
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEarth-abundant photocatalytic systems for the visible-light-driven reduction of CO2 to CO†
Alonso
Rosas-Hernández‡
,
Christoph
Steinlechner‡
,
Henrik
Junge
and
Matthias
Beller
 *
*
Leibniz Institute for Catalysis at the University of Rostock, Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: matthias.beller@catalysis.de
First published on 24th February 2017
Abstract
Herein, we report a highly selective photocatalytic system, based on an in situ copper photosensitizer and an iron catalyst, for the reduction of CO2 to CO. Turnover numbers (TON) up to 487 (5 h) with selectivities up to 99% and ΦCO = 13.3% were observed. Stern–Volmer analysis allowed us to establish a reductive quenching mechanism between the Cu PS and electron donor.
Over the last 50 years, the use of fossil fuels has increased the concentration of greenhouse gas carbon dioxide in the atmosphere.1 Technologies to control CO2 production and reduce its accumulation in the atmosphere are highly desired. In this context, photochemical reduction of CO2 provides a pathway to convert a waste product into more valuable molecules whereby the energy from sunlight is stored.2 Thus, not only carbon dioxide can be used as C1-feedstock but also this approach allows to harvest the energy from sunlight, helping the transition towards a more sustainable energy source.
Inspired by the seminal work of Lehn and co-workers,3 several molecularly defined systems have been developed to mediate the photochemical reduction of CO2.2a In these works usually a multicomponent system is used employing a photosensitizer (PS) and a catalyst (Cat) which is responsible for CO2-coordination and its subsequent reduction. Ru(II) polypyridyl and cyclometalated Ir(III) complexes are often used as PS due to their strong oxidation and reduction power and extended lifetimes of their excited states.4 In addition, transition metal complexes based on Ru,5 Re,6 and Ir7 have been successfully applied as Cat. Due to the obvious advantages, the use of more earth-abundant metals for both PS and Cat is desired in these materials.
Recently, several complexes based on non-precious transition metals have been described as Cat in the photoreduction of CO2.8 For instance, Bonin, Robert et al. reported the use of an iron tetraphenylporphyrin complex together with an iridium-based PS to reduce CO2 selectively to CO with a turnover number (TON) of 140.8g The substitution of the Ir PS by an organic dye decreased the activity of the catalytic system by half. A CoII complex supported by a cyclic pentadentate ligand has been also used as Cat in the presence of an Ir PS, driving a selective CO2-to-CO conversion with a TON of 270.8e A photocatalytic system comprising a quaterpyridine CoII or FeII complex as Cat and a ruthenium-based PS led to CO2 reduction into CO with high selectivity (98%) and TONs between 497 and 2660.8a When an organic dye is used instead of the Ru PS, the system converted CO2 to CO with a TON of 1365 (based on Cat, c = 0.005 mM) and a quantum yield ΦCO = 1.1%. In addition, Ishitani and co-workers recently reported the use of a dimeric CuI complex, supported by a tetradentate ligand, as PS for the CO2 reduction to CO with a selectivity of 78% over H2 production.9 The catalytic process was performed in the presence of an FeII complex as Cat and 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole (BIH) as sacrificial electron donor (SD). The efficiency and durability for CO formation were high, with ΦCO = 6.7% and TONCO = 270. Apart from all these interesting developments, the use of abundant-metal complexes for efficient and durable photocatalytic systems for selective CO2 reduction with visible light still remains rare.
In our research group, several heteroleptic CuI complexes were developed in the last years and used as PS in photocatalytic water reduction.10 To render an operationally simpler process, we also established a photocatalytic system where the synthesis and isolation of the corresponding Cu PS is bypassed.11 Accordingly, the heteroleptic emissive Cu complex is formed in situ, allowing a facile screening of a broad range of ligands in order to easily correlate the structural features of the PS with the observed catalytic activity.
Here, we describe for the first time the application of this process to the photochemical reduction of CO2. More specifically, we employed cyclopentadienone iron complexes as Cat,12 generating a fully earth-abundant catalytic system.
