
Emerging investigators series: prioritization of suspect hits in a sensitive suspect screening workflow for comprehensive micropollutant characterization in environmental samples†
Amy L.
Pochodylo
and
Damian E.
Helbling
*
School of Civil and Environmental Engineering, Cornell University, 220 Hollister Hall, Ithaca, NY, USA. E-mail: damian.helbling@cornell.edu; Fax: +1 607 255 9004; Tel: +1 607 255 5146
First published on 24th November 2016
Abstract
The emergence of suspect screening has enabled the comprehensive characterization of micropollutants in water systems. In this work, we developed a sensitive suspect screening workflow and applied it to characterize the occurrence of micropollutants in eighteen water samples collected from an urban water system in New York State. We used high-resolution mass spectrometry to collect full-scan and data-dependent tandem mass spectra from the water samples and compiled a suspect database that contained 1113 chemical substances including pesticides, pharmaceuticals, personal care products, and industrial chemicals. The suspect screening workflow included peak picking, suspect database matching, isotopic pattern scoring, a replication filter, blank subtraction and artifact removal, and clustering of suspect hits. Each step in the workflow relied only on the quality of the analytical data, and was optimized and validated using a set of compounds that covered a broad range of physicochemical properties. After applying the optimized suspect screening workflow to the data acquired from the water samples, we developed a series of prioritization strategies that ranked the resulting suspect hits according to metrics that we hypothesized would favor true positive detections. We then acquired authentic standards for suspect hits based on their ranking on the priority lists to confirm or reject their occurrence. With this approach, we confirmed the presence of 112 micropollutants in at least one of the eighteen water samples. Comparing these results to the scope of conventional micropollutant monitoring methods, we approximate that our suspect screening approach more than doubled the number of micropollutants that may otherwise have been identified.
Water impactMan-made chemicals such as pesticides, pharmaceuticals, and personal care products have been measured in water resources around the world. This work introduces a new approach to comprehensively characterize the occurrence of these so-called micropollutants in environmental samples. With this approach, we identified 112 micropollutants occurring in at least one sample collected from drinking water, wastewater, and surface water systems. |
Introduction
Micropollutants can be defined as synthetic organic chemicals that have been measured in water and wastewater systems at trace concentrations.1–3 Despite our growing understanding of micropollutant occurrence in water systems around the world, analytical methods that can be applied to evaluate micropollutant occurrence in water quality monitoring campaigns are still developing. Quantitative target screening remains the standard approach, wherein a sensitive analytical method is developed and validated, typically with liquid chromatography and mass spectrometry, and is applied to quantify the occurrence of a finite set of micropollutants in different types of aquatic matrices.4,5 Advances in target screening have led to the development of broad, multi-residue analytical methods that enable quantification of over one hundred micropollutants at low ng L−1 concentrations in a single analysis.6,7Despite the clear value of the data acquired during target screening, there remain limitations to this approach. First, target screening focuses water quality monitoring on a fixed set of micropollutants. However, the numbers and types of micropollutants that may occur in a water system are dependent on a variety of local features such as land use,8 proximity to industry,9 type of sewer system,10 type of wastewater treatment system,10 and population demographics.11 These factors suggest that analytical methods need to be flexible and easily adaptable to address the numbers and types of micropollutants that may occur in any region of interest. Second, target screening requires the use of authentic standards for the identification and quantification of target analytes. In addition to being laborious and economically inefficient, the process of selecting analytes for a target screening method is particularly challenging when the types of micropollutants that may be present in a water system are unknown. Finally, even the broadest multi-residue target screening methods only evaluate the occurrence of a fraction of the micropollutants that are expected to occur in any water system. As a result, risk assessments based on the results of target screening methods can significantly underestimate the chemical risk associated with micropollutant occurrence.12
Recent advances in high-resolution mass spectrometry (HRMS) have enabled the development of more comprehensive and versatile analytical methods for water quality monitoring without the need for authentic standards.13–16 Suspect screening is an emerging approach that relies on the high mass accuracy and high mass resolution afforded by HRMS to link features in mass spectral acquisitions to suspect chemicals that may occur in a sample.17 The general approach involves peak picking in the full-scan mass spectral acquisition and matching the accurate masses of the picked peaks to the exact masses of the major adducts (e.g., [M + H]+ or [M − H]−) and the theoretical isotopic patterns of suspect chemicals. Each match is a tentative detection of a suspect chemical and is referred to subsequently as a “suspect hit.” Suspect screening methods have been described for identifying suspect chemicals in a variety of matrices including water and wastewater,12 lake sediments,18 urine,19 and processed animal products.20
There are at least two important considerations that must be addressed prior to developing a new suspect screening workflow for a particular application. First, it is important to consider the numbers and types of chemical substances to be included as suspect chemicals. One approach is to “screen smart”, where a relatively small number of suspect chemicals is selected whose presence will provide key insights into a particular problem. For example, suspect screening has been applied to identify putative transformation products of sulfonamide antibiotics,21 photo-degradation products of iodinated contrast media,22 and biotransformation products of structurally-similar chemical substances.23,24 Another approach is to “screen big”, where a suspect database that includes thousands of suspect chemicals is built to represent the universe of likely chemical substances that may be present in a particular sample.13,16,25 This latter approach may be the most appropriate when the goal is comprehensive characterization of micropollutants in environmental samples, though large suspect databases should be applied with caution as more suspect chemicals are likely to result in more false positive detections. Second, it is important to consider how the suspect screening workflow should be optimized. Much recent research has focused on improving the false positive rate of suspect screening methods. Some commonly explored strategies include in silico prediction of the retention times26–28 or tandem mass spectral (MS2) fragments of suspect chemicals,13,29 data which can be incorporated into suspect screening workflows to further evaluate suspect hits. Others have considered intensity-dependent mass error adjustments30 or statistical rejection filters.31,32 Whereas these techniques have led to successful suspect screening discoveries and a general reduction in false positive rates, those benefits come at the expense of higher false negative rates, which narrow the comprehensiveness of the suspect screening. Another optimization approach is to balance the false positive and false negative rates,14,33,34 though the concession made in balancing the error rates may also lead to less comprehensive coverage of the suspect screening method. To the best of our knowledge, no suspect screening method has been described that explicitly aims to minimize the false negative rate to enable the most comprehensive characterization of micropollutant occurrence in a water system.
