
H2S-tolerant oxygen-permeable ceramic membranes for hydrogen separation with a performance comparable to those of palladium-based membranes†
Wenping
Li
ab,
Zhongwei
Cao
a,
Lili
Cai
ab,
Lixiao
Zhang
ab,
Xuefeng
Zhu
 *a and
Weishen
Yang
*a
*a and
Weishen
Yang
*a
aState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China. E-mail: zhuxf@dicp.ac.cn; yangws@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100049, China
First published on 14th November 2016
Abstract
A new method for hydrogen separation to acquire high-purity hydrogen using an oxygen-permeable ceramic membrane is proposed and verified in this work. A high hydrogen separation rate of up to 16.3 mL cm−2 min−1 was achieved on an asymmetric dual-phase membrane at 900 °C. No performance degradation was observed in a long-term operation with the feed gas containing 200 ppm H2S.
Broader contextHydrogen, as a high-quality, clean and sustainable energy carrier, has been extensively studied over the last two decades. The techniques related to hydrogen separation and purification are significantly important for the application of hydrogen in various fields. In this study, we propose a new method for hydrogen separation to acquire high-purity hydrogen using an oxygen-permeable ceramic membrane. A high hydrogen separation rate of up to 16.3 mL cm−2 min−1 was achieved at 900 °C with a separation factor up to >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. This separation rate is 2–3 orders of magnitude higher than those of proton conducting membranes and is comparable to those of Pd based metallic membranes. No performance degradation was observed during a long-term operation with a feeding gas containing 200 ppm H2S. This type of membrane can be used to produce high-purity or ultra-high-purity hydrogen for fuel cells, semiconductor manufacturing, photovoltaic cell production, etc. The experiments presented in this study indicate that this new method has a bright future for hydrogen separation to acquire high-purity hydrogen in view of its high selectivity, high hydrogen separation rate and high stability under a H2S-containing atmosphere. 000. This separation rate is 2–3 orders of magnitude higher than those of proton conducting membranes and is comparable to those of Pd based metallic membranes. No performance degradation was observed during a long-term operation with a feeding gas containing 200 ppm H2S. This type of membrane can be used to produce high-purity or ultra-high-purity hydrogen for fuel cells, semiconductor manufacturing, photovoltaic cell production, etc. The experiments presented in this study indicate that this new method has a bright future for hydrogen separation to acquire high-purity hydrogen in view of its high selectivity, high hydrogen separation rate and high stability under a H2S-containing atmosphere.
|
Hydrogen, as a clean and sustainable energy carrier, is a key element of the energy matrix, and the related aspects, such as production,1–6 separation,7 storage and utilization,8,9 have been extensively studied over the last two decades. Hydrogen is not only an ideal fuel for fuel cells,10,11 but is also widely used in many important industries, such as semiconductor manufacturing, photovoltaic cell production, chemical industries and national defence industries.9,12,13 Fossil fuels or other carbon-based fuels are likely to be the main sources for hydrogen production in the short to medium term,9 because, currently, the cost of H2 generated from renewable energy sources (biomass, solar energy, etc.) is too high to be accepted by markets.14 However, hydrogen produced via steam reforming of fossil fuels or other carbon-based fuels contains many impurities, such as CH4, CO, CO2 and traces of H2S.15–17 The purity of the acquired hydrogen is too low to be directly used for many industrial processes. The CO concentration in hydrogen gas needs to be decreased to <10 ppm, as it is strongly poisonous to the Pt anode of fuel cells.18,19 In addition, ultra-high-purity hydrogen, >6 N, is needed for the production of polycrystalline silicon.20 Therefore, the separation process is significantly important for hydrogen applications in various fields.
Compared to other methods for hydrogen separation, methods based on inorganic dense hydrogen-permeable membranes are regarded to be some of the most promising methods for the production of high-purity hydrogen and ultra-high-purity hydrogen in industry due to their high separation factors (>1000).21–23 There are two prerequisites for the industrial application of these inorganic dense hydrogen-permeable membranes, i.e. both high hydrogen permeation flux and high stability under the operation conditions are required, especially in the presence of H2S.
