Na2S/Na2SO3 sacrificial reagents are 13.26%, 19.70%, 5.31% and 16.52%, respectively, which are much larger than those measured for pristine twinned Zn0.5Cd0.5S solid solution under the same conditions. This result suggests also that Zn0.5Cd0.5S/PdP∼0.33S∼1.67 is a much better catalyst than the twinned Zn0.5Cd0.5S solid solution no matter what kind of sacrificial reagent is used.
The stability of the catalyst was tested in a long-term operation. The results are shown in Fig. 4B. After 50 hours of irradiation, almost constant H2 production rates of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 were measured in different sacrificial reagents. The measured TON values of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 based on PdP∼0.33S∼1.67, with lactic acid, ascorbic acid, TEOA and Na2S/Na2SO3 sacrificial reagents, are 62025, 114557, 25399 and 73418, respectively. The optimal TON (114557) and TOF (2291) for PdP∼0.33S∼1.67 were achieved in a 50 hours reaction under visible light illumination in ascorbic acid aqueous solution (Table S6, ESI†). Contrarily, the pristine twinned Zn0.5Cd0.5S solid solution is much more unstable than Zn0.5Cd0.5S/PdP∼0.33S∼1.67, especially in ascorbic acid, twinned Zn0.5Cd0.5S solid solution lost completely its catalytic activity after only 1.5 hour reaction (Fig. S11, ESI†).
The stabilities of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 in different sacrificial reagents were also evaluated by repeating the experiments under the same conditions and measuring the H2 evolution rate for each circle. The results are shown in Fig. S9B (ESI†) and Table 1. After six cycles of experiments, the H2 evolution rates of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 in different sacrificial reagents show a small decrease in comparison with that of the first cycle. Zn0.5Cd0.5S/PdP∼0.33S∼1.67 presents the best stability in ascorbic acid solution with 95.5% of the H2 evolution rate of the first cycle remaining after six cycles. Zn0.5Cd0.5S/PdP∼0.33S∼1.67 seems to be most unstable in TEOA solution, but there still remain 90.56% of the catalytic activity after six cycles.
| LA | AA | TEOA | Na2S/Na2SO3 | |
|---|---|---|---|---|
| Zn0.5Cd0.5S/PdP∼0.33S∼1.67 | 153.03 | 324.44 | 61.63 | 188.29 |
| Loss (%) | 8.14 | 4.52 | 9.44 | 8.71 |
Moreover, the improved stability of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 in different sacrificial reagents in comparison with that of pristine twinned Zn0.5Cd0.5S was also supported by the TEM examination. After irradiation for six cycles in 0.75 M ascorbic acid aqueous solution, the images of the pristine twinned Zn0.5Cd0.5S showed a shell on the surface of the catalyst (Fig. 5A), which can be attributed to the photocorrosion. The high resolution TEM images showed also distortions on the parallel twinned structures of the Zn0.5Cd0.5S solid solution (Fig. 5B). But the TEM images of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 did not show any changes after irradiation for six cycles compared with that before the photocatalytic reaction (Fig. 5C). Besides, XRD patterns of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 before and after the photocatalytic reaction confirmed the structural stability of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 (Fig. 5D). These results suggested that the stability of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 has been improved significantly because of the deposition of PdP∼0.33S∼1.67.