The goal of this research was to develop and apply a suspect screening workflow to comprehensively characterize the occurrence of micropollutants in an urban water system in New York State. To meet this goal, we collected water samples at the intake and from the finished water of a drinking water treatment plant (DWTP), at the influent and effluent of a wastewater treatment plant (WWTP), and from a surface water system that receives the effluent of the WWTP. We then: (i) developed and optimized a novel suspect screening workflow; (ii) validated the performance of the suspect screening workflow in each of the matrices; and (iii) applied the suspect screening workflow to the set of water samples to identify suspect micropollutants. Our approach was to “screen smart” while remaining comprehensive because the occurrence of micropollutants had never been assessed in the study area. Therefore, the suspect database contained 1113 chemical substances that have been reported as water-relevant micropollutants in water systems around the world and are likely to be detected by our HRMS analytical method. Additionally, we systematically optimized the suspect screening workflow to minimize the false negative rate. We then developed a series of novel prioritization strategies to rank suspect hits in a way that we expected would give priority to true positive detections. Authentic standards were acquired to confirm or reject the occurrence of all prioritized suspect chemicals.
Methods
Standards and reagents
All authentic standards were acquired from Sigma Aldrich except for emtricitabine and N,N-didesmethyl-venlafaxine which were acquired from Toronto Research Chemicals. Stock solutions of each chemical were prepared at a concentration of 0.5 or 1 g L−1 using 100% HPLC-grade methanol (EMD Millipore), 100% anhydrous ethanol (Decon Labs), ACS-grade dimethyl sulfoxide (Macron Fine Chemicals), HPLC-grade acetonitrile (Fisher Chemical), or nanopure water produced by a Milli-Q system (EMD Millipore) depending on the solubility of the chemical. The stock solutions were used to prepare authentic standards at a concentration of 100 μg L−1 or a chemical mix at a concentration of 5 mg L−1 using nanopure water. The stock solutions were stored in a freezer at −20 °C while the chemical standards and mix were stored in a refrigerator at 4 °C.Environmental sample collection
Water samples were collected from an urban water system in New York State during four sampling events in May, July, September, and December 2015. We collected time-proportional, 24 hour composite samples at the raw water intake and from a continuously-flowing stream of the finished drinking water of a DWTP and at the influent and effluent of a WWTP. We did not collect a sample of the finished drinking water in May 2015 due to logistical challenges. ISCO automatic samplers were used with a 15 minute sampling interval for all 24 hour composite samples. Samples were cooled on ice during sampling and during transportation to the laboratory for analysis. In addition to samples collected at the DWTP and the WWTP, a grab sample was collected in May, July, and September from a freshwater lake that receives the effluent of the WWTP; grab samples were collected at a depth of approximately 180 centimeters below the lake surface. A December sample was not collected from the freshwater lake due to ice cover on the lake surface.Sample enrichment
Within 48 hours of sample collection, 500 mL (May and July) or 1 L samples (September and December) were filtered (GF/F; 0.7 μm, Whatman) and pH-adjusted to 6.3–6.7 (using formic acid and ammonia solutions). We adapted a previously reported offline solid phase extraction (SPE) method for sample enrichment.35 The mixed bed cartridge was designed to enrich neutral, cationic, and anionic species with a broad range of polarities, though we acknowledge that some types of chemicals may not be adequately recovered with even broad SPE methods. More details on the SPE method are provided in the ESI.†Analytical method
We adapted the analytical method from one previously reported for the non-target and suspect screening of transformation products of organic micropollutants by means of high-performance liquid chromatography (HPLC) coupled with high-resolution mass spectrometry (HRMS, quadrupole-orbitrap, QExactive, Thermo Fisher Scientific).36,37 Briefly, the mobile phase consisted of LC/MS-grade water (A, Fisher Scientific) and HPLC-grade methanol (B, Fisher Scientific), each amended with 0.1% (volume) MS-grade formic acid (Fisher Scientific). The mobile phase was pumped to a reversed-phase analytical column (XBridge C18 column, 2.1 × 50 mm, particle size 3.5 μm, Waters) at a flow rate of 200 μL min−1. Other separation techniques including hydrophilic interaction liquid chromatography (HILIC) could enhance the breadth of chemicals that are separated into clearly identifiable peaks,15 though the majority of the chemicals included in this suspect screening are expected to be adequately separated by reversed-phase chromatography.12,14 The mass spectrometer was coupled with electrospray ionization and acquired full-scan mass spectra at an m/z (hereafter referred to as “mass”) range of 100 to 1000 in positive and negative polarity modes. Data-dependent tandem mass spectra (MS2) were acquired in separate experiments at the exact masses of all investigated micropollutants. MS2 experiments were performed on all authentic chemical standards to identify diagnostic MS2 fragments and on each of the samples following analysis and interpretation of the full-scan data to develop an inclusion list. Inclusion lists contained the exact masses of the [M + H]+ and [M − H]− adducts of all micropollutants detected in the suspect screening. We used XCalibur v3.1.66.10 (Thermo Fisher Scientific) software for analysis and interpretation of extracted ion chromatograms (EICs), mass spectra (MS), and tandem mass spectra (MS2). Further details on the instrument method and parameters for the MS and MS2 acquisitions are available in the ESI.†Compilation of a suspect database
The chemical substances included in the suspect database were collected from three sources: the “Eawag Compounds in MassBank” database, which contains 815 micropollutants known to occur in water resources around the world;38 the New York State Pesticide Product, Ingredient and Manufacturer System (PIMS) database, which contains all pesticides previously or currently registered for use in New York State;39 and the pharmaceutical compounds included in the United States Geological Survey (USGS) Wastewater Methods, which contain a number of pharmaceuticals that are particularly relevant for the study area.4,5 After compiling the chemical substances from these sources, we manually trimmed the list to include only those substances that are expected to be amenable to analysis by means of our HPLC HRMS method, as has been previously described.14 We removed all compounds with a mass less than 100 Da or greater than 1000 Da, all compounds that contained no carbon atoms, all compounds that contained no heteroatoms, and all compounds that incorporated a metallic element such as copper or iron. The final suspect database contained 1113 chemical substances including pesticides (e.g., herbicides, insecticides, biocides; 524), pharmaceuticals (423), lifestyle chemicals (e.g., personal care products, food additives; 30) and “other” compounds such as industrial chemicals and naturally produced chemicals (136). The database included the names, the SMILES notation, the molecular formula, and the exact masses of the neutral molecule and [M + H]+ and [M − H]− adducts for each of the 1113 chemical substances.Dilution series of validation compounds
We selected 45 chemical substances from the suspect database to serve as validation compounds in the optimization and validation of the suspect screening workflow. The 45 validation compounds included 25 pharmaceuticals or pharmaceutical transformation products (TPs), 17 pesticides or pesticide TPs, 2 lifestyle chemicals, and 1 industrial chemical. The 45 chemical substances were selected to represent chemicals with a broad range of physicochemical properties and ionizable functional groups. More details on the 45 validation compounds are provided in the ESI.† An analytical mix containing the 45 validation compounds was prepared at a concentration of 5 mg L−1 and diluted in nanopure water to generate a dilution series at volumes of 1 L and individual compound concentrations of 0 ng L−1, 5 ng L−1, 25 ng L−1, 50 ng L−1, 100 ng L−1, 250 ng L−1, 350 ng L−1, 500 ng L−1, and 750 ng L−1. Each sample in the dilution series was concentrated by means of SPE and analyzed by means of HPLC HRMS as described in the preceding.Results and discussion
The goal of this research was to develop a suspect screening workflow and apply it to comprehensively characterize the occurrence of micropollutants in an urban water system in New York State. To meet this goal, we first developed and optimized a suspect screening workflow and then validated its performance in a variety of environmental matrices. We finally applied the validated suspect screening workflow to characterize the occurrence of micropollutants in eighteen water samples. The steps involved in the development and optimization, validation, and application of the suspect screening workflow are provided schematically in Fig. 1 and described in the following. | ||
| Fig. 1 Schematic of the steps involved in the development and optimization, validation, and application of the suspect screening workflow. | ||
Development and optimization of suspect screening workflow
We used the full-scan mass spectra acquired from the dilution series containing the 45 validation compounds to develop and optimize a suspect screening workflow, and used the full suspect database containing 1113 suspect chemicals to identify suspect hits. We evaluated a number of potential steps to be included in the suspect screening workflow. After application of each step, we identified the number of suspect hits that matched one of the 45 validation compounds (true positives, TR), the number of suspect hits that did not match one of the 45 validation compounds (false positives, FP), and the number of validation compounds for which there were no matching suspect hits (false negatives, FN). We then calculated method sensitivity as TR/(TR+FN) and method selectivity as TR/(TR+FP). The workflow was systematically optimized with the primary objective of maximizing method sensitivity and a secondary objective of maximizing method selectivity. As such, we didn't include any steps in the final suspect screening workflow that relied on in silico predictions of the properties of the suspect chemicals13,26–29 that increased the false negative rate. Instead, we included a series of conservative suspect screening steps that relied solely on the quality of the analytical data and were systematically optimized to meet our objectives. The steps included in the final suspect screening workflow were peak picking, suspect database matching, isotope pattern scoring, a replication filter, blank subtraction and artifact removal, and clustering of suspect hits.We used the TraceFinder v3.1 software for peak picking, though a number of open source software packages are available for peak picking within high-resolution mass spectra.40,41 Peak picking algorithms rely on a number of user-defined peak picking parameters that determine how mass spectra are clustered and whether or not a cluster of mass spectra will be defined as a peak. We systematically adjusted the magnitude of each peak picking parameter to investigate its effect on the results. We then compared the accurate masses of each of the picked peaks with the exact masses of the [M + H]+ and [M − H]− adducts of each of the 1113 chemicals in the suspect database to identify suspect hits. Other major adducts such as [M + Na]+ or [M + NH4]+ were not considered during suspect database matching because our analyses demonstrated that inclusion of other adducts did not improve the method sensitivity but significantly lowered the method selectivity. We optimized each peak picking parameter by identifying the parameter value that resulted in identification of all 45 validation compounds while minimizing the total number of suspect hits identified in representative high (750 ng L−1) and low (25 ng L−1) concentration samples of the dilution series. The parameters that had the largest influence on the results of peak picking were the area noise factor, the peak noise factor, the baseline window, the peak area threshold, and the signal-to-noise ratio. Details on the optimization of each of these parameters are provided in the ESI.†
The optimized peak picking and suspect database matching routine was applied to the full-scan mass spectra acquired from the representative high and low concentration samples from the dilution series. A total of 893 and 647 suspect hits were identified in each of the samples, respectively, as shown in Fig. 2. Peaks representing each of the 45 validation compounds were picked in both samples reflecting a method sensitivity of 100%. However, the large number of suspect hits yielded a poor method selectivity of 6.0% across the dilution series. Therefore, additional suspect screening workflow steps were developed to reduce false positive suspect hits and improve the method selectivity.
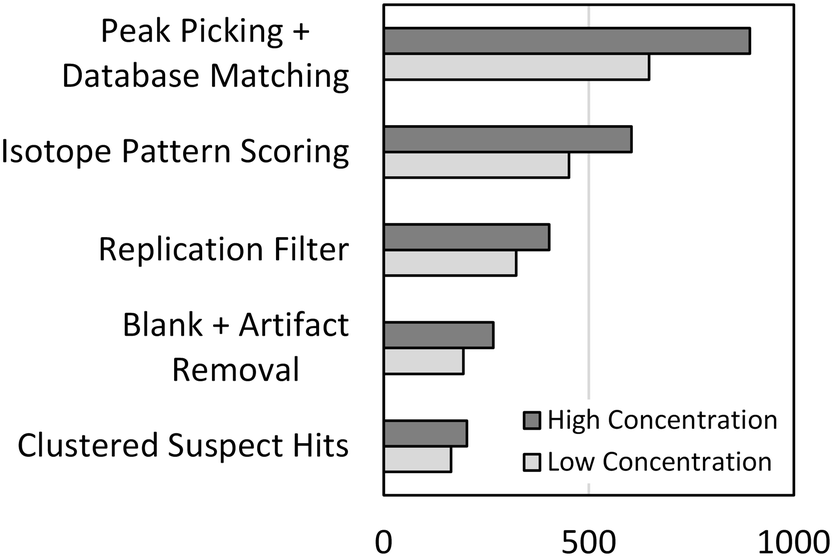 | ||
| Fig. 2 The total number of suspect hits identified in the high and low concentration samples from the dilution series after peak picking and suspect database matching and the number of suspect hits remaining after each step of the suspect screening workflow. | ||
Isotopic pattern scoring can be applied to suspect screening workflows to remove suspect hits that do not contain an isotopic pattern matching the theoretical isotopic pattern of the suspect chemical. Isotopic pattern scores can be assigned in TraceFinder or other software packages based on deviations between the measured and predicted masses and intensities of the isotopic pattern. We optimized the isotopic pattern scoring in the same way that we optimized the peak picking parameters as described in the preceding, details of which are available in the ESI.† After applying the optimized isotopic pattern scoring routine to the high and low concentration samples of the dilution series, the total number of suspect hits was reduced to 604 and 452, respectively, as shown in Fig. 2. Isotopic pattern scoring had no effect on method sensitivity, but the reduction in the total number of suspect hits resulted in an improved method selectivity of 8.8%.
The remaining steps in the suspect screening workflow were developed to remove false positive suspect hits resulting from analytical noise or matrix constituents. First, the replication filter was developed to remove suspect hits that were not detected robustly over replicate analytical injections from the same sample. We reasoned that the peak picking algorithm may pick peaks related to noise or other transient substances in any single analytical injection, but chemical substances that are present and stable in the sample should generate a robust series of picked peaks across a set of multiple injections. We determined that three replicate analytical injections was sufficient to eliminate a significant number of false positive suspect hits resulting from analytical noise, as detailed in the ESI.† Second, the blank subtraction and artifact removal step was added to remove matrix constituents from the list of suspect hits. For blank subtraction, we removed suspect hits from a sample if a suspect hit was present in the 0 ng L−1 sample (the blank) and had a peak area greater than or equal to the peak area measured for that suspect hit in the sample. This is a conservative approach that does not incorporate a peak area amplifier into blank subtraction as has been reported elsewhere.12 Instead, we developed artifact removal which removed all suspect hits that were identified in every sample of the dilution series and had peak areas that did not vary significantly over the dilution series. Artifact removal enables removal of matrix constituents that may be present in the blank at slightly lower peak areas than in the sample without the need for applying an arbitrary amplifier. Finally, the clustering of suspect hits removes all extra annotations of a single suspect chemical to generate a list of unique suspect hits identified in each sample. The effects that each of these steps had on reducing the total number of suspect hits are presented in Fig. 2. After applying the full suspect screening workflow, the total number of suspect hits was reduced to 203 and 164 in the high and low concentration samples from the dilution series, respectively. One of the validation compounds (primidone) was lost from the low concentration sample during the blank subtraction step resulting in a method sensitivity of 98.9%. However, the continued reduction in the total number of suspect hits resulted in an improved method selectivity of 24.8%. It is important to note that selectivity is a function of the number of chemical substances contained in the suspect database. For example, if the suspect database contained only the 45 validation compounds, the selectivity of the suspect screening workflow would be 100%. Therefore, the optimization of the suspect screening workflow resulted in a highly sensitive method that has a method selectivity of approximately 25% when the suspect database contains 1113 chemical substances.
Validation of suspect screening workflow
The goal of the method validation was to evaluate the performance of the suspect screening workflow when applied to water samples with varying matrices. Therefore, we applied the optimized suspect screening workflow to the full-scan mass spectra acquired from the eighteen environmental samples and used a suspect database containing only the 45 validation compounds to identify suspect hits. Simultaneously, we manually inspected extracted ion chromatograms (EICs) at an extraction width of 5 ppm, MS data, and MS2 data from each of the eighteen water samples to confirm or reject the occurrence of each of the 45 validation compounds. A validation compound was confirmed to occur in a sample when the EIC contained a chromatographic peak containing at least five MS scans, and the retention time, MS spectra, and MS2 fragments matched those of the authentic standard. Suspect hits that were confirmed to occur following manual inspection of the analytical data were defined as true positives. Suspect hits that were rejected following manual inspection of the analytical data were defined as false positives. Validation compounds that were confirmed to occur following manual inspection of the analytical data but were not identified as suspect hits were defined as false negatives.An accounting of the true positive and false negative detections is presented in Fig. 3. Twenty four of the 45 validation compounds were confirmed to occur in at least one of the eighteen water samples. The suspect screening yielded a total of 349 suspect hits among the eighteen water samples; 203 of those were confirmed as true positives and the remaining 146 were false positives. The majority of false positives were identified as such based on non-matching retention times when compared to the authentic standard. While an in silico retention time prediction step may have eliminated many of the false positive hits, the increased selectivity would have come at the expense of reduced sensitivity due to the uncertainty inherent in retention time prediction. This observation highlights the need for improved in silico tools for the prediction of retention times of suspect chemicals in liquid chromatography applications. Some false positive suspect hits were also rejected following inspection of MS or MS2 spectra. These were substances that were measured with very low intensities that either did not yield strong MS signals or did not trigger the dd-MS2 experiment. These substances could be considered as true positives that were below the limit of detection of the suspect screening workflow, but were conservatively identified as false positives here.
 | ||
| Fig. 3 An accounting of the 24 validation compounds that were confirmed to occur in at least one of the eighteen water samples. TR indicates that the detection was a true positive in the suspect screening workflow; FN indicates that the detection was a false negative in the suspect screening workflow and only identified following manual inspection of the analytical data. 5-Methyl-1H-benz is short for 5-methyl-1H-benzotriazole. | ||
There were also 24 instances in which validation compounds were confirmed to occur following manual inspection of the analytical data but were not identified as suspect hits and are therefore false negatives. The majority of the compounds that were identified as false negatives were filtered out of the suspect screening workflow during the isotopic pattern scoring step, though the isotopic pattern was clear upon manual inspection. Adjusting the isotopic pattern scoring threshold or the allowable mass and intensity deviations did not improve the performance of the suspect screening method, so no modifications were made to the suspect screening workflow based on this observation. Other validation compounds were lost during the replication filter, particularly substances that were present at low intensity and in complex wastewater influent or effluent matrices. Together, these observations suggest that the limit of detection of our suspect screening workflow will not be as low as the limit of detection of an analogous targeted analytical method. Nevertheless, the method sensitivity across varying matrices based on these results was 89.4% and the method selectivity was 58.2%.
Application of suspect screening workflow
We applied the optimized suspect screening workflow to the full-scan mass spectra acquired from the eighteen water samples collected from an urban catchment in New York State and used the full suspect database containing 1113 chemical substances. The results of peak picking and suspect database matching yielded an average of 2000 suspect hits per sample, reflecting the chemical complexity of the environmental samples. It is important to note that these 2000 suspect hits are not necessarily unique suspect chemicals. Rather, some suspect chemicals were counted multiple times because multiple peaks were picked at a single accurate mass or were detected in each polarity mode, allowing the total number of suspect hits counted at this step to be greater than the actual number of chemical substances contained in the suspect database. However, the average percent reduction in suspect hits was greater after each step of the suspect screening workflow for the environmental samples than the optimization samples as detailed in the ESI,† though the difference was not always significant. The average number of suspect hits remaining for each of the eighteen water samples following the clustering of suspect hits was 200, which compares well in magnitude to the number of suspect hits remaining following method optimization. This average number of suspect hits ranged between 99 in the May DWTP influent to 344 in the September WWTP effluent. A total of 534 unique suspect hits were identified among the eighteen water samples. Based on the criteria established during the development and optimization of the suspect screening workflow, each of the suspect hits had: a chromatographic peak that was picked by the TraceFinder software with a peak area greater than 1E6 (arbitrary units) and an accurate mass that matched the exact mass of a major adduct of a suspect chemical within a mass tolerance of 5 ppm; a TraceFinder isotopic pattern score greater than 65% with mass deviations of theoretical isotopes within 10 ppm and intensity deviations of theoretical isotopes within 10%; presence of these analytical features in triplicate measurements of the same sample; and either absence of these analytical features in the blank or absence from a list of analytical artifacts.Prioritization of suspect hits for confirmation
The challenge that remains following application of a sensitive suspect screening workflow is defining a means to prioritize the suspect hits in a way that favors true positive detections. For example, a recent study described a method to prioritize suspect hits based on an expected threshold of toxicological concern.42 The authors approximated the concentration and toxicological impact of each suspect hit based on the relative height of the picked peak. While the prioritization was biased towards suspect hits with larger peak heights, this prioritization strategy enabled the authors to confirm 24 of the suspect hits.42We developed two groups of novel prioritization strategies that ranked the resulting suspect hits according to metrics that we hypothesized would favor true positive detections. We then acquired authentic standards (when available) for suspect hits in the order in which they were ranked on the priority list and collected analytical data for confirmation of the suspect hits. We continued evaluating suspect hits on each priority list until we investigated the top 30 suspect hits or the running selectivity of the prioritization dropped below 60%, whichever came later. We selected 60% as the running selectivity threshold based on the results of method validation; we reasoned that attaining the selectivity obtained when applying the suspect screening method with a suspect list that contained only 45 chemical substances would be an ambitious benchmark to achieve with a larger suspect database. The running selectivity was calculated as the selectivity of the method as a function of the number of true positives and false positives identified as we evaluated the priority list. Suspect hits for which authentic standards were not acquired were included in the calculation of running selectivity and were assigned a selectivity of 25%, which is based on the conservative assumption that only one out of four suspect hits for which authentic standards were not acquired is a true positive, as was observed during method optimization.
The first group of prioritization strategies was based on Web of Science (WOS) searches for each of the 534 suspect hits using the search string “environment* AND water AND [name of suspect hit]”. We ranked each of the suspect hits based on the number of WOS search returns that were received for each suspect hit as of February 2016. We reasoned that suspect hits with more WOS search returns would be more likely to occur in our water samples. As is summarized in Table 1, the WOS prioritization resulted in the investigation of 36 suspect hits with an authentic standard and 22 of those were confirmed for a confirmation rate of 61%. The running selectivity of the method dropped to 60% after 38 suspect hits were investigated, and there were two suspect hits for which authentic standards could not be acquired. We then coupled the WOS ranking with other metrics aiming to further refine the prioritization of the suspect hits. For example, we developed a priority list based on the WOS rankings and considered only suspect hits that were present in both the WWTP influent and effluent samples during at least two sampling events (WOS + WWTPs). We reasoned that this strategy would prioritize persistent wastewater-derived micropollutants. We investigated 63 suspect hits based on this prioritization and confirmed 46 of them, 36 of which were additional unique confirmations beyond the WOS priority list alone. We also prioritized suspect hits based on the WOS ranking of suspect hits present in all lake samples (WOS + lake), present in all DWTP intake samples (WOS + DWTP), containing at least one chlorine atom (WOS + Cl), contained in the USGS wastewater methods (WOS + USGS), contained in the New York State PIMS database (WOS + PIMS), and pharmaceuticals that were present in the WWTP influent and effluent during at least two sampling events (WOS + WWTPs + pharmaceuticals). All combinations resulted in the confirmed identification of unique compounds beyond the WOS priority list alone. The results of these prioritization strategies are summarized in Table 1.
| Length of list | Cmpds Investigated | Confirmed Cmpds | % confirmed | Unique confirmationsa | % unique confirmations | |
|---|---|---|---|---|---|---|
| a Unique confirmations are confirmations made beyond the WOS prioritization alone. For the WOS + WWTPs + Pharms prioritization strategy, unique confirmations are confirmations beyond the WOS + WWTPs prioritization. | ||||||
| Web of Science (WOS) | 38 | 36 | 22 | 61% | — | — |
| WOS + WWTPs | 87 | 63 | 46 | 73% | 36 | 78% |
| WOS + lake | 30 | 23 | 15 | 65% | 12 | 80% |
| WOS + DWTP | 30 | 18 | 14 | 78% | 12 | 86% |
| WOS + Cl | 33 | 26 | 18 | 69% | 12 | 67% |
| WOS + USGS | 89 | 57 | 45 | 79% | 36 | 80% |
| WOS + PIMS | 30 | 25 | 12 | 48% | 5 | 42% |
| WOS + WWTPs + Pharms | 97 | 69 | 51 | 74% | 24 | 47% |
The second group of prioritization strategies was based on the maximum peak area recorded for each suspect hit. We reasoned that there would be greater confidence in the results of each of the steps in the suspect screening workflow for suspect hits with larger peak areas. Further, this was a means to prioritize suspect hits in a way that is independent of whether or not the suspect chemical has been previously reported as a water pollutant. As is summarized in Table 2, the peak area prioritization resulted in 44 suspect hits investigated with an authentic standard and 38 of those were confirmed for a confirmation rate of 86%. The running selectivity of the method reached 60% after 77 suspect hits were investigated. We coupled the peak area metric to the same metrics described for the WOS search and those results are presented in Table 2.
| Length of list | Cmpds investigated | Confirmed Cmpds | % confirmed | Unique confirmationa | % unique confirmations | |
|---|---|---|---|---|---|---|
| a Unique confirmations are confirmations made beyond the peak area prioritization alone. For the peak area + WWTPs + Pharms prioritization strategy, unique confirmations are confirmations beyond the peak area + WWTPs prioritization. | ||||||
| Peak area | 77 | 44 | 38 | 86% | — | — |
| Peak area + WWTPs | 78 | 44 | 38 | 86% | 3 | 8% |
| Peak area + lake | 32 | 19 | 16 | 84% | 1 | 6% |
| Peak area + DWTP | 30 | 18 | 14 | 78% | 2 | 14% |
| Peak area + Cl | 42 | 26 | 21 | 81% | 20 | 95% |
| Peak area + USGS | 97 | 60 | 49 | 82% | 22 | 45% |
| Peak area + PIMS | 30 | 15 | 9 | 60% | 7 | 78% |
| Peak area + WWTPs + Pharms | 81 | 48 | 40 | 83% | 17 | 43% |
In total, the WOS group of prioritization strategies enabled the confirmation of 103 suspect hits and the peak area group of prioritization strategies enabled the confirmation of 92 suspect hits. Many suspect hits were prioritized and confirmed in both strategies, but 20 suspect hits were only prioritized and confirmed in the WOS group and 9 suspect hits were only prioritized and confirmed in the peak area group. Plots of the running selectivity for each of the prioritization strategies and a complete list of all suspect hits compared with an authentic standard are provided in the ESI.† Suspect hits that were not confirmed or rejected with an authentic standard are not discussed in this manuscript.
Confirmed compounds
The application of our suspect screening workflow and subsequent prioritization of suspect hits resulted in the confirmation of 112 micropollutants in at least one of the eighteen water samples. An accounting of the 88 micropollutants that were confirmed beyond the 24 confirmed during method validation are presented in Fig. 4. In addition, 58 suspect hits were evaluated and identified as false positives following comparison with an authentic standard. Therefore, when considering only the suspect hits that were investigated based on their inclusion on a priority list, the final suspect screening method had a selectivity of 65.9%. This selectivity is significantly greater than the selectivity observed during method optimization and validation, demonstrating the efficacy of our prioritization strategies for ranking suspect hits in a way that favors true positives. | ||
| Fig. 4 An accounting of the 88 suspect chemicals that were confirmed to occur in at least one of the eighteen water samples. All confirmed suspect chemicals are true positives (TR).1 Carbamazepine-10,11-epoxide;2 dextromethorphan;3 ethyl 3-(N-butylacetamido) propionate;4 hydrochlorothiazide; 5N4-acetylsulfamethoxazole;6 perfluorobutyric acid;7 perfluorooctanoic acid;8 tris(1,3-dichloro-2-propyl)phosphate;9 tris(2-chloro-ethyl) phosphate. | ||
We compared the numbers of micropollutants confirmed by our suspect screening approach to the numbers that may otherwise have been identified using more conventional target screening approaches. The USGS National Water Quality Laboratory maintains an index of target screening methods for micropollutants in water and wastewater matrices. When five of the most comprehensive methods are combined,4,5,43–45 they enable target screening for over 250 micropollutants amenable to analysis by HPLC HRMS including pharmaceuticals, pesticides, personal care products and industrial chemicals. Of the 112 micropollutants identified in this research, 54 of them are included in these target screening methods. The fractions of micropollutants identified in each of our water samples that are or are not included in these target screening methods is provided in Fig. 5. Based on this comparison, we approximate that our suspect screening approach more than doubled the number of micropollutants that may have otherwise been identified, even with very a comprehensive target screening approach.
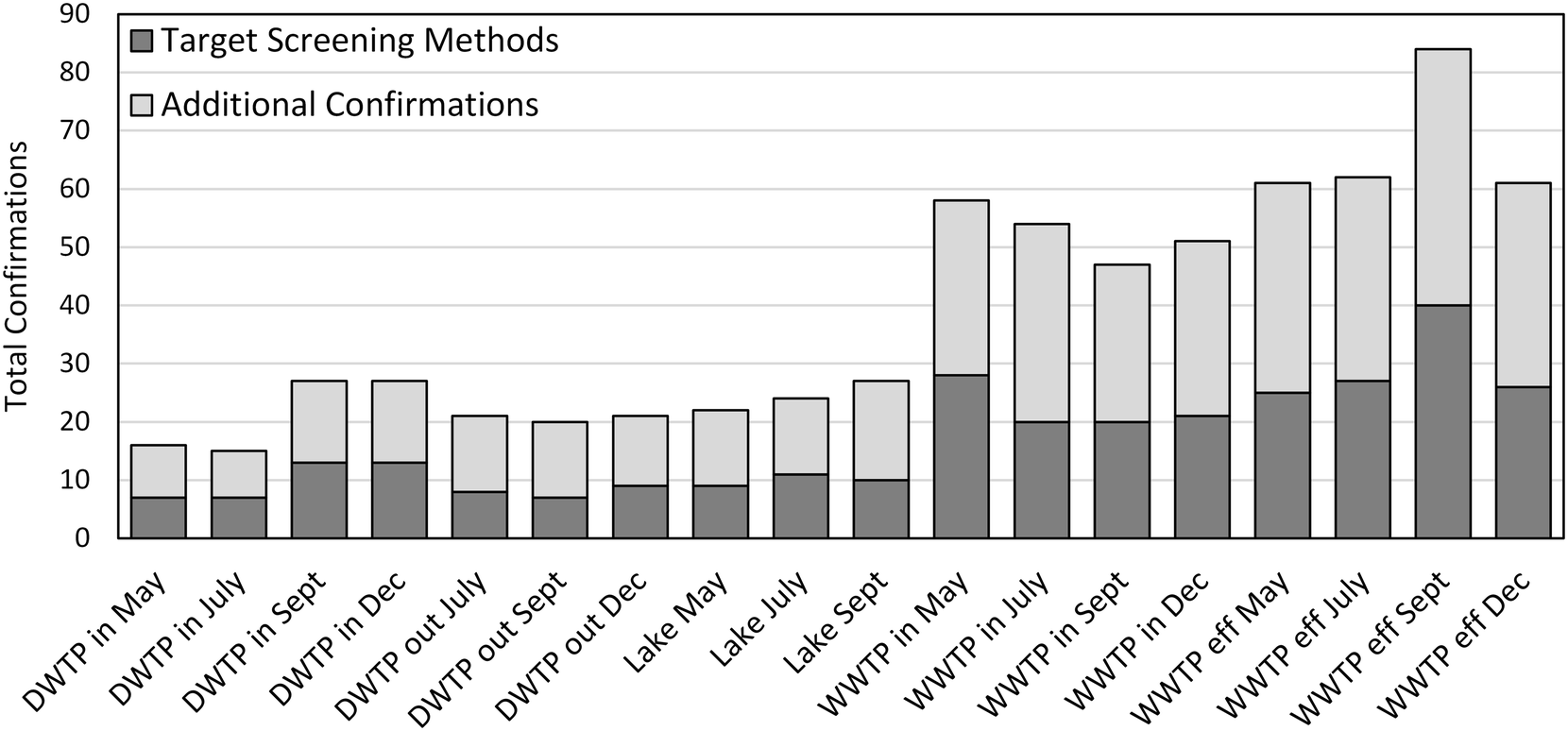 | ||
| Fig. 5 An accounting of the number of suspect hits confirmed in each of the eighteen water samples. The darker bars represent the number of confirmed suspect hits that are included in five comprehensive target screening methods. The lighter bars represent additional confirmed suspect hits that are not included in the in five comprehensive target screening methods. | ||
An exhaustive discussion of the types of micropollutants that we identified in this work is beyond the scope of this manuscript. However, there are several observations worth noting. First, there were 8 micropollutants that were present in every WWTP effluent sample and in every lake sample: 5-methyl-1H-benzotriazole, atenolol acid, caffeine, DEET, gabapentin, metformin, saccharin, and sucralose. The importance of each of these persistent micropollutants as indicators of anthropogenic influence and concerns over their respective toxicities has been discussed elsewhere,46–49 though we are unaware of any previous work that has identified this mixture of micropollutants in a single water system or with a single analytical approach. Second, some micropollutants that were detected have received attention for their putative or known health effects on exposed ecosystems or human populations. Two perfluorinated alkyl substances (PFASs) were confirmed to occur in wastewater and surface water samples (PFOA and PFBA) in the study area. There has been increasing concern over the occurrence of PFASs in water, particularly in areas adjacent to military installations or chemical manufacturing industries.9 The study area is not situated near these types of sources, but the trace detection of two PFASs demonstrates their prevalence in the environment. Additionally, a number of the pesticides (e.g., 2,4-d, atrazine, metolachlor, simazine) identified in the surface water samples are known or putative endocrine disruptors and their occurrence must be noted.50 Third, most of the micropollutants that were detected in this research are polar chemicals that are expected to favor partitioning to water, but some have exhibited the potential for bioaccumulation including the PFASs and the UV filters benzophenone and benzophenone-3.51,52 Finally, many of the micropollutants confirmed in our study have frequently been reported to occur in water resources around the world. However, some of the micropollutants have rarely been reported as water contaminants or are believed to be reported here for the first time. These include the anticonvulsant levetiracetam, the antihistamine fexofenadine, the antiviral drug emtricitabine, the cough suppressant dextromethorphan, the diuretic triamterene, the fungicide iodocarb, the insect repellant ethyl butylacetylaminopropionate, and the muscle relaxants carisoprodol, metaxalone, and methocarbamol. These results are not only interesting from a novelty perspective, but also demonstrate the breadth of chemical coverage that suspect screening affords, as these chemical substances represent a broad range of chemical structures and physicochemical properties and are unlikely to be included together in conventional target screening methods.
Conclusions
One of the major challenges in addressing the micropollutant problem in water resources is the incredible number of chemical substances that may be present in a water system at any point in time or space. The aggregate of all of these chemical substances makes up the environmental exposome, or the complete set of chemicals to which an individual or an ecosystem is exposed in a lifetime. There is considerable interest in characterizing the environmental exposome, as the majority of cases of human morbidity and mortality are caused by exposures to toxic substances.53 Suspect screening offers a step forward in the characterization of the environmental exposome. The suspect screening approach described in this manuscript is novel in at least two ways. First, the suspect screening workflow was developed and optimized to maximize sensitivity. In other words, we explicitly aimed to maximize the likelihood of characterizing suspect chemicals that were actually in the samples. Second, maximizing method sensitivity consequently resulted in a relatively low method selectivity; we addressed the low method selectivity by exploring a series of novel prioritization strategies that aimed to rank the suspect hits based on their likelihood of being true positives. The result of this approach enabled us to confirm the identity of 112 micropollutants in the study area. While this remains a small fraction of the environmental exposome, we approximate that this suspect screening approach more than doubled the number of micropollutants that would have otherwise been identified using a more conventional approach. As suspect screening and other similar environmental forensics tools develop, the environmental exposome will become more fully elucidated which will enable more comprehensive risk assessments and the development of concomitant water pollution prevention or remediation practices.Acknowledgements
We thank Patrick Phillips (USGS) and Tia-Marie Scott (USGS) for coordinating the sampling campaign. We thank Jose Lozano, Chris Bordlemay, and Susan Allen-Gil for useful discussions. This work was supported by the College of Engineering and the School of Civil and Environmental Engineering at Cornell University. A. L. P. acknowledges NSF GRFP grant no. 1144153.References
- S. D. Richardson and S. Y. Kimura, Anal. Chem., 2016, 88, 546–582 Search PubMed
.
- R. P. Schwarzenbach, B. I. Escher, K. Fenner, T. B. Hofstetter, C. A. Johnson, U. von Gunten and B. Wehrli, Science, 2006, 313, 1072–1077 Search PubMed
- D. W. Kolpin, E. T. Furlong, M. T. Meyer, E. M. Thurman, S. D. Zaugg, L. B. Barber and H. T. Buxton, Environ. Sci. Technol., 2002, 36, 1202–1211 Search PubMed
- E. T. Furlong, M. C. Noreiga, C. J. Kanagy, L. K. Kanagy, L. J. Coffey and M. R. Burkhardt, Determination of Human-Use Pharmaceuticals in Filtered Water by Direct Aqueous Injection–High-Performance Liquid Chromatography/Tandem Mass Spectrometry, 2014 Search PubMed.
- E. T. Furlong, S. L. Werner, B. D. Anderson and J. D. Cahill, Determination of human-health pharmaceuticals in filtered water by chemically modified styrene-divinylbenzene resin-based solid-phase extraction and high-performance liquid chromatography/mass spectrometry, 2008 Search PubMed.
- S. Huntscha, H. P. Singer, C. S. McArdell, C. E. Frank and J. Hollender, J. Chromatogr. A, 2012, 1268, 74–83 Search PubMed
- C. Jansson and J. Kreuger, J. AOAC Int., 2010, 93, 1732–1747 Search PubMed
- I. K. Wittmer, R. Scheidegger, H.-P. Bader, H. Singer and C. Stamm, Sci. Total Environ., 2011, 409, 920–932 Search PubMed
- X. Zhang, R. Lohmann, C. Dassuncao, X. C. Hu, A. K. Weber, C. D. Vecitis and E. M. Sunderland, Environ. Sci. Technol. Lett., 2016, 3, 316–321 Search PubMed
- P. J. Phillips, A. T. Chalmers, J. L. Gray, D. W. Kolpin, W. T. Foreman and G. R. Wall, Environ. Sci. Technol., 2012, 46, 5336–5343 Search PubMed
- P. J. Phillips, C. Schubert, D. Argue, I. Fisher, E. T. Furlong, W. Foreman, J. Gray and A. Chalmers, Sci. Total Environ., 2015, 512, 43–54 Search PubMed
- C. Moschet, I. Wittmer, J. Simovic, M. Junghans, A. Piazzoli, H. Singer, C. Stamm, C. Leu and J. Hollender, Environ. Sci. Technol., 2014, 48, 5423–5432 Search PubMed
- F. Wode, P. van Baar, U. Duennbier, F. Hecht, T. Taute, M. Jekel and T. Reemtsma, Water Res., 2015, 69, 274–283 Search PubMed
- C. Moschet, A. Piazzoli, H. Singer and J. Hollender, Anal. Chem., 2013, 85, 10312–10320 Search PubMed
- P. Gago-Ferrero, E. L. Schymanski, A. A. Bletsou, R. Aalizadeh, J. Hollender and N. S. Thomaidis, Environ. Sci. Technol., 2015, 49, 12333–12341 Search PubMed
- R. Bade, L. Bijlsma, T. H. Miller, L. P. Barron, J. V. Sancho and F. Hernández, Sci. Total Environ., 2015, 538, 934–941 Search PubMed
- M. Krauss, H. Singer and J. Hollender, Anal. Bioanal. Chem., 2010, 397, 943–951 Search PubMed
- A. C. Chiaia-Hernandez, M. Krauss and J. Hollender, Environ. Sci. Technol., 2013, 47, 976–986 Search PubMed
- J. A. Baz-Lomba, M. J. Reid and K. V. Thomas, Anal. Chim. Acta, 2016, 914, 81–90 Search PubMed
- J. Nacher-Mestre, M. Ibanez, R. Serrano, C. Boix, L. Bijlsma, B. T. Lunestad, R. Hannisdal, M. Alm, F. Hernandez and M. H. G. Berntssen, Chemosphere, 2016, 154, 231–239 Search PubMed
- M. Majewsky, T. Glauner and H. Horn, Anal. Bioanal. Chem., 2015, 407, 5707–5717 Search PubMed
- B. Zonja, A. Delgado, S. Pérez and D. Barceló, Environ. Sci. Technol., 2015, 49, 3464–3472 Search PubMed
- R. Gulde, U. Meier, E. L. Schymanski, H.-P. E. Kohler, D. E. Helbling, S. Derrer, D. Rentsch and F. Fenner, Environ. Sci. Technol., 2016, 50, 2908–2920 Search PubMed
- D. E. Helbling, J. Hollender, H.-P. E. Kohler and K. Fenner, Environ. Sci. Technol., 2010, 44, 6628–6635 Search PubMed
- E. Pitarch, M. Ines Cervera, T. Portoles, M. Ibanez, M. Barreda, A. Renau-Prunonosa, I. Morell, F. Lopez, F. Albarran and F. Hernandez, Sci. Total Environ., 2016, 548, 211–220 Search PubMed
- R. Bade, L. Bijlsma, J. V. Sancho and F. Hernández, Talanta, 2015, 139, 143–149 Search PubMed
- T. Letzel, A. Bayer, W. Schulz, A. Heermann, T. Lucke, G. Greco, S. Grosse, W. Schüssler, M. Sengl and M. Letzel, Chemosphere, 2015, 137, 198–206 CrossRef CAS PubMed
- M. Ibáñez, V. Borova, C. Boix, R. Aalizadeh, R. Bade, N. S. Thomaidis and F. Hernández, J. Hazard. Mater., 2017, 323, 26–35 Search PubMed
- R. Bade, N. I. Rousis, L. Bijlsma, E. Gracia-Lor, S. Castiglioni, J. V. Sancho and F. Hernandez, Anal. Bioanal. Chem., 2015, 407, 8979–8988 Search PubMed
- L. Vergeynst, H. Van Langenhove, P. Joos and K. Demeestere, Anal. Bioanal. Chem., 2014, 406, 2533–2547 Search PubMed
- J. J. Lohne, S. B. Turnipseed, W. C. Andersen, J. Storey and M. R. Madson, J. Agric. Food Chem., 2015, 63, 4790–4798 Search PubMed
- M. Solliec, A. Roy-Lachapelle and S. Sauvé, Rapid Commun. Mass Spectrom., 2015, 29, 2361–2373 Search PubMed
- H. G. J. Mol, P. Zomer and M. de Koning, Anal. Bioanal. Chem., 2012, 403, 2891–2908 Search PubMed
- L. Vergeynst, H. Van Langenhove and K. Demeestere, Anal. Chem., 2015, 87, 2170–2177 Search PubMed
- S. Kern, K. Fenner, H. P. Singer, R. P. Schwarzenbach and J. Hollender, Environ. Sci. Technol., 2009, 43, 7039–7046 Search PubMed
- M. Wang and D. E. Helbling, Water Res., 2016, 102, 241–251 Search PubMed
- D. E. Helbling, J. Hollender, H.-P. E. Kohler, H. Singer and K. Fenner, Environ. Sci. Technol., 2010, 44, 6621–6627 Search PubMed
- M. A. Stravs, E. L. Schymanski, H. P. Singer and J. Hollender, J. Mass Spectrom., 2013, 48, 89–99 Search PubMed
- New York State Department of Environmental Conservation (NYSDEC), PIMS - Pesticide Product, Ingredient, and Manufacturer System, 2016 Search PubMed.
- C. A. Smith, E. J. Want, G. O'Maille, R. Abagyan and G. Siuzdak, Anal. Chem., 2006, 78, 779–787 Search PubMed
- M. Loos, enviMass v3.1, Zurich, Switzerland, 2016 Search PubMed.
- R. M. A. Sjerps, D. Vughs, J. A. van Leerdam, T. L. ter Laak and A. P. van Wezel, Water Res., 2016, 93, 254–264 Search PubMed
- E. T. Furlong, B. D. Anderson, S. L. Werner, P. P. Soliven, L. J. Coffey and M. R. Burkhardt, Determination of Pesticides in Water by Graphatized Carbon-Based Solid-Phase Extraction and High-Performance Liquid Chromatography/Mass Spectrometry, 2001 Search PubMed.
- W. T. Foreman, J. L. Gray, R. C. ReVello, C. E. Lindley, S. A. Losche and L. B. Barber, Determination of Steroid Hormones and Related Compounds in Filtered and Unfiltered Water by Solid-Phase Extraction, Derivatization, and Gas Chromatography with Tandem Mass Spectrometry, 2012 Search PubMed.
- S. D. Zaugg, S. G. Smith, M. P. Schroeder, L. B. Barber and M. R. Burkhardt, Methods of Analysis by the U.S. Geological Survey National Water Quality Laboratory—Determination of Wastewater Compounds by Polystyrene-Divinylbenzene Solid-Phase Extraction and Capillary-Column Gas Chromatography/Mass Spectrometry, 2007 Search PubMed.
- M. Ruff, M. S. Mueller, M. Loos and H. P. Singer, Water Res., 2015, 87, 145–154 Search PubMed
- M. E. Balmer, H. R. Buser, M. D. Muller and T. Poiger, Environ. Sci. Technol., 2005, 39, 953–962 Search PubMed
- M. Scheurer, H. Brauch and F. T. Lange, Anal. Bioanal. Chem., 2009, 394, 1585–1594 Search PubMed
- B. Kasprzyk-Hordern, R. M. Dinsdale and A. J. Guwy, Water Res., 2008, 42, 3498–3518 Search PubMed
- W. Mnif, A. I. H. Hassine, A. Bouaziz, A. Bartegi, O. Thomas and B. Roig, Int. J. Environ. Res. Public Health, 2011, 8, 2265–2303 Search PubMed
- P. Gago-Ferrero, M. S. Diaz-Cruz and D. Barcelo, Sci. Total Environ., 2015, 518, 518–525 Search PubMed
- M. Houde, J. W. Martin, R. J. Letcher, K. R. Solomon and D. C. G. Muir, Environ. Sci. Technol., 2006, 40, 3463–3473 Search PubMed
- D. Wishart, D. Arndt, A. Pon, T. Sajed, A. C. Guo, Y. Djoumbou, C. Knox, M. Wilson, Y. Liang, J. Grant, Y. Liu, S. A. Goldansaz and S. M. Rappaport, Nucleic Acids Res., 2015, 43, D928–D934 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ew00248j |
| This journal is © The Royal Society of Chemistry 2017 |