
Development of electro-active forward osmosis membranes to remove phenolic compounds and reject salts†
Jingguo
Li
a,
Qing
Liu
a,
Yanbiao
Liu
 *b and
Jianping
Xie
*a
*b and
Jianping
Xie
*a
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, 117585 Singapore. E-mail: chexiej@nus.edu.sg; Fax: +65 6516 1936; Tel: +65 6516 1067
bNUS Environmental Research Institute, National University of Singapore, 5A Engineering Drive 1, #02-01, 117411 Singapore. E-mail: eriliuyb@nus.edu.sg; Fax: +65 6872 1320; Tel: +65 6601 3302
First published on 17th November 2016
Abstract
Forward osmosis (FO) is a promising membrane technology with good salt selectivity and high water permeability. However, it shows limited rejection of certain small organic molecules such as phenolic compounds. Here we developed a composite membrane resulting from the integration of FO with an electro-oxidation process to achieve both effective removal of phenolic compounds (>92% at 2.5 V) and good salt rejection (>98% for Na2SO4). In addition, this composite membrane can be readily integrated into the currently used membrane system. The design presented in this study could provide an advanced solution to the critical demand for water and be used in the next generation of water treatment technologies.
Water impactHere, we report a promising electro-active membrane designed to address a long-standing problem (specifically the poor retention rate of small organic molecules) in the emerging forward osmosis (FO) membrane technology. By integrating FO with an electro-oxidation process, the as-fabricated electro-active FO membranes showed good rejection of both inorganic ions (salts) and organic molecules (phenolic compounds). |
Introduction
The rapid development of modern industry has caused the serious environmental issues that we are now facing. These important issues have motivated society to explore sustainable solutions for preserving clean water.1,2 One of the environmental concerns is related to organic pollutants such as phenol and phenolic compounds. These compounds are highly used in various industrial sectors such as petrochemical plants, oil refineries, phenolic resin and pharmaceutical industries; and they have posed a serious problem for the environment by contaminating ground water and surface water.3–5 Some technologies including adsorption, extraction and oxidation have been developed to address this problem.6–8 Among the newly investigated technologies, electrochemical systems are particularly attractive.9 The electrolytic process is generally operated under ambient temperature and pressure, and this process can be controlled by setting the applied voltage or current.10–14 The advanced electrochemical filtration process is one good example. This technology has been recently developed and has shown good potential in several laboratory-scale applications, including oxidation of organics/ions15,16 and inactivation of bacteria/viruses.17 It is thus worthwhile to further extend this technology to other industrial sectors by integrating it with other materials to generate a wider usage. For example, a combination of electrochemistry and filtration processes can have synergistic effects, which could effectively promote both the electrochemical oxidation kinetics and the electro-osmotic flow efficiency.18Of all the filter materials recently developed, carbon-based materials such as carbon nanotubes (CNTs)19–26 have attracted particularly extensive attention mainly due to their high surface area, good conductivity, and high chemical/mechanical stability.27–29 There are several successful CNT filter designs. For example, an electrochemical CNT filter was recently reported to achieve 95% oxidation of methyl orange.18,30 In addition, these porous CNT filters can also serve as effective flow-through electrodes for reduction–oxidation reactions (e.g., reduction of nitrobenzene to aniline, followed by its oxidation to non-aromatic products).31 Several surface modification methods have been used to further improve the current efficiency. For example, doping with tin oxide or other functional nanomaterials has been shown to help mitigate corrosion of the filter and/or tune the oxygen evolution over-potential (OEP) of the CNT filters.32–34 Indeed, the CNT-based electrochemical filter has achieved a certain level of success. However, in order for this technology to be usable for practical applications, three limitations of this filter must be addressed: i) its high cost, ii) its poor durability and mechanical strength, and iii) the difficulty in fabricating large sheets from this filter.
Besides organic compounds, the removal of dissolved salts from the wastewater matrix is another equally important issue.3,6,7,35 Pressure-driven membrane-based processes, such as forward osmosis (FO), have been recently developed to remove dissolved salts and organic compounds.1,36–41 The FO process utilizes osmotic pressure to achieve good salt selectivity and high water permeability; however, it has shown limited success in rejecting certain small organic molecules such as phenolic compounds. An efficient strategy that has been used to solve this problem is the combining of FO with another technology. For example, adsorption technology was incorporated into the FO system to achieve satisfactory rejection of both salts and organic compounds.3,42 However, further optimization of the membrane materials is required for the composite membranes to be sufficiently effective in practical applications.43
Here we designed such composite membranes by incorporating a commercialized carbon nanofiber (CNF) into an FO membrane. Similar to the process used to fabricate thin film composite (TFC) membranes, the CNF-imbedded FO membrane (hereafter described as a CNF-TFC membrane) was here prepared by casting a thin layer of a polymeric membrane onto a pre-designed CNF surface, followed by an in situ interfacial polymerization of a polyamide layer on the composite substrate. The proposed fabrication process was shown to effectively improve the processability of CNF as well as the mechanical strength of the polymeric membrane. In addition, the unique porous structure of the resultant TFC membranes was also able to facilitate mass transfer of organic molecules towards the CNF surface, leading to better electrochemical reactivity of the as-fabricated composite membranes. Such a composite TFC membrane has shown good anti-fouling properties.44 However, it should be mentioned that the FO membrane can only displays high salt rejection, and now the incorporated electro-active CNF layer could help achieve effective removal of phenolic compounds. To demonstrate the efficiency of the as-designed composite membranes, we chose phenol as a model small organic compound because the current membrane separation processes (e.g., reverse osmosis (RO) and FO) only showed very limited retention rates (e.g., <50%) for phenol. Presented below are the details of this investigation.