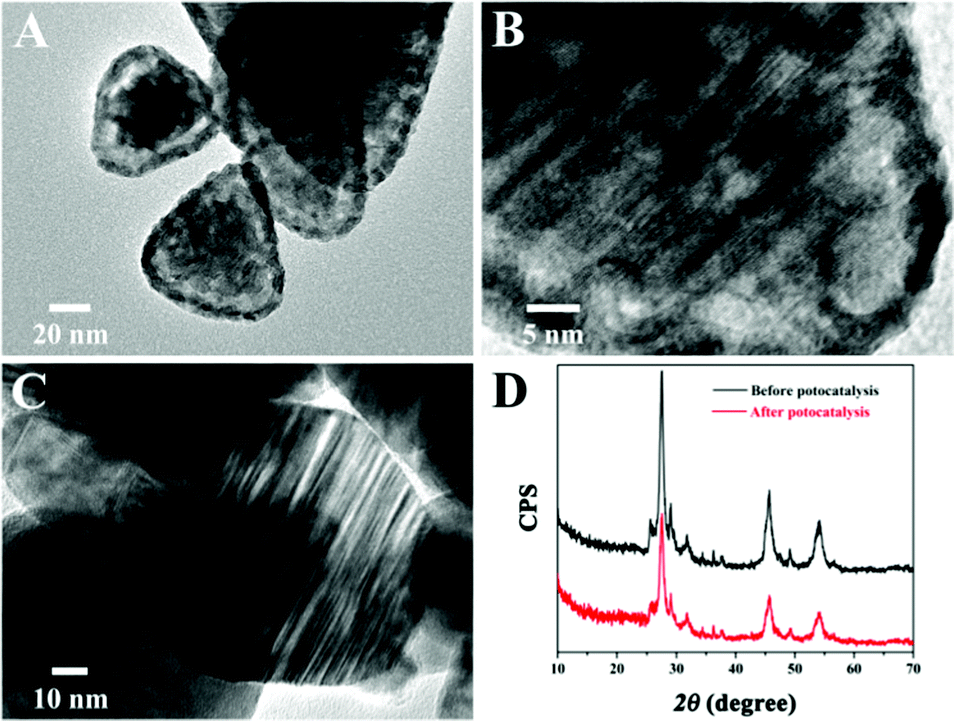 | ||
| Fig. 5 (A and B) TEM images of the pristine Zn0.5Cd0.5S catalyst after six cycles of catalytic reaction in 0.75 M ascorbic acid aqueous solution. (C) TEM image of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 after six cycles of catalytic reactions in 0.75 M ascorbic acid aqueous solution. (D) Comparison between the XRD patterns of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 before and after six cycles of catalytic reactions in 0.75 M ascorbic acid aqueous solution. | ||
In addition, no hydrogen production is detected in the absence of either irradiation or a photocatalyst, suggesting that hydrogen is produced by the photocatalytic reaction. When Pd(PPh3)2Cl2 or its residue after heating at 370 °C for 12 h in Ar flow (30 mL min−1) was used alone as a catalyst in different sacrificial reagents, no hydrogen evolution is detected, suggesting that Pd(PPh3)2Cl2 or its residue after 370 °C calcination is not active for photocatalytic H2 evolution under the experimental conditions.68,69 Moreover, catalytic activity of the pristine twinned Zn0.5Cd0.5S solid solution after heating at 370 °C for 13 h was also tested, but no obvious changes were identified in the H2 production rate. This result suggests that the improved catalytic activity of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 was really caused by the deposition of PdP∼0.33S∼1.67, not by the heating.
All these results as mentioned above indicate that Zn0.5Cd0.5S/PdP∼0.33S∼1.67 is an excellent photocatalyst with remarkable catalytic activity and stability for H2 production under visible light irradiation under both acidic and alkaline conditions. The deposited PdP∼0.33S∼1.67 on the surface of twinned Zn0.5Cd0.5S solid solution has not only promoted the catalytic activity, but also improved significantly the stability of the catalyst under acidic conditions.