We started our investigations using iron cyclopentadienone complex 1 as CO2-reduction catalyst. In situ generation of the Cu PS was performed with equimolar quantities of a simple copper salt ([Cu(MeCN)4]PF6), a bidentate phosphine ligand (xantphos), and a phenantroline derivate (bathocuproine, 5); see Scheme 1. We observed that upon dissolution of all components in a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) mixture of N-methyl-2-pyrrolidone (NMP) and triethanolamine (TEOA), with the addition of a SD (1-benzyl-1,4-dihydrinicotinamide, BNAH) and subsequent visible light irradiation for 5 h, CO2 was reduced to CO with a TON of 245 and a selectivity of 86% over H2 production (Table S3,† entry 1). This result suggests that the copper-based PS is easily formed in situ and it is able to mediate a photoinduced electron transfer from the SD to the CO2-reduction catalyst 1 (vide infra).
:1 (v/v) mixture of N-methyl-2-pyrrolidone (NMP) and triethanolamine (TEOA), with the addition of a SD (1-benzyl-1,4-dihydrinicotinamide, BNAH) and subsequent visible light irradiation for 5 h, CO2 was reduced to CO with a TON of 245 and a selectivity of 86% over H2 production (Table S3,† entry 1). This result suggests that the copper-based PS is easily formed in situ and it is able to mediate a photoinduced electron transfer from the SD to the CO2-reduction catalyst 1 (vide infra).
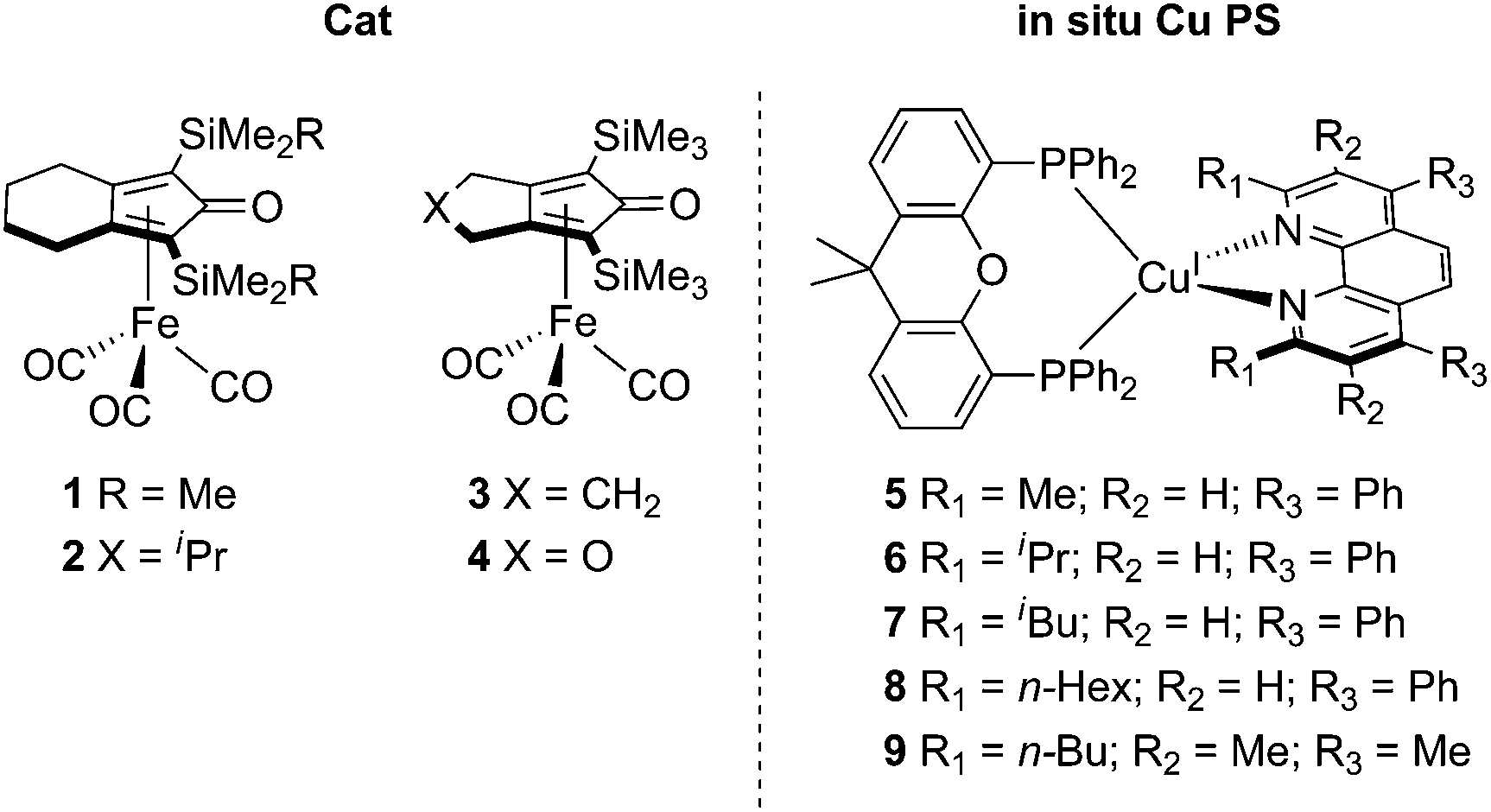 | ||
| Scheme 1 Molecular structure of the cyclopentadienone iron complexes used as Cat and the in situ formed Cu PS. | ||
It is worthy to point out that an amine (BNAH) has been employed as sacrificial electron donor. In a large-scale use of this technology, photochemical reduction of CO2 needs to be coupled with water oxidation reaction. So water can be used as an electron source, just as in the case of photosynthesis. Since the oxidation of water is also a challenging reaction by itself,13 it is a common practice to optimize both reactions separately. Thus, sacrificial donors such as amines are used to explore the activity of potential catalysts in CO2 photoreduction.
