
The marriage and integration of nanostructures with different dimensions for synergistic electrocatalysis†
Han
Zhu
 a,
Li
Gu
a,
Danni
Yu
a,
Yingjun
Sun
bc,
Meng
Wan
a,
Ming
Zhang
a,
Lei
Wang
c,
Lina
Wang
a,
Weiwei
Wu
a,
Juming
Yao
a,
Mingliang
Du
*a and
Shaojun
Guo
*bde
a,
Li
Gu
a,
Danni
Yu
a,
Yingjun
Sun
bc,
Meng
Wan
a,
Ming
Zhang
a,
Lei
Wang
c,
Lina
Wang
a,
Weiwei
Wu
a,
Juming
Yao
a,
Mingliang
Du
*a and
Shaojun
Guo
*bde
aCollege of Materials and Textiles, Key Laboratory of Advanced Textile Materials and Manufacturing Technology of the Ministry of Education, Zhejiang Sci-Tech University, 310018, P. R. China. E-mail: du@zstu.edu.cn
bDepartment of Materials Science and Engineering, College of Engineering, Peking University, 100871, P. R. China. E-mail: guosj@pku.edu.cn
cCollege of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, P. R. China
dBIC-ESAT, College of Engineering, Peking University, Beijing 100871, China
eKey Laboratory of Theory and Technology of Advanced Batteries Materials, College of Engineering, Peking University, Beijing 100871, China
First published on 1st December 2016
Abstract
The search for new ways to make inexpensive and efficient electrocatalysts to replace precious-metal platinum catalysts for oxygen reduction and water splitting is still a great challenge. Here, we report a facile and effective strategy for the rational design and construction of three-dimensional (3D) architectures for superior electrocatalysis through the integration of one-dimensional (1D) electrospun carbon nanofibers (CNFs), 1D carbon nanotubes (CNTs) and 0D oxygen-deficient Mn3Co7–Co2Mn3O8 nanoparticles (NPs). The rationale behind the marriage and integration of nanostructures with different dimensions presented in this work is that during heat treatment, the in situ-produced CoMnO NPs are partly reduced to Co2Mn3O8 by a carbon precursor with an amount of metallic Mn3Co7 formed at the interface between the Co2Mn3O8 NPs and carbon, which can act as the catalysts for the growth of 3D CNT forests. The 3D CoMnO@CNT/CNF architectures exhibit superior electrocatalytic activity and stability for the oxygen reduction, oxygen evolution and hydrogen evolution reactions. The remarkable electrochemical properties are mainly attributed to the synergistic effects from the engineering of oxygen-deficient binary CoMn oxide NPs with exposed active sites and 3D hierarchical porous structures consisting of branched CNTs and interconnected CNFs. The present work demonstrates the first example of integrating multiple active catalytic centers onto/into 3D architectures for developing highly efficient non-precious metal nanocatalysts for electrochemical energy devices.
Broader contextElectrocatalytic water splitting is a practical and environmentally friendly approach to generate hydrogen for powering fuel cells. On the journey to pursue the replacement of noble metals as electrocatalysts with earth-abundant elements, carbon-based nanomaterials incorporated with transition metals have attracted growing attention. Hierarchical carbon nanomaterials are highly suitable as electrocatalysts and electrocatalyst supports in electrocatalytic conversion devices. The intrinsic kinetics of an electrocatalyst are associated with the nanostructure of the active phase and the support, while the overall properties are also affected by the mesostructure. Here, we provide a new design of three-dimensional architectures for superior electrocatalysis through the integration of electrospun carbon nanofibers (CNFs), carbon nanotubes (CNTs) and oxygen-deficient CoMnO nanoparticles. This strategy involves new transition metal oxide catalysts for CNT growth using polymer nanofibers as carbon resources. The morphology, nanostructure, crystal phase and electrocatalytic activity of the CoMnO@CNT/CNF architecture can be controlled by adjusting the graphitization process and conditions. The remarkable electrochemical properties are mainly attributed to the synergistic effects obtained from the engineering of oxygen-deficient Mn3Co7–Co2Mn3O8 with exposed active sites and the 3D hierarchical porous structure, which consists of branched CNTs and interconnected CNFs. This work demonstrates a novel integration-and-marriage concept for developing superior catalysts for electrochemical energy devices. |
Introduction
The rise of clean and renewable hydrogen offers mankind a promising opportunity to overcome our reliance on fossil fuels and to address the challenges of global warming, environmental pollution and the energy crisis.1–3 Electrocatalytic water splitting through the two half-reactions, hydrogen evolution reaction (HER) and oxygen evolution reaction (OER), is a clean and sustainable approach to generate molecular hydrogen (H2).4–6 In addition, hydrogen is supposedly an economic fuel used in electrochemical cells for electric devices, such as fuel cells and metal–air batteries.7,8 Oxygen reduction reaction (ORR) and OER are the key reactions for the important energy conversion and storage processes in electrochemical devices.9 These important electrochemical reaction processes need effective catalysts to minimize reaction kinetic barriers, expedite the reactions, reduce the overpotential, and thus improve the energy conversion efficiency. To achieve these targets, substantial efforts have been devoted to designing low-cost earth-abundant transition metal-based electrocatalysts to replace the state-of-the-art Pt and IrO2 catalysts for ORR, HER and OER.9–11 A series of affordable, sustainable, and efficient catalysts, such as transition metal sulfides,12 carbides,13 nitrides,14 and phosphides,15 show great potential for enhancing ORR, HER and OER catalysis, but their performance is still far from that of the commercial Pt/C catalyst. This is because only controlling the chemical composition of the transition metal-based materials cannot provide enough electrocatalytic active centers for achieving highly efficient nanoelectrocatalysts.One of the most promising ways for addressing this issue is by controlling three-dimensional (3D) nanoarchitectures using hierarchical meso- and/or macro-porosity and adequate catalytic centers to improve the kinetics of the catalysis process.16,17 Oxygen vacancies in nanocrystals were shown to be the effective electrocatalytic centers due to their good/optimized affinity for O2.18–20 However, achieving 3D carbon nanotubes/transition metal oxide-based architectures with an adequate amount of oxygen vacancies that function as effective active centers has received very limited attention. Herein, we report the first example of attaching carbon nanotubes/oxygen-deficient CoMnO nanoparticle (NP) hybrids onto carbon nanofibers (CNFs) to form 3D nanoarchitectures for boosting ORR, HER and OER electrocatalysis. The unique part of the synthesis presented is that the CoMnO NPs were gradually reduced from CoMn2O4 crystals to oxygen-deficient Co2Mn3O8 NPs with metallic Co3Mn7 surfaces during the heat treatment, and the oxygen-deficient Co2Mn3O8 NPs formed were used as catalysts to grow the branched CNTs, which then formed 3D nanoarchitectures.
We found that the oxygen-deficient Co2Mn3O8 NPs with metallic Co3Mn7 surfaces are encapsulated at the tips of CNTs and in the channels of the porous graphitic CNF matrix. By integrating the catalytically active Co2Mn3O8 NPs, high nitrogen doping and 3D CNTs, the 3D architectures exhibit superior electrocatalytic activity and stability for ORR with onset and half-wave potentials of 0.87 and 0.81 V vs. reversible hydrogen electrode (RHE), HER with a low overpotential of 175 mV vs. RHE at a current density of 10 mA cm−2 and a Tafel slope of 101 mV decade−1, and OER with an overpotential of 370 mV vs. RHE at 10 mA cm−2 and a Tafel slope of 66 mV decade−1, which are among the best results reported to date for multifunctional catalysts. These remarkable electrochemical properties are mainly attributed to the synergistic effects from the engineering of oxygen-deficient Mn3Co7–Co2Mn3O8 NPs with exposed active sites and 3D hierarchical porous structures consisting of branched CNTs and interconnected CNFs. The Mn3Co7–Co2Mn3O8@CNTs/CNFs exhibit remarkable stability under continuous operation for 10 h during the electrocatalysis process. This facile strategy for the marriage and integration of 1D CNTs, 1D CNFs and 0D oxygen-deficient Mn3Co7–Co2Mn3O8 NPs provides new prospects in the development of highly efficient multifunctional nanomaterials for electrochemical energy devices.
