
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photovoltaic mixed-cation lead mixed-halide perovskites: links between crystallinity, photo-stability and electronic properties†
Waqaas
Rehman
,
David P.
McMeekin
,
Jay B.
Patel
,
Rebecca L.
Milot
,
Michael B.
Johnston
 ,
Henry J.
Snaith
and
Laura M.
Herz
*
,
Henry J.
Snaith
and
Laura M.
Herz
*
Department of Physics, University of Oxford, Clarendon Laboratory, Parks Road, Oxford OX1 3PU, UK. E-mail: laura.herz@physics.ox.ac.uk
First published on 15th December 2016
Abstract
Lead mixed halide perovskites are highly promising semiconductors for both multi-junction photovoltaic and light emitting applications due to their tunable band gaps, with emission and absorption energies spanning the UV-visible to near IR regions. However, many such perovskites exhibit unwanted halide segregation under photo-illumination, the cause of which is still unclear. In our study, we establish crucial links between crystal phase stability, photostability and optoelectronic properties of the mixed-cation lead mixed-halide perovskite CsyFA(1−y)Pb(BrxI(1−x))3. We demonstrate a region for caesium content between 0.10 < y < 0.30 which features high crystalline quality, long charge-carrier lifetimes and high charge-carrier mobilities. Importantly, we show that for such high-quality perovskites, photo-induced halide segregation is strongly suppressed, suggesting that high crystalline quality is a prerequisite for good optoelectronic quality and band gap stability. We propose that regions of short-range crystalline order aid halide segregation, possibly by releasing lattice strain between iodide rich and bromide rich domains. For an optimized caesium content, we explore the orthogonal halide-variation parameter space for Cs0.17FA0.83Pb(BrxI(1−x))3 perovskites. We demonstrate excellent charge-carrier mobilities (11–40 cm2 V−1 s−1) and diffusion lengths (0.8–4.4 μm) under solar conditions across the full iodide–bromide tuning range. Therefore, the addition of caesium yields a more photo-stable perovskite system whose absorption onsets can be tuned for bandgap-optimized tandem solar cells.
Broader contextHybrid metal halide perovskites have emerged as an important new class of materials for photovoltaics. Their integration in tandem with other photovoltaic technologies such as silicon cells is highly desirable, allowing low-cost cells with efficiencies in excess of the Shockley–Queisser single-junction limit. Optimized photocurrent matching between top and bottom cells requires careful control over band-gap energy of the perovskite, for which tunable mixed iodide–bromide lead perovskites are the most prominent contenders. However, photo-stability across the full halide mixture has proven elusive for single-cation methylammonium or formamidinium perovskites, with the material segregating into iodide-rich and bromide-rich regions under illumination. Here we examine the highly tunable, mixed-cation, mixed-halide perovskite CsyFA(1−y)Pb(BrxI(1−x))3 to identify regions of structural instability and their prominent causes. Crucially, we show that materials in regions of enhanced crystallinity are also more stable against photo-induced halide segregation. Our findings demonstrate that crystal interfaces in the perovskite play a determining role in nucleating such halide segregation. We demonstrate that once the material contains a suitable fraction of caesium, excellent charge-carrier mobilities and diffusion lengths can be obtained across the full iodide–bromide tuning range. Careful control over crystallinity and alloying can therefore yield photo-stable perovskites with band gaps optimized for tandem solar cells. |
Introduction
In recent years, single junction perovskite solar cells (PSC) have attracted considerable attention, with power conversion efficiencies (PCE) increasing substantially from 3.8% in 20091 to the current world record of 22.1%.2–5 In addition, tandem architectures such as perovskite-on-perovskite and perovskite-on-silicon have been reported with efficiencies ranging between 19–23%6–9 and stipulated potential for reaching 35%,10 far in excess of the Shockley–Queisser limit for conventional single junction solar cells.11 Such extraordinary performance has been attributed to excellent optoelectronic and material-specific properties of hybrid perovskites which make them well suited for photovoltaics. Hybrid metal halide perovskites have been shown to feature high optical absorption in the visible region,12,13 sharp band edges, low trap densities14 and desirable charge transport properties such as high charge-carrier diffusion lengths coupled with long charge-carrier lifetimes.15–20 In addition, these materials allow for a range of versatile processing techniques including spin coating,21–23 2-step interdiffusion,24 chemical vapour deposition,25 spray pyrolysis,26 atomic layer deposition,27 dip coating,28 and thermal evaporation,29,30 making perovskites a top contender for low-cost solar energy conversion. The variability of the perovskite ABX3 crystal structure accommodates a relatively wide material space to be explored. Efficient perovskite photovoltaics have incorporated methylammonium (MA+, CH3NH3+), formamidinium (FA+, CH3(NH2)2+) or cesium (Cs+) as the A cation.13,28,31 The most popular divalent B metal cations32 are typically Pb2+ or Sn2+, and the final component is a halide anion X = (Cl−; Br−; I−).15,33 By intermixing these components the bandgap of perovskites can be tuned widely, which allows attractive coloration and most importantly, bandgap engineering for tandem solar cell design.Despite the success story of perovskite solar cells in the research lab, arguably the most pressing issue to address before commercialisation is long-term stability. Here, the challenge is threefold, since instabilities may arise from changes in crystal structure, chemical decomposition, or de-mixing (such as halide segregation). For instance, the most widely studied MAPbI3 undergoes a structural phase transition above 55 °C,34 and degrades quickly upon exposure to moisture, air and heat above 85 °C.35 Alternatively, FAPbI3 offers improved thermal stability and a reduced band gap closer to the optimum value for single junction solar cells.11 However, FAPbI3 is prone to a structural instability at room temperature, transforming from the photo-active “black” perovskite α-phase into a photo-inactive “yellow” non-perovskite hexagonal δ-phase (δH-phase) aided by the presence of solvents and humidity.12,34,36 Alternatively, all-inorganic perovskite solar cells employing caesium (Cs+) as the A cation have been explored.31,37 Because of the smaller ionic radius of the Cs+ cation38 – 1.81 Å compared to 2.70 Å for MA+ and 2.79 Å for FA+ – the bandgap of CsPbI3 widens to 1.73 eV, within the optimum range of 1.7 to 1.8 eV for a top cell material in tandem with a crystalline Si bottom solar cell.39 However, the photo-active black α-phase of CsPbI3 only crystallizes above 300 °C while at lower temperatures the material reverts back to a photo-inactive yellow orthorhombic δ-phase (δO-phase).37,40 Nonetheless, mixed halide CsPb(BrxI(1−x))3 materials show significantly better resistance under heat stress41 and enhanced photo-stability42 compared to their MA+ and FA+-based counterparts.
Such differing causes of instability triggered an intense research strand to create stable alloys, that is, to use A-cation intermixing to create hybrid perovskites devoid of thermal or structural instabilities.43–46 For example, initial attempts to substitute small amounts of FA+ with MA+ in FAPbI3 were shown to stabilize the perovskite phase and yield higher PCEs, resulting from the stronger interaction between the more polar MA+ and the PbI6 octahedral cage.47 However, mixed-cation FA+/MA+ perovskites still retained a propensity towards decomposition and degradation under heat stress and humid environments,36,48 which is probably caused by the relatively volatile nature of methylammonium.35 A promising recent avenue has been the partial substitution of Cs+ for FA+, MA+ or MA+/FA+ for which substantial enhancements of the PCE and photo- and moisture stability were found by a range of groups.44–46 Li et al. argued that the effective ionic radius of the Cs+/FA+ mixture can be used to fine-tune the Goldschmidt tolerance factor towards structurally more stable regions.49 Saliba et al. demonstrated triple-cation perovskite solar cells with stabilized efficiencies to above 21%, suggesting that the addition of Cs+ to MA+/FA+ mixed cation perovskites further eliminates remnant yellow phase impurities.43
Such attempts to stabilise the crystal structure of hybrid perovskites through cation intermixing could potentially also have the beneficial side effects of preventing halide segregation. Mixed-halide iodide–bromide perovskites are an important material class that can allow a 1.75 eV bandgap crucial for integration with silicon in tandem cells.6 However, both MA+ and FA+ versions of lead mixed-halide perovskites have been found to be unstable under illumination, with the material segregating into iodide-rich and bromide-rich regions.15,50 Recently, McMeekin et al. demonstrated a mixed-cation Cs0.17FA0.83Pb(Br0.4I0.6)3 perovskite with 1.74 eV bandgap that allowed for 17% PCE in a single-junction device and exhibited low disorder, long charge-carrier lifetimes and high mobilities.6 These mixed-cation mixed-halide perovskites could be crystallised at much higher temperatures than previously reported hybrid perovskites, which lead to materials with high levels of crystallinity, as judged by X-ray diffraction measurements. Importantly, this material appeared not to suffer from halide-segregation, which had previously marred prospects for wide-band gap mixed halide perovskites. This finding, along with observations by Zhou et al.,51 suggests that enhanced crystallinity may open a pathway towards suppressing halide segregation and creating a perovskite absorber material which can sustain a wide band gap and high open-circuit voltage.