Notably, when dissolving a heteroleptic CuI complex with both diimine and diphosphine ligands an equilibrium with the homoleptic CuI(N–N)2 complex (N–N = diimine ligand) is established.10d Evidently, this equilibrium can be affected by the ratio between the diimine and diphosphine ligand in the in situ formation of the Cu PS, thus influencing the catalytic activity of the system as well. In fact, when the reaction was performed in the absence of a source of diphosphine ligand, the generation of CO dropped dramatically (Table S2,† entry 2). In this case, only a homoleptic CuI(N–N)2 complex can be formed, suggesting that the photophysical properties of such copper complex are not adequate to drive the photochemical reduction of CO2.14 Accordingly, when an excess of diphosphine ligand was used (i.e. 2 or 3 equiv.) the catalytic activity of the system was improved (TONCO = 271 and 270 and selectivity = 95% and 97%, respectively; Table S2,† entries 5 and 6). Under these experimental conditions, the presence of the heteroleptic Cu PS is favoured over the inactive homoleptic species.
Considering the limitations of BNAH as SD (i.e. inner-filter effect and rapid back-electron transfer from oxidized products),15 we turned our attention to BIH, a suitable electron donor recently used in photocatalytic reactions.8a,9 In fact, applying BIH, the photocatalytic activity of the system raised up to a TONCO = 487 with a selectivity of 99% (Table 1, entry 1). Thus, BIH was used as SD in the following photocatalytic investigations.
|
|
|||||
|---|---|---|---|---|---|
| Entry | N–N ligand | [Fe] complex | TONb (CO) | TONb (H2) | Selec. (CO) |
|
a Reaction conditions: NMP/TEOA (5:1, v/v) 7.5 mL; 5 μmol [Cu(MeCN)4]PF6; 15 μmol xantphos P–P ligand; 5 μmol N–N ligand; 1 μmol Fe catalyst; BIH 0.1 M; Hg-lamp (light output 1.5 W) equipped with a 400–700 nm filter.
b Determined using calibrated GC; TON = n(CO)/n(catalyst).
c Reaction performed using MeCN/TEOA (5:1, v/v) as solvent mixture.