Pd and Pd alloy membranes with excellent hydrogen permeability have been extensively studied.21,24–29 Although Pd alloy membranes have been commercialized for hydrogen separation, the problem of instability under an atmosphere containing H2S is still not well resolved.30 Besides, the scarce resources limit large scale applications of Pd and Pd alloy membranes. The dense ceramic hydrogen-permeable membrane (also known as proton conducting membrane) materials consist of earth abundant elements; however, these ceramic membranes exhibit low hydrogen permeability and are easily poisoned by H2S, even at the level of tens of ppm.15,31–34
Here, we propose a new method for hydrogen separation to acquire high-purity hydrogen using an oxygen-permeable ceramic membrane with a high hydrogen separation rate comparable to those of Pd and Pd alloy membranes and excellent stability under a H2S-containing atmosphere. The oxygen-permeable ceramic membrane is a mixed ionic–electronic conductor, in which the oxygen ions can directionally diffuse from one side to the other side under the driving force of the chemical potential gradient of oxygen.35–37 To separate hydrogen, low-purity hydrogen is fed into side I, as shown in Fig. 1, and steam is fed into side II. In side II, water combines with electrons and splits into hydrogen and oxygen ions at an elevated temperature, then oxygen ions diffuse to side I and react with low-purity hydrogen to produce water and electrons. Electrons go back to side II, which creates electrical neutrality in the membrane. Based on the permeation principle of the oxygen-permeable ceramic membrane, the ideal separation selectivity for hydrogen against other gases, such as CH4, CO, CO2, H2S etc., should be 100%, because only oxygen ions can permeate through the membrane. Therefore, high-purity hydrogen is acquired after condensation and drying of the outlet gas of side II. The amount of hydrogen produced on side II is equivalent to that consumed on side I. As a whole, no net chemical reaction occurs in the above-mentioned process in the oxygen-permeable ceramic membrane reactor, but a mass exchange is achieved between the two sides. Thus, it is reasonable to regard this process as a hydrogen separation process with the help of steam.
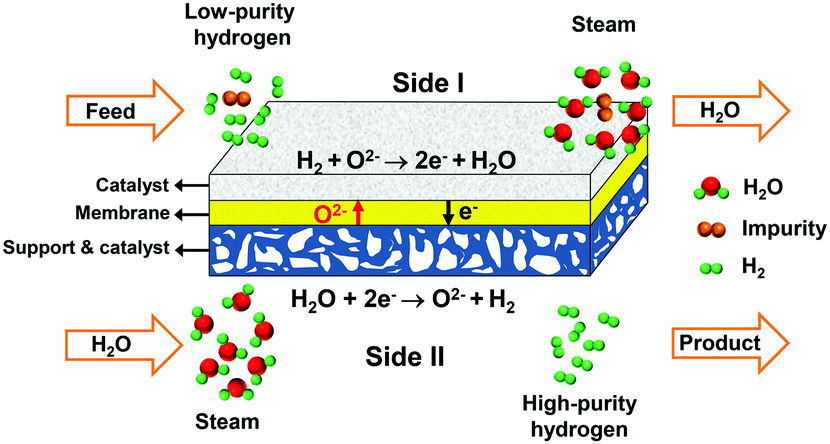 | ||
| Fig. 1 The concept of hydrogen separation using an oxygen-permeable ceramic membrane. | ||
From the view point of hydrogen separation, using an oxygen-permeable ceramic membrane reactor to efficiently upgrade hydrogen purity is of great significance due to its high selectivity, high hydrogen separation rate and exceptional stability. To the best of our knowledge, there is no report that explicitly proposes the concept of hydrogen separation to acquire high-purity hydrogen using oxygen-permeable ceramic membranes.
In this study, we chose a dual-phase membrane of 75 wt% Sm0.15Ce0.85O1.925–25 wt% Sm0.6Sr0.4Al0.3Fe0.7O3−δ (SDC–SSAF) to illustrate the hydrogen separation concept, because this membrane has a high oxygen permeation flux and excellent stability under CO2 and a strong reducing atmosphere.38–40 An asymmetric SDC–SSAF membrane was prepared for the hydrogen separation experiment. A procedure involving slurry preparation, tape-casting, lamination and co-sintering was used to fabricate the asymmetric dual-phase membrane. The membrane has a 40 μm-thick dense layer as the separation layer and a 600 μm-thick porous layer as the support layer with a porosity of approximately 40%, as shown in Fig. 2a–c. To speed up the reactions in side I and II, the asymmetric SDC–SSAF membranes were coated with a 1 wt% Ru/Sm0.15Ce0.85O1.925 (1 wt% Ru/SDC) catalyst on side I (dense layer surface) and infiltrated with a Ru-based catalyst in side II (porous support layer) (Fig. 2c and d and ESI,† Fig. S1a). Before being sealed in a membrane reactor using a silver ring (ESI,† Fig. S2), the asymmetric SDC–SSAF membranes were subjected to nitrogen permeation experiments under a pressure gradient of 1 atm to make sure that they were gas-tight.
 | ||
| Fig. 2 SEM images of asymmetric SDC–SSAF membranes. (a) Dense layer surface of the fresh membrane. (b) Cross-view of the fresh membrane. The inset image shows the cross-view of the porous support layer at high resolution. (c and d) Cross-views of the membrane infiltrated with a catalyst and its porous support layer, respectively. (e and f) Cross-views of the spent SDC–SSAF membrane operated under an atmosphere without H2S and its porous support layer, respectively. (g and h) Cross-views of the spent SDC–SSAF membrane operated under a H2S-containing atmosphere and its porous support layer, respectively. The thickness of the dense layer of the membrane is ∼40 μm. | ||
Assuming bulk diffusion to be the rate determining step in hydrogen separation, from Wagner equation (eqn (1)) and the hydrogen separation principle (eqn (2)),41 the equation for hydrogen separation rates can be derived as shown in eqn (3). The hydrogen separation rate was greatly affected by temperature and the oxygen partial pressure gradient across the oxygen-permeable ceramic membrane.