3.6 Surface areas and pore-size distributions of twinned Zn0.5Cd0.5S solid solution and Zn0.5Cd0.5S/PdP∼0.33S∼1.67
The textural characteristics of twinned Zn0.5Cd0.5S solid solution and Zn0.5Cd0.5S/PdP∼0.33S∼1.67 have been measured by N2 adsorption/desorption isotherms. As shown in Fig. 6, a well-defined step at P/P0 of 0.8–1.0 was observed on the N2 adsorption/desorption isotherms of these samples, which are typical type IV isotherms. The BET surface areas calculated for the pristine twinned Zn0.5Cd0.5S solid solution and Zn0.5Cd0.5S/PdP∼0.33S∼1.67 are 20.74 m2 g−1 and 16.52 m2 g−1, respectively, whereas the average pore size distributions are all around 233.89 nm (Fig. 6 and Table S7, ESI†). The slightly smaller surface area of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 compared with that of pristine twinned Zn0.5Cd0.5S solid solution may be attributed to the coverage of PdP∼0.33S∼1.67, which suggests that the improved catalytic activity of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 is not caused by the changes in the specific surface area.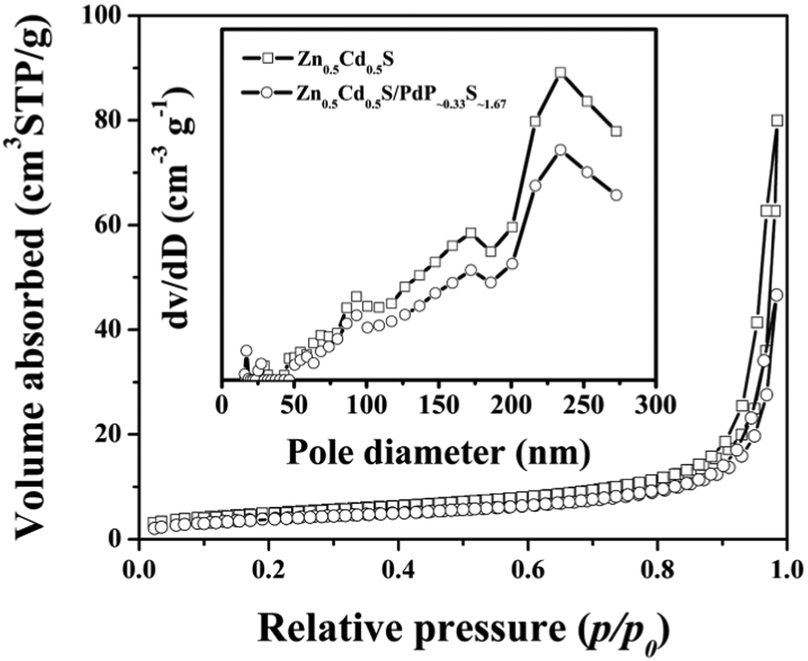 | ||
| Fig. 6 Nitrogen adsorption–desorption isotherms and the corresponding pore size distribution curves (inset) of twinned Zn0.5Cd0.5S (hollow square) and Zn0.5Cd0.5S/PdP∼0.33S∼1.67 (hollow circle). | ||
3.7 The transient photocurrent responses of twinned Zn0.5Cd0.5S solid solution and Zn0.5Cd0.5S/PdP∼0.33S∼1.67
To find out the reasons for the enhancement of the photocatalytic activity of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 over twinned Zn0.5Cd0.5S solid solution in either acidic or alkaline sacrificial reagent solutions, the transient photocurrent responses of twinned Zn0.5Cd0.5S solid solution and Zn0.5Cd0.5S/PdP∼0.33S∼1.67 electrodes towards visible light irradiation are measured. As shown in Fig. 7, the transient photocurrent response of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 in aqueous solution of ascorbic acid and Na2S/Na2SO3 is remarkably larger (8.2 times and 5.1 times) than that of twinned Zn0.5Cd0.5S solid solution. This result indicates that the hole and electron separation in Zn0.5Cd0.5S/PdP∼0.33S∼1.67 is much more efficient than that in twinned Zn0.5Cd0.5S solid solution, which might be responsible for the improvement of the photocatalytic activity according to the previous studies.57,70,71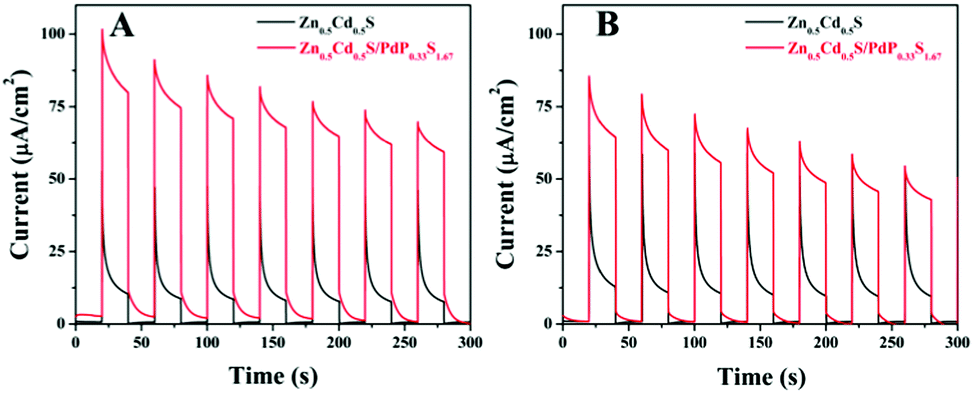 | ||
| Fig. 7 Transient photocurrent responses of twinned Zn0.5Cd0.5S (black) and Zn0.5Cd0.5S/PdP∼0.33S∼1.67 (red) photocatalysts in the 0.75 M ascorbic acid (pH = 2.39) (A) and 0.7 M Na2S and 0.5 M Na2SO3 (pH = 13.86) (B) electrolytes under visible light irradiation. | ||
3.8 The absorption and photoluminescence spectra of twinned Zn0.5Cd0.5S and Zn0.5Cd0.5S/PdP∼0.33S∼1.67