Results and discussion
A two-step strategy was used to fabricate a 3D oxygen-deficient Mn3Co7–Co2Mn3O8@CNT/CNF architecture. In the first step, polyacrylonitrile (PAN) nanofibers were prepared via electrospinning the mixture containing a certain amount of Co(NO3)2 and Mn(Ac)2. As-made Co–Mn/PAN nanofibers exhibit a fiber morphology with a uniform diameter of approximately 560 nm (Fig. S1, ESI†). After pre-oxidation at 400 °C for 5 h under air conditions and thermal treatment at 1000 °C for 6 h in an inert atmosphere (Ar/NH3), the Co–Mn/PAN polymer nanofibers were converted into 3D nanoarchitectures containing porous CNFs, binary cobalt and manganese oxide NPs, and branched CNT forests. Large amounts of CNTs spring up densely from the CNFs, and are vertically grown around the CNFs to form novel 3D nanoarchitectures (Fig. 1a). The binary CoMnO NPs either exist at the tips of the CNTs or are enclosed inside the CNTs, where the NPs are in intimate contact with the carbon shell (Fig. 1b).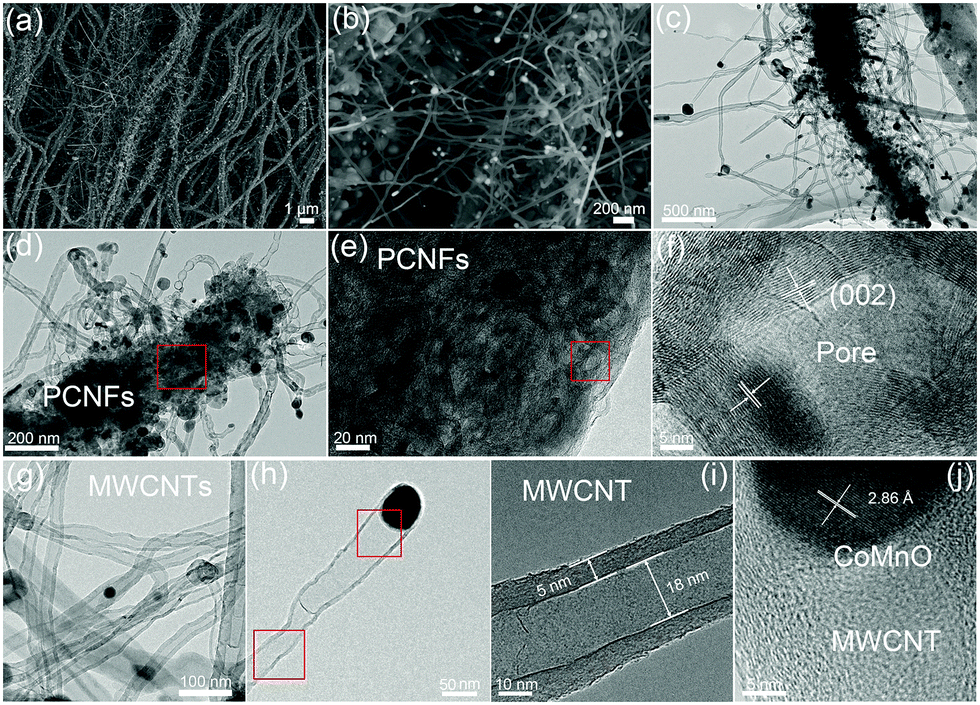 | ||
| Fig. 1 (a and b) FE-SEM and (c and d) TEM images of the 3D oxygen-deficient Mn3Co7–Co2Mn3O8@CNTs/CNFs architecture. (e) High-magnification TEM image of the porous graphitic CNFs matrix. (f) High-resolution TEM (HRTEM) image of the fullerene-like graphitic carbon pores catalyzed by the CoMnO NPs. (g) TEM image of the CNTs and (h) single CNTs, with each NT tip containing one CoMnO NP. (i) HRTEM image of the CNTs. (j) HRTEM image of the CoMnO NPs encapsulated into CNTs. | ||
Unlike the chemical vapor deposition (CVD) method for the growth of CNTs in a vapor–liquid–solid process, the CoMnO NPs were first formed inside the nanofibers during the oxidation process at 400 °C. And then the as-formed CoMnO NPs catalyze the conversion from the amorphous organic nanofibers to the graphitic porous CNFs along with the growth of CNTs. Fig. 1c and d show the hierarchical structure of the porous CNFs with CoMnO@CNT forests. We found that in addition to the CoMnO NPs encapsulated in CNTs, there are certain amounts of CoMnO NPs embedded in the CNFs. The encapsulated CoMnO NPs can catalyze the conversion from amorphous carbon to graphitic carbon, which leads to the formation of porous graphitic CNFs. Because of the strong catalytic effect of the CoMnO NPs, the CNF matrix consists of a large amount of fullerene-like graphitic carbon pores catalyzed by the CoMnO NPs (Fig. 1e). The fullerene-like pores clearly exhibit well-ordered graphene layers with a d-spacing of 3.4 Å, which corresponds to the (002) planes of carbon (Fig. 1f). The graphitic wall of the fullerene-like pore ranges between approximately 10 and 40 graphene layers. The graphene layers not only hold the NPs but also improve the electrical conductivity of the carbon matrix. Fig. 1g and h show the TEM images of CNTs catalyzed by the CoMnO NPs. Most of the CoMnO NPs are located at the tips of the CNTs, and the length of the CNTs ranges from 0.5 μm to 5 μm. The as-formed CNTs are multi-walled with a diameter of approximately 10–50 nm and a wall thickness of 5–15 nm (Fig. 1g–i). From Fig. 1i, the graphene planes are parallel to the tube axis with a layer distance of 3.40 Å. At the tips of the multi-walled CNTs (MWCNTs) (Fig. 1j), the CoMnO NPs exhibit a d-spacing of 2.86 Å, which corresponds to the [200] plane of Co2Mn3O8 NPs.
We further employ a high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM)-energy-dispersive X-ray spectroscopy (EDX) mapping technique to investigate the elemental distribution of the 3D CoMnO@CNT/CNF architecture. Uniform and distinct MWCNTs along with the CoMnO NPs are clearly observed (Fig. 2a and b). There are strong K-edge signals of carbon and nitrogen elements in the nanotubes, which confirms the formation of N-doped CNTs. Furthermore, the signals of carbon and nitrogen appear around the NP area, indicating that the CoMnO NPs were encapsulated into the CNTs. The NPs have strong K-edge signals of cobalt, manganese and oxygen elements, confirming the successful formation of binary CoMnO NPs. Furthermore, there are signals of manganese beyond the CoMnO boundary, which is attributed to the metallic manganese on the surface of the CoMnO NPs (Fig. 2c). The HRTEM image of the CoMnO NPs at the tip of CoMnO@CNTs also provides the evidence of the formation of metallic manganese at the interface between the surfaces of CoMnO NPs and the surrounding fullerene carbon layers. These results indicate that the binary CoMn oxide NPs encapsulated into the CNTs are not single-phase NPs, and an amount of metallic Mn is formed around the CoMnO NPs.
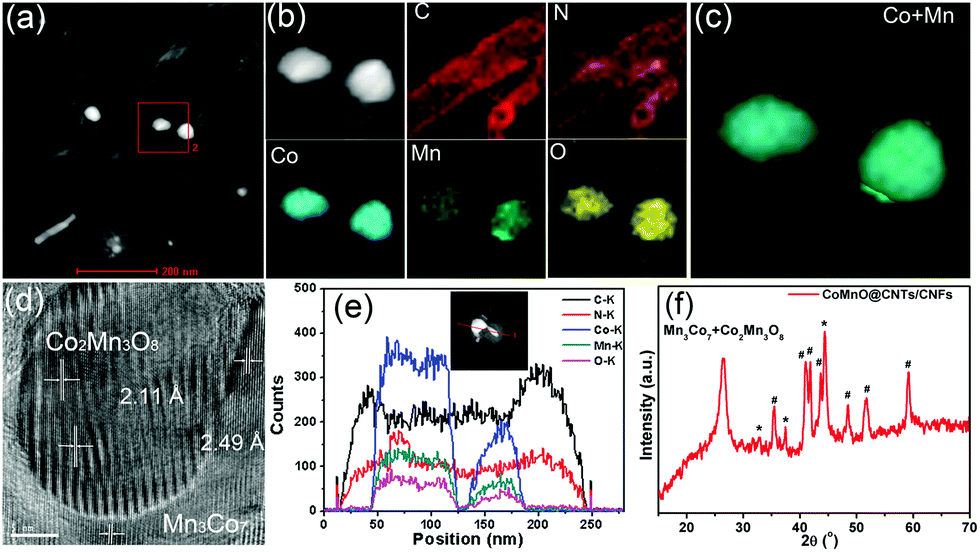 | ||
| Fig. 2 (a) HAADF-STEM image of the oxygen-deficient Mn3Co7–Co2Mn3O8@CNTs/CNFs. (b) STEM-EDX mapping image of the selected area of Mn3Co7–Co2Mn3O8@CNTs/CNFs. (c) The overlay between Co and Mn in the STEM-EDX mapping area. (d) HRTEM image of a single oxygen-deficient Mn3Co7–Co2Mn3O8 NP at the tip of a CNT. (e) Line-scan EDX spectra of the oxygen-deficient Mn3Co7–Co2Mn3O8@CNTs/CNFs, and the inset in (e) is the line-scan area. (f) XRD pattern of the Mn3Co7–Co2Mn3O8@CNT/CNF architecture. | ||
As shown in Fig. 2d, the marked area of the CoMnO NP exhibits distinct lattice fringes with an interlayer distance of 2.11 Å, corresponding to the (122) plane of Co2Mn3O8 NPs. There is also another lattice fringe outside CoMnO with an interlayer distance of 2.49 Å, which belongs to the metallic Mn3Co7. This is because the CoMnO NPs, directly in contact with carbon, were partly reduced to Co2Mn3O8 with an amount of metallic Mn3Co7 formed at the interface between the Co2Mn3O8 NPs and carbon. The line-scan STEM-EDS spectra provide more evidence of the formation of CoMnO NPs. As revealed in Fig. 2h, the K-edge signals of the Co, Mn and O elements are mainly centered at the bright spot, which corresponds to the CoMnO NPs. The surrounding of the CoMnO nanoparticles exhibits an even distribution of carbon and nitrogen signals, which indicates the successful formation of N-doped CNTs.