Here we examine such links by exploring a wide parameter space in the mixed-cation, mixed-halide perovskite CsyFA(1−y)Pb(BrxI(1−x))3. We identify boundaries and transitions between different alloying regions with structural instability and identify their prominent causes. Our investigation highlights a strong link between crystallinity and optoelectronic properties such as trap-related recombination and charge-carrier mobility. Crucially, we show that materials in regions of enhanced crystallinity are also more stable against photo-induced halide segregation. Our findings demonstrate that crystal interfaces in the perovskite play a determining role in nucleating such halide segregation. Careful control over grain boundaries may therefore be sufficient to eliminate such effects, giving rise to photo-stable materials.
Results & discussion
We commence our study by first characterizing the structural stability of the mixed-cation series CsyFA(1−y)Pb(Br0.4I0.6)3 with Cs+ content y varying between 0 and 1. Fig. 1 displays sample X-ray diffraction (XRD) patterns for four CsyFA(1−y)Pb(Br0.4 I0.6)3 films with different Cs+ fractions y = 0.01, 0.05, 0.20, 0.60; further spectra covering the full range of y are shown in the ESI† (Fig. S1 & S2). The (100) and (200) reflections off the CsyFA(1−y)Pb(Br0.4I0.6)3 perovskite are clearly visible near 2θ ∼ 14–15° and 28–30°. However, additional diffraction peaks can also be observed, in particular for low and high caesium content. We identify these peaks as belonging to the yellow hexagonal δH-phase, which features for the FA+ rich samples, and to the orthorhombic δO-phase which contributes at the Cs+ rich end of the series. For ease of identification, Fig. S1 in the ESI† presents XRD patterns of tri-iodide and mixed-halide FAPbX3 and CsPbX3 films that have converted into the yellow δ-phase. Increasing the Cs+ content from 0 to 5% (Fig. 1b) leads to a marked reduction in such inclusions while at 20% Cs+ content (Fig. 1c) they are mostly, but not entirely, suppressed, and the XRD spectrum exhibits strong, sharp perovskite α-phase peaks. By examining in suitably small steps the full Cs+ concentration range we find that the (100) reflection has the highest intensity for Cs0.2FA0.8Pb(Br0.4I0.6)3 (y = 0.2), indicating the highly crystalline nature at this particular caesium fraction (Fig. S3, ESI†). Beyond y = 0.2, the perovskite diffraction amplitude declines, and reflections assigned to the yellow δO-phase, associated with Cs+ rich films, appear. At y = 0.60, the crystal structure is still relatively strongly oriented and ordered, however, the δO phase emerges and adversely affects the crystallinity of the material (Fig. 1d). This structural instability can be understood in terms of the Goldschmidt tolerance factor for such compounds. While the FAPbI3 perovskite α-phase exists at the upper end of the tolerance range due to the large ionic radius of FA, the CsPbI3 perovskite α-phase forms at the lower end; hence both compounds have a tendency to deteriorate into competing δ-phases.49 Mixing Cs+ and FA+ yields structurally stable compounds because the effective ionic A cation radius lies closer to that of MA+ and therefore is more amenable to the perovskite structure promoting higher crystallinity. Yi et al.45 theoretically calculated that the mixed Cs+/FA+ cation perovskites should be structurally stable over the entire phase composition. However, our experimental results here suggest that good structural stability is only achieved within the range of y = 0.1 to 0.3.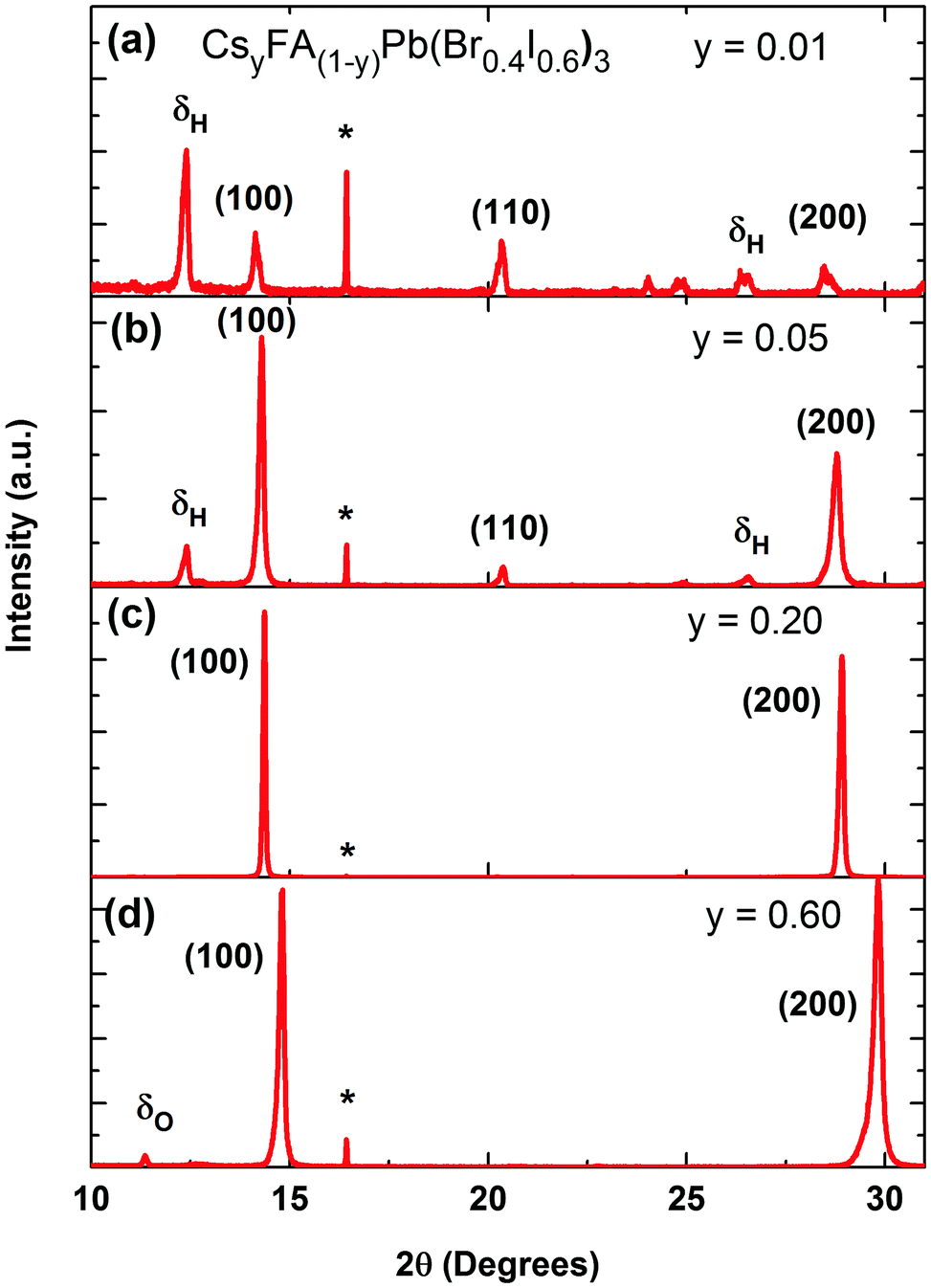 | ||
| Fig. 1 X-ray diffraction (XRD) pattern of four different CsyFA(1−y)Pb(Br0.4I0.6)3 perovskite films with Cs+ content varying as (a) y = 0.01, (b) y = 0.05, (c) y = 0.20 and (d) y = 0.60. δH and δO denote diffraction peaks from the hexagonal δ-phase of FA-rich CsyFA(1−y)Pb(Br0.4I0.6)3 and the orthorhombic δ-phase of Cs-rich CsyFA(1−y)Pb(Br0.4I0.6)3, while (*) marks diffraction patterns related to the z-cut quartz substrates. | ||
An additional factor influencing mixed Cs+/FA+ perovskites is the internal distortions that may arise when ions of such differing ionic radii are distributed across the A cation sites. We discover clear indications for such distortion, as for example in the slight splitting of the perovskite reflection peak visible at ∼29.8° for 80% Cs+ inclusion (see Fig. S2, ESI†).6,38 In addition, we find that the pseudo-cubic lattice parameter derived from the peak shift of the (100) XRD reflection changes in a non-linear manner with caesium content. Fig. 2a displays the observed (100) peak shift from ∼14.2° to 14.9°, and the associated (Bragg's law) lattice parameter reduction from 6.25 to 5.96 Å over the entire compositional range under study. The reduction of the pseudo-cubic cell volume with increasing incorporation of the smaller Cs+ cation results from a stronger interaction between the A cation and X halides.46 In addition, it may increase the cationic charge of Pb2+ ions, thus changing the bonding nature between Pb2+ and halides in the 3D perovskite.52 Interestingly, the (100) diffraction peak shifts with Cs+ content in a distinctly nonlinear manner. The associated lattice parameter (Fig. 2a, inset) first decreases relatively gently towards 30%, but then drops abruptly beyond. This behaviour is markedly different from MA+-to-FA+ organic cation and I−-to-Br− halide substitutions6,53 for which linear shifts in lattice parameter (Vegard's law) are generally observed. We attribute the non-linear shift observed here to the substantial mismatch between the ionic radii of Cs+ and FA+ (0.98 Å), which is an order of magnitude larger than that between MA+ and FA+ (0.09 Å) and approximately five times larger than the difference between Br− and I− (0.21 Å).38,54 As a result, the lattice may become extremely distorted in particular for the central regions of relatively even Cs+ and FA+ contributions, leading to rapid changes in lattice parameter and potentially a change in space group. This observation is corroborated by photoluminescence (PL) and absorption measurements (Fig. 2b), which display a similar trend, namely abruptly shifting PL peaks (red dots) and absorption onsets (black dots) between 30–60% Cs+ content. Across the full compositional range, the PL peak energy EPL tunes broadly from 1.72 eV to 2.05 eV (Fig. 2b). The absorption onsets EAbs are only slightly blue-shifted (by ∼10–40 meV) which suggests that the PL mainly arises from band-edge emission rather than from trap states, or impurity regions rich in iodide.