In the photoluminescence spectrum of twinned Zn0.5Cd0.5S, an emission band at 469 nm can be identified, which can be assigned to the band gap emission of twinned Zn0.5Cd0.5S upon excitation at 350 nm (Fig. S12, ESI†).72 The broad emission band in the longer-wavelength region may be attributed to the emission of stacking defect (twin crystal) or surface states.72,73 The emission of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 is extremely weak. The absolute fluorescence quantum yield (Φf) of twinned Zn0.5Cd0.5S is 5.7%, which is much higher than that of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 (∼0%, Table S8, ESI†), indicating the more efficient separation of electron–hole pairs in Zn0.5Cd0.5S/PdP∼0.33S∼1.67 than that in twinned Zn0.5Cd0.5S. This result is in good agreement with the transient photocurrent measurements as mentioned above.The UV-vis diffuse reflection spectra of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 and pristine twinned Zn0.5Cd0.5S solid solution are shown in Fig. 8A. The absorption edge of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 is similar to that of pure Zn0.5Cd0.5S. The absorption of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 in the visible region can be attributed to the absorption of PdP∼0.33S∼1.67.53,74
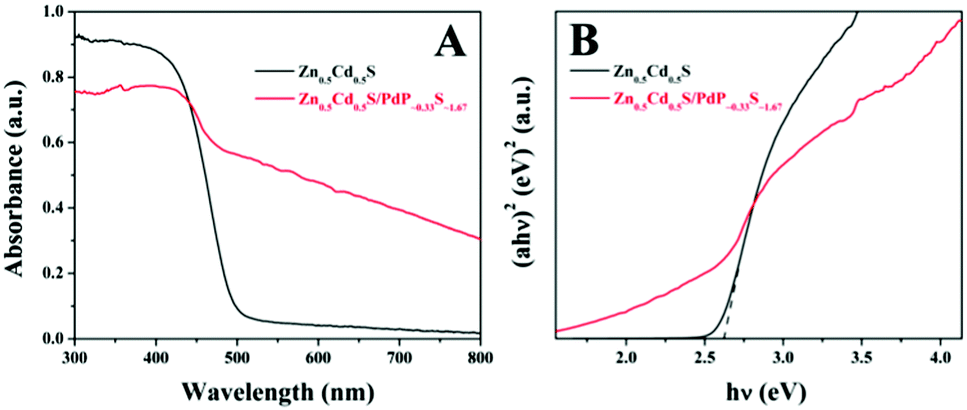 | ||
| Fig. 8 UV-vis diffuse reflection spectra (A) and band-gap calculation (B) of twinned Zn0.5Cd0.5S (black) and Zn0.5Cd0.5S/PdP∼0.33S∼1.67 (red) photocatalysts. | ||
3.9 The photocatalytic H2 production mechanism of Zn0.5Cd0.5S/PdP∼0.33S∼1.67
As mentioned above, three pure compounds, which have a formula of PdPxS2−x, can be formed, they are PdPS, PdP0.63S1.37, and PdP0.33S1.67. Compound PdPS is a semiconductor with a narrow band gap, but the other two compounds are all metallic solids with very good conductivities.53 So the function of PdP∼0.33S∼1.67 in the nanocomposite of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 should be like that of a metal, an electron collector and a site of H+ reduction.33,75 The CB and VB levels can be estimated by following empirical formulas: ECB = χ − 0.5Eg − EC; EVB = χ + 0.5Eg − EC.76 where ECB is the CB edge potential. χ is the electronegativity of the semiconductor, expressed as the geometric mean of the absolute electronegativity of the constituent atoms. The absolute electronegativity of an individual atom is defined as the arithmetic mean of the atomic electron affinity and the first ionization energy. EC is the energy of free electrons on the hydrogen scale, and the value is ∼4.5 eV.23 Following the literature,77,78 the CB and VB of the twinned Zn0.5Cd0.5S are calculated to be −0.34 eV and 2.28 eV, respectively.At the interface between the ZB and WZ segment in the twin crystal Zn0.5Cd0.5S solid solution, the CB and VB energy levels of the ZB segment upshifted a bit with respect to those of the WZ segment, which could simultaneously lead to efficient separation of photogenerated electrons and holes.79 The electrons are then collected by metallic PdP∼0.33S∼1.67, where the reduction of H+ takes place. This quick and efficient removing of the photogenerated electrons from the twinned Zn0.5Cd0.5S solid solution have stabilized the charge separated states and thus promoted the catalytic activity. It is worth noting that the PdP∼0.33S∼1.67 particles deposited not only on the surface of the WZ segment, but also on the surface of the ZB segment. Therefore, it is possible for the PdP∼0.33S∼1.67 particles to collect electrons directly from the ZB segment too, which could also lead to an efficient separation of the photoinduced charges and improvement in the catalytic activity.