The crystalline structure, surface chemical state and chemical composition of the CoMnO@CNTs/CNFs were further investigated by powder X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS) measurements. The XRD pattern of the Mn3Co7–Co2Mn3O8@CNTs/CNFs prepared at 1000 °C exhibits a broad band at 25.2°, which corresponds to the (002) plane of the graphitic carbon of CNFs catalyzed by Mn3Co7–Co2Mn3O8 NPs (Fig. 2f).21 A sharp and distinct peak located at 26.5° corresponds to the CNTs and indicates the high degree of graphitic carbon.22 A series of other XRD peaks located at 32.7°, 44.4° and 48.5° are characteristic of the (103), (122) and (220) planes of Co2Mn3O8 nanocrystals (labeled as *, JPCDS card #30-0446).23 Furthermore, some peaks emerged at 35.4°, 41.0°, 41.8°, 43.7°, 48.4°, 51.4° and 59.0°, which correspond to the Mn3Co7 nanocrystals (labeled as #, PDF#18-0407); this further confirms the formation of the metallic Mn3Co7 phase.24
XRD patterns of the CoMnO@CNTs/CNFs heated at different temperatures (400, 600 and 800 °C) were obtained to further investigate the structure of the 3D CoMnO@CNT/CNF architectures (Fig. 3a). The XRD pattern of the CoMnO@CNTs/CNFs prepared at 400 °C (Fig. 3a) shows a peak at approximately 24.2°, which is attributed to the (002) plane of carbon. The other peaks at 29.8°, 43.9°, 64.4° and 77.4°, respectively, can be indexed to the (202), (400), (440), (622) planes of spinel-type (Co,Mn)(Mn,Co)2O4 crystals (PDF#18-0409).23 Therefore, at the initial stage of 400 °C, the CoMnO NPs are mainly spinel-type CoMn2O4 crystals formed on CNFs (Fig. 3a). The XRD pattern of the CoMnO@CNTs/CNFs prepared at 600 °C indicates that the CoMn2O4 and Co2Mn3O8 NPs co-exist, which demonstrates the phase transition from CoMn2O4 to Co2Mn3O8 as a result of the surrounding carbon. From the XRD pattern of the CoMnO@CNTs/CNFs prepared at 800 °C, it was found that the previous CoMn2O4 NPs are completely transformed into Co2Mn3O8. By increasing the temperature from 400 to 1000 °C, the CoMnO NPs evolved from CoMn2O4 to oxygen-vacant Co2Mn3O8 with separated metallic Mn3Co7 at the surface. The reason for the formation of such a unique structure is that during the growth of CNTs under an Ar/NH3 condition at 1000 °C, the as-formed CoMn2O4 NPs were reduced to Co2Mn3O8 with a partial reduction of metallic Mn3Co7 on the surfaces surrounded by carbon shells; this reduction is required for the formation of the CNTs.
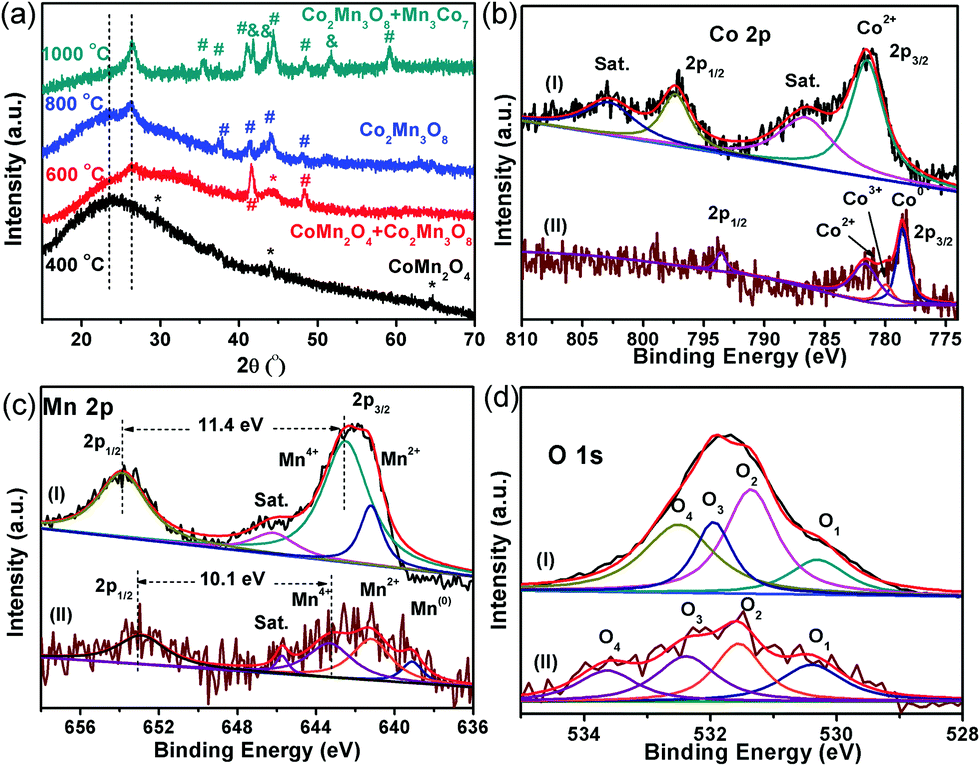 | ||
| Fig. 3 (a) XRD pattern of the CoMnO@CNT/CNF architecture prepared at different temperatures. The (b) Co 2p, (c) Mn 2p and (d) O 1s XPS spectra of (curve I) pre-oxidation CoMn2O4/CNFs prepared at 400 °C and (curve II) oxygen-deficient Mn3Co7–Co2Mn3O8@CNTs/CNFs prepared at 1000 °C. | ||
The XPS spectra of the CoMn2O4/CNFs prepared at 400 °C and the oxygen-deficient Mn3Co7–Co2Mn3O8@CNTs/CNFs prepared at 1000 °C were obtained to further investigate the change in the surface chemical state (Fig. 3). The XPS survey spectra of the CoMn2O4/CNFs and Mn3Co7–Co2Mn3O8@CNT/CNF hybrids both exhibit C, N, O, Co and Mn elements (Fig. S2 and S3, ESI†). The C 1s spectrum of CoMn2O4/CNFs shows four peaks with the binding energies (BEs) at 284.6, 285.3, 286.7 and 288.9 eV, which correspond to the –C–C/H, –C–N, –C–OH and –O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O functional groups, respectively (Fig. S4, ESI†).25 The CoMn2O4/CNFs prepared at 400 °C exhibit abundant oxygen groups. For the oxygen-deficient Mn3Co7–Co2Mn3O8@CNTs/CNFs prepared at 1000 °C, the C 1s spectra (Fig. S5, ESI†) show three peaks with BEs at 284.7, 285.2 and 285.8 eV, corresponding to the sp2-hybridized graphitic carbon, –C–N and –C–OH/CO groups, respectively.26 Compared with the CoMn2O4/CNF hybrid, the percentage of the oxygen groups of the Mn3Co7–Co2Mn3O8@CNTs/CNFs decreases significantly.
C–O functional groups, respectively (Fig. S4, ESI†).25 The CoMn2O4/CNFs prepared at 400 °C exhibit abundant oxygen groups. For the oxygen-deficient Mn3Co7–Co2Mn3O8@CNTs/CNFs prepared at 1000 °C, the C 1s spectra (Fig. S5, ESI†) show three peaks with BEs at 284.7, 285.2 and 285.8 eV, corresponding to the sp2-hybridized graphitic carbon, –C–N and –C–OH/CO groups, respectively.26 Compared with the CoMn2O4/CNF hybrid, the percentage of the oxygen groups of the Mn3Co7–Co2Mn3O8@CNTs/CNFs decreases significantly.
The Co 2p spectrum of CoMn2O4/CNFs (Fig. 3b, curve I) includes two main peaks with BEs at 781.6 and 797.4 eV, corresponding to Co 2p3/2 and 2p1/2 of the Co2+ ions. The satellite peaks for the Co2+ ions are located at 786.6 and 802.9 eV, respectively, which is in agreement with the previous results reported in the literature for spinel-type CoMn2O4 crystals.27 However, compared with those of CoMn2O4/CNFs, the main peaks in the Co 2p XPS spectra of Mn3Co7–Co2Mn3O8@CNTs/CNFs (Fig. 3b, curve II) move to lower BEs. Three main peaks with BEs at 778.6, 780.0 and 781.6 eV are attributed to Co 2p3/2 for Co0, Co3+ and Co2+, respectively, and another peak at 793.6 eV is mainly assigned to Co 2p1/2 for Co0.28 The shifting of the Co 2p region of the Mn3Co7–Co2Mn3O8@CNTs/CNFs to lower BEs can be attributed to the surface reduction of CoMn2O4 by the surrounding carbon during the growth process of the CNTs. The appearance of metallic Co (778.6 eV) confirms that there are amounts of metallic Co separated out from the spinel CoMn2O4 and formed at the interface between the surrounding carbon and the surface of CoMn2O4. Because of the strong reduction ability of the carbon precursor at 1000 °C, the CoMn2O4 NPs formed on the CNFs surfaces were converted into Mn3Co7–Co2Mn3O8 NPs with oxygen vacancies.
The Mn 2p XPS spectra of the CoMn2O4/CNFs and Mn3Co7–Co2Mn3O8@CNTs/CNFs are shown in Fig. 3c. Three distinct spin–orbit peaks centered at 642.5, 653.9 and 646.3 eV are observed with a spin-energy separation of 11.4 eV, corresponding to the Mn 2p3/2, Mn 2p1/2 and satellite peaks of the Mn(IV) state.29,30 In addition, the Mn2+ state is also present, as indicated by the existence of a less prominent peak at 641.2 eV. The Mn 2p XPS spectra of the Mn3Co7–Co2Mn3O8@CNTs/CNFs show the predominant peaks shifting to lower BEs, and the peaks for Mn 2p3/2 and Mn 2p1/2 of the Mn4+ states shift to 643.3 and 653.4 eV a much weaker satellite peak of Mn4+. Therefore, the peaks of Mn4+ for the Mn3Co7–Co2Mn3O8@CNTs/CNFs indicate a different CoMn oxide state with a spin-energy separation of 10.1 eV, which is in the form of Co2Mn3O8 crystals. In addition, a new emerged peak located at 539.0 eV corresponds to the metallic Mn(0) state, further demonstrating the existence of metallic Mn on the surface of Co2Mn3O8 NPs.