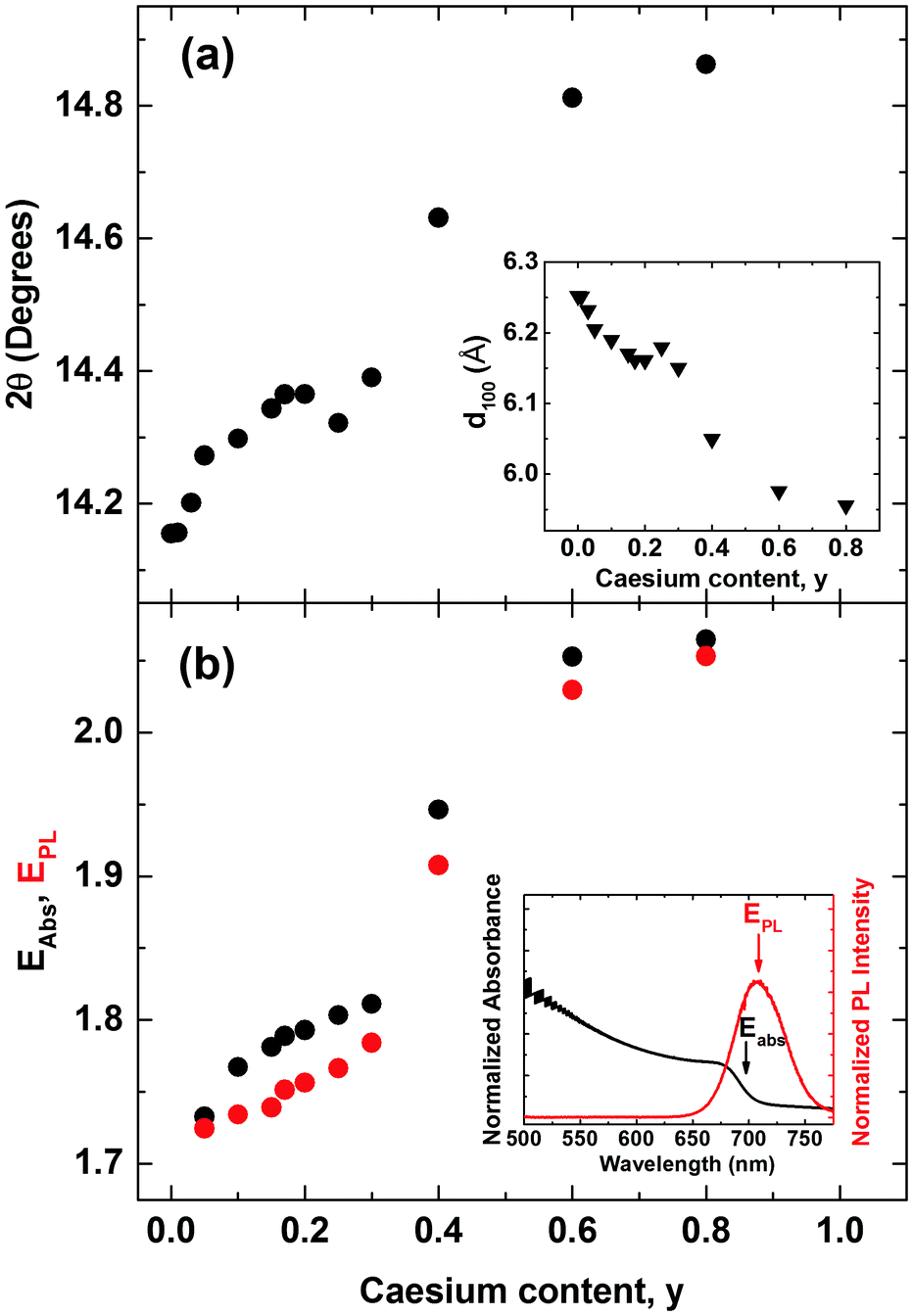 | ||
| Fig. 2 (a) Shift of the cubic (100) reflection peak in CsyFA(1−y)Pb(Br0.4I0.6)3 perovskite with varying Cs+ content (0 < y < 0.8) taken from XRD spectra in Fig. 1. Inset: Value of the (100) crystallographic plane separation with varying Cs+ content derived from the scattering angle (Bragg's law) (b) absorbance onset values EAbs (black dots), determined from the maximum of the differential of the absorbance spectra near the absorption edge taken with Fourier transform infrared spectroscopy (shown in Fig. S5, ESI†). PL peak positions EPL (red dots) were taken from in situ steady-state PL spectra (shown in Fig. S4, ESI†) following excitation at 400 nm with an intensity of 14.9 mW cm−2 (fluence of 13.5 μJ cm−2, pulse duration 40 fs, repetition rate 1.1 kHz). | ||
Overall, our investigations demonstrate that the structurally most stable compositions in the CsyFA(1−y)Pb(Br0.4I0.6)3 series are found in the region for Cs+ content of 0.1 < y < 0.3, since within this range we observe little evidence for Cs+ of FA+ yellow phase inclusions.
We illustrate the influence of Cs+/FA+ composition upon the crystallinity for the CsyFA(1−y)Pb(Br0.4I0.6)3 perovskite, in Fig. 3a, where we display the FWHM of the (100) cubic perovskite peaks as a function of Cs+ content. According to the Scherrer equation, the broadening of peak widths from XRD patterns are inversely proportional to the mean crystallite size in the films and/or the microstrain.55–57 We find that on adding Cs+ to FAPb(Br0.4I0.6)3, the FWHM decreases by 62% until a global minimum is reached at y = 0.2. At the same time, we also observe a significant increase in the scattering peak intensity with the addition of caesium, up to x = 0.2, with a subsequent, consistent reduction in scattering intensity with further increases in caesium content (Fig. S3, ESI†). Both of these observations, i.e. the narrower peak widths and significant increase in scattering intensity, show that the addition of a small fraction of caesium results in an increased fraction of crystalline material and/or larger crystalline domains. The narrower peak widths may also indicate reduced microstrain, another indicator of high crystalline quality.