Materials and methods
Materials
Polyethersulfone (PES) was purchased from Solvay Co., Ltd. N-methyl-2-pyrrolodinone (NMP, >99.5%) and polyethylene glycol 400 (PEG, Mn = 400 g mol−1) were purchased from Merck. Deionized water used in the experiments was produced by using a Milli-Q ultrapure water system (Millipore, USA). CNF was purchased from CeTech Co., Ltd. All chemicals and materials were used as received.Membrane fabrication
The CNF-imbedded PES (CNF-PES) membrane was prepared by using the Loeb–Sourirajan wet-phase inversion method. The PES polymers were first dried overnight in a vacuum oven to remove moisture that was present. The composition of the casting solution was PES/PEG-400/NMP/water (wt%) = 20/37.9/37.9/4.2. The casting solutions were degassed for 24 h, followed by casting onto the CNF surface using a 150 μm casting knife. The casted membrane was then immediately immersed into a water coagulation bath at room temperature, where it was kept for 24 h to ensure complete precipitation.In a typical TFC FO membrane fabrication, the membrane substrate was first immersed into a solution of 2.0 wt% m-phenylenediamine (MPD, >99%) in deionized water for 1 min. The top surface of the membrane was then brought into contact with a solution of 0.05 wt% 1,3,5-benzenetricarbonyl trichloride (TMC, 98%) in n-hexane for 30 s, which led to the formation of a polyamide thin film layer. Finally, these membrane samples were stored in deionized water at room temperature until they were used.
Morphology and contact angle of the CNF-TFC membranes
The membrane morphology was examined using field emission scanning electric microscopy (FESEM, JEOL JSM-6700F). The water contact angle was measured by using a contact angle goniometer (Rame Hart) at room temperature with deionized water as the probe liquid to determine the surface hydrophilicity of the membranes. The contact angle was recorded and calculated by the software immediately once the water drop touched the membrane surface. We obtained ten readings, randomly at different locations, and an average contact angle was calculated to minimize the experimental errors. The mechanical properties of these membranes, including Young's modulus, tensile strength and elongation at break, were measured by using an Instron tensiometer (model 5542, Instron Corp.). A constant elongation rate of 10 mm min−1 with a starting gauge length of 30 mm was applied.Determination of membrane porosity
Membrane porosity ε was defined as the total volume of the pores divided by the volume of the membrane. The porosity of the support was determined by using the gravimetric method according to the equation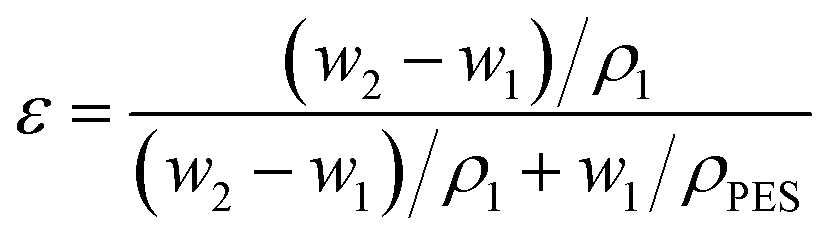 | (1) |
Determination of mass transport performance of the membranes
Water permeability (A), salt permeability (B), and salt rejection (R) of the as-fabricated TFC membranes were determined by using a stainless steel dead-end stirred cell under RO mode. All experiments were conducted three times and the average values were obtained. The effective membrane area was 19.5 cm2, and all tests were performed at room temperature. Water permeability, A, was determined based on eqn (2), where Jw is the volumetric water flux and ΔP is the applied pressure.| A = Jw/ΔP | (2) |
Salt rejection, R, was determined by using eqn (3), where Cp and Cf are the salt concentrations in the permeate and feed solutions, respectively.
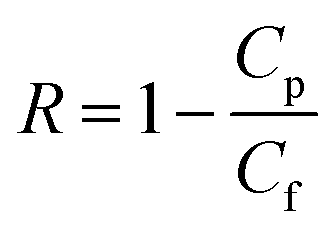 | (3) |
Salt permeability, B, was calculated from eqn (4) based on the solution-diffusion model, where Δπ is the osmotic pressure across the membrane.
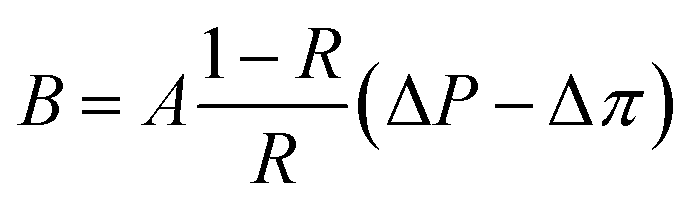 | (4) |
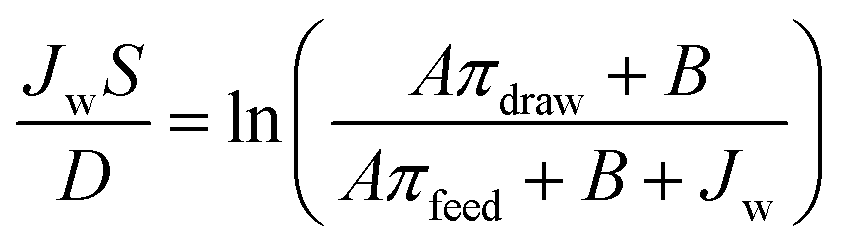
The membrane structure parameter (S) was determined by fitting the A, B and R values into eqn (5), where πdraw and πfeed are the osmotic pressures of the draw and feed solutions, respectively, and D is the salt diffusion coefficient.
 | (5) |
Determination of FO performance and phenol selectivity
FO experiments were conducted according to a previous study.45 The membrane test module consisted of one water channel on each side of the membrane with a dimension of 2.0 cm in length and 1.0 cm in width. The effective membrane area was 2.0 cm2 and no spacer was used in the testing. All of the measurements were taken with an initial volume of 500 mL for the feed solution (5 mmol L−1 phenol buffered with 10 mmol L−1 Na2SO4) and 200 mL for the draw solution (1 mol L−1 Na2SO4). The draw solution and feed solution flowed through the filtration cell countercurrently at the same volumetric flow rate of 0.3 L min−1 and they were recirculated. All of the experiments were carried out at room temperature, with the polyamide layer facing the draw solution (pressure-retarded osmosis, PRO, mode).The water permeation flux, Jw (L m−2 h−1, LMH), was determined by using eqn (6) based on the absolute weight change of the feed and the effective membrane area, Am (m2).
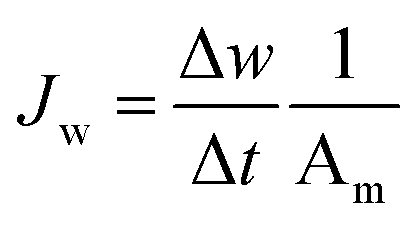 | (6) |
In this equation, Δw (kg) is the absolute change of weight of the water that permeated across the TFC-FO membrane over a predetermined time Δt (h) during the FO test.
The reverse salt flux, Js (g m−2 h−1, gMH), was determined from the increase in conductivity of the feed when deionized water was used as the feed solution according to the equation
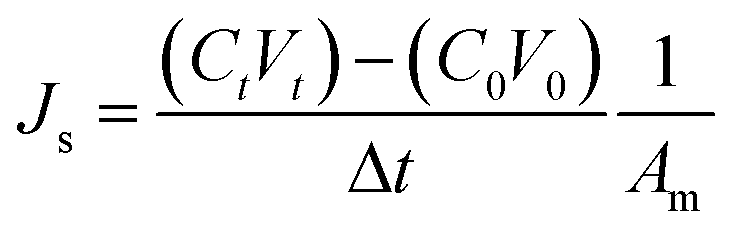 | (7) |
Retention of phenol was recorded at a fixed interval, Rn, based on the phenol concentration in both feed and draw solutions, and the retention was determined by the equation
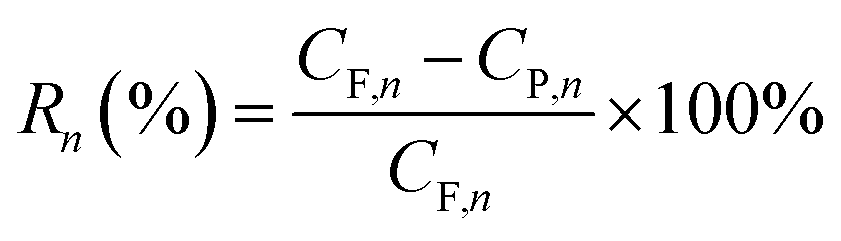 | (8) |
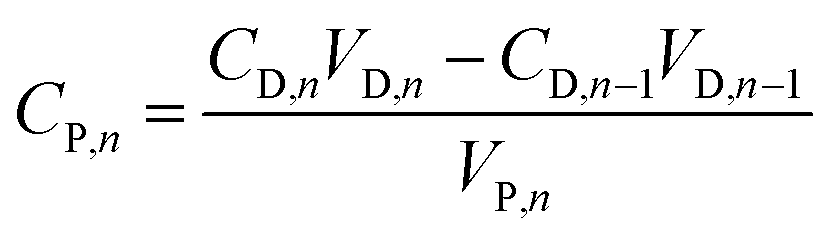 | (9) |
Salt rejection in the FO process was recorded based on the salt concentration in both feed and draw solutions, and the retention was determined by applying the equation
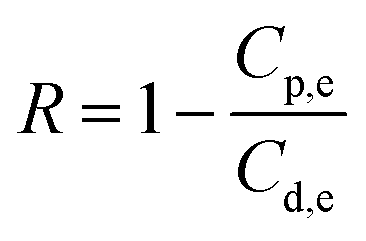 | (10) |
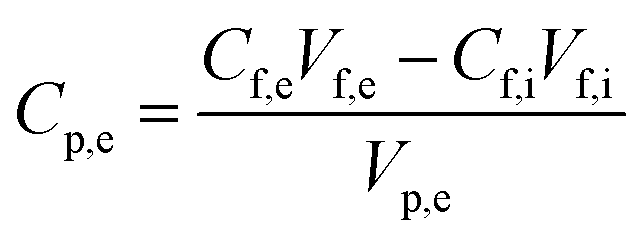 | (11) |
Results and discussion
The preparation of the CNF-TFC membranes followed a well-developed fabrication technique for commercial TFC membranes. This salient point also suggests a good potential of the proposed composite membranes for large-scale production and industry application. In a typical laboratory-scale study, an A4-sized CNF-TFC membrane was prepared, as shown in Fig. S1.† Interestingly, after being combined with polyethersulfone (PES), the fragile CNF film became much more flexible (Fig. 1a and S1†). This property is attractive, as a flexible film facilitates the transport and operation of the as-designed composite membranes. In addition, the CNF integration was shown to significantly enhance the mechanical strength of traditional TFC membranes. Compared to the tensile strength and Young's modulus of the pristine TFC membranes, those of a typical CNF-TFC prototype membrane (i.e., CNF-TFC-1) were 7- and 40-fold better, respectively (Table 1). The enhancement in mechanical strength was attributed to the synergistic effect of the polymer and the imbedded carbon materials. A similar improvement in mechanical strength of polymeric materials has been reported in a previous study.46 More interestingly, the incorporation of a CNF layer also converted the TFC membranes from being non-conductive to being highly conductive with a typical conductivity of 1050 S m−1. | ||
| Fig. 1 Characterizations of the as-fabricated CNF-TFC composite membrane (a–d) and the pristine TFC membrane (e–h). Optical properties (a and e) and FESEM images of cross-section (b and f), top (c and g), and bottom (d and h) surfaces. | ||
| Membrane | Thickness (μm) | Tensile strength (MPa) | Young's modules (MPa) | Maximum tensile strain (%) | Conductivity (S m−1) |
|---|---|---|---|---|---|
| TFC | 150 ± 5 | 2.6 ± 0.8 | 90.4 ± 5.6 | 25.6 ± 2.4 | — |
| CNF-TFC | 190 ± 10 | 27.7 ± 1.5 | 6123.6 ± 15.3 | 0.68 ± 0.3 | 1050 ± 41 |
To understand how the CNF layer was incorporated into a PES support membrane, we used field emission scanning electron microscopy (FESEM) to examine the microstructures of the as-fabricated CNF-TFC membranes and the pristine TFC membrane. Compared to the traditional PES membrane, which has been observed to feature a large finger-like macro-void cross-sectional structure (Fig. 1e and f), the CNF-PES membrane was observed to have a dense PES skin layer and a composite sub-layer (Fig. 1b) with thicknesses of 35 μm and 165 μm, respectively. The CNF layer was well imbedded in the polymer phase, which was uniformly distributed across the sub-layer and served as the backbone of the composite system. Meanwhile, the morphology of the pristine TFC membrane (Fig. 1g) appeared very similar to that of the top surface of the selective polyamide layer when TFC was combined with CNF (Fig. 1c), which indicated that the top skin layer of the CNF-TFC membranes remained unchanged after the incorporation of CNF.
We also used atomic force microscopy (AFM) to evaluate the surface morphology of the polyamide layer. As shown in Fig. S2,† no obvious differences were seen for the mean surface roughness (Ra) values between the membranes with and without CNF incorporated. These data suggested that the top polyamide layer was not affected by the CNF incorporation. We also measured the water contact angles for both the membranes with and without CNF incorporated, and no obvious differences were observed, indicating similar surface properties for both membranes. However, in contrast to the porous structure of the PES membrane (Fig. 1h), the bottom section of the CNF-TFC membrane (Fig. 1d) was influenced by the incorporation of CNF, which was uniformly distributed in the porous PES matrix. It should be noted that the casting and precipitation process should be controlled well to prevent the following two situations: i) complete imbedding of CNF into the PES matrix (which would totally block the electrocatalytic active centers on the CNF layer); and ii) incomplete binding of the PES layer to the CNF layer (in this situation, the layers would be separated, which would deteriorate the overall membrane performance). To further understand the impact of CNF incorporation on the support membrane structure, we recorded the structure parameters of both CNF-incorporated PES (CNF-PES, 543 ± 35 μm) and the pristine PES (485 ± 50 μm) membranes. A slight deterioration of the structure parameter was observed, which could be ascribed to the reduction of membrane porosity (from 75.2 ± 2.3 to 62.4 ± 2.4) and water permeability (from 420 ± 15 to 406 ± 12), as shown in Table S1.† However, the as-modified membranes inherited the skin layer structure, and hence showed promise for the traditional FO membrane process.
To demonstrate the possible usefulness of the as-fabricated membranes for FO applications, we modified the conventional FO set-up. As shown in Fig. 2 and S3,† we incorporated a constant power supply into the FO set-up, with the anode and cathode connected to the CNF-TFC membrane and the feed solution, respectively. The membrane performance was assessed under pressure-retarded osmosis (PRO) mode (selective layer facing draw solution). Due to the osmotic pressure difference between the feed and draw solutions, water tended to flow from the feed to the draw side. First, we varied the applied voltage exerted on the as-fabricated membranes (anode), for which the Ti plate was employed as the counter electrode (cathode). The CNF-TFC membranes showed a lower water flux than did the pristine TFC membrane (Fig. 3). This lower flux was due to the incorporation of the hydrophobic carbon layer with decreased membrane porosity. In particular, the average water flux of the CNF-TFC membranes over 24 h was decreased by ∼11.8% to 35.2% when compared with that of the pristine TFC membrane (11.2 ± 1.0 L m−2 h−1). However, once the testing period was prolonged to 24 h, a similar water flux was obtained for all of these membranes and the water flux was also quite stable in the long run, showing a standard deviation ≤1.5 for all of the CNF-TFC membranes. Meanwhile, the as-fabricated membranes still displayed high selectivity (>98%, Fig. 4) for salt rejection (determined from eqn (10) and (11)), which also suggested that the polyamide layer was stable at various applied voltages. Both the stable water flux and salt selectivity are encouraging for the usability of the membranes for FO applications. Note that we also monitored the current flow during the operation. Application of the power supply yielded a 1–2 mA current in the first few seconds of operation, and this current corresponded to the movement of cations (Na+) and anions (SO42−) in the feed solution. The current then dropped to zero and remained constant at zero during the prolonged operation (24 h). The low current density may have been due to the small effective area (2 cm2) of the membrane used for the measurement.
 | ||
| Fig. 2 Schematic illustration of the electro-active FO set-up. | ||
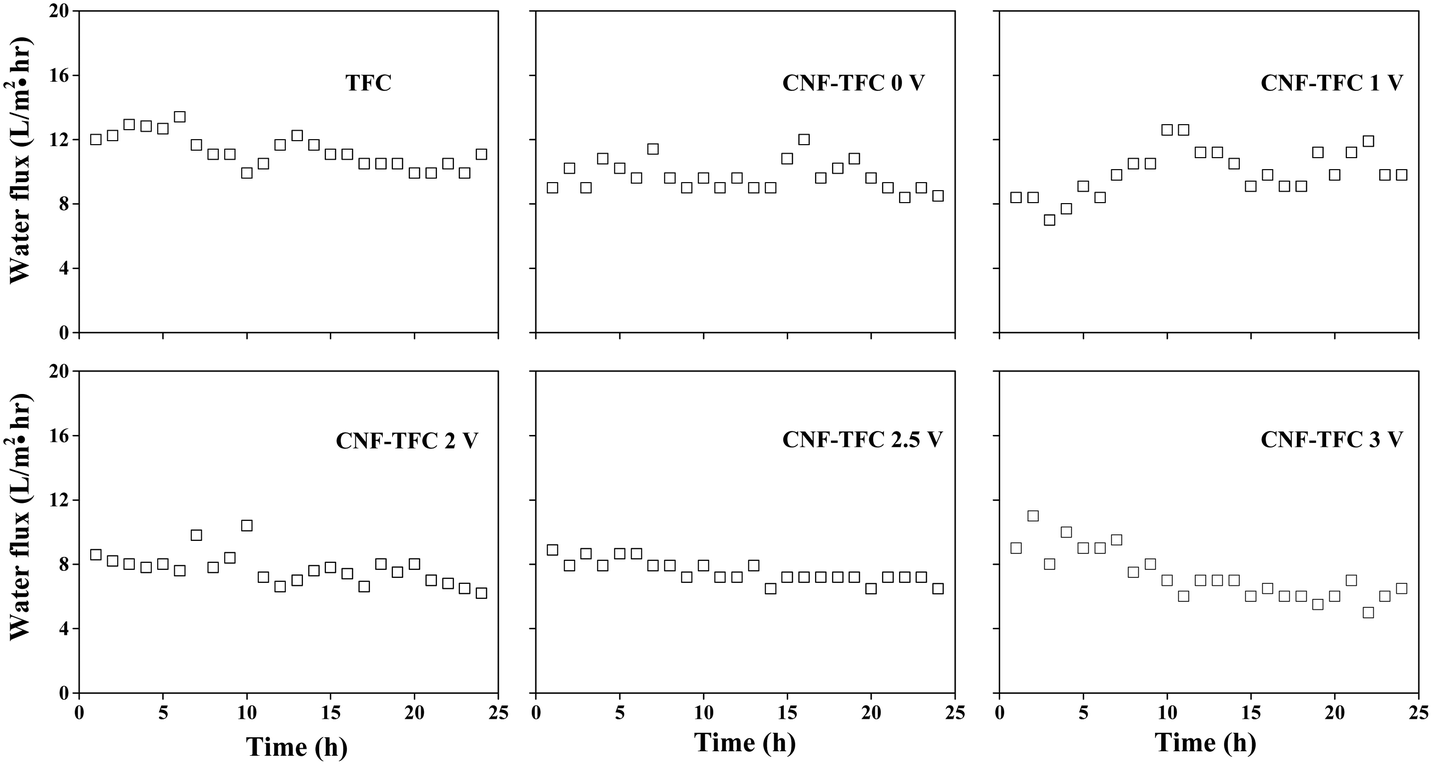 | ||
| Fig. 3 Water flux as a function of time and applied voltage for the as-fabricated membranes (experimental conditions: 5 mmol L−1 phenol (feed solution) and 1 mol L−1 Na2SO4 (draw solution)). | ||
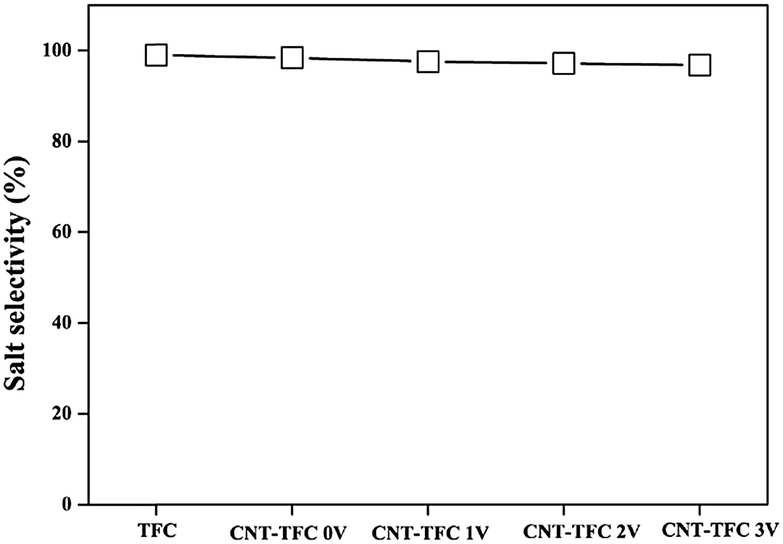 | ||
| Fig. 4 Salt rejection of the as-fabricated CNF-TFC membranes as a function of applied voltage (experimental conditions: 5 mmol L−1 phenol (feed solution) and 1 mol L−1 Na2SO4 (draw solution)). | ||
Along with the flow of water from the feed solution to the draw solution, phenol (whose concentration was high in the feed solution) also tended to flow from the feed solution through the semi-permeable membrane to the draw solution. Although some recent efforts have been devoted to purifying phenol-contaminated water using commercially available membrane technologies such as RO and FO, the average retention for phenol was low (e.g., ∼45%). The low retention of phenol was related to its molecular properties, specifically its small size and its charge neutrality in aqueous solutions. In our design, the phenol retention performance was assessed using sodium sulfate (Na2SO4) as the draw solution (Fig. 5). We chose the inert Na2SO4 rather than the common NaCl as the draw solution in order to eliminate the production of aqueous oxidants, specifically Cl2˙−, caused by anodic chloride oxidation under a high applied voltage (i.e., >2.5 V).47 The pristine TFC membrane only showed a limited retention for phenol (∼45% after 6 h), consistent with the reported data.6
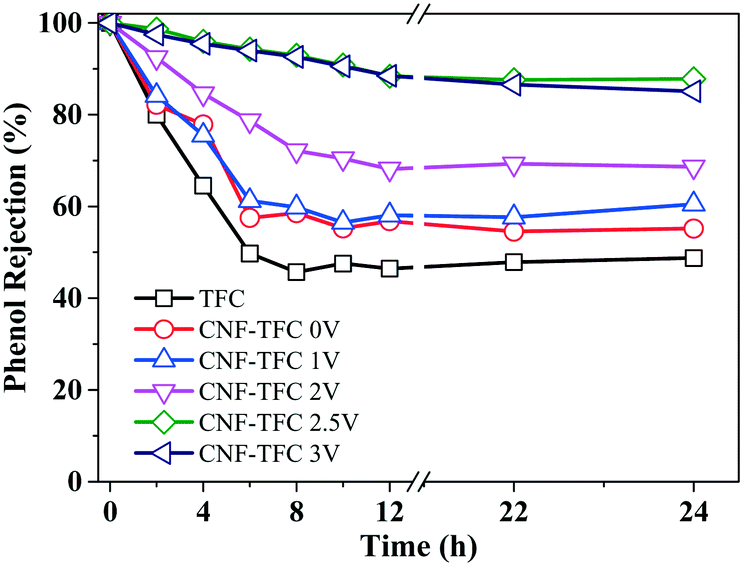 | ||
| Fig. 5 Phenol retention as a function of applied voltage (0–3 V) and operation time (0–24 h) for the as-fabricated membranes. | ||
Similarly, the CNF-TFC membranes subjected to a relatively low voltage (0–2 V) also showed a low rate of phenol retention over a continuous 24 h run (<60%), indicating that these applied voltages were not sufficient for the electro-active CNF-TFC membranes to effect phenol oxidation or rejection. However, once the applied voltage was increased to 2.5 V, the CNF-TFC membranes did reject more than 92% of the phenol over a 24 h continuous operation. This significant improvement in the removal of phenol by the CNF-TFC membranes was largely due to the accelerated rate of phenol oxidation at 2.5 V.
At an even higher voltage of 3 V, the retention of phenol slightly decreased. This decrease may have been due to the oxidation potential of phenol being compromised by the oxidation of water. In addition, the increased formation of gas bubbles, specifically H2 at the cathode and O2 at the anode, at this high applied voltage of 3 V may have further increased the pressure within the membrane and blocked certain electro-active sites, thus contributing negatively to the membrane performance and water flux. This hypothesis was supported by the observation of a slight reduction in phenol retention efficiency (92.0 ± 4.10% at 2.5 V vs. 91.2 ± 4.36% at 3 V) and in water flux (7.54 ± 0.68 L m−2 h−1 at 2.5 V vs. 7.31 ± 1.55 L m−2 h−1 at 3 V) as the applied voltage was increased from 2.5 V to 3 V. At the optimum applied voltage of 2.5 V, the desirable retention of phenol was attributed to the polyamide selectivity and the electro-oxidation from the CNF layer. In particular, at low or zero voltage, the polyamide selectivity was the dominant contributor, where slow or no oxidation could occur under this operation condition. The physical adsorption capability of phenol was further evaluated for both TFC and CNF-TFC membranes, and their equilibrium adsorptions of phenol were 0.65 and 0.72 mg g−1, respectively (Fig. S4†), which clearly suggested the electro-oxidation to be responsible for the promotion of phenol rejection rather than the physical absorption in our system.
To further illustrate the phenol removal mechanism, we analyzed the water samples in the draw solution side. We first measured their total organic carbon (TOC) values. Unlike the RO membrane process, the actual permeate in the FO process was diluted by the draw solution, for which there was a larger quantity than there was for the permeate solution; therefore, the draw solution showed a much lower TOC value than did the feed solution. All of the CNF-TFC membranes operating under an applied voltage clearly showed lower TOC values than did that operating without an applied voltage (Fig. S5†). The total or partial oxidation of phenol may have occurred at the CNF-TFC surface, which could have further prevented the transport of this organic molecule through the membrane. The lowest TOC was achieved at an applied voltage of 2.5 V. This result was also consistent with the data for phenol selectivity. However, one major by-product (with an absorption peak at 247 nm, see Fig. S6†) appeared when the applied voltage was above 2 V. This new component may have been benzoquinone, a dominant by-product of phenol electro-oxidation, as reported in several previous studies.14,48,49 Further combining the current design with another RO process may achieve a complete rejection of the as-produced charged and relatively large (in molecular size) by-products.
We further used SEM to monitor evolution of the morphology of the as-fabricated CNF-TFC membranes during the operation. As shown in Fig. 6, the polyamide selective layer was maintained very well. This observation suggested that the electrochemical operation would not affect the selective layer. However, the CNF incorporated in the support layer was decorated with several polydispersed nanoparticles, which may have been polyphenol formed during the electro-oxidation process or inorganic sodium persulfate from the oxidation of the electrolyte. The buildup of these polymers/precipitates may cause adverse effects on the electrochemistry by increasing the resistance to electron transfer. This problem could be addressed by using certain engineering methods. For example, washing a CNT filter with an acid (e.g., mixture of 50% ethanol with 50% 1 M HCl) was used to effectively remove the major buildup on the filter.14,16 We further investigated the effects of changing the concentration of phenol in the feed solution at a fixed voltage of 2.5 V. The retention of phenol was apparently generally increased at the lower concentrations of phenol in the feed solution (Fig. S7†). Therefore, our proposed technology may perform relatively well for the treatment of wastewater, which typically contains low concentrations of phenol.
 | ||
| Fig. 6 FESEM images, with different magnifications (×5500, a; ×9000, b), of the as-fabricated CNF-TFC membranes after 24 h of continuous operation (experimental conditions: 2.5 V applied voltage, 5 mmol L−1 phenol (feed solution) and 1 mol L−1 Na2SO4 (draw solution)). | ||
Conclusions
In this study, we have integrated the electro-oxidation process into the well-developed FO process, leading to a promising water purification system that could simultaneously remove phenolic compounds and soluble salts. In particular, the incorporated carbon layer led to an efficient electro-oxidation of phenolic compounds, while the traditional FO membrane was responsible for salt rejection. The proposed composite membranes could be readily integrated into a commercially available water purification system.Acknowledgements
This work is supported by GE-NUS research grant (WBS number of R-706-005-004-592). We thank Professor Neal Tai-Shung Chung (Department of Chemical & Biomolecular Engineering, NUS) and Professor Choon Nam Ong (NUS Environmental Research Institute, NERI) for their great help on this project.Notes and references
- A. Subramani and J. G. Jacangelo, Water Res., 2015, 75, 164 CrossRef CAS PubMed.
- M. R. Wiesner and S. Chellam, Environ. Sci. Technol., 1999, 33, 360A CrossRef CAS PubMed.
- A. Bódalo, J. L. Gómez, M. Gómez, G. León, A. M. Hidalgo and M. A. Ruíz, Desalination, 2008, 223, 323 CrossRef.
- S. Mohammadi, A. Kargari, H. Sanaeepur, K. Abbassian, A. Najafi and E. Mofarrah, Desalin. Water Treat., 2014, 53, 2215 CrossRef.
- M. Xiao, J. Zhou, Y. Tan, A. Zhang, Y. Xia and L. Ji, Desalination, 2006, 195, 281 CrossRef CAS.
- B. D. Coday, B. G. Yaffe, P. Xu and T. Y. Cath, Environ. Sci. Technol., 2014, 48, 3612 CrossRef CAS PubMed.
- R. W. Holloway, J. Regnery, L. D. Nghiem and T. Y. Cath, Environ. Sci. Technol., 2014, 48, 10859 CrossRef CAS PubMed.
- Y. Park, A. H. Skelland, L. J. Forney and J. H. Kim, Water Res., 2006, 40, 1763 CrossRef CAS PubMed.
- B. P. Chaplin, Environ. Sci.: Processes Impacts, 2014, 16, 1182 CAS.
- G. Gao and C. D. Vecitis, Environ. Sci. Technol., 2011, 45, 9726 CrossRef CAS PubMed.
- C. F. de Lannoy, D. Jassby, D. D. Davis and M. R. Wiesner, J. Membr. Sci., 2012, 415–416, 718 CrossRef CAS.
- X. Sun, X. Su, J. Wu and B. J. Hinds, Langmuir, 2011, 27, 3150 CrossRef CAS PubMed.
- D. K. Hubler, J. C. Baygents, B. P. Chaplin and J. Farrell, J. Electrochem. Soc., 2014, 161, E182 CrossRef.
- Y. Liu, J. Xie, C. N. Ong, C. D. Vecitis and Z. Zhou, Environ. Sci.: Water Res. Technol., 2015, 1, 769 CAS.
- A. M. Zaky and B. P. Chaplin, Environ. Sci. Technol., 2014, 48, 5857 CrossRef CAS PubMed.
- Y. Liu, H. Liu, Z. Zhou, T. Wang, C. N. Ong and C. D. Vecitis, Environ. Sci. Technol., 2015, 49, 7974 CrossRef CAS PubMed.
- F. Yi, S. Chen and C. Yuan, J. Hazard. Mater., 2008, 157, 79 CrossRef CAS PubMed.
- Y. Liu, J. H. Dustin Lee, Q. Xia, Y. Ma, Y. Yu, L. Y. Lanry Yung, J. Xie, C. N. Ong, C. D. Vecitis and Z. Zhou, J. Mater. Chem. A, 2014, 2, 16554 CAS.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282 CrossRef CAS PubMed.
- G. S. Ajmani, D. Goodwin, K. Marsh, D. H. Fairbrother, K. J. Schwab, J. G. Jacangelo and H. Huang, Water Res., 2012, 46, 5645 CrossRef CAS PubMed.
- D. G. Goodwin Jr., Z. Xia, T. B. Gordon, C. Gao, E. J. Bouwer and D. H. Fairbrother, Environ. Sci.: Nano, 2016, 3, 545 RSC.
- K. Zuo, H. Liu, Q. Zhang, P. Liang, C. D. Vecitis and X. Huang, Electrochim. Acta, 2016, 211, 199 CrossRef CAS.
- R. Sengur, C. F. de Lannoy, T. Turken, M. Wiesner and I. Koyuncu, Desalination, 2015, 359, 123 CrossRef CAS.
- D. Shan, S. Deng, T. Zhao, G. Yu, J. Winglee and M. R. Wiesner, Chem. Eng. J., 2016, 294, 353 CrossRef CAS.
- K. Goh, H. E. Karahan, L. Wei, T.-H. Bae, A. G. Fane, R. Wang and Y. Chen, Carbon, 2016, 109, 694 CrossRef CAS.
- Y. Jiang, P. Biswas and J. Fortner, Environ. Sci.: Water Res. Technol., 2016, 2, 915–922 CAS.
- B. Hinds, Curr. Opin. Solid State Mater. Sci., 2012, 16, 1 CrossRef CAS.
- B. J. Hinds, N. Chopra, T. Rantell, R. Andrews, V. Gavalas and L. G. Bachas, Science, 2004, 303, 62 CrossRef CAS PubMed.
- A. W. Carpenter, C. F. De Lannoy and M. R. Wiesner, Environ. Sci. Technol., 2015, 49, 5277 CrossRef CAS PubMed.
- H. Liu, J. Liu, Y. Liu, K. Bertoldi and C. D. Vecitis, Carbon, 2014, 80, 651 CrossRef CAS.
- G. Gao, Q. Zhang and C. D. Vecitis, J. Mater. Chem. A, 2014, 2, 6185 CAS.
- H. Liu, A. Vajpayee and C. D. Vecitis, ACS Appl. Mater. Interfaces, 2013, 5, 10054 CAS.
- M. Panizza and G. Cerisola, Chem. Rev., 2009, 109, 6541 CrossRef CAS PubMed.
- Z. Wen, Q. Wang, Q. Zhang and J. Li, Adv. Funct. Mater., 2007, 17, 2772 CrossRef CAS.
- C. H. Tan and H. Y. Ng, Desalin. Water Treat., 2012, 13, 356 CrossRef.
- K. Lutchmiah, A. R. D. Verliefde, K. Roest, L. C. Rietveld and E. R. Cornelissen, Water Res., 2014, 58, 179 CrossRef CAS PubMed.
- N. T. Hancock, P. Xu, D. M. Heil, C. Bellona and T. Y. Cath, Environ. Sci. Technol., 2011, 45, 8483 CrossRef CAS PubMed.
- M. Xie, L. D. Nghiem, W. E. Price and M. Elimelech, Water Res., 2012, 46, 2683 CrossRef CAS PubMed.
- D. L. Shaffer, J. R. Werber, H. Jaramillo, S. Lin and M. Elimelech, Desalination, 2015, 356, 271 CrossRef CAS.
- F. Perreault, H. Jaramillo, M. Xie, M. Ude, L. D. Nghiem and M. Elimelech, Environ. Sci. Technol., 2016, 50, 5840 CrossRef CAS PubMed.
- Q. Liu, Z. Zhou, G. Qiu, J. Li, J. Xie and J. Y. Lee, ACS Sustainable Chem. Eng., 2015, 3, 2959 CrossRef CAS.
- I. Ali, Chem. Rev., 2012, 112, 5073 CrossRef CAS PubMed.
- S. Zhao, L. Zou, C. Y. Tang and D. Mulcahy, J. Membr. Sci., 2012, 396, 1 CrossRef CAS.
- Q. Liu, G. Qiu, Z. Zhou, J. Li, G. L. Amy, J. Xie and J. Y. Lee, Environ. Sci. Technol., 2016, 50, 10596 CrossRef CAS PubMed.
- G. Han, S. Zhang, X. Li, N. Widjojo and T.-S. Chung, Chem. Eng. Sci., 2012, 80, 219 CrossRef CAS.
- D. Li and H. Wang, J. Mater. Chem., 2010, 20, 4551 RSC.
- C. D. Vecitis, M. H. Schnoor, M. S. Rahaman, J. D. Schiffman and M. Elimelech, Environ. Sci. Technol., 2011, 45, 3672 CrossRef CAS PubMed.
- M. Basu, S. Sarkar, S. Pande, S. Jana, A. Kumar Sinha, S. Sarkar, M. Pradhan, A. Pal and T. Pal, Chem. Commun., 2009, 14, 7191 RSC.
- T. Wilke, M. Schneider and K. Kleinermanns, Open J. Phys. Chem., 2013, 3, 97 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ew00275g |
| This journal is © The Royal Society of Chemistry 2017 |