As shown in curve II in Fig. 3c, the peak belonging to Mn2+ shifts to 640.7 eV, which indicates the strong reduction of Mn from CoMn2O4 to Co2Mn3O8 and the formation of metallic Mn3Co7 on the Co2Mn3O8 surface caused by the reduction of the surrounding carbon.31 The Mn/Co atomic ratio in the CoMn2O4/CNFs is 1.74, while the Mn/Co atomic ratio in the Co2Mn3O8/CNTs/CNFs is reduced to 0.67, which indicates the phase transformation of CoMn oxides during the CNT growth process. In addition, the Mn/Co ratio decreases from 1.74 to 0.67, which indicates that there are a certain amounts of metallic Mn3Co7 separated out from the Co2Mn3O8 phase and formed at the surface between the Co2Mn3O8 NPs and the carbon shell. The strong shifts of the Mn2+ oxidation state of the Mn3Co7–Co2Mn3O8@CNTs/CNFs to lower BEs is inevitable due to the valence degeneracy and strong charge transfer from the surrounding carbon.32 As is known, the BEs of Mn 2p3/2 can increase by increasing its valence state. The reduction of CoMn2O4 by the surrounding carbon in an Ar/NH3 atmosphere at 1000 °C leads to a shift of the Mn 2p spectrum to lower BEs, which suggests a decrease in the valence state of Mn ions with oxygen vacancies.33
The shifts of the BEs in the Co 2p and Mn 2p XPS spectra are possibly related to the partial removal of oxygen from the lattice of CoMn2O4, and indeed, there is also a decrease in the atomic ratio [O]/[Co + Mn] from 3.97 to 2.55. This means that the reduction certainly leads to the formation of oxygen vacancies in the CoMn2O4 structure. The O 1s spectra (curve a, Fig. 3d) of the CoMn2O4/CNFs contain four oxygen contributions. The BEs at 531.3, 531.9 and 532.5 eV are the characteristic peaks of hydroxyl groups and surface-adsorbed oxygen groups on the CNFs.34 The other peak at 530.3 eV corresponds to the lattice oxygen in the metal oxide (metal–O–metal) of CoMn2O4.35 The O 1s region of the Mn3Co7–Co2Mn3O8@CNTs/CNFs also shows four chemical states with BEs at 530.4, 531.5, 532.4 and 533.7 eV. The BE at 530.4 eV belongs to the oxygen of the Co2Mn3O8 nanocrystals. The other peaks with BEs at 531.5, 532.4 and 533.7 eV are assigned to the coordinatively unsaturated oxygen species, oxygen groups on the carbon surface and adsorbed water molecules.36 The results from the O 1s XPS spectra demonstrate that the surfaces of CNTs and CNFs possess some oxygen groups, which make the CNTs and CNFs hydrophilic. The results from XRD and XPS reveal the formation of oxygen-deficient Co2Mn3O8 nanostructures after the 1000 °C heating treatment.
Fig. 4 shows the evolution of the morphologies of the CoMnO@CNT/CNF nanostructures prepared at different temperatures. Fig. 4a and b display the FE-SEM and TEM images of the CoMn2O4/CNFs treated at 400 °C. Small CoMn2O4 NPs are formed uniformly on the surfaces of pre-oxidized CNFs. The size of the CoMn2O4 NPs ranges from 3 to 10 nm. The HRTEM image (the inset in Fig. 4b) clearly shows the typical lattice distance of the (400) planes of CoMn2O4 NPs. After the pre-oxidation process, the CoMn2O4/CNFs formed were further graphitized under Ar/NH3 conditions. Fig. 4c and d shows the morphology of the CoMnO/CNFs heated at 600 °C. We found that the CNFs became rough and porous as the size of CoMnO NPs (consisting of CoMn2O4 and Co2Mn3O8, Fig. 4c) increased. In addition, the CoMnO NPs were surrounded by several carbon layers, which resulted in the formation of CoMnO/carbon core/shell structures (Fig. 4d). This occurred because the CoMnO NPs could catalyze the graphitization of the surrounding carbon during the graphitization process. It should be noted that the treatment at 600 °C only leads to the formation of CoMnO/carbon core/shell structures.
 | ||
| Fig. 4 FE-SEM and TEM images (a and b) of the CoMn2O4/CNFs prepared by pre-oxidation at 400 °C under atmospheric conditions, (c and d) CoMn2O4–Co2Mn3O8/CNFs prepared by pre-oxidation and then graphitization at 600 °C under Ar/NH3 conditions, (e and f) the Co2Mn3O8@CNTs/CNFs prepared by pre-oxidation and then graphitization at 800 °C under Ar/NH3 conditions, and (g and h) the oxygen-deficient Mn3Co7–Co2Mn3O8@CNTs/CNFs prepared by pre-oxidation and then graphitization at 1000 °C under Ar/NH3 conditions. Insets in (a), (c), (e) and (g) are the corresponding high-magnification FE-SEM images and insets in (b), (d), (f) and (h) are the corresponding HRTEM images. | ||
When the graphitization temperature increases to 800 °C, small CNTs with Co2Mn3O8 NPs located at their tips are vertically grown on porous CNFs (Fig. 4e and f). The length of the CNTs grown ranges from 50 to 400 nm while the size of the encapsulated Co2Mn3O8 NPs ranges from 10 to 50 nm. Because of the high graphitization temperature, large amounts of the onion-like carbon are formed in the CNFs due to the catalytic effects of Co2Mn3O8 NPs. The encapsulated Co2Mn3O8 NPs are also surrounded by multilayer carbon, and the layers of carbon increase as the temperature increases. The HRTEM image (the inset in Fig. 4f) illustrates that the interlayer distance of the surrounding carbon is 3.6 Å, which corresponds to the (002) planes of carbon. Upon increasing the temperature to 1000 °C, large amounts of CNTs were grown densely on the CNFs to form the CNT forest architectures. The length of the grown CNTs increases to hundreds of micrometers. The graphitic porous CNFs encapsulated with oxygen-deficient Mn3Co7–Co2Mn3O8/carbon core/shell and onion-like carbon structures were also obtained.
HAADF-STEM and STEM-EDX mapping images further confirmed the structure and morphology evolutions and the CNT growth process. As shown in Fig. 5a and b, the STEM-EDS mapping images show distinct nanofibers decorated with small nanoparticles. Fig. 5b exhibits C, N, Co, Mn and O elements, demonstrating the formation of CoMn2O4 NPs on O-rich CNFs. As shown in Fig. 5c, the CoMnO/CNFs prepared at 600 °C display porous structures and the CoMnO nanoparticles were confirmed by the STEM-EDS mapping images (Fig. 5d). As shown in the STEM mapping images (Fig. 5e), the Co2Mn3O8@CNT/CNF architectures consist of branched N-doped CNTs with the encapsulated Co2Mn3O8 NPs at their tips and porous graphitic CNF hosts with some Co2Mn3O8@carbon core–shell structures and onion-like carbon nanostructures. Notably, the STEM-EDS mapping images of oxygen-deficient Mn3Co7–Co2Mn3O8@CNTs/CNFs prepared at 1000 °C also indicate that the CoMnO NPs at the tips of the N-doped CNTs serve as the standard catalyst for the growth of CNTs.