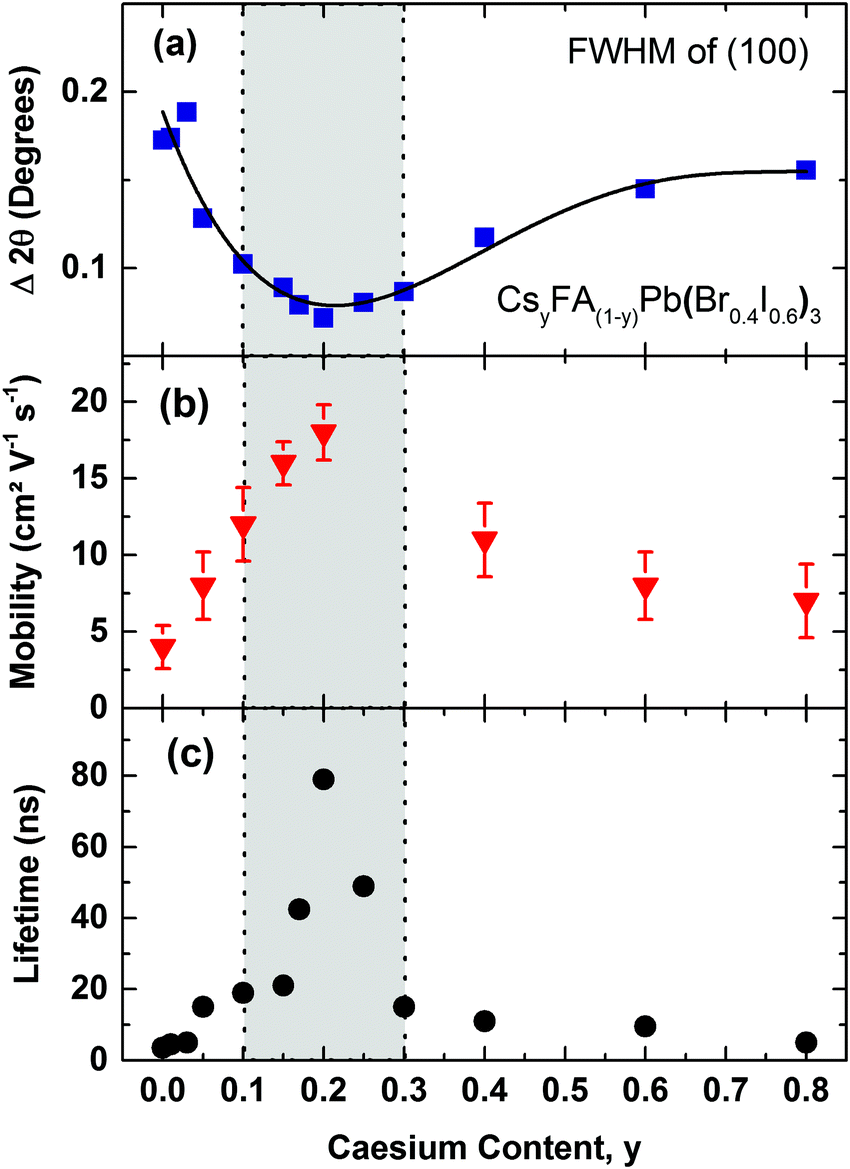 | ||
| Fig. 3 (a) Full width at half maximum (FWHM) of the (100) XRD peak in CsyFA(1−y)Pb(Br0.4I0.6)3 perovskites, with varying Cs+ content (0.0 < y < 0.8) taken from the respective XRD spectra (Fig. S2, ESI†). The solid line is a guide to the eye. (b) Effective charge-carrier mobilities of CsyFA(1−y)Pb(Br0.4I0.6)3 with varying Cs+ content y. Error bars indicate the range of values obtained. (c) Charge-carrier lifetimes of CsyFA(1−y)Pb(Br0.4I0.6)3 films with varying Cs+ content obtained from TCSPC measurements at low excitation fluence (Fig. S6, ESI†). The films were excited at a wavelength of 400 nm. The grey shaded area bound by dotted vertical lines marks the region of relative Cs+ content with high crystallinity and stability. | ||
To elucidate the link between crystalline quality and optoelectronic properties, we proceed by determining charge-carrier mobilities and trap-related recombination rates in CsyFA(1−y)Pb(Br0.4I0.6)3 films. Fig. 3b presents effective charge-carrier mobilities measured using optical-pump, THz-probe (OPTP) spectroscopy (see ESI† for details). Under the reasonable assumption that excitonic effects are weak at room temperature16,17 and hence the free-charge conversion is close to unity, these values represent charge-carrier mobilities at THz frequencies. We find excellent correlation between charge-carrier mobility and crystallinity (Fig. 3a & b). Adding Cs+ to the poorly crystalline FAPb(Br0.4I0.6)3 film results in a steep rise in charge-carrier mobility up to a relative Cs+ content of 0.2 and is then followed by a plunge to much lower values in the 0.3 < y < 0.8 region. Consequently, the material with the sharpest XRD diffraction peaks, Cs0.2FA0.8Pb(Br0.4I0.6)3 (y = 0.2), also exhibits the highest charge-carrier mobility of (18 ± 2) cm2 V−1 s−1, consistent with our previous report for a similar composition,6 and signifying a striking improvement compared to the earlier reported15 highly disordered FA+ cation, mixed-halide FAPb(Br0.4I0.6)3 perovskite (1 cm2 V−1 s−1).
Another factor that may be strongly influenced by crystal grain size is trap-mediated recombination, considering that a number of studies suggest increased trap density at grain boundaries.58,59 According to DFT studies, such traps could comprise elemental defects such as lead vacancies (electron traps) or interstitial organic cation FA+ (hole traps).60 In addition, grain boundaries may reduce the bandgap and increase electron–phonon coupling, thus accelerating the non-radiative charge recombination.61 Crystal defects, such as dislocations and terraces may also lead to microstrain in the perovskite and be responsible for the formation of trap sites.57Fig. 3c displays time-resolved PL lifetimes derived from time-correlated single photon counting (TCSPC) (traces shown in Fig. S6, ESI†) under low-intensity pulsed excitation for which decay dynamics mostly reflect non-radiative trap-assisted recombination. PL lifetimes again inversely correlate with the XRD peak width, displaying a peak value for y = 0.20 (79 ns), with strongly diminishing numbers for compositions in the range of y < 0.1 (4–15 ns) and y > 0.30 (5–11 ns) (Fig. 3c). For CsyFA(1−y)Pb(Br0.4I0.6)3 we hence identify the region of 0.1 < y < 0.3 Cs+ content as yielding the most stable crystal structure, highest crystalline quality and optimized optoelectronic properties for photovoltaics.
The crucial question to be answered for these mixed-halide perovskites is whether such systems can be stabilized against photo-induced halide segregation, which has been observed in a number of FA+ and MA+ based lead mixed halide perovskites.15,42,50 The light-induced mechanisms driving the segregation of such perovskites into iodide-rich and bromide-rich domains are still poorly understood. We therefore use the now well-characterized CsyFA(1−y)Pb(Br0.4I0.6)3 system to investigate the photo-stability of the material against halide segregation and to explore the mechanisms driving this effect. In Fig. 4 we present photo-induced changes to the PL emission wavelength as a function of time under excitation, for CsyFA(1−y)Pb(Br0.4I0.6)3 films, using Cs+ concentration levels from three distinct regions of high FA+ (y = 0.05) and high Cs+ (y = 0.60) materials, and for the most crystalline material (y = 0.20). Over the course of 30 minutes the films are excited with light at 400 nm with an intensity of 100 mW cm−2 (equiv. 1 Sun), and the average energy of the emitted photons 〈Eph〉 is extracted from the measured PL spectra (provided in Fig. S7, ESI†). We find that 〈Eph〉 shows only a marginal change for the y = 0.20 film, while conversely, on the compositional edges, both the low Cs+y = 0.05 and high Cs+y = 0.60 films exhibit a decline in their average photon energy 〈Eph〉 in accordance with halide segregation, which results in charge-carrier funnelling into low-band-gap iodide-rich domains.
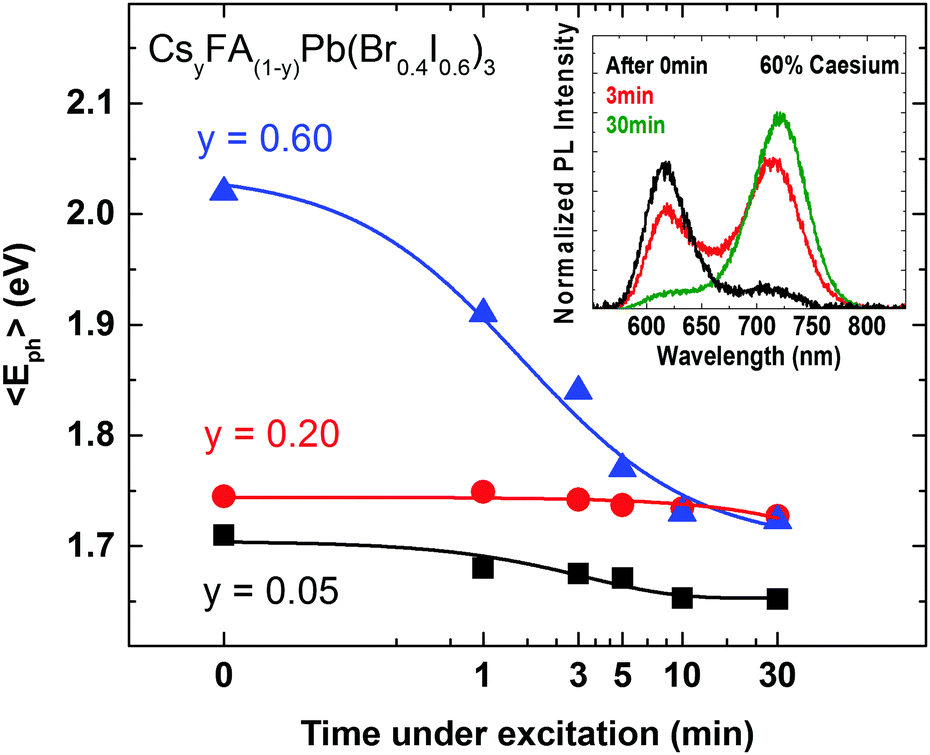 | ||
| Fig. 4 Change of the average PL photon energy 〈Eph〉 for emission from three different CsyFA(1−y)Pb(Br0.4I0.6)3 films with y = 0.05, y = 0.20 and y = 0.60. The films were exposed to continuous illumination over the course of 30 minutes and excited at 400 nm with an intensity of 100 mW cm−2 (fluence of 0.1 μJ cm−2, pulse duration 100 fs, repetition rate 80 MHz). Blue, red and black solid lines are guides to the eye. Inset: Spectral changes in emission from a Cs0.6FA0.4Pb(Br0.4I0.6)3 film over 30 minutes of continuous illumination. | ||
These observations demonstrate a strong correlation between material crystalline quality and stability towards photo-induced halide segregation. We note that Hu et al. similarly found that films of MAPbBr0.8I2.2 displayed strongly enhanced photo-stability when processed on a non-wetting hole transporting layer that yielded larger crystallites.62 Our results hence demonstrate that the coveted scenario of stable mixed-halide perovskites with 1.7 to 1.8 eV band gap can be achieved provided the crystalline quality of the material is sufficiently high.