|
|||||
| 1 | 5 | 1 | 487 | 7 | 99% |
| 2c | 5 | 1 | 64 | 15 | 81% |
| 3 | 6 | 1 | 360 | 17 | 95% |
| 4 | 7 | 1 | 310 | 8 | 97% |
| 5 | 8 | 1 | 404 | 26 | 94% |
| 6 | 9 | 1 | 467 | 31 | 94% |
| 7 | 5 | 2 | 254 | 9 | 97% |
| 8 | 5 | 3 | 260 | 16 | 94% |
| 9 | 5 | 4 | 80 | 11 | 88% |
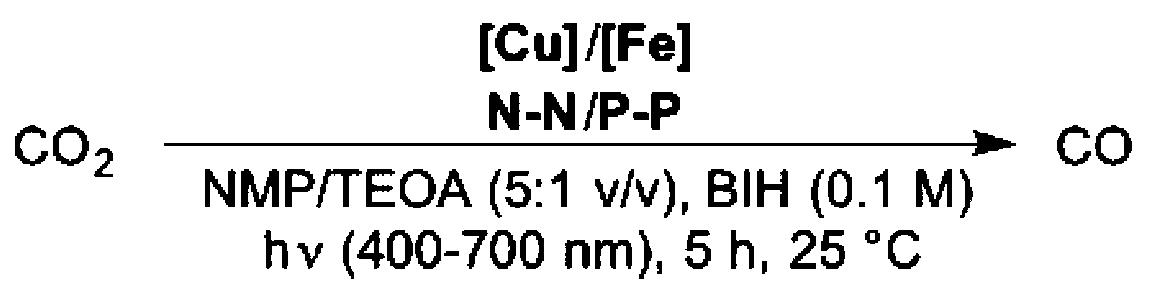
Notably, using an isolated sample of Cu complex 5 instead of the in situ generated PS, the catalytic activity was slightly lower (Table S4,† entry 2), which proves that the heteroleptic Cu PS is effectively assembled in situ.
Excited state lifetimes of heteroleptic CuI PS are heavily influenced by exciplex quenching of the resulted flattened CuII complex upon light irradiation.14 Commonly, steric effects of the diimine ligand influence the degree of excited state distortion. Thus, aiming to increase the excited state lifetime of the Cu PS and the catalytic activity of the system as well, we tested several sterically demanding 1,10-phenantroline derivates as ligands (Table 1, entries 1–6). However, when diimine 7 – containing iBu groups in the 2 and 9 positions of the phenantroline moiety – was used instead of bathocuproine 5, the activity of CO2 photoreduction was diminished (entry 1 vs. entry 4, Table 1). Interestingly, when applying ligand 9 instead of bathocuproine, the catalytic activity is almost similar (Table 1, entry 6). We assume that not only the steric effects of the substituents in position 2 and 9 of the phenantroline play a major role, but also the electronic properties of the various substituents in the remaining positions.
Steric effects caused by bidentate phosphine ligands also influence the degree of excited state distortion as well as the equilibrium between homoleptic and heteroleptic species.10d,11 For instance, if the flexible diphosphine ligand DPEPhos 11 was used instead of xantphos (Table S1,† entry 1), the catalytic activity of the system decreased (TONCO = 340). On the other hand, applying the sterically demanding thixantphos 10 led to catalytic activities comparable to xantphos (Table S1,† entry 2). Since it has been previously reported that the ratio between homoleptic and heteroleptic species is comparable for all three diphosphines,11 the observed differences in activity are mainly due to the restrictions imposed by such ligands during the flattening distortion of the copper complexes. Obviously, the lower catalytic activity displayed by 11 is due to the less steric hindrance of the substituents on the phosphorous atom and the smaller bite angle (102°), which both are responsible for preventing flattening distortion and exciplex quenching by nucleophilic attack of a solvent molecule.
Notably, we observed that the activity of the photocatalytic system is strongly influenced by the structure of the cyclopentadienone iron catalyst. Comparing the activities of complexes 1 and 2 (Table 1, entries 1 and 7), it becomes evident that increasing the steric hindrance of the substituents at the silicon atom has a negative influence on the activity of the system. In addition, when a heteroatom (oxygen) is present in the saturated cycle of the ligand (Table 1, entry 9), the catalytic performance decreased dramatically. Specifically, the TON is reduced from 487 to 80. Attempts to increase the catalyst productivities (TON) by elongation of irradiation time failed, suggesting that the photocatalytic system is no longer active after 5 h.