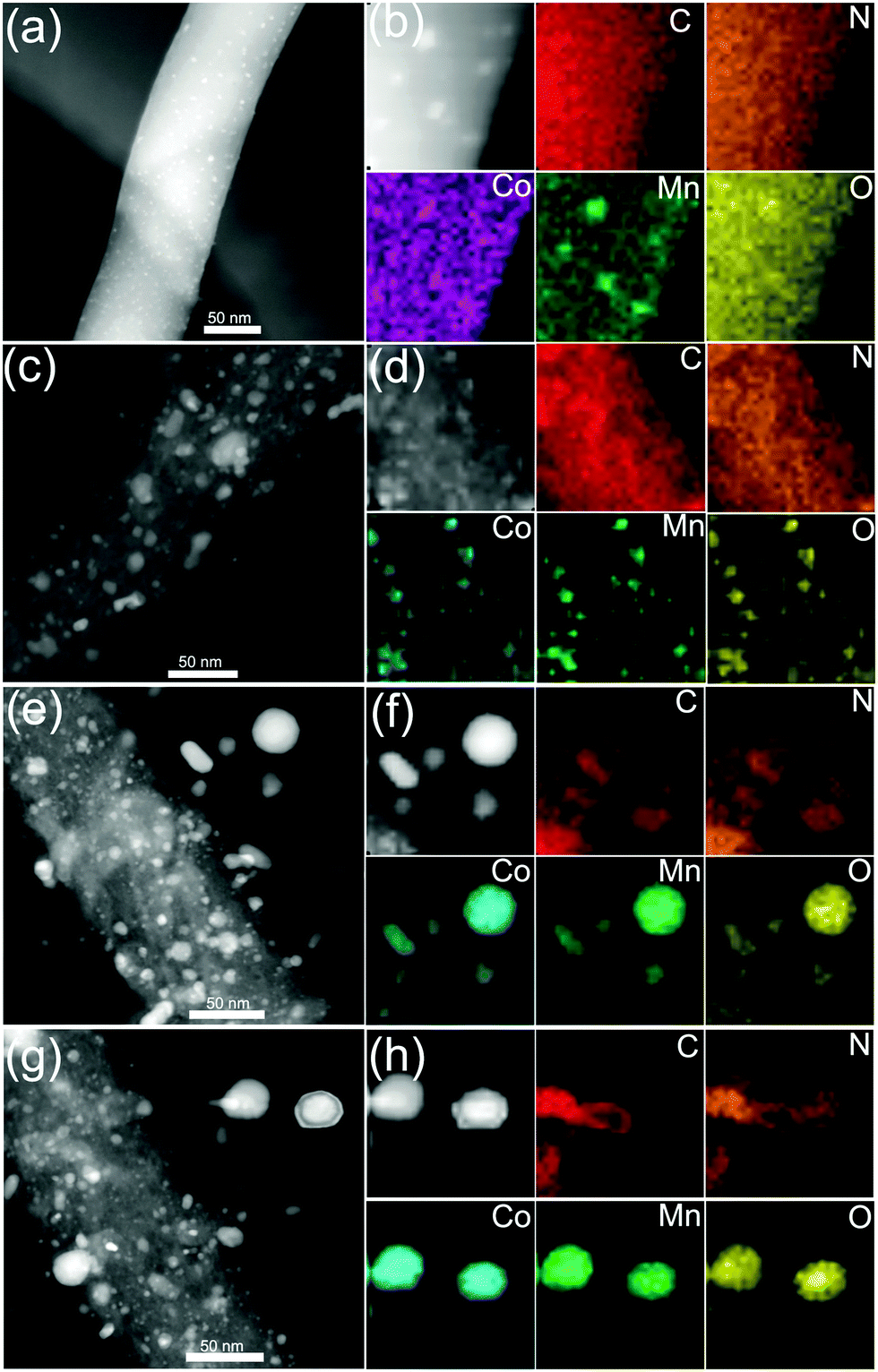 | ||
| Fig. 5 HAADF-STEM and STEM-EDX mapping images (a and b) of the CoMn2O4/CNFs prepared by pre-oxidation at 400 °C under atmospheric conditions, (c and d) CoMn2O4–Co2Mn3O8/CNFs prepared by pre-oxidation and then graphitization at 600 °C under Ar/NH3 conditions, (e and f) the Co2Mn3O8@CNTs/CNFs prepared by pre-oxidation and then graphitization at 800 °C under Ar/NH3 conditions, and (g and h) the oxygen-deficient Mn3Co7–Co2Mn3O8@CNTs/CNFs prepared by pre-oxidation and then graphitization at 1000 °C under Ar/NH3 conditions. | ||
We also made individual Co oxides/CNFs and Mn oxides/CNF hybrids under the same synthesis conditions. Here, no CNTs are observed on the CNFs, indicating that the individual Co or Mn oxide NPs cannot catalyze the growth of CNTs under the same conditions (Fig. S6 and S7, ESI†). Fig. S8 (ESI†) confirms that the as-formed Co oxides on the CNFs consist of metallic Co and CoO nanocrystals, indicating the formation of Co oxide structures with oxygen vacancies during the heat treatment process. Similarly, the Mn oxides/CNFs also show the existence of Mn, MnO2 and Mn3O4 nanocrystals (Fig. S9, ESI†). Both the Co–CoO/CNFs and Mn–MnOx/CNFs have large amounts of fullerene-like graphitic carbon pores in their CNFs, which indicate that the individual oxygen-vacancy Co and Mn oxides can only catalyze the graphitization of CNFs without supporting the growth of CNTs. Therefore, the oxygen-deficient Mn3Co7–Co2Mn3O8 NPs play an important role in the growth of CNTs and the graphitization of CNFs. The growth of CNTs begins with the phase transitions from CoMn2O4 to Co2Mn3O8, and the oxygen-vacancy Co2Mn3O8 NPs with Mn3Co7 on their surfaces can work well as catalysts for the growth of CNT forests. Due to the catalytic effects of Mn3Co7–Co2Mn3O8 NPs, the amorphous carbon can themselves form fullerene-like graphitic carbon layers surrounding the Mn3Co7–Co2Mn3O8 NPs. Compared with the traditional metal catalysts for the growth of CNTs, the oxygen atoms in Co2Mn3O8 NPs can improve the capture of carbon species and consequently facilitate the growth of CNTs on the surface of Mn3Co7–Co2Mn3O8 NPs. These findings demonstrate a new kind of transition metal oxide catalyst for the growth of CNTs and also highlight the importance of oxygen in the growth of CNTs.
The HER and OER activity of the CoMnO/CNTs/CNFs were investigated to illustrate the structure-dependent behavior (Fig. 6). The CoMn2O4/CNFs prepared at 400 °C without the CNT growth show low HER activity with a η−10 value (the overpotential at a current density of −10 mA cm−2) of 343 mV and a high Tafel slope of 245 mV decade−1, which indicates the poor HER activity of the CoMn2O4/CNFs (Fig. 6a and b). The CoMnO@CNTs/CNFs with small CNT tips (consisting of CoMn2O4 and Co2Mn3O8) prepared at 600 °C exhibit a smaller η−10 value of 232 mV and a Tafel slope of 139 mV decade−1 (Fig. 6a and b). When the temperature increases further to 800 °C, the Co2Mn3O8@CNTs/CNFs formed with small CNTs show a lower η−10 value of 218 mV and a Tafel slope of 136 mV decade−1, which demonstrates the improved HER activity (Fig. 6a and b). The electrocatalytic parameters are summarized in Table 1. The oxygen-deficient Mn3Co7–Co2Mn3O8@CNTs/CNFs with CNT forests (prepared at 1000 °C) exhibit the lowest η−10 value of 175 mV and a Tafel slope of 98 mV decade−1 (Fig. 6a and b), indicating that the growth of CNTs and encapsulated oxygen-deficient Co2Mn3O8 NPs with separated Mn3Co7 on their surfaces can greatly improve the HER activity. The electrocatalytic HER activity obtained is lower than that of recently reported transition metal oxide/carbon catalysts (Table S1, ESI†).37–39 The Nyquist plots of the CoMnO@CNT/CNF catalysts with various morphologies are obtained in 0.5 M H2SO4, shown in Fig. S10 (ESI†). The EIS results reveal dramatically decreased charge transfer resistance (RCT) for the Mn3Co7–Co2Mn3O8@CNTs/CNFs (∼12 Ω), revealing their more facile electrode kinetics and a faster diffusion rate of reactants for enhancing the catalytic activity (Fig. S10, ESI†). The 3D Mn3Co7–Co2Mn3O8@CNT/CNF membrane was further tailored into a 1 × 1 cm area and used directly as the electrode for the stability test. Fig. 6c shows that the Mn3Co7–Co2Mn3O8@CNT/CNF electrode exhibits a smaller decrease (11.8%) in the current density for HER under continuous operation for 10 h, whereas the Pt/C catalyst shows a relatively larger decrease (19.5%) during the continuous operation for 10 h.
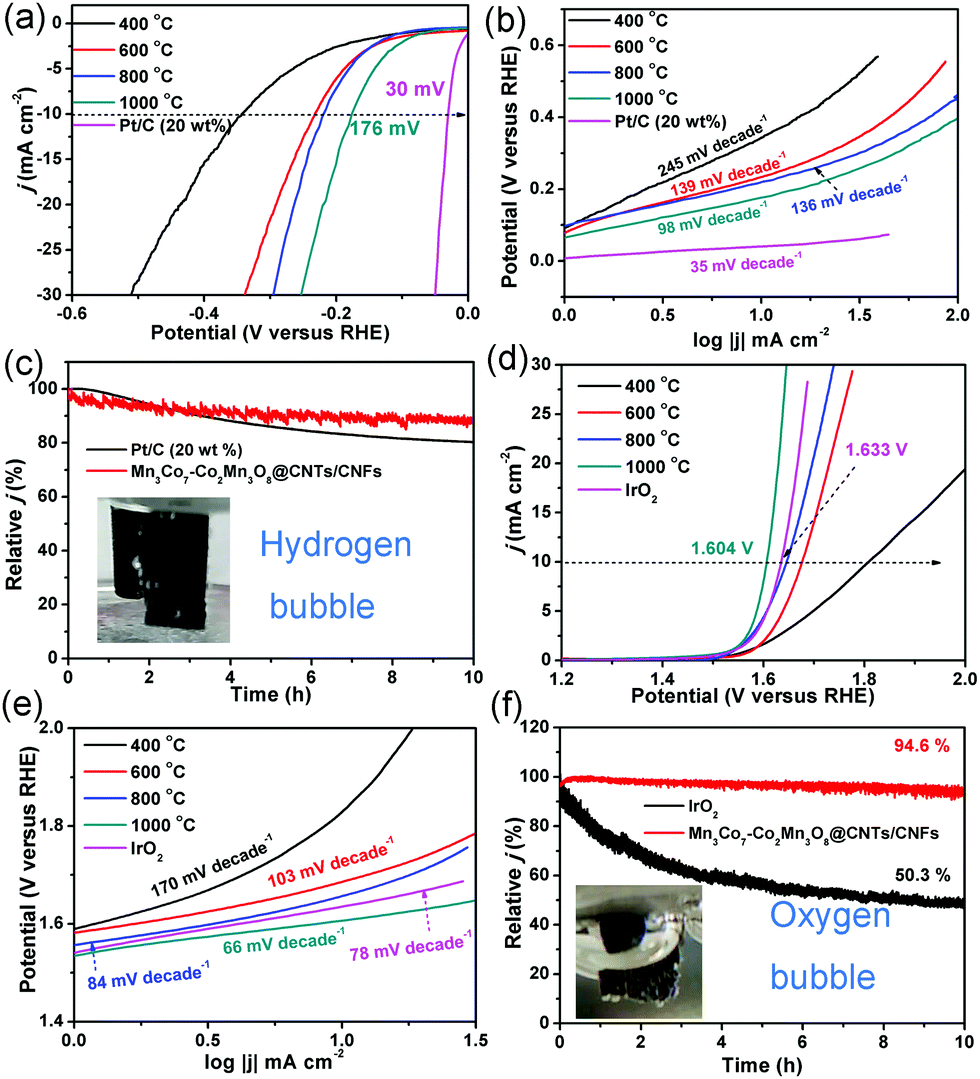 | ||
| Fig. 6 (a) HER polarization curves and corresponding (b) Tafel slopes of the CoMnO@CNT/CNF catalysts prepared at different temperatures (400–1000 °C) and commercial Pt/C (20 wt%) catalysts in 0.5 M H2SO4 solution. (c) The chronoamperometric responses at a constant potential of −0.20 V vs. RHE for the oxygen-deficient Mn3Co7–Co2Mn3O8@CNT/CNF electrode (prepared at 1000 °C) and commercial Pt/C catalysts. The inset in (c) is the photograph of the generation of H2 bubbles on the Mn3Co7–Co2Mn3O8@CNT/CNF membrane electrode. (d) OER polarization curves and corresponding (e) Tafel slopes of the CoMnO@CNT/CNF catalysts prepared at different temperatures (400–1000 °C) and commercial IrO2 (20 wt%) catalysts in 1 M KOH solution. The inset in (f) is the photograph of the generation of O2 bubbles on the Mn3Co7–Co2Mn3O8@CNT/CNF membrane electrode. | ||
| Reactions | Catalysts | ||||||
|---|---|---|---|---|---|---|---|
| CoMn2O4/CNFs (400 °C) | CoMn2O4–Co2Mn3O8/CNFs (600 °C) | Co2Mn3O8@CNTs/CNFs (800 °C) | Mn3Co7–Co2Mn3O8@CNTs/CNFs (1000 °C) | Pt/C | IrO2 | ||
| HER | η at 10 mA cm−2 (mV) | 345 | 233 | 220 | 176 | 30 | — |
| Tafel slope (mV dec−1) | 245 | 139 | 136 | 98 | 35 | — | |
| OER | η at 10 mA cm−2 (mV) | 577 | 447 | 410 | 374 | — | 404 |
| Tafel slope (mV dec−1) | 170 | 103 | 84 | 66 | — | 78 | |
| ORR | Onset potential (V) | — | — | — | 0.89 | 0.87 | — |
| Tafel slope (mV dec−1) | — | — | — | 70 | 70 | — | |