These findings also shed light on the fundamental mechanisms underlying the photo-induced halide segregation in mixed iodide–bromide perovskites. Recent theoretical work has suggested that the MAPb(BrxI(1−x))3 system suffers from a general miscibility gap in the central region (0.3 < x < 0.6) because of the mismatch between the two different halide anion radii.63 However, photo-induced halide segregation in MAPb(BrxI(1−x))3 has been found to reverse spontaneously over the time scale of a few minutes after the illumination has been removed. Hence such experimental studies do not point towards a miscibility gap being primarily responsible for halide segregation, but rather towards a photo-induced, reversible migration of ions. Our findings demonstrate that such photo-induced ion migration is aided in materials of low crystalline quality. There are two plausible explanations for why such correlation may occur. The first is that high material crystallinity and photo-stability against halide segregation simply derive from the same effect: that is, a perovskite crystal structure that lies well within the Goldschmidt tolerance factor provides high barriers against deterioration into both alternative crystal phases and halide segregation. However, we consider this scenario unlikely as it cannot explain why one given mixed-halide perovskite can acquire higher photo-stability against halide segregation when it is processed into films of higher crystallinity.62 We therefore propose the second alternative as the more likely scenario, which is that regions of short-range crystalline order aid the photo-induced segregation of mixed bromide–iodide perovskites. Such effects could for example derive from the large mismatch in lattice constant between iodide-rich and bromide-rich perovskites (see Fig. 2a). Halide segregation within a single-crystal mixed-halide structure would cause sizeable lattice strain at the interfaces between iodide-rich and bromide-rich domains and may therefore not be energetically feasible. Near a grain boundary, or at the boundary between regions of short-range crystalline order, such lattice strain could be more easily accommodated without a penalty, facilitating halide segregation in materials of low crystallinity. An alternative possibility is that halide segregation is nucleated at already existing defect sites or amorphous regions which are present at grain surfaces in high density.58,59 Hence control over grain surfaces and crystallinity appear to be key to photo-stable mixed halide perovskites.
Having established that highly crystalline CsyFA(1−y)Pb(Br0.4I0.6)3 perovskites are feasible in the region of 20% Cs+ inclusion, we now explore the effect of varying halide composition. Mixed lead halide perovskites APb(BrxI(1−x))3 offer excellent band gap tunability that lends itself to light-emitting applications and optimized bandgap materials for tandem solar cells. While for a Si–perovskite tandem a 30–40% Br− perovskite top cell is required, alternative perovskite–perovskite tandem solar cells with MAPbI3 require a top cell with higher than 60% Br− fraction.53 However, photo-stable tunability across the full bromide–iodide range has proven difficult for single-cation MA+, FA+ or Cs+ perovskites, with crystallinity and charge-carrier mobility ‘gaps’ appearing in the central region.15,42,50 Consequently, we unravel the optoelectronic properties of the Cs0.17FA0.83Pb(BrxI(1−x))3 perovskite series with varying Br content x.
We first examine trap-related monomolecular charge-carrier recombination channels by analysing the photoluminescence decay transients under low excitation fluences (Fig. S8, ESI†). From stretched exponential fits we extract charge-carrier lifetimes τ across the series, which vary from 22 ns for 100% bromide to 191 ns for the 100% iodide film. Fig. 5a displays the inverse value τ−1, that is, the mono-molecular decay rates k1, as a function of bromide content for the whole series of 11 samples. Trap-related charge-carrier recombination increases monotonically with increasing bromide content, in particular beyond 60% bromide fraction.
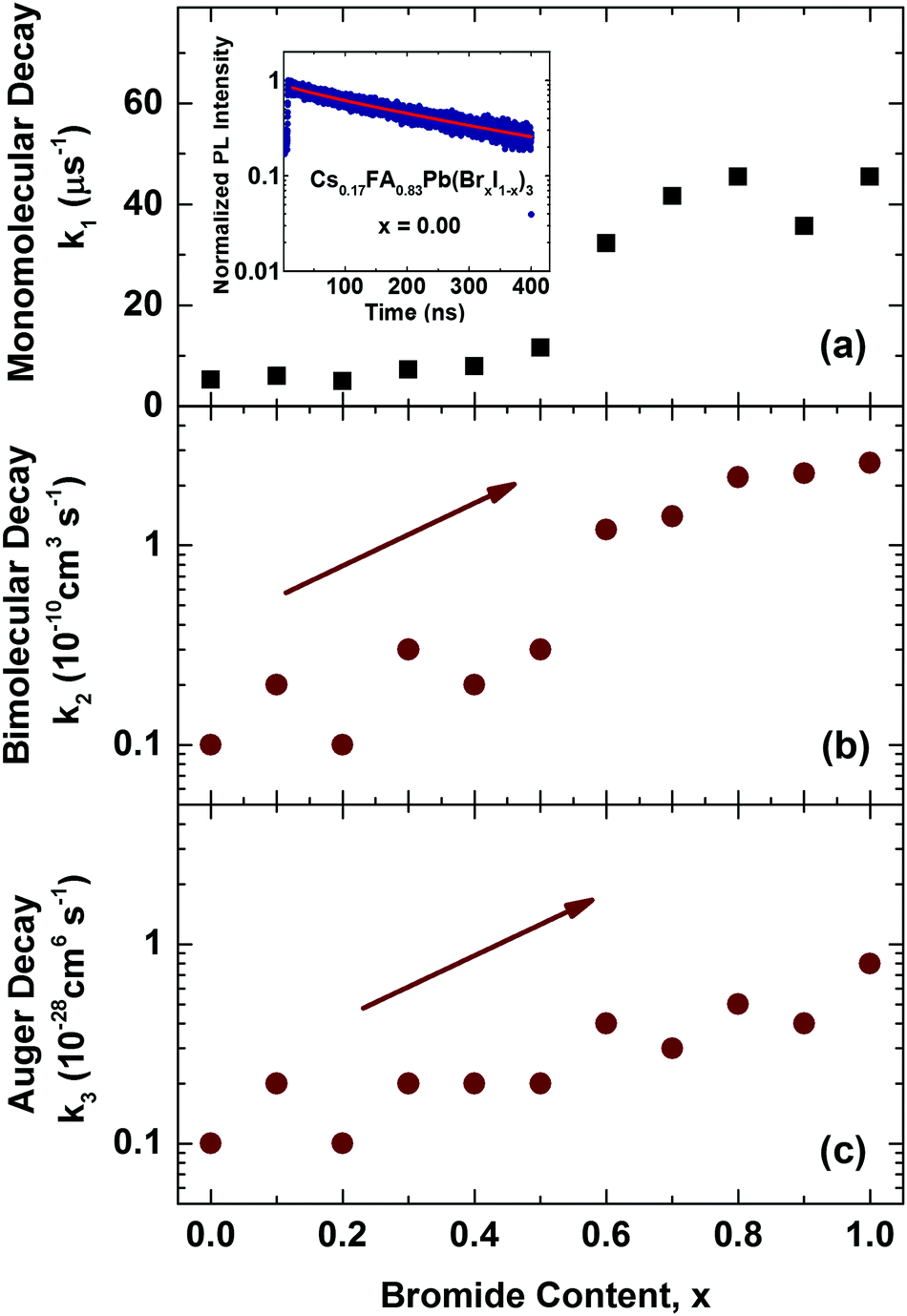 | ||
| Fig. 5 (a) Mono-molecular, (b) bi-molecular and (c) Auger recombination rate constants, k1, k2 and k3 for mixed-halide Cs0.17FA0.83Pb(BrxI(1−x))3 perovskites with varying Br− content x. Values for k1 were extracted from stretched exponential fits to PL decay traces from time-resolved PL measurements. The inset to (a) shows an example PL decay trace for the iodide-only mixed cation Cs0.17FA0.83PbI3 film, including a stretched exponential fit (red line). Values for k2 and k3 were extracted from fits to THz photoconductivity transients which are shown in the ESI† (Fig. S9). | ||
This effect most likely results from larger trap state densities in Br−-rich films, which could again be related to lower crystallinity, in accordance with changes in charge-carrier mobility (vide infra). It has been proposed that for very high Br− content, the thermodynamic favourability of the perovskite α-phase in Cs+/FA+ lead mixed-halide perovskites may decrease45 which could explain the observed trend. However, despite this, the stabilizing effect of Cs+ incorporation material series is a remarkable success, since it results in long charge-carrier recombination lifetimes in excess of several tens of nanoseconds even for the intermediate mixed-halide region (0.3 < x < 0.5), in marked contrast to FA+-only perovskites.15