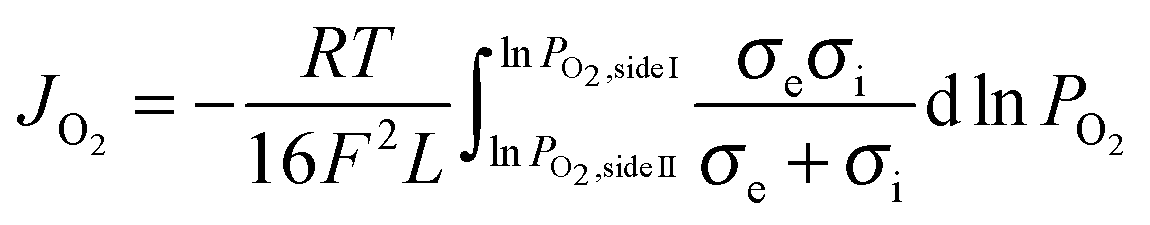 | (1) |
| FH2,sep = 2JO2 | (2) |
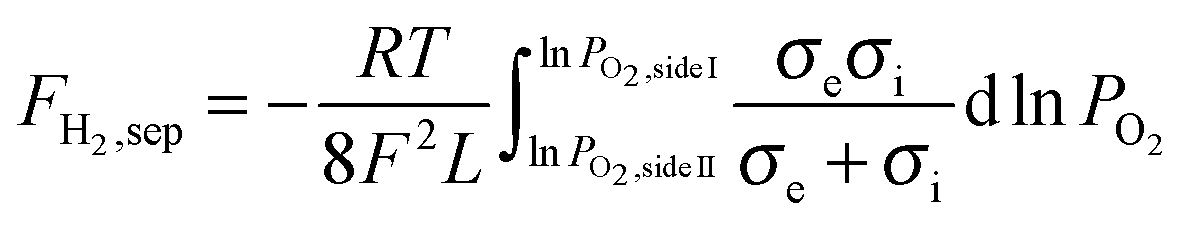 | (3) |
I and PO2,sideII are the gas constant, Faraday constant, temperature, membrane thickness, electronic conductivity, ionic conductivity, and oxygen partial pressures of side I and side II, respectively.
Here, the factors, i.e. H2 concentration and the flow rate of the H2/N2 mixed gas (H2/N2 = 1:1) on side I as well as the flow rate of the steam/He mixed gas (90% H2O with He balance) on side II, have a direct influence on the oxygen partial pressure gradient across the asymmetric SDC–SSAF membrane. So these factors were extensively studied to explore their effects on the hydrogen separation rate. As shown in Fig. 3a and b, the H2 concentration and the flow rate of the H2/N2 mixed gas (H2/N2 = 1:1) on side I have an obvious influence on the hydrogen separation rate. When the hydrogen concentration on side I is 10%, the corresponding hydrogen separation rate is 5.6 mL cm−2 min−1, but it is tripled (i.e. 16.3 mL cm−2 min−1) with 100% hydrogen fed into side I. The hydrogen separation rate increases from 6.3 to 15.3 mL cm−2 min−1 as the flow rate of the H2/N2 mixed gas increases from 20 to 200 mL min−1. However, the flow rate of the steam/He mixed gas on side II has a weaker influence on the hydrogen separation rate compared to the above-mentioned two factors. For example, as the flow rate of the steam/He mixed gas on side II increases from 30 to 400 mL min−1, the hydrogen separation rate slightly increases from 9.0 to 13.4 mL cm−2 min−1. In the above experiments, the oxygen partial pressure gradient across the membrane is in the range of 102–104 and its variation trend remains in step with that of the hydrogen separation rate. This result indicates that a high oxygen partial pressure gradient across the membrane results in a high hydrogen separation rate. In theory, the separation factor should be infinite due to the 100% permeation selectivity towards oxygen for the oxygen-permeable ceramic membrane. However, in practical operations, perfect sealing is unachievable and thus leakage is unavoidable. As a result, in the process of varying the gas flow rates, the separation factors, as shown in the ESI,† Tables S1 and S2, are in the order of 103–104. Nevertheless, the separation factors achieved in this study are comparable to those of Pd-based membranes reported in the literature, and can still be considered as high values in the area of separation. Therefore, it is feasible to use an oxygen-permeable ceramic membrane to upgrade low-purity hydrogen to high-purity hydrogen.