The OER activity of the CoMnO@CNTs/CNFs prepared at temperatures from 400 to 1000 °C is shown in Fig. 6d and e. With increased temperatures, the η10 values decrease from 591 to 374 mV and the Tafel slopes also decrease from 170 to 66 mV decade−1. The oxygen-deficient Mn3Co7–Co2Mn3O8@CNTs/CNFs prepared at 1000 °C display a η10 value of only 374 mV, which is over 160 mV lower than that of IrO2 catalysts (1.633 V, η10 = 403 mV). The OER activity achieved by the oxygen-deficient Mn3Co7–Co2Mn3O8@CNTs/CNFs compares favorably to that of recently reported OER catalysts, including N–Fe–CNT,25 Mn–Co/CNT,40 CP/CTs/Co–S,41 Co@Co3O4–CNTs,16 NiFe LDH,42 and La0.8Sr0.2MnO3−δ43 (Table S2, ESI†). The Tafel slope of the Mn3Co7–Co2Mn3O8@CNTs/CNFs is only 66 mV decade−1, which is lower than that of IrO2 (78 mV decade−1), further confirming the superior OER performance. The Mn3Co7–Co2Mn3O8@CNT/CNF electrode is very stable under continuous operation for 10 h, while the IrO2 catalyst displays a continuous loss in the current density during the OER operation (Fig. 6f). The results indicate that the change in temperature leads to the evolution of the morphology and structure of the CoMnO@CNTs/CNFs, and the oxygen-deficient structures exhibit enhanced OER and HER activity. Fig. S11 (ESI†) displays the morphologies and structures of the Mn3Co7–Co2Mn3O8@CNT/CNF electrode after continuous stability tests for HER and OER, respectively. The electrocatalysts can still keep the 3D architectures with branched CNTs and the Mn3Co7–Co2Mn3O8 did not dissolve in the acid or alkaline solution due to the protection of carbon shells in CNTs and CNFs, indicating their superior stability.
Based on these exciting OER results, the ORR activity of the oxygen-deficient Mn3Co7–Co2Mn3O8@CNTs/CNFs was also determined. As shown in Fig. 7a, a well-defined cathodic peak is clearly observed at ∼0.81 V for the Mn3Co7–Co2Mn3O8@CNTs/CNFs in O2-saturated 0.1 M KOH solution while no redox peaks were observed in Ar-saturated 0.1 M KOH solution, indicating their excellent ORR activity. The LSV of the Mn3Co7–Co2Mn3O8@CNTs/CNFs exhibits a half-wave potential (E1/2) of 0.82 V for ORR (Fig. 7b), which is better than that of Pt/C (0.81 V). The Tafel slope of the Mn3Co7–Co2Mn3O8@CNTs/CNFs is 70 mV decade−1, which is close to that of the Pt/C catalyst (70 mV decade−1) and indicates their excellent electrocatalytic activity for ORR. The LSVs of the Mn3Co7–Co2Mn3O8@CNTs/CNFs were further recorded at different rotating rates from 400 rpm to 3200 rpm (Fig. 7d). The electron-transfer numbers (n) of the Mn3Co7–Co2Mn3O8@CNTs/CNFs at different potentials are analyzed by the slopes of the linearly fitted Koutecky–Levich (K–L) plots using the K–L equation. The K–L plots exhibit first-order reaction kinetics with regard to the concentration of dissolved oxygen at various potentials (Fig. 7e). The value of n is calculated to be in the range of 3.41–3.73, which is close to the theoretical value of 4.00 for Pt/C and indicates a high selectivity for the four-electron-dominated ORR pathway. The ORR activity of Mn3Co7–Co2Mn3O8@CNTs/CNFs was also compared with those of recently reported state-of-art catalysts (Table S3, ESI†), further revealing their excellent ORR electrocatalytic performance. The stability test results (Fig. 7f) indicate that the Mn3Co7–Co2Mn3O8@CNT/CNF catalyst is very stable for the ORR (85.3% of current density remain) under continuous operation for more than 10 h, whereas the Pt/C exhibits a continuous current loss (54.2% of the initial current density remains) during the continuous operation.
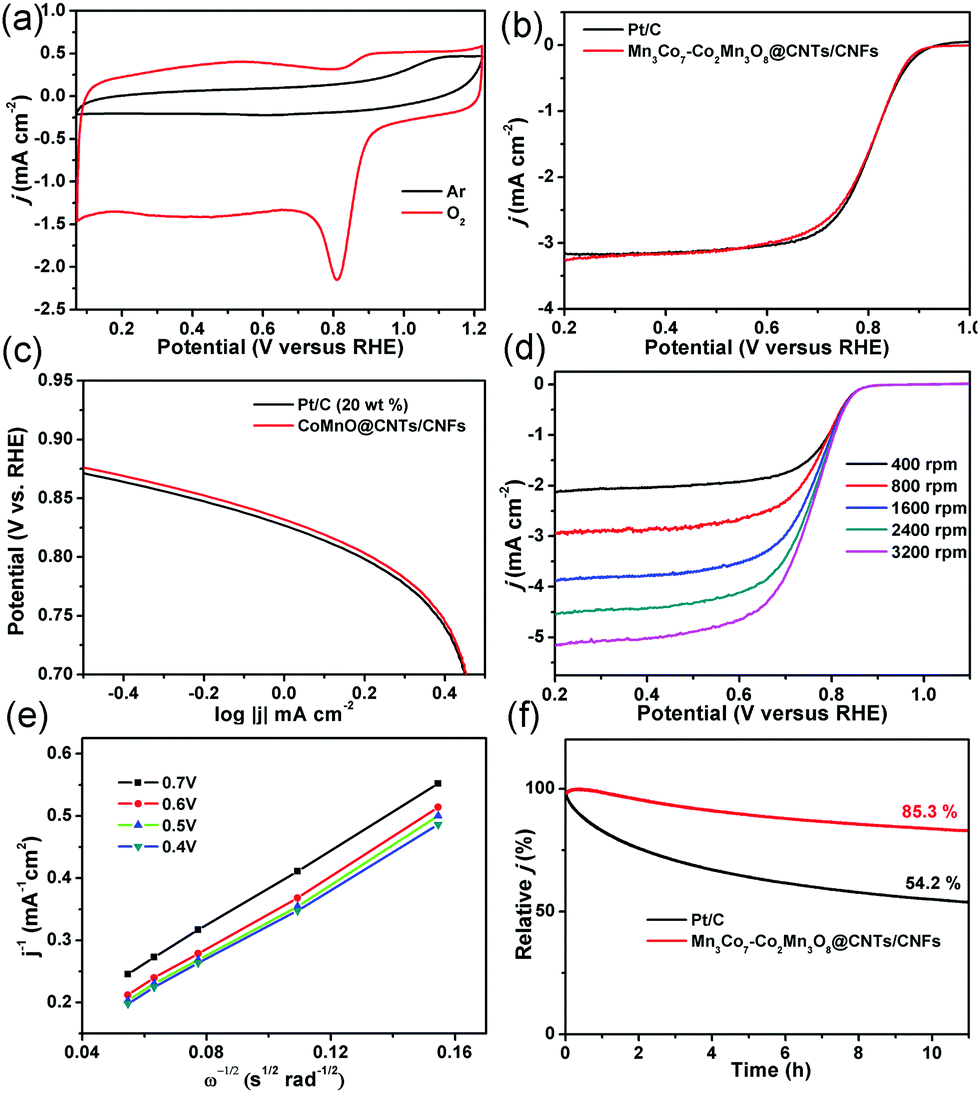 | ||
| Fig. 7 (a) CVs of the oxygen-deficient Mn3Co7–Co2Mn3O8@CNT/CNF catalyst (prepared at 1000 °C) in Ar- and O2-saturated 0.1 M KOH. (b) ORR polarization curves of the oxygen-deficient Mn3Co7–Co2Mn3O8@CNTs/CNFs and Pt/C (20 wt%) catalysts. (c) Tafel slopes of Pt/C (20 wt%) and Mn3Co7–Co2Mn3O8@CNTs/CNFs. (d) Polarization curves of oxygen-deficient Mn3Co7–Co2Mn3O8@CNT/CNF catalysts at different rotation rates. (e) The K–L plots of oxygen-deficient Mn3Co7–Co2Mn3O8@CNT/CNF catalysts at various potentials. (f) The chronoamperometric responses of the oxygen-deficient Mn3Co7–Co2Mn3O8@CNT/CNF electrode and Pt/C catalysts for ORR. | ||
The oxygen-deficient Co2Mn3O8@CNTs/CNFs with metallic Mn3Co7 on the Co2Mn3O8 surfaces exhibit excellent electrocatalytic activity and good stability for HER, OER and ORR. The integration of 1D CNTs, 1D CNFs and 0D oxygen-deficient Mn3Co7–Co2Mn3O8 along with their synergetic effect is vital for improving the kinetics of ORR, OER and HER. (a) Small Mn3Co7 alloy NPs are formed at the interfaces between the Co2Mn3O8 NPs and carbon shells. The oxygen vacancies can enhance the electron mobility and make the Co2Mn3O8 NPs easily accessible to O2, which enables them to serve as active sites for ORR and OER. The large amounts of O2 adsorbed by the oxygen vacancies in the Co2Mn3O8@CNTs/CNFs can react directly with the separated Mn3Co7 on the surfaces to form strong metal–O2 bonds, which leads to the elongation of the O–O bond and lowers the O2 reduction barrier.43–49 Therefore, by promoting the partial dissociation of metallic Mn3Co7 to form CoMn oxides and exposing more Mn/Co active centers, the oxygen-deficient Mn3Co7–Co2Mn3O8@CNTs/CNFs lead to a highly efficient ORR and OER activity. (b) During the HER process, the metallic Mn3Co7 on the surfaces can lower the free energy of adsorption of the oxides for hydrogen ions and enhance the hydrogen adsorption, which results in the low overpotential values. The 3D framework structures and oxygen-deficient Co2Mn3O8 can lower the gas desorption energy barrier for higher HER activity. (c) The surrounding carbon shells not only improve the conductivity of Mn3Co7–Co2Mn3O8 NPs but they also stabilize the oxygen vacancies during the electrocatalysis process. Moreover, the branched CNTs grown on CNFs possess a 3D hierarchical porous structure and a high surface area, which facilitates the diffusion of active species and enhances the mass transport during the electrocatalytic process. The unique 3D hierarchical framework structure formed by the interconnected CNTs provides excellent conductivity, stability and smooth pathways to facilitate the penetration of electrolytes and enlarge the contact surface between reactants and active sites. (d) Furthermore, the 3D hierarchical structure formed by the 1D CNTs and CNFs enables a facile release of evolved gas bubbles to further improve the reaction kinetics of the ORR, OER and HER. The surrounding carbon shells of the CoMnO NPs provide remarkable corrosion resistance in a harsh operating environment. Benefiting from the synergistic effects of the unique 3D hierarchical structures, the excellent conductivity of the CNTs and CNFs, and the oxygen vacancy structures of the Mn3Co7–Co2Mn3O8 NPs, the Mn3Co7–Co2Mn3O8@CNTs/CNFs work as highly efficient electrocatalysts for oxygen reduction reaction and water splitting. In addition, this strategy can be applied for constructing other organic materials. As shown in Fig. S12 (ESI†), the silk fiber can also be used as the fiber reactor to grow the CNTs with CoMnO as catalysts, leading to the formation of a 3D nanoarchitecture. The silk derived carbon fibers are densely covered by branched CNTs without any additional carbon resources. This work demonstrates a novel integration-and-marriage concept for developing superior catalysts for electrochemical energy devices.