In order to get insights into the mechanism of the photochemical CO2 reduction catalyzed by iron complex 1 in the presence of in situ Cu PS 5, the quantum yield of the overall catalytic photoredox cycle reduction was determined by using the following equation:
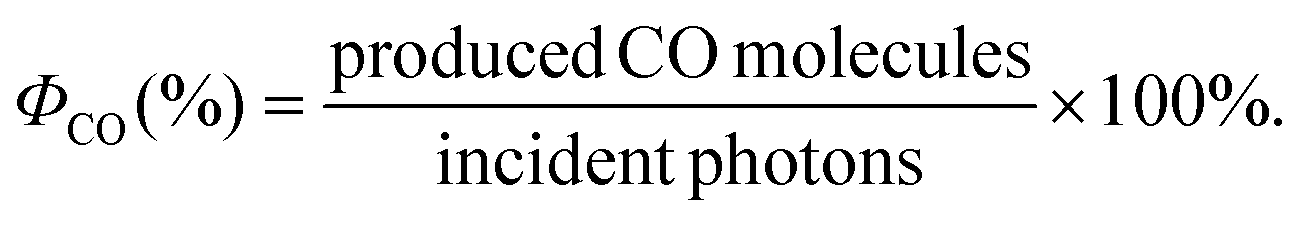
The number of incident photons was determined using an iron actinometer (see ESI† for details). After 2 h of irradiation using monochromatic light (415 nm), 2.5 × 10−4 mol of CO were quantified. Hence, the calculated quantum yield for this molecular system is 13.3%, which is twice as high as the previously reported catalytic system for CO2 photoreduction based on a dimeric Cu PS and an Fe phenanthroline catalyst.8
To probe the mechanism of photoinduced electron transfer in this catalytic system, we further carried out quenching experiments of the in situ Cu PS 5 excited state by the iron catalyst 1 and by the electron donor (BIH) using Stern–Volmer analysis (see ESI† for details). At an excitation wavelength of 450 nm, the luminescence at 550 nm of the Cu PS was effectively quenched by BIH with an apparent quenching rate constant of 1.1 × 108 M−1 s−1 (Fig. 1a). For comparison, typical quenching constants for Ru PS are in the order of 107–1010 M−1 s−1, suggesting that reductive quenching of Cu PS with the electron donor BIH can reasonably operate in this photocatalytic system. On the other hand, the fluorescence of Cu PS 5 was not attenuated by iron catalyst 1. That excludes the possibility of an oxidative quenching mechanism (Fig. 1b) by the catalyst itself. These observations are in agreement with the reported redox potentials of the involved species. For instance, whereas an electron transfer from BIH (Eox = 0.33 V vs. SCE)15 to the excited state of the PS (Cu PS−/Cu PS*, E0 = 1.03 V)10b is thermodynamically feasible, an electron transfer from the excited state of the PS (Cu PS*/Cu PS+, E0 = −1.35 V vs. SCE)10b to the iron complex 1 (Ered = −1.65 V vs. SCE) is not possible.