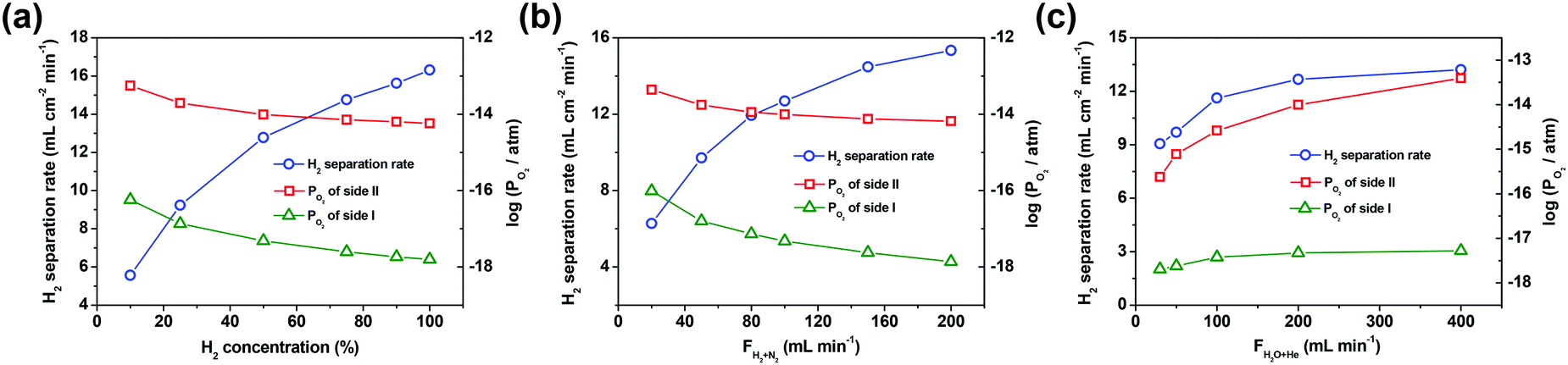 | ||
| Fig. 3 The effect of H2 concentration (a) and the flow rate of H2/N2 mixed gas (H2/N2 = 1:1) (b) on side I, and the flow rate of the steam/He mixed gas (90% H2O with He balance) (c) on side II on the hydrogen separation rate and the corresponding PO2 for both sides of the SDC–SSAF membrane at 900 °C. For (a) and (c), the gas flow rate fed into side I was 100 mL min−1 (N2 as the balance gas); for (c), H2/N2 = 1:1. For (a) and (b), the gas flow rate fed into side II was 200 mL min−1 (90% H2O with He balance). | ||
Temperature is another factor that has a great influence on the hydrogen separation rate, due to the fact that several steps involved in the hydrogen separation process, such as water splitting, hydrogen oxidation and oxygen permeation across the oxygen-permeable ceramic membrane, are all thermally activated. Thus, the influences of temperature on the hydrogen separation rate and the separation factor were studied in a wide temperature range of 600–950 °C. The hydrogen separation rate enhances one order of magnitude when the temperature increases from 600 to 950 °C, as shown in Fig. 4(a). It should be pointed out that even at a temperature as low as 600 °C, the hydrogen separation rate still reaches 1.7 mL cm−2 min−1, which is higher than the hydrogen separation rate of all proton conducting membranes acquired at 900 °C. Although the separation factor decreases as the temperature decreases (ESI,† Table S3), it still reaches about 2000 at 600 °C, indicating that the asymmetric SDC–SSAF membrane reactor has an excellent hydrogen separation performance even at a low temperature. Although the driving force, i.e. the oxygen partial pressure gradient across the membrane, decreases with the increase of temperature, the membrane still provides a higher hydrogen separation rate at a higher temperature. The reason for this is that all the steps involved in the hydrogen separation process heavily depend on temperature. Therefore, in this membrane reactor, the hydrogen separation rate is so strongly accelerated by temperature that the influence of the oxygen partial pressure gradient on the hydrogen separation rate is masked by the action of temperature. The activation engery of the asymmetric SDC–SSAF membrane for hydrogen separation is 56.3 ± 0.8 kJ mol−1 (Fig. 4b), which is slightly lower than that of the asymmetric SDC–SSAF membrane for oxygen permeation (70.2 ± 1.7 kJ mol−1),38 but is similar to that of the symmetric SDC–SSAF membrane (53.8 ± 1 kJ mol−1) with both sides coated with catalysts for oxygen exchange reactions.40 Besides, when varying the H2 concentration and the flow rate of the H2/N2 mixed gas (H2/N2 = 1:1) on side I, as well as the flow rate of the steam/He mixed gas on side II, FH2,sep and log(PO2,sideI/PO2,sideII) have a good linear relationship (ESI,† Fig. S3). These results indicate that the hydrogen separation process is dominated by the bulk diffusion of oxygen ions and electrons; in other words, the water splitting and hydrogen oxidation steps on the membrane surfaces are fast enough and are thus not the rate determining steps.
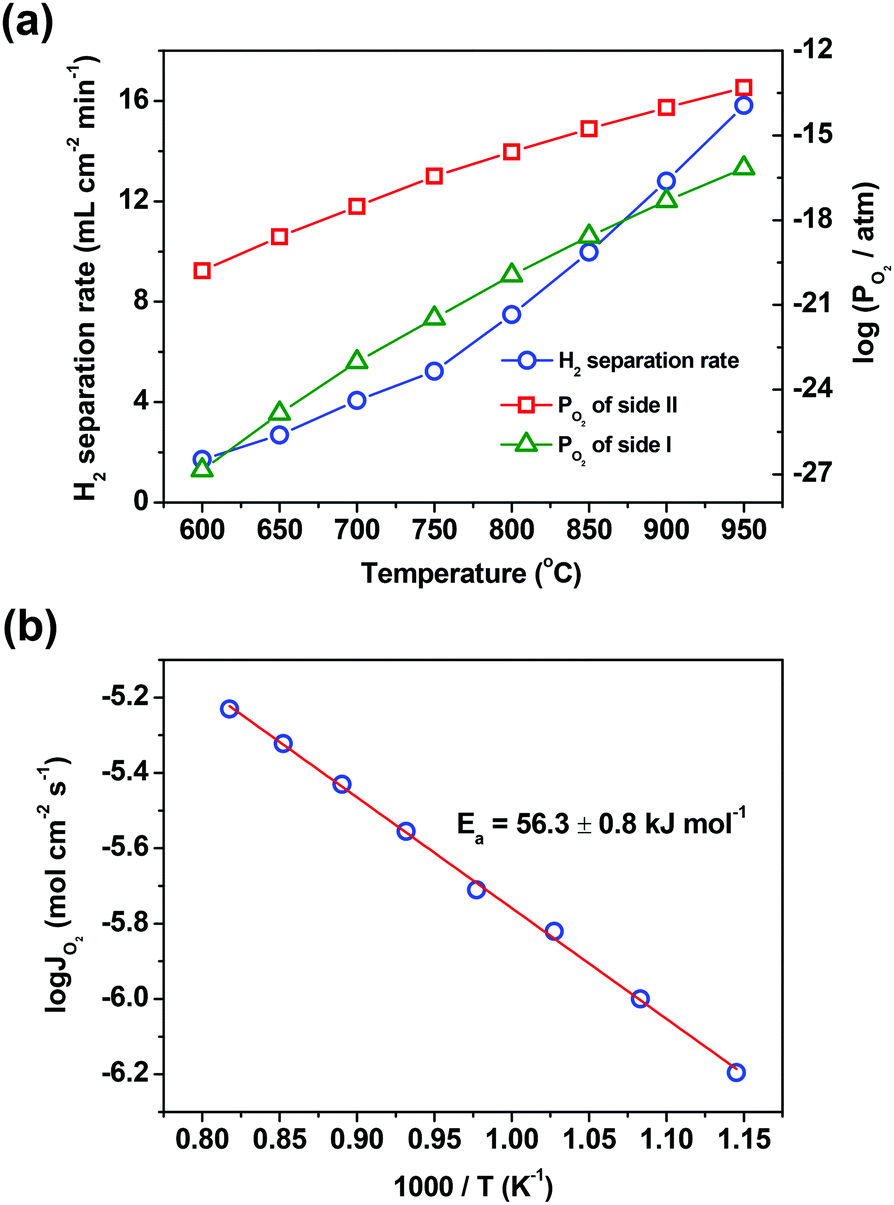 | ||
| Fig. 4 (a) The relationship between temperature and H2 separation rate and the corresponding PO2 for both sides of the asymmetrical SDC–SSAF membrane. (b) The Arrhenius plots of oxygen permeation fluxes. The gas flow rates are 100 mL min−1 (H2/N2 = 1:1) for side I and 200 mL min−1 (90% H2O with He balance) for side II. | ||
Except for the hydrogen separation rate, the stability of the membrane under the operation conditions must be taken into consideration, since it is the most important prerequisite for practical applications. At 900 °C, the long-term stability of the asymmetric SDC–SSAF membrane for hydrogen separation was tested with 50 mL min−1 H2 + 50 mL min−1 N2 fed into side I and 180 mL min−1 H2O + 20 mL min−1 He fed into side II for 100 h after 200 h of operation at different hydrogen partial pressures, flow rates and temperatures. The hydrogen separation rate remained at 12.6 mL cm−2 min−1 (Fig. 5a) and the SEM characterization of the membrane morphology showed no obvious change after the 300 h test (Fig. 2e and f and ESI,† Fig. S1b). In a practical application, low-purity hydrogen ready for purification usually contains H2S, which is poisonous for Pd and Pd alloy membranes and proton conducting membranes. Therefore, the performance of the asymmetric SDC–SSAF membrane reactor for hydrogen separation was evaluated under an atmosphere with different H2S concentrations (Fig. 5b). Once 20 ppm H2S was introduced into the membrane reactor, the hydrogen separation rate suddenly decreased from 12.8 to 10.8 mL cm−2 min−1, then gradually recovered to its original value in the following steps with enhancing H2S concentration. The asymmetric SDC–SSAF membrane reactor was also exposed to an environment with 200 ppm H2S to evaluate its long-term stability under an atmosphere with a higher H2S concentration (Fig. 5c). There was no obvious change in the hydrogen separation rate in the 100 h experiment. Compared to the membrane operated without H2S, the microstructures of the membrane and catalyst were not influenced by the presence of 200 ppm of H2S gas (Fig. 2g and h and ESI,† Fig. S1c). The EDS analysis reveals that no sulphur element was enriched on the surface of side I (ESI,† Fig. S4). This result indicates that the asymmetric SDC–SSAF membrane is resistant to the poisoning of high concentrations of H2S.
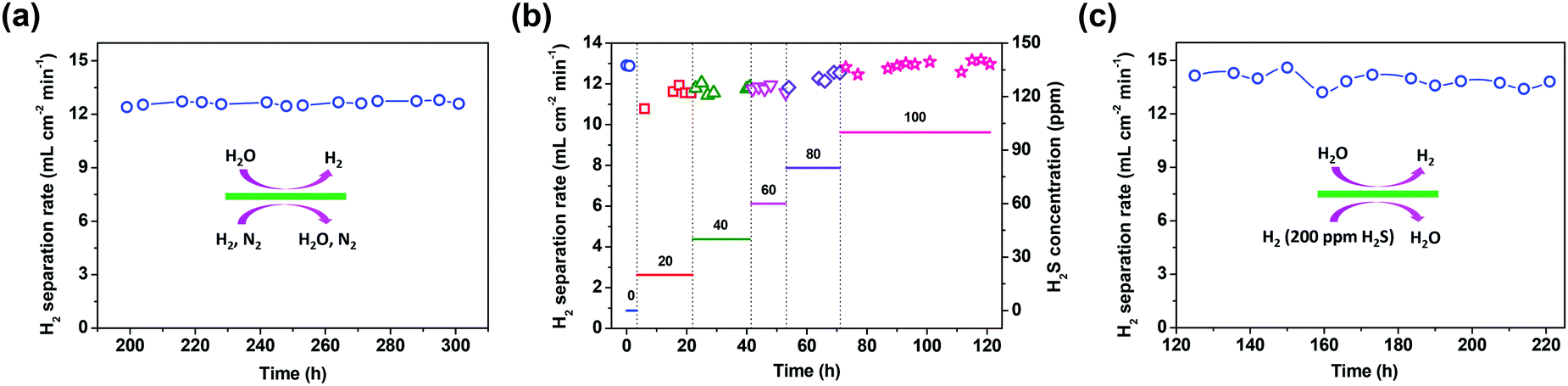 | ||
| Fig. 5 Long-term operation of the membrane reactor for hydrogen separation at 900 °C. (a) A stable operation of the asymmetric SDC–SSAF membrane for hydrogen separation. (b) Effect of H2S concentration of membrane side I on the hydrogen separation rate. (c) A stable operation of the asymmetric SDC–SSAF membrane for hydrogen separation with 200 ppm H2S on side I. For (a), (b) and (c), the gas flow rate fed into side II was 200 mL min−1 (90% H2O with He balance). Side I: for (a), 50 mL min−1 H2 + 50 mL min−1 N2; for (b), 50 mL min−1 H2 + 50 mL min−1 N2 + ppm level H2S; and for (c), 50 mL min−1 H2 + 200 ppm H2S. | ||
Usually, when exposed to a H2S-containing atmosphere, for Pd and Pd alloy membranes, the formation of palladium sulphide (Pd4S) on the membrane surface prohibits hydrogen permeation through the membrane;42 for proton conducting membranes, H2S can deteriorate the performance of the proton conducting membrane through the adsorption of sulfur on the membrane surface as well as reacting with membrane materials to form sulphur-containing compounds.17 During the hydrogen separation process in the asymmetric SDC–SSAF membrane reactor, steam is produced from the reaction between oxygen ions and hydrogen in side I. The existence of steam is beneficial to the oxidation of H2S. Besides, the oxidation of part of the H2S can effectively reduce the concentration of H2S.42–44 In this study, the concentration of steam on side I of the asymmetric SDC–SSAF membrane (about 13 vol%) is so high that the 200 ppm H2S gas has no obvious influence on the hydrogen separation rate.
Hydrogen separation via an oxygen-permeable ceramic membrane is an extremely promising method for upgrading hydrogen purity with a high hydrogen separation rate and excellent stability under a H2S-containing atmosphere. Table S4 (ESI†) compares the hydrogen separation rates of several inorganic dense membranes, including Pd and Pd alloy membranes, proton conducting ceramic membranes and the oxygen-permeable ceramic membrane of this study. As shown in Table S4 (ESI†), the hydrogen separation rate of the asymmetric SDC–SSAF membrane is one to two orders of magnitude higher than those of the proton conducting ceramic membranes and is close to those of the Pd and Pd alloy membranes.
Conclusions
In this work, we have proposed and verified a new method for hydrogen separation to acquire high-purity hydrogen using an oxygen-permeable ceramic membrane. This method has a high hydrogen separation rate comparable to those of Pd and Pd alloy membranes. In addition, the oxygen-permeable ceramic membrane is stable under a H2S-containing atmosphere without showing performance degradation.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (21476225, U1508203 and 91545202), the Youth Innovation Promotion Association of the Chinese Academy of Sciences, the Strategic Priority Research Program of the Chinese Academy of Sciences (grant no. XDB17020400) and the Dalian Institute of Chemical Physics (DICP DMTO201503).Notes and references
- F. Wei, F. Steinbach, Z. W. Cao, X. F. Zhu and A. Feldhoff, Angew. Chem., Int. Ed., 2016, 55, 8648–8651 CrossRef PubMed.
- L. C. Mu, Y. Zhao, A. L. Li, S. Y. Wang, Z. L. Wang, J. X. Yang, Y. Wang, T. F. Liu, R. T. Chen, J. Zhu, F. T. Fan, R. G. Li and C. Li, Energy Environ. Sci., 2016, 9, 2463–2469 CAS.
- X. L. Dong, Z. K. Liu, W. Q. Jin and N. P. Xu, J. Power Sources, 2008, 185, 1340–1347 CrossRef CAS.
- A. Thursfield, A. Murugan, R. Franca and I. S. Metcalfe, Energy Environ. Sci., 2012, 5, 7421–7459 CAS.
- W. P. Li, X. F. Zhu, Z. W. Cao, W. P. Wang and W. S. Yang, Int. J. Hydrogen Energy, 2015, 40, 3452–3461 CrossRef CAS.
- E. Rebollo, C. Mortalò, S. Escolástico, S. Boldrini, S. Barison, J. M. Serra and M. Fabrizio, Energy Environ. Sci., 2015, 8, 3675–3686 CAS.
- C. Yacou, S. Smart and J. C. Diniz da Costa, Energy Environ. Sci., 2012, 5, 5820–5832 CAS.
- S. Dutta, J. Ind. Eng. Chem., 2014, 20, 1148–1156 CrossRef CAS.
- K. Mazloomi and C. Gomes, Renewable Sustainable Energy Rev., 2012, 16, 3024–3033 CrossRef CAS.
- X. M. Zhang, L. Liu, Z. Zhao, B. F. Tu, D. R. Ou, D. A. Cui, X. M. Wei, X. B. Chen and M. J. Cheng, Nano Lett., 2015, 15, 1703–1709 CrossRef CAS PubMed.
- C. C. Duan, J. H. Tong, M. Shang, S. Nicodemski, M. Sanders, S. Ricote, A. almansoori and R. O'Hayre, Science, 2015, 349, 1321–1326 CrossRef CAS PubMed.
- M. Momirlan and T. N. Veziroglu, Renewable Sustainable Energy Rev., 2002, 6, 141–179 CrossRef CAS.
- R. Ramachanran and R. K. Menon, Int. J. Hydrogen Energy, 1998, 23, 593–598 CrossRef.
- S. S. Penner, Energy, 2006, 31, 33–43 CrossRef CAS.
- Z. T. Tao, L. T. Yan, J. L. Qiao, B. L. Wang, L. Zhang and J. J. Zhang, Prog. Mater. Sci., 2015, 74, 1–50 CrossRef CAS.
- N. A. Al-Mufachi, N. V. Rees and R. Steinberger-Wilkens, Renewable Sustainable Energy Rev., 2015, 47, 540–551 CrossRef CAS.
- S. M. Fang, L. Bi, X. S. Wu, H. Y. Gao, C. S. Chen and W. Liu, J. Power Sources, 2008, 183, 126–132 CrossRef CAS.
- X. Cheng, Z. Shi, N. Glass, L. Zhang, J. J. Zhang, D. T. Song, Z. S. Liu, H. J. Wang and J. Shen, J. Power Sources, 2007, 165, 739–756 CrossRef CAS.
- G. L. Zhou, Y. Jiang, H. M. Xie and F. L. Qiu, Chem. Eng. J., 2005, 109, 141–145 CrossRef CAS.
- J. Miyazaki, T. Kajiyama, K. Matsumoto, H. Fujiwarat and M. Yatabe, Int. J. Hydrogen Energy, 1996, 21, 335–341 CrossRef CAS.
- M. E. Ayturk, N. K. Kazantzis and Y. H. Ma, Energy Environ. Sci., 2009, 2, 430–438 CAS.
- S. Adhikari and S. Fernando, Ind. Eng. Chem. Res., 2006, 45, 875–881 CrossRef CAS.
- X. Y. Tan and K. Li, Inorganic membrane reactors: fundamentals and applications, Wiley, West Sussex, 2015 Search PubMed.
- J. R. Young, Rev. Sci. Instrum., 1963, 34, 891–892 CrossRef CAS.
- W. P. Wang, X. L. Pan, X. L. Zhang, W. S. Yang and G. X. Xiong, Sep. Purif. Technol., 2007, 54, 262–271 CrossRef CAS.
- H. Li, A. Caravella and H. Y. Xu, J. Mater. Chem. A, 2016, 4, 14069–14094 CAS.
- K. Zhang, H. Y. Gao, Z. B. Rui, P. Liu, Y. D. Li and Y. S. Lin, Ind. Eng. Chem. Res., 2009, 48, 1880–1886 CrossRef CAS.
- S. Yun and S. Ted Oyama, J. Membr. Sci., 2011, 375, 28–45 CrossRef CAS.
- H. Lu, L. L. Zhu, W. P. Wang, W. S. Yang and J. H. Tong, Int. J. Hydrogen Energy, 2015, 40, 3548–3556 CrossRef CAS.
- M. Kajiwara, S. Uemiya and T. Kojima, Int. J. Hydrogen Energy, 1999, 24, 839–844 CrossRef CAS.
- J. Song, B. Meng, X. Y. Tan and S. M. Liu, Int. J. Hydrogen Energy, 2015, 40, 6118–6127 CrossRef CAS.
- H. Yoon, S.-J. Song, T. Oh, J. L. Li, K. L. Duncan and E. D. Wachsman, J. Am. Ceram. Soc., 2009, 92, 1849–1852 CrossRef CAS.
- Y. Y. Wei, J. Xue, H. H. Wang and J. Caro, J. Membr. Sci., 2015, 488, 173–181 CrossRef CAS.
- Z. W. Zhu, W. P. Sun, L. T. Yan, W. F. Liu and W. Liu, Int. J. Hydrogen Energy, 2011, 36, 6337–6342 CrossRef CAS.
- X. F. Zhu and W. S. Yang, AIChE J., 2008, 54, 665–672 CrossRef CAS.
- Z. P. Shao, W. S. Yang, Y. Cong, H. Dong, J. H. Tong and G. X. Xiong, J. Membr. Sci., 2000, 172, 177–188 CrossRef CAS.
- K. Zhang, Z. P. Shao, C. Z. Li and S. M. Liu, Energy Environ. Sci., 2012, 5, 5257–5264 CAS.
- Z. W. Cao, X. F. Zhu, W. P. Li, B. Xu, L. N. Yang and W. S. Yang, Mater. Lett., 2015, 147, 88–91 CrossRef CAS.
- X. F. Zhu, Q. M. Li, Y. Cong and W. S. Yang, Catal. Commun., 2008, 10, 309–312 CrossRef CAS.
- X. F. Zhu, Y. Liu, Y. Cong and W. S. Yang, Solid State Ionics, 2013, 253, 57–63 CrossRef CAS.
- C. Wagner, Prog. Solid State Chem., 1975, 10, 3–16 CrossRef.
- S.-Y. Jeon, M.-B. Choi, B. Singh and S.-J. Song, J. Membr. Sci., 2013, 428, 46–51 CrossRef CAS.
- L. Yang, S. Z. Wang, K. Blinn, M. F. Liu, Z. Liu, Z. Cheng and M. L. Liu, Science, 2009, 326, 126–129 CrossRef CAS PubMed.
- J.-H. Wang and M. L. Liu, J. Power Sources, 2008, 176, 23–30 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ee02967a |
| This journal is © The Royal Society of Chemistry 2017 |