Conclusions
In summary, by starting with the PAN nanofibers containing Co–Mn salts as the precursor, we report an effective strategy for the rational design and construction of 3D framework architectures based on the integration of 0D oxygen-deficient Mn3Co7–Co2Mn3O8 NPs, 1D N-doped CNTs and 1D graphitic porous CNFs. The optimized 3D nanostructure exhibits superior electrocatalytic activity and stability for ORR, OER and HER. The remarkable electrochemical properties are mainly attributed to the synergistic effects obtained from the engineering of oxygen-deficient Mn3Co7–Co2Mn3O8 with exposed active sites and the 3D hierarchical porous structure, which consists of branched CNTs and interconnected CNFs. This work demonstrates a novel integration-and-marriage concept for developing superior catalysts for electrochemical energy devices.Experimental
Fabrication of a 3D oxygen-deficient Mn3Co7–Co2Mn3O8@CNT/CNF architecture
In a typical procedure, 0.15 g Co(NO3)2 and 0.15 g Mn(CH3COO)2 were firstly dissolved in 13.5 g N,N-dimethylformamide (DMF) solution containing 10 wt% PAN, and then stirred for 10 h to get a homogeneous solution. The homogeneous solution was immediately transferred into a plastic syringe attached to a 23 gauge needle that was electrically connected to a high voltage power supply. The applied voltage was 10 kV, the needle-to-collector distance was 15 cm, and the flow rate of the solution was 0.6 mL h−1. The electrospun nanofibrous membranes were carefully detached from the home-built collector, and then dried at 80 °C for 3 h in an oven to vaporize the residual solvent. The direct growth of the Mn3Co7–Co2Mn3O8@CNT/CNF hybrid was carried out in a home-built chemical vapor deposition (CVD) furnace. The as-collected Co–Mn–PAN precursor nanofibrous membrane was placed in a ceramic boat located at the center of the heating zone of the furnace. The Co–Mo–PAN precursor nanofibrous membranes were firstly heated to 400 °C in air at a rate of 5 °C min−1 and maintained for 6 h for stabilization. Then, the furnace was heated up to 1000 °C at a rate of 5 °C min−1 and maintained under Ar/NH3 conditions for 6 h.Material characterization
Field emission scanning electron microscopy (FE-SEM) images were characterized using a JSM-6700F FE-SEM (JEOL, Japan) at an accelerating voltage of 3 kV. Transmission electron microscopy (TEM) images were taken using a JSM-2100 transmission electron microscope (JEOL, Japan) at an accelerating voltage of 200 kV. The high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images, STEM mapping, and line-scan energy dispersive X-ray spectroscopy (EDX) were recorded using a STEM (Tecnai G2 F30S-Twin, Philips-FEI) at an accelerating voltage of 300 kV. X-ray diffraction (XRD) patterns were analyzed using a Bruker AXS D8 DISCOVER X-ray diffractometer with Cu Kα radiation (λ = 1.5406 Å) at a scanning rate of 0.02 2θ s−1 in the 2θ range of 10–80°. X-ray photoelectron spectra (XPS) of the products were recorded using an X-ray photoelectron spectrometer (Kratos Axis Ultra DLD) with an Al (mono) Kα source (1486.6 eV). The Al Kα source was operated at 15 kV and 10 mA.Electrochemical characterization
The electrochemical experiments were performed in a three-electrode cell at 25 °C. The graphite rod was used as a counter electrode. The saturated calomel electrode (SCE) and Hg/HgO (1 M KOH) electrodes served as the reference electrodes for the 0.5 M H2SO4 and 1 M KOH electrolytes, respectively. The polarization curves were obtained at a sweep rate of 2 mV s−1 after the initial cyclic voltammograms (CVs) (Autolab potentiostat/galvanostat (Model PGSTAT302N) workstation) for 1000 cycles at a scan rate of 100 mV s−1 between 0 and 1.20 V vs. reversible hydrogen electrode (RHE). The potentials were referenced to RHE by adding a value of (0.244 + 0.059 pH) V for SCE and (0.99 + 0.059 pH) for Hg/HgO (1 M KOH). The Ohmic potential drop (iR) losses that arise from the solution resistance were all corrected. Tafel plots were recorded with the linear portions at low overpotential fitted to the Tafel equation (η = a + b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logj, where η is the overpotential, j is the cathodic current density, and b is the Tafel slope).
logj, where η is the overpotential, j is the cathodic current density, and b is the Tafel slope).
The catalyst ink was prepared by dispersing a 3 mg catalyst in 1 mL isopropanol/water solution (75/25 volumetric ratio). Next, 25 μL Nafion solution (5 wt%) was added to the ink as a proton conducting binder to ensure good adhesion onto the electrode. The catalyst ink was ultrasonicated for 30 min to generate a homogeneous ink. 5 μL of the catalyst ink was transferred onto a glassy carbon electrode (∅ 3 mm) that was previously buff polished using a suspension of alumina. The catalyst loading amount on the working electrode was 215 μg cm−2. The polarization curves were obtained at a sweep rate of 2 mV s−1 after the initial cyclic voltammograms for 1000 cycles at a scan rate of 100 mV s−1 between 0 and 1.20 V vs. RHE. The EIS was carried out in a potentiostatic mode in the frequency range of 105 to 0.1 Hz with an amplitude of 10 mV at −0.25 V vs. RHE in 0.5 M H2SO4. The stability test of the oxygen-deficient Mn3Co7–Co2Mn3O8@CNTs/CNFs was also used in the typical three-electrode system. The Mn3Co7–Co2Mn3O8@CNT/CNF membrane was tailored into a neat square (1 × 1 cm) and directly utilized as the working electrode in 0.5 M H2SO4 and 1 M KOH electrolytes. The CV profiles were obtained in N2- or O2-saturated 0.1 M KOH solution at a scan rate of 20 mV s−1. The RDE tests for ORR are measured in O2-saturated 0.1 M KOH solution at different rotation rates at a scan rate of 10 mV s−1.
Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (51373154, 51472182, 21571112, 51372125 and 51671003), the National Key Research and Development Program of China (No. 2016YFB0100201), the start-up supports from Peking University, Young Thousand Talented Program, the Program for Innovative Research Team of Zhejiang Sci-Tech University, and the 521 Talent Project of Zhejiang Sci-Tech University.Notes and references
- J. Luo, J. H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N. G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- C. Liu, B. C. Colón, M. Ziesack, P. A. Silver and D. G. Nocera, Science, 2016, 352, 1210–1213 CrossRef CAS PubMed.