We may further examine higher order recombination processes, such as bi-molecular (k2) and Auger recombination (k3), which typically17 only contribute significantly at charge-carrier densities in excess of 1018 cm−3. We extract these rate constants from THz photoconductivity transients obtained from ultrafast optical-pump THz-probe spectroscopy (Fig. S9, ESI†) which reflect the decay of charge-carriers following photo-excitation with an intense pulse at 400 nm. Fits of the solutions to the rate equation dn(t)/dt = −k3n3 − k2n2 − k1n, modified to account for the excitation density profile (see ESI† for details) yield bi-molecular (k2) and Auger recombination (k3) rate constants as a function of bromide fraction, as shown in Fig. 5b & c. In excellent agreement with our previous study,15 we find that increasing the bromide fraction from 0 to 1 leads to an increase by roughly an order of magnitude for both k2 (0.1–2.6 × 10−10 cm3 s−1) and k3 (0.1–0.8 × 10−28 cm6 s−1). Such correlations confirm that these processes are intrinsic to band structure changes encountered across the series, rather than crystallinity or disorder.15
Finally, we investigate the charge-carrier scattering mechanisms in Cs0.17FA0.83Pb(BrxI(1−x))3 which may be probed by measurements of the PL linewidth and charge-carrier mobility across the series (Fig. 6). Across the full range from 100% iodide (x = 0) to 100% bromide (x = 1) the FWHM of the PL spectra exhibits a continuous increase up until x < 0.8 beyond which a slight decrease is recorded. Two factors are likely to influence the observed trends here. On the one hand, lowered crystallinity and disordered alloy formation are associated with higher energetic disorder, which yields broader PL spectra. This effect has been observed through correlations between the FWHM and the Urbach energy associated with absorption tails,64 and with charge-carrier mobility.15 Such disorder may be more prevalent for the central region where even bromide/iodide content supports higher alloying disorder,63 and towards higher bromide fraction because of the slightly decreased thermodynamic favourability of perovskite α-phases in Br−-rich films.45 In addition to these extrinsic, disorder-related effects, intrinsic factors such as changes in electron–phonon coupling can influence PL line shape broadening and charge-carrier mobilities.65 It has recently been shown that FAPbBr3 supports stronger electron–phonon coupling via a dominant Fröhlich interaction of charge-carriers with LO phonons than FAPbI3 because the former is associated with a smaller value of the high-frequency dielectric function.65 Hence the increase in electron–phonon interactions can contribute to the trend of increasing PL broadening with increasing bromide content.
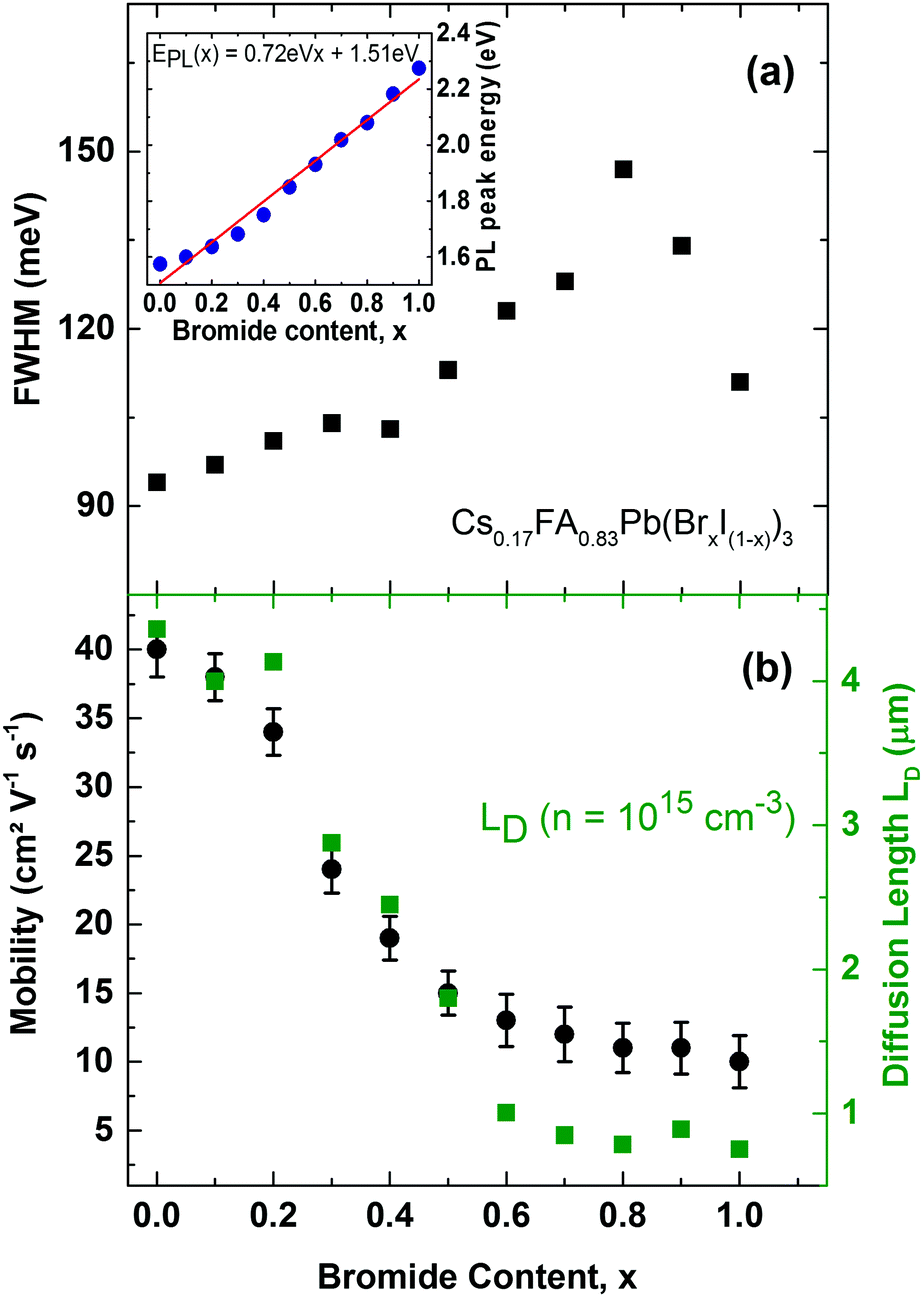 | ||
| Fig. 6 (a) FWHM of the PL emission spectra of mixed-halide Cs0.17FA0.83Pb(BrxI(1−x))3 perovskites as function of Br− content x. Inset: Correlation of the PL peak energy with changing Br− content. The red solid line is a linear fit yielding EPL(x) = 0.72 eVx + 1.51 eV. (b) Effective charge-carrier mobilities (black dots) and charge-carrier diffusion length (green squares) of mixed-halide Cs0.17FA0.83Pb(BrxI(1−x))3 with varying Br− content. Error bars indicate the range of values obtained. | ||
Charge-carrier scattering caused by extrinsic disorder or intrinsic interactions with phonons will also adversely affect the charge-carrier mobility. To further investigate such effects, we display in Fig. 6b charge-carrier mobility values as a function of Br− content, as extracted from THz photo-conductivity measurements (see ESI† for details). We find a continuous decrease of the mobility with increasing Br− content, in accordance with the increase in charge-carrier scattering already evident in the PL emission linewidth. Hence the highest charge-carrier mobility values are found for iodide-rich films (0 < x < 0.2), and reach excellent values between 34–40 cm2 V−1 s−1. The presence of 17% caesium in Cs0.17FA0.83PbI3 thus increases the charge-carrier mobility by almost 50% with respect to FAPbI3 for which we have previously reported15 a THz mobility of 27 cm2 V−1 s−1. Importantly, we find that caesium clearly stabilizes the charge-carrier mobilities in the intermediate mixed-halide regime (0.3 < x < 0.5) where values for Cs0.17FA0.83Pb(BrxI(1−x))3 now range between 15–24 cm2 V−1 s−1, in contrast to the ∼1 cm2 V−1 s−1 previously reported15 for FAPbBr0.4I0.6. The addition of caesium hence leads to a stabilization of the crystal structure against both structural changes and halide segregation that is in turn reflected in drastically improved charge-carrier motion through the perovskite. High carrier mobilities within the active layer are crucial for charge-carrier extraction from the complete photovoltaic cell. To illustrate this point, Fig. 6b also displays charge-carrier diffusion lengths LD calculated from the Einstein relation using the determined charge-carrier recombination constants (k1, k2, k3) and mobilities (see ESI† for details). At a charge-carrier density typical17 for photovoltaic device operation (n ∼ 1015 cm−3), we find charge-carrier diffusion lengths in the micron range tuning monotonically from 0.8 μm for Cs0.17FA0.83PbBr3 to 4.4 μm for Cs0.17FA0.83PbI3 without any sign of the disorder and instability gap previously observed for the FA+-only system. In addition, all values significantly exceed the optical absorption depth, demonstrating that mixed-halide perovskites can clearly serve as materials in efficient planar-heterojunction solar cells across the full halide tuning range thus enabling a variety of tandem combinations.