4. Conclusions
In summary, we have successfully prepared a novel hybrid catalyst composed of twinned Zn0.5Cd0.5S solid solution and PdP∼0.33S∼1.67 for H2 evolution from water by a template etching method. The tetragonal phase PdP∼0.33S∼1.67 was vectorially deposited on to the surface of the twinned Zn0.5Cd0.5S solid solution and thus heterojunctions formed between the PdP∼0.33S∼1.67 and the twinned Zn0.5Cd0.5S solid solution. The synergistic effects of both homojunctions and heterojunctions have suppressed the charge recombination dramatically and thus the activities of the catalyst are significantly improved. The H2 production rate of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 under visible light illumination (λ > 420 nm) in 0.75 M ascorbic acid (pH = 2.39) aqueous solution is 372.12 μmol h−1 mg−1, which is 67 times larger than that of pristine twinned Zn0.5Cd0.5S solid solution under the same conditions, which is the highest value ever reported so far for sulfide photocatalysts in the presence of acidic sacrificial reagents. In 0.7 M Na2S/0.5 M Na2SO3 (pH = 13.86) solution, the H2 production rate of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 is 246.04 μmol h−1 mg−1, which is 5.8 times larger than that of pristine twinned Zn0.5Cd0.5S solid solution. Based on the fact that the twinned Zn0.5Cd0.5S solid solution is the best pristine sulfide catalyst for hydrogen evolution up to date,42,43 the further improved catalytic activity of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 is significant. By combining homojunctions and heterojunctions with carefully designed band alignment in one catalyst, the charge separation can be improved and thus excellent catalytic activities can be achieved. This strategy might be useful in the design of a new photocatalyst.Acknowledgements
This work was financially supported by the Natural Science Foundation of China (Grant No. 21173136, and 91233108).Notes and references
- H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS PubMed.
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- Y. Shi and B. Zhang, Chem. Soc. Rev., 2016, 45, 1529–1541 RSC.
- H. Kato, H. Kobayashi and A. Kudo, J. Phys. Chem. B, 2002, 106, 12441–12447 CrossRef CAS.
- D. J. Martin, N. Umezawa, X. Chen, J. Ye and J. Tang, Energy Environ. Sci., 2013, 6, 3380–3386 CAS.
- G. Hitoki, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, Chem. Commun., 2002, 1698–1699 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- H. Kato and A. Kudo, J. Phys. Chem. B, 2002, 106, 5029–5034 CrossRef CAS.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082–3089 CrossRef CAS PubMed.
- M. Liu, W. You, Z. Lei, G. Zhou, J. Yang, G. Wu, G. Ma, G. Luan, T. Takata, M. Hara, K. Domen and C. Li, Chem. Commun., 2004, 2192–2193 RSC.