- M. J. Kenney, M. Gong, Y. Li, J. Z. Wu, J. Feng, M. Lanza and H. Dai, Science, 2013, 342, 836–840 CrossRef CAS PubMed.
- J. Zhang, Z. Xia and L. Dai, Sci. Adv., 2015, 1, e1500564 Search PubMed.
- B. Zhang, X. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. García-Melchor, L. Han, J. Xu, M. Liu, L. Zheng, F. P. G. de Arquer, C. T. Dinh, F. Fan, M. Yuan, E. Yassitepe, N. Chen, T. Regier, P. Liu, Y. Li, P. D. Luna, A. Janmohamed, H. L. Xin, H. Yang, A. Vojvodic and E. H. Sargent, Science, 2016, 352, 333–337 CrossRef CAS PubMed.
- L. Bu, S. Guo, X. Zhang, X. Shen, D. Su, G. Lu, X. Zhu, J. Yao, J. Guo and X. Huang, Nat. Commun., 2016, 7, 11850 CrossRef CAS PubMed.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- H. B. Yang, J. Miao, S. F. Hung, J. Chen, H. B. Tao, X. Wang, L. Zhang, R. Chen, J. Gao, H. M. Chen, L. Dai and B. Liu, Sci. Adv., 2016, 2, e1501122 Search PubMed.
- J. Miao, F. X. Xiao, H. B. Yang, S. Y. Khoo, J. Chen, Z. Fan, Y. Y. Hsu, H. M. Chen, H. Zhang and B. Liu, Sci. Adv., 2015, 1, e1500259 Search PubMed.
- H. Fei, J. Dong, M. J. Arellano-Jiménez, G. Ye, N. D. Kim, E. L. G. Samuel, Z. Peng, Z. Zhu, F. Qin, J. Bao, M. J. Yacaman, P. M. Ajayan, D. Chen and J. M. Tour, Nat. Commun., 2015, 6, 8668 CrossRef CAS PubMed.
- H. Zhu, J. Zhang, R. Yanzhang, M. Du, Q. Wang, G. Gao, J. Wu, G. Wu, M. Zhang, B. Liu, J. Yao and X. Zhang, Adv. Mater., 2015, 27, 4752–4759 CrossRef CAS PubMed.
- H. Lin, N. Liu, Z. Shi, Y. Guo, Y. Tang and Q. Gao, Adv. Funct. Mater., 2016, 26, 5590–5598 CrossRef CAS.
- V. W. Lau, I. Moudrakovski, T. Botari, S. Weinberger, M. B. Mesch, V. Duppel, J. Senker, V. Blum and B. V. L. Lau, Nat. Commun., 2016, 7, 12165 CrossRef CAS PubMed.
- D. Li, H. Baydoun, C. N. Verani and S. L. Brock, J. Am. Chem. Soc., 2016, 138, 4006–4009 CrossRef CAS PubMed.
- A. Aijaz, J. Masa, C. Rçsler, W. Xia, P. Weide, A. J. R. Botz, R. A. Fischer, W. Schuhmann and M. Muhler, Angew. Chem., Int. Ed., 2016, 55, 4087–4091 CrossRef CAS PubMed.
- B. Y. Xia, Y. Yan, N. Li, H. B. Wu, X. W. Lou and X. Wang, Nat. Energy, 2016, 1, 15006 CrossRef.
- R. Gao, Z. Li, X. Zhang, J. Zhang, Z. Hu and X. Liu, ACS Catal., 2015, 6, 400–406 CrossRef.
- T. Xu, T. Shimada, Y. Araki and J. Wang, Nano Lett., 2015, 16, 454–458 CrossRef PubMed.
- R. Wu, J. Zhang, Y. Shi, D. Liu and B. Zhang, J. Am. Chem. Soc., 2015, 137, 6983–6986 CrossRef CAS PubMed.
- Y. Shen, L. Yan, H. Song, J. Yang, G. Yang, X. Chen, J. Zhou, Z. Z. Yu and S. Yang, Angew. Chem., Int. Ed., 2012, 51, 12202–12205 CrossRef CAS PubMed.
- W. Yang, X. Liu, X. Yue, J. Jia and S. Guo, J. Am. Chem. Soc., 2015, 137, 1436–1439 CrossRef CAS PubMed.
- K. N. Jung, S. M. Hwang, M. S. Park, K. J. Kim, J. G. Kim, S. X. Dou, J. H. Kim and J. W. Lee, Sci. Rep., 2015, 5, 7665 CrossRef CAS PubMed.
- H. Yan, C. F. Blanford, W. H. Smyrl and A. Stein, Chem. Commun., 2000, 1477–1478 RSC.
- H. T. Chung, J. H. Won and P. Zelenay, Nat. Commun., 2013, 4, 1922 CrossRef PubMed.
- Y. Qiu, G. Li, Y. Hou, Z. Pan, H. Li, W. Li, M. Liu, F. Ye, X. Yang and Y. Zhang, Chem. Mater., 2015, 27, 1194–1200 CrossRef CAS.
- J. Kang, A. Hirata, L. Chen, S. Zhu, T. Fujita and M. Chen, Angew. Chem., Int. Ed., 2015, 54, 8100–8104 CrossRef CAS PubMed.
- Y. J. Cho, C. H. Kim, H. S. Kim, W. S. Lee, S. H. Park and J. Park, Chem. Mater., 2008, 20, 4694–4702 CrossRef CAS.
- J. Masa, W. Xia, I. Sinev, A. Zhao, Z. Sun, S. Grützke, P. Weide, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2014, 53, 8508–8512 CrossRef CAS PubMed.
- X. Ge, Y. Liu, F. W. T. Goh, T. S. A. Hor, Y. Zong, P. Xiao, Z. Zhang, S. H. Lim, B. Li, X. Wang and Z. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 12684–12691 CAS.
- M. Ghiasi, M. U. Delgado-Jaime, A. Malekzadeh, R. P. Wang, P. S. Miedema, M. Beye and F. M. F. de Groot, J. Phys. Chem. C, 2016, 120, 8167–8174 CAS.
- J. Duan, S. Chen, S. Dai and S. Qiao, Adv. Funct. Mater., 2014, 24, 2072–2078 CrossRef CAS.
- D. Jeong, K. Jin, S. E. Jerng, H. Seo, D. Kim, S. H. Nahm, S. H. Kim and K. T. Nam, ACS Catal., 2015, 5, 4624–4628 CrossRef CAS.
- Y. Zhu, W. Zhou, J. Yu, Y. Chen, M. Liu and Z. Shao, Chem. Mater., 2016, 28, 1691–1697 CrossRef CAS.
- E. McCalla, A. M. Abakumov, M. Saubanère, D. Foix, E. J. Berg, G. Rousse, M. L. Doublet, D. Gonbeau, P. Novák, G. V. Tendeloo, R. Dominko and J. M. Tarascon, Science, 2015, 350, 1516–1521 CrossRef CAS PubMed.
- S. Chen, J. Duan, M. Jaroniec and S. Qiao, Adv. Mater., 2014, 26, 2925–2930 CrossRef CAS PubMed.
- Z. L. Wang, X. F. Hao, Z. Jiang, X. P. Sun, D. Xu, J. Wang, H. X. Zhong, F. L. Meng and X. B. Zhang, J. Am. Chem. Soc., 2015, 137, 15070–15073 CrossRef CAS PubMed.
- X. Xu, Y. Chen, W. Zhou, Z. Zhu, C. Su, M. Liu and Z. Shao, Adv. Mater., 2016, 28, 6442–6448 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, K. C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- A. Zhao, J. Masa, W. Xia, A. Maljusch, M. G. Willinger, G. Clavel, K. Xie, R. Schlögl, W. Schuhmann and M. Muhler, J. Am. Chem. Soc., 2014, 136, 7551–7554 CrossRef CAS PubMed.
- J. Wang, H. Zhong, Z. Wang, F. Meng and X. Zhang, ACS Nano, 2016, 10, 2342–2348 CrossRef CAS PubMed.
- M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef CAS PubMed.
- M. Risch, K. A. Stoerzinger, S. Maruyama, W. T. Hong, I. Takeuchi and Y. Shao-Horn, J. Am. Chem. Soc., 2014, 136, 5229–5232 CrossRef CAS PubMed.
- T. Y. Ma, Y. Zheng, S. Dai, M. Jaroniec and S. Z. Qiao, J. Mater. Chem. A, 2014, 2, 8676–8682 CAS.
- Q. Zhang, Z. Li, S. Wang, R. Li, X. Zhang, Z. Liang, H. Han, S. Liao and C. Li, ACS Catal., 2016, 6, 2182–2191 CrossRef CAS.
- F. M. Toma, A. Sartorel, M. Iurlo, M. Carraro, P. Parisse, C. Maccato, S. Rapino, B. R. Gonzalez, H. Amenitsch, T. D. Ros, L. Casalis, A. Goldoni, M. Marcaccio, G. Scorrano, G. Scoles, F. Paolucci, M. Prato and M. Bonchio, Nat. Chem., 2010, 2, 826–831 CrossRef CAS PubMed.
- H. B. Tao, L. Fang, J. Chen, H. B. Yang, J. Gao, J. Miao, S. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 9978–9985 CrossRef CAS PubMed.
- M. A. Ha and A. N. Alexandrova, J. Chem. Theory Comput., 2016, 12, 2889–2895 CrossRef CAS PubMed.
- H. A. Tahini, X. Tan, U. Schwingenschlögl and S. C. Smith, ACS Catal., 2016, 6, 5565–5570 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ee03054h |
| This journal is © The Royal Society of Chemistry 2017 |