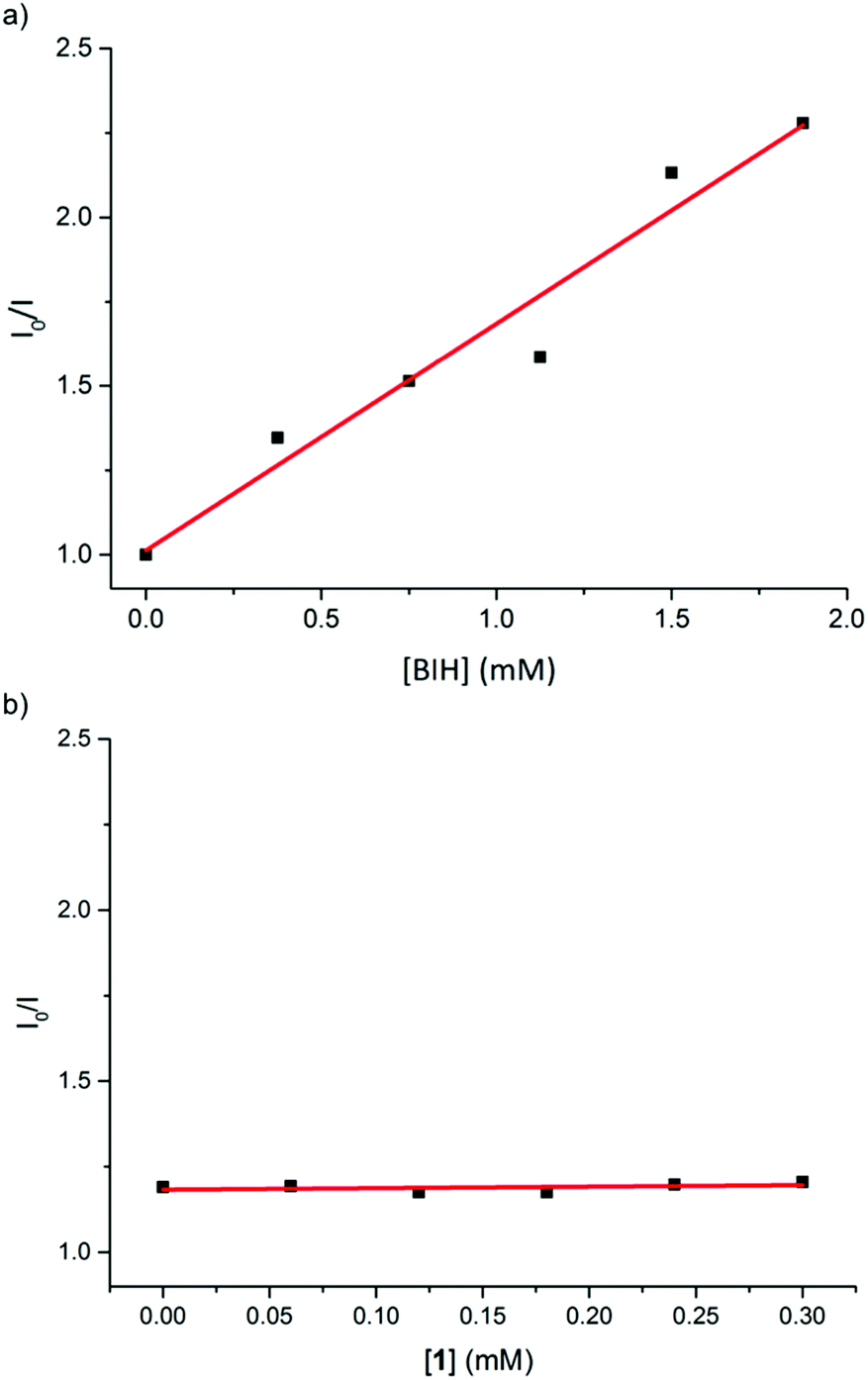 | ||
| Fig. 1 Linear plots of ratio of fluorescence intensities of in situ Cu PS 5 in the absence and presence of (a) BIH (y = 0.672x + 1.013, R2 = 0.94) and (b) 1, according to the Stern–Volmer equation. | ||
Clearly, a reductive quenching mechanism operates in the photocatalytic CO2 reduction mediate by complex 1 and Cu PS 5. Accordingly, a photoinduced electron transfer between the Cu PS and BIH takes place first, resulting in the generation of the reduced state of PS (Cu PS−). Subsequent reduction of the iron catalyst by Cu PS−, generates the active catalyst species that mediates the photoreduction of CO2 to CO, as previously observed when an Ir PS was used.12
Conclusions
In summary, we have described a novel selective photocatalytic system for CO2 reduction based on earth-abundant elements, combining a copper-based photosensitizer and a molecularly-defined iron catalyst. The Cu PS was easily generated in situ using a copper salt, bidentate phosphines, and easily available diimine ligands. When such a luminescent Cu complex was combined with an iron cyclopentadienone catalyst in the presence of light and a suitable electron donor, carbon dioxide is selectively reduced to CO with a TON up to 487 and a ΦCO = 13.3%. According to mechanistic investigations, the photocatalytic reduction of CO2 proceeded via reductive quenching of the excited Cu PS with BIH and further electron transfer from the resulting reduced species of Cu PS to the iron catalyst.Acknowledgements
This work has been supported by the state of Mecklenburg-Vorpommern, the BMBF, and the EU fund H2020-MSCA-ITN-2015 in Horizon 2020 as part of the project NoNoMeCat (675020). A. R.-H. acknowledges financial support from the DAAD.Notes and references
- M. Aresta, in Carbon Dioxide as Chemical Feedstock, ed. M. Aresta, Wiley-VCH, Weinheim, 2010, pp. 1–14 Search PubMed.
- (a) Y. Yamazaki, H. Takeda and O. Ishitani, J. Photochem. Photobiol., C, 2015, 25, 106–137 CrossRef CAS; (b) T. Yui, Y. Tamaki, K. Sekizawa and O. Ishitani, Top. Curr. Chem., 2011, 303, 151–184 CrossRef CAS PubMed; (c) A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed.
- (a) J. Hawecker, J.-M. Lehn and R. Ziessel, Helv. Chim. Acta, 1986, 69, 1990 CrossRef CAS; (b) J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536 RSC.
- (a) A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS; (b) A. Ito and T. J. Meyer, Phys. Chem. Chem. Phys., 2012, 14, 13731–13745 RSC; (c) K. A. King, P. J. Spellane and R. J. Watts, J. Am. Chem. Soc., 1985, 107, 1431–1432 CrossRef CAS; (d) A. Kapturkiewicz and G. Angulo, Dalton Trans., 2003, 3907–3913 RSC; (e) P. J. Hay, J. Phys. Chem. A, 2002, 106, 1634–1641 CrossRef CAS.
- (a) Y. Kuramochi, J. Itabashi, K. Fukaya, A. Enomoto, M. Yoshida and H. Ishida, Chem. Sci., 2015, 6, 3063–3074 RSC; (b) Y. Kuramochi, M. Kamiya and H. Ishida, Inorg. Chem., 2014, 53, 3326–3332 CrossRef CAS PubMed; (c) H. Ishida, T. Terada, K. Tanaka and T. Tanaka, Inorg. Chem., 1990, 29, 905–911 CrossRef CAS; (d) J.-M. Lehn and R. Ziessel, J. Organomet. Chem., 1990, 382, 157–173 CrossRef CAS; (e) H. Ishida, K. Tanaka and T. Tanaka, Chem. Lett., 1988, 17, 339–342 CrossRef; (f) I. Hitoshi, T. Koji and T. Toshio, Chem. Lett., 1987, 16, 1035–1038 CrossRef.
- (a) G. Sahara and O. Ishitani, Inorg. Chem., 2015, 54, 5096 CrossRef CAS PubMed; (b) A. Nakada, K. Koike, T. Nakashima, T. Morimoto and O. Ishitani, Inorg. Chem., 2015, 54, 1800 CrossRef CAS PubMed; (c) A. Nakada, K. Koike, K. Maeda and O. Ishitani, Green Chem., 2016, 18, 139 RSC; (d) E. Kato, H. Takeda, K. Koike, K. Ohkubo and O. Ishitani, Chem. Sci., 2015, 6, 3003 RSC; (e) C. Liu, K. D. Dubois, M. E. Louis, A. S. Vorushilov and G. Li, ACS Catal., 2013, 3, 655 CrossRef CAS; (f) J. Schneider, K. Q. Vuong, J. A. Calladine, X.-Z. Sun, A. C. Whitwood, M. W. George and R. N. Perutz, Inorg. Chem., 2011, 50, 11877 CrossRef CAS PubMed; (g) J. Ettedgui, Y. Diskin-Posner, L. Weiner and R. Neumann, J. Am. Chem. Soc., 2011, 133, 188 CrossRef CAS PubMed; (h) H. Takeda, K. Koike, H. Inoue and O. Ishitani, J. Am. Chem. Soc., 2008, 130, 2023 CrossRef CAS PubMed.
- (a) R. O. Reithmeier, S. Meister, A. Siebel and B. Rieger, Dalton Trans., 2015, 44, 6466 RSC; (b) R. O. Reithmeier, S. Meister, B. Rieger, A. Siebel, M. Tschurl, U. Heiz and E. Herdtweck, Dalton Trans., 2014, 43, 13259 RSC; (c) S. Sato, T. Morikawa, T. Kajino and O. Ishitani, Angew. Chem., Int. Ed., 2013, 52, 988 CrossRef CAS PubMed.
- (a) Z. Guo, S. Cheng, C. Cometto, E. Anxolabéhère-Mallart, S.-M. Ng, C.-C. Ko, G. Liu, L. Chen, M. Robert and T.-C. Lau, J. Am. Chem. Soc., 2016, 138, 9413–9416 CrossRef PubMed; (b) P. L. Cheung, C. W. Machan, A. Y. S. Malkhasian, J. Agarwal and C. P. Kubiak, Inorg. Chem., 2016, 55, 3192–3198 CrossRef CAS PubMed; (c) P. G. Alsabeh, A. Rosas-Hernández, E. Barsch, H. Junge, R. Ludwig and M. Beller, Catal. Sci. Technol., 2016, 6, 3623–3630 RSC; (d) F. Wang, B. Cao, W.-P. To, C.-W. Tse, K. Li, X.-Y. Chang, C. Zang, S. L.-F. Chan and C.-M. Che, Catal. Sci. Technol., 2016, 6, 7408–7420 RSC; (e) L. Chen, Z. Guo, X.-G. Wei, C. Gallenkamp, J. Bonin, E. Anxolabéhère-Mallart, K.-C. Lau, T.-C. Lau and M. Robert, J. Am. Chem. Soc., 2015, 137, 10918–10921 CrossRef CAS PubMed; (f) S. L.-F. Chan, T. L. Lam, C. Yang, S.-C. Yan and N. M. Cheng, Chem. Commun., 2015, 51, 7799–7801 RSC; (g) J. Bonin, M. Robert and M. Routier, J. Am. Chem. Soc., 2014, 136, 16768–16771 CrossRef CAS PubMed; (h) H. Takeda, H. Koizumi, K. Okamoto and O. Ishitani, Chem. Commun., 2014, 50, 1491–1493 RSC; (i) V. S. Thoi, N. Kornienko, C. G. Margarit, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2013, 135, 14413–14424 CrossRef CAS PubMed.
- H. Takeda, K. Ohashi, A. Sekine and O. Ishitani, J. Am. Chem. Soc., 2016, 138, 4354–4357 CrossRef CAS PubMed.
- (a) S.-P. Luo, E. Mejía, A. Friedrich, A. Pazidis, H. Junge, A.-E. Surkus, R. Jackstell, S. Denurra, S. Gladiali, S. Lochbrunner and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 419–423 CrossRef CAS PubMed; (b) E. Mejía, S.-P. Luo, M. Karnahl, A. Friedrich, S. Tschierlei, A.-E. Surkus, H. Junge, S. Gladiali, S. Lochbrunner and M. Beller, Chem. – Eur. J., 2013, 19, 15972–15978 CrossRef PubMed; (c) M. Karnahl, E. Mejía, N. Rockstroh, S. Tschierlei, S.-P. Luo, K. Grabow, A. Kruth, V. Brüser, H. Junge, S. Lochbrunner and M. Beller, ChemCatChem, 2014, 6, 82–86 CrossRef CAS; (d) S. Fischer, D. Hollmann, S. Tschierlei, M. Karnahl, N. Rockstroh, E. Barsch, P. Schwarzbach, S.-P. Luo, H. Junge, M. Beller, S. Lochbrunner, R. Ludwig and A. Brückner, ACS Catal., 2014, 4, 1845–1849 CrossRef CAS.