- A. Mukherji, R. Marschall, A. Tanksale, C. Sun, S. C. Smith, G. Q. Lu and L. Wang, Adv. Funct. Mater., 2011, 21, 126–132 CrossRef CAS.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed.
- K. Maeda, T. Takata, M. Hara, N. Saito, Y. Inoue, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2005, 127, 8286–8287 CrossRef CAS PubMed.
- K. Zhang and L. Guo, Catal. Sci. Technol., 2013, 3, 1672–1690 CAS.
- Q. Lu, Y. Yu, Q. Ma, B. Chen and H. Zhang, Adv. Mater., 2016, 28, 1917–1933 CrossRef CAS PubMed.
- J. Liu, M. S. Kelley, W. Wu, A. Banerjee, A. P. Douvalis, J. Wu, Y. Zhang, G. C. Schatz and M. G. Kanatzidis, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5530–5535 CrossRef CAS PubMed.
- Q. Li, H. Meng, J. Yu, W. Xiao, Y. Zheng and J. Wang, Chem. – Eur. J., 2014, 20, 1176–1185 CrossRef CAS PubMed.
- C. Xing, Y. Zhang, W. Yan and L. Guo, Int. J. Hydrogen Energy, 2006, 31, 2018–2024 CrossRef CAS.
- Q. Li, H. Meng, P. Zhou, Y. Zheng, J. Wang, J. Yu and J. Gong, ACS Catal., 2013, 3, 882–889 CrossRef CAS.
- J. A. Villoria, R. M. Navarro Yerga, S. M. Al-Zahrani and J. L. G. Fierro, Ind. Eng. Chem. Res., 2010, 49, 6854–6861 CrossRef CAS.
- X. Wang, G. Liu, Z.-G. Chen, F. Li, G. Q. Lu and H.-M. Cheng, Electrochem. Commun., 2009, 11, 1174–1178 CrossRef CAS.
- L. Wang, W. Wang, M. Shang, W. Yin, S. Sun and L. Zhang, Int. J. Hydrogen Energy, 2010, 35, 19–25 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- F. E. Osterloh, Chem. Mater., 2008, 20, 35–54 CrossRef CAS.
- S. R. Lingampalli, U. K. Gautam and C. N. R. Rao, Energy Environ. Sci., 2013, 6, 3589–3594 CAS.
- X. Zong, H. Yan, G. Wu, G. Ma, F. Wen, L. Wang and C. Li, J. Am. Chem. Soc., 2008, 130, 7176–7177 CrossRef CAS PubMed.
- N. Li, B. Zhou, P. Guo, J. Zhou and D. Jing, Int. J. Hydrogen Energy, 2013, 38, 11268–11277 CrossRef CAS.
- J. Zhang, Q. Xu, S. Z. Qiao and J. Yu, ChemSusChem, 2013, 6, 2009–2015 CrossRef CAS PubMed.
- H. Yan, J. Yang, G. Ma, G. Wu, X. Zong, Z. Lei, J. Shi and C. Li, J. Catal., 2009, 266, 165–168 CrossRef CAS.
- Z. Sun, H. Zheng, J. Li and P. Du, Energy Environ. Sci., 2015, 8, 2668–2676 CAS.
- Z. Sun, Q. Yue, J. Li, J. Xu, H. Zheng and P. Du, J. Mater. Chem. A, 2015, 3, 10243–10247 CAS.
- Z. Sun, H. Chen, Q. Huang and P. Du, Catal. Sci. Technol., 2015, 5, 4964–4967 CAS.
- Q. Yue, Y. Wan, Z. Sun, X. Wu, Y. Yuan and P. Du, J. Mater. Chem. A, 2015, 3, 16941–16947 CAS.
- S. Cao, Y. Chen, C.-C. Hou, X.-J. Lv and W.-F. Fu, J. Mater. Chem. A, 2015, 3, 6096–6101 CAS.
- S. Cao, Y. Chen, C.-J. Wang, X.-J. Lv and W.-F. Fu, Chem. Commun., 2015, 51, 8708–8711 RSC.
- P. Kalisman, Y. Nakibli and L. Amirav, Nano Lett., 2016, 16, 1776–1781 CrossRef CAS PubMed.
- K. Wu, Z. Chen, H. Lv, H. Zhu, C. L. Hill and T. Lian, J. Am. Chem. Soc., 2014, 136, 7708–7716 CrossRef CAS PubMed.
- M. Liu, L. Wang, G. Lu, X. Yao and L. Guo, Energy Environ. Sci., 2011, 4, 1372–1378 CAS.
- M. Liu, D. Jing, Z. Zhou and L. Guo, Nat. Commun., 2013, 4, 2278–2285 Search PubMed.
- A. Litke, J. P. Hofmann, T. Weber and E. J. M. Hensen, Inorg. Chem., 2015, 54, 9491–9498 CrossRef CAS PubMed.
- Y.-Y. Hsu, N.-T. Suen, C.-C. Chang, S.-F. Hung, C.-L. Chen, T.-S. Chan, C.-L. Dong, C.-C. Chan, S.-Y. Chen and H. M. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 22558–22569 CAS.
- R. S. Barbiéri, C. R. Belatto and A. C. Massabni, J. Therm. Anal., 1995, 44, 903–909 CrossRef.
- J. Wang, Q. Yang, Z. Zhang, T. Lia and S. Zhang, Dalton Trans., 2010, 39, 227–233 RSC.
- K. Zhang, Z. Zhou and L. Guo, Int. J. Hydrogen Energy, 2011, 36, 9469–9478 CrossRef CAS.
- J. Zhang, J. Yu, Y. Zhang, Q. Li and J. R. Gong, Nano Lett., 2011, 11, 4774–4779 CrossRef CAS PubMed.
- D.-H. Ha, A. H. Caldwell, M. J. Ward, S. Honrao, K. Mathew, R. Hovden, M. K. A. Koker, D. A. Muller, R. G. Hennig and R. D. Robinson, Nano Lett., 2014, 14, 7090–7099 CrossRef CAS PubMed.
- F. Dawood and R. E. Schaak, J. Am. Chem. Soc., 2009, 131, 424–425 CrossRef CAS PubMed.
- J. Park, H. Zheng, Y. Jun and A. P. Alivisatos, J. Am. Chem. Soc., 2009, 131, 13943–13945 CrossRef CAS PubMed.
- T. A. Bither, P. C. Donohue and H. S. Young, J. Solid State Chem., 1971, 3, 300–307 CrossRef CAS.
- H. Li, M. Zanella, A. Genovese, M. Povia, A. Falqui, C. Giannini and L. Manna, Nano Lett., 2011, 11, 4964–4970 CrossRef CAS PubMed.
- J. Zhang, L. Qi, J. Ran, J. Yu and S. Z. Qiao, Adv. Energy Mater., 2014, 1301925 CrossRef.
- J. Song, Q. Tian, J. Gao, H. Wu, Y. Chen and X. Li, CrystEngComm, 2014, 16, 1277–1286 RSC.
- L. Wang, Z. Yao, F. Jia, B. Chen and Z. Jiang, Dalton Trans., 2013, 42, 9976–9981 RSC.
- S. N. Guo, Y. L. Min, J. C. Fan and Q. J. Xu, ACS Appl. Mater. Interfaces, 2016, 8, 2928–2934 CAS.
- R. Bhatt, S. Bhattacharya, R. Basu, A. Singh, U. Deshpande, C. Surger, S. Basu, D. K. Aswal and S. K. Gupta, Thin Solid Films, 2013, 539, 41–46 CrossRef CAS.
- NIST X-ray photoelectron spectroscopic data, NIST, USA, Web page: http://srdata.nist.gov/xps/.
- S. Sarkar and S. Sampath, Chem. Commun., 2014, 50, 7359–7362 RSC.
- S. Zhao, J. Huang, Q. Huo, X. Zhou and W. Tu, J. Mater. Chem. A, 2016, 4, 193–199 CAS.
- X. Wang, H. Tian, X. Cui, W. Zheng and Y. Liu, Dalton Trans., 2014, 43, 12894–12903 RSC.
- J. Yu, J. Zhang and M. Jaroniec, Green Chem., 2010, 12, 1611–1614 RSC.
- W. Zhang, Y. Wang, Z. Wang, Z. Zhong and R. Xu, Chem. Commun., 2010, 46, 7631–7633 RSC.
- Z. Sun, H. Chen, Q. Huang and P. Du, Catal. Sci. Technol., 2015, 5, 4964–4967 CAS.
- Q. Yue, Y. Wan, Z. Sun, X. Wu, Y. Yuan and P. Du, J. Mater. Chem. A, 2015, 3, 16941–16947 CAS.
- A. A. Lutich, G. Jiang, A. S. Susha, A. L. Rogach, F. D. Stefani and J. Feldmann, Nano Lett., 2009, 9, 2636–2640 CrossRef CAS PubMed.
- S. Chen, D. L. Jacobs, J. K. Xu, Y. X. Li, C. Y. Wang and L. Zang, RSC Adv., 2014, 4, 48486–48491 RSC.
- Z. Yan, X. Yu, A. Han, P. Xu and P. Du, J. Phys. Chem. C, 2014, 118, 22896–22903 CAS.
- S. Cao, Y. Chen, C.-J. Wang, P. He and W.-F. Fu, Chem. Commun., 2014, 50, 10427–10429 RSC.
- H. S. Chen, S. R. Chung, T. Y. Chen and K. W. Wang, J. Mater. Chem. C, 2014, 2, 2664–2667 RSC.
- B. Gong, Y. Lu, P. Wu, Z. Huang, Y. Zhu, Z. Dang, N. Zhu, G. Lu and J. Huang, Appl. Surf. Sci., 2016, 365, 280–290 CrossRef CAS.
- A. Hamidani and B. Bennecer, Comput. Mater. Sci., 2010, 48, 115–123 CrossRef CAS.
- K. Chang, Z. Mei, T. Wang, Q. Kang, Sh. Ouyang and J. Ye, ACS Nano, 2014, 7078–7087 CrossRef CAS PubMed.
- F. Hulliger, J. Phys. Chem. Solids, 1965, 26, 639–645 CrossRef CAS.
- X. Dai, M. Xie, S. Meng, X. Fu and S. Chen, Appl. Catal., B, 2014, 158–159, 382–390 CrossRef CAS.
- J. Zhang and X. Liu, Dalton Trans., 2014, 43, 9296–9302 RSC.
- T. Kida, G. Guan, Y. Minami, T. Ma and A. Yoshida, J. Mater. Chem., 2003, 13, 1186–1191 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The SEM, TEM, HRTEM, SAED, ICP and EDS results of twinned Zn0.5Cd0.5S solid solution; TG and DTA curves of Pd(PPh3)2Cl2; IR spectra of Zn0.5Cd0.5S, Zn0.5Cd0.5S/Pd(PPh3)2Cl2, and Zn0.5Cd0.5S/PdP∼0.33S∼1.67; STEM images of Zn0.5Cd0.5S/PdP∼0.33S∼1.67; X-ray diffraction patterns of twinned Zn0.5Cd0.5S calcined at 372 °C; the overall survey XPS spectrum of twinned Zn0.5Cd0.5S and Zn0.5Cd0.5S/PdP∼0.33S∼1.67 nanocomposites; the rate of H2 evolution of Zn0.5Cd0.5S/PdP∼0.33S∼1.67 (1–6) samples loaded with various amounts of PdP∼0.33S∼1.67 as cocatalysts; the rate of H2 evolution under visible light irradiation after 3 hours using 1 mg and 10 mg photocatalysts for twinned Zn0.5Cd0.5S and Zn0.5Cd0.5S/PdP∼0.33S∼1.67 in different sacrificial reagents; images of twinned Zn0.5Cd0.5S and Zn0.5Cd0.5S/PdP∼0.33S∼1.67 nanocomposites before and after the photocatalytic H2 evolution test; photoluminescence spectra, apparent quantum yields (QEs) of twinned Zn0.5Cd0.5S and Zn0.5Cd0.5S/PdP∼0.33S∼1.67; the turnover number (TON) and turnover frequency (TOF) of twinned Zn0.5Cd0.5S and Zn0.5Cd0.5S/PdP∼0.33S∼1.67 in different sacrificial reagents. See DOI: 10.1039/c6ee02414a |
| This journal is © The Royal Society of Chemistry 2017 |