Conclusions
In summary, we have revealed pertinent links between crystal phase stability, photo-stability and optoelectronic properties in mixed-cation lead mixed-halide CsyFA(1−y)Pb(BrxI(1−x))3 perovskite films. By finely tuning the Cs+ content y in the perovskite, we have ascertained a stable region between 0.10 < y < 0.30 of high crystalline quality, high photo-stability against halide segregation and excellent charge-carrier lifetimes and mobilities. Within this stable region, the effective cation radius of the FA+/Cs+ mixture is located well within the Goldschmidt tolerance factor and hence stable formation of a perovskite α-phase is preferred over deterioration into a photo-inactive yellow non-perovskite δ-phases. Within the CsyFA(1−y)Pb(Br0.4I0.6)3 series we find optimized optoelectronic properties for y = 0.20, which exhibits high charge-carrier mobilities of (18 ± 2) cm2 V−1 s−1 and charge-carrier lifetimes of ∼80 ns. Importantly, we demonstrate that for materials with high crystallinity, photo-induced halide segregation is strongly suppressed. We propose that regions of short-range crystalline order, or grain boundaries, enable halide segregation, possibly by releasing lattice strain between iodide rich and bromide rich domains, or by providing nucleation sites through defects or amorphous material accumulated near grain boundaries. Such correlations also suggest that the stoichiometry of the perovskite alone may not be the only factor determining photo-stability. For example, even within the parameter space explored here, changes in processing conditions or ambient moisture could erode crystallinity, making these same materials susceptible to photo-induced halide segregation. In addition, we postulate that A-cation segregation could also trigger halide segregation; a normally stable material with stoichiometry Cs0.2FA0.8Pb(Br0.4I0.6)3 could still fall outside of the stability zone normally attained for 10–30% Cs+ if it was actually composed of segregated domains with 0% (FA+ only) and 40% Cs, each of which falls outside the stable range. These examples highlight that in order to attain materials that are photo-stable against halide segregation, processing routes must yield large grain sizes and even alloying. As we show here, these targets are highly feasible. Finally, we explore the orthogonal halide-variation parameter space for Cs0.17FA0.83Pb(BrxI(1−x))3 perovskites over the full compositional range (0 < x < 1). We demonstrate that once the material contains a sufficient fraction of caesium, excellent charge-carrier mobilities and diffusion lengths can be obtained across the full iodide–bromide tuning range. Band gaps tune linearly with Vegard's law across the whole range, and unlike previously observed for FAPb(BrxI(1−x))3, there are no effects of lowered optoelectronic properties or stability in the central region. Therefore, the addition of some caesium yields an otherwise elusive perovskite system whose absorption onsets can be tuned for optimized, photo-stable perovskite–perovskite and silicon–perovskite tandem solar cells.Acknowledgements
The authors acknowledge support by the Engineering and Physical Sciences Research Council UK (EPSRC). W. R. thanks the Hans-Böckler-Foundation for support through a doctoral scholarship.Notes and references
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- http://www.nrel.gov/ncpv/images/efficiency_chart.jpg, accessed 7/28/2016.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338(6107), 643–647 CrossRef CAS PubMed.
- J. Burschka, N. Pellet, S. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Grätzel, Nature, 2013, 499(7458), 316–319 CrossRef CAS PubMed.
- W. S. Yang, J. H. Noh, N. J. Jeon, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Science, 2015, 348(6240), 1234–1237 CrossRef CAS PubMed.
- G. E. Eperon, T. Leijtens, K. A. Bush, T. Green, J. T.-W. Wang, D. P. McMeekin, G. Volonakis, R. L. Milot, D. J. Slotcavage, R. Beslile, J. B. Patel, E. S. Parrott, R. J. Sutton, W. Ma, F. Moghadam, B. Conings, A. Babayigit, H.-G. Boyen, F. Giustino, L. M. Herz, M. B. Johnston, M. D. McGehee and H. J. Snaith, Science, 2016, 354, 861–865 CrossRef CAS PubMed.
- D. P. McMeekin, G. Sadoughi, W. Rehman, G. E. Eperon, M. Saliba, M. T. Horantner, A. Haghighirad, N. Sakai, L. Korte, B. Rech, M. B. Johnston, L. M. Herz and H. J. Snaith, Science, 2016, 351, 151–155 CrossRef CAS PubMed.
- B. Chen, Y. Bai, Z. Yu, T. Li, X. Zheng, Q. Dong, L. Shen, M. Boccard, A. Gruverman, Z. Holman and J. Huang, Adv. Energy Mater., 2016, 6, 1601128 CrossRef.
- S. Albrecht, M. Saliba, J. P. C. Baena, F. Lang, L. Kegelmann, M. Mews, L. Steier, A. Abate, J. Rappich and L. Korte, Energy Environ. Sci., 2016, 9, 81–88 CAS.
- M. Anaya, J. P. Correa-Baena, G. Lozano, M. Saliba, P. Anguita, B. Roose, A. Abate, U. Steiner, M. Grätzel and M. E. Calvo, J. Mater. Chem. A, 2016, 4, 11214–11221 CAS.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- G. E. Eperon, S. D. Stranks, C. Menelaou, M. B. Johnston, L. M. Herz and H. J. Snaith, Energy Environ. Sci., 2014, 7, 982–988 CAS.
- T. M. Koh, K. Fu, Y. Fang, S. Chen, T. Sum, N. Mathews, S. G. Mhaisalkar, P. P. Boix and T. Baikie, J. Phys. Chem. C, 2013, 118, 16458–16462 Search PubMed.
- D. Shi, V. Adinolfi, R. Comin, M. Yuan, E. Alarousu, A. Buin, Y. Chen, S. Hoogland, A. Rothenberger, K. Katsiev, Y. Losovyj, X. Zhang, P. A. Dowben, O. F. Mohammed, E. H. Sargent and O. M. Bakr, Science, 2015, 347, 519–522 CrossRef CAS PubMed.
- W. Rehman, R. L. Milot, G. E. Eperon, C. Wehrenfennig, J. L. Boland, H. J. Snaith, M. B. Johnston and L. M. Herz, Adv. Mater., 2015, 27, 7938–7944 CrossRef CAS PubMed.
- L. M. Herz, Annu. Rev. Phys. Chem., 2016, 67, 65–89 CrossRef CAS PubMed.
- M. B. Johnston and L. M. Herz, Acc. Chem. Res., 2016, 49, 146–154 CrossRef CAS PubMed.
- C. Ponseca Jr and V. Sundström, Nanoscale, 2016, 8, 6249–6257 RSC.
- R. L. Milot, G. E. Eperon, H. J. Snaith, M. B. Johnston and L. M. Herz, Adv. Funct. Mater., 2015, 25, 6218–6227 CrossRef CAS.
- C. Wehrenfennig, M. Liu, H. J. Snaith, M. B. Johnston and L. M. Herz, APL Mater., 2014, 2, 081513 CrossRef.
- H. Kim, C. Lee, J. Im, K. Lee, T. Moehl, A. Marchioro, S. Moon, R. Humphry-Baker, J. Yum and J. E. Moser, Sci. Rep., 2012, 2, 591 Search PubMed.
- J. H. Heo, S. H. Im, J. H. Noh, T. N. Mandal, C. Lim, J. A. Chang, Y. H. Lee, H. Kim, A. Sarkar and M. K. Nazeeruddin, Nat. Photonics, 2013, 7, 486–491 CrossRef CAS.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. I. Seok, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS PubMed.
- Z. Xiao, C. Bi, Y. Shao, Q. Dong, Q. Wang, Y. Yuan, C. Wang, Y. Gao and J. Huang, Energy Environ. Sci., 2014, 7, 2619–2623 CAS.
- Q. Chen, H. Zhou, Z. Hong, S. Luo, H. Duan, H. Wang, Y. Liu, G. Li and Y. Yang, J. Am. Chem. Soc., 2013, 136, 622–625 CrossRef PubMed.
- A. T. Barrows, A. J. Pearson, C. K. Kwak, A. D. F. Dunbar, A. R. Buckley and D. G. Lidzey, Energy Environ. Sci., 2014, 7, 2944–2950 CAS.
- B. R. Sutherland, S. Hoogland, M. M. Adachi, P. Kanjanaboos, C. T. Wong, J. J. McDowell, J. Xu, O. Voznyy, Z. Ning, A. J. Houtepen and E. H. Sargent, Adv. Mater., 2015, 27, 53–58 CrossRef CAS PubMed.
- G. E. Eperon, C. E. Beck and H. J. Snaith, Mater. Horiz., 2016, 3, 63–71 RSC.
- M. Liu, M. B. Johnston and H. J. Snaith, Nature, 2013, 501, 395–398 CrossRef CAS PubMed.
- O. Malinkiewicz, A. Yella, Y. H. Lee, G. M. Espallargas, M. Graetzel, M. K. Nazeeruddin and H. J. Bolink, Nat. Photonics, 2014, 8, 128–132 CrossRef CAS.
- R. J. Sutton, G. E. Eperon, L. Miranda, E. S. Parrott, B. A. Kamino, J. B. Patel, M. T. Hörantner, M. B. Johnston, A. A. Haghighirad, D. T. Moore and H. J. Snaith, Adv. Energy Mater., 2016, 6, 1502458 CrossRef.
- F. Hao, C. C. Stoumpos, D. H. Cao, R. P. Chang and M. G. Kanatzidis, Nat. Photonics, 2014, 8, 489–494 CrossRef CAS.
- C. Wehrenfennig, G. E. Eperon, M. B. Johnston, H. J. Snaith and L. M. Herz, Adv. Mater., 2014, 26, 1584–1589 CrossRef CAS PubMed.
- C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Inorg. Chem., 2013, 52, 9019–9038 CrossRef CAS PubMed.
- B. Conings, J. Drijkoningen, N. Gauquelin, A. Babayigit, J. D'Haen, L. D'Olieslaeger, A. Ethirajan, J. Verbeeck, J. Manca and E. Mosconi, Adv. Energy Mater., 2015, 5, 1500477 CrossRef.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Nature, 2015, 517, 476–480 CrossRef CAS PubMed.
- Q. A. Akkerman, V. D’Innocenzo, S. Accornero, A. Scarpellini, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2015, 137, 10276–10281 CrossRef CAS PubMed.
- A. Amat, E. Mosconi, E. Ronca, C. Quarti, P. Umari, M. K. Nazeeruddin, M. Grätzel and F. De Angelis, Nano Lett., 2014, 14, 3608–3616 CrossRef CAS PubMed.
- C. D. Bailie and M. D. McGehee, MRS Bull., 2015, 40, 681–686 CrossRef CAS.
- G. E. Eperon, G. M. Paternò, R. J. Sutton, A. Zampetti, A. A. Haghighirad, F. Cacialli and H. J. Snaith, J. Mater. Chem. A, 2015, 3, 19688–19695 CAS.
- M. Kulbak, D. Cahen and G. Hodes, J. Phys. Chem. Lett., 2015, 6, 2452–2456 CrossRef CAS PubMed.
- R. E. Beal, D. J. Slotcavage, T. Leijtens, A. R. Bowring, R. A. Belisle, W. H. Nguyen, G. F. Burkhard, E. T. Hoke and M. D. McGehee, J. Phys. Chem. Lett., 2016, 7, 746–751 CrossRef CAS PubMed.
- M. Saliba, T. Matsui, J. Seo, K. Domanski, J. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Energy Environ. Sci., 2016, 9, 1989–1997 CAS.
- H. Choi, J. Jeong, H. Kim, S. Kim, B. Walker, G. Kim and J. Y. Kim, Nano Energy, 2014, 7, 80–85 CrossRef CAS.
- C. Yi, J. Luo, S. Meloni, A. Boziki, N. Ashari-Astani, C. Grätzel, S. M. Zakeeruddin, U. Röthlisberger and M. Grätzel, Energy Environ. Sci., 2016, 9, 656–662 CAS.
- J. Lee, D. Kim, H. Kim, S. Seo, S. M. Cho and N. Park, Adv. Energy Mater., 2015, 5, 1501310 CrossRef.
- A. Binek, F. C. Hanusch, P. Docampo and T. Bein, J. Phys. Chem. Lett., 2015, 6, 1249–1253 CrossRef CAS PubMed.
- N. Pellet, P. Gao, G. Gregori, T. Yang, M. K. Nazeeruddin, J. Maier and M. Grätzel, Angew. Chem., Int. Ed., 2014, 53, 3151–3157 CrossRef CAS PubMed.
- Z. Li, M. Yang, J. Park, S. Wei, J. J. Berry and K. Zhu, Chem. Mater., 2015, 28, 284–292 CrossRef.
- E. T. Hoke, D. J. Slotcavage, E. R. Dohner, A. R. Bowring, H. I. Karunadasa and M. D. McGehee, Chem. Sci., 2015, 6, 613–617 RSC.
- Y. Zhou, M. Yang, O. S. Game, W. Wu, J. Kwun, M. A. Strauss, Y. Yan, J. Huang, K. Zhu and N. P. Padture, ACS Appl. Mater. Interfaces, 2016, 8(3), 2232–2237 CAS.
- R. Lindblad, N. K. Jena, B. Philippe, J. Oscarsson, D. Bi, A. Lindblad, S. Mandal, B. Pal, D. D. Sarma, O. Karis, H. Siegbahn, E. M. J. Johansson, M. Odelius and H. M. Rensmo, J. Phys. Chem. C, 2015, 119, 1818–1825 CAS.
- T. J. Jacobsson, J. Correa-Baena, M. Pazoki, M. Saliba, K. Schenk, M. Grätzel and A. Hagfeldt, Energy Environ. Sci., 2016, 9, 1706–1724 Search PubMed.
- http://abulafia.mt.ic.ac.uk/shannon/ptable.php, accessed 8/7/2016.
- P. Scherrer, Kolloidchemie Ein Lehrbuch, Springer, 1912, pp. 387–409 Search PubMed.
- J. I. Langford and A. Wilson, J. Appl. Crystallogr., 1978, 11, 102–113 CrossRef CAS.
- J. T.-W. Wang, Z. Wang, S. Pathak, W. Zhang, D. W. deQuilettes, F. Wisnivesky-Rocca-Rivarola, J. Huang, P. K. Nayak, J. B. Patel, H. A. Mohd Yusof, Y. Vaynzof, R. Zhu, I. Ramirez, J. Zhang, C. Ducati, C. Grovenor, M. B. Johnston, D. S. Ginger, R. J. Nicholas and H. J. Snaith, Energy Environ. Sci., 2016, 9, 2892–2901 CAS.
- D. W. deQuilettes, S. M. Vorpahl, S. D. Stranks, H. Nagaoka, G. E. Eperon, M. E. Ziffer, H. J. Snaith and D. S. Ginger, Science, 2015, 348, 683–686 CrossRef CAS PubMed.
- W. Nie, H. Tsai, R. Asadpour, J. C. Blancon, A. J. Neukirch, G. Gupta, J. J. Crochet, M. Chhowalla, S. Tretiak, M. A. Alam, H. L. Wang and A. D. Mohite, Science, 2015, 347, 522–525 CrossRef CAS PubMed.
- J. Kim, S. Lee, J. H. Lee and K. Hong, J. Phys. Chem. Lett., 2014, 5, 1312–1317 CrossRef CAS PubMed.
- R. Long, J. Liu and O. V. Prezhdo, J. Am. Chem. Soc., 2016, 138, 3884–3890 CrossRef CAS PubMed.
- M. Hu, C. Bi, Y. Yuan, Y. Bai and J. Huang, Adv. Sci., 2015, 3, 1500301 CrossRef PubMed.
- F. Brivio, C. Caetano and A. Walsh, J. Phys. Chem. Lett., 2016, 7, 1083–1087 CrossRef CAS PubMed.
- A. Sadhanala, F. Deschler, T. H. Thomas, S. E. Dutton, K. C. Goedel, F. C. Hanusch, M. L. Lai, U. Steiner, T. Bein, P. Docampo, D. Cahen and R. H. Friend, J. Phys. Chem. Lett., 2014, 5, 2501–2505 CrossRef CAS PubMed.
- A. D. Wright, C. Verdi, R. L. Milot, G. E. Eperon, M. A. Pérez-Osorio, H. J. Snaith, F. Giustino, M. B. Johnston and L. M. Herz, Nat. Commun., 2016, 7, 11755 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, synthesis and film fabrication, experimental techniques and analysis, further characterization data for CsyFA(1−y)Pb(Br0.4I0.6)3 films, further characterization data for Cs0.17FA0.83Pb(BrxI(1−x))3 films. See DOI: 10.1039/c6ee03014a |
| This journal is © The Royal Society of Chemistry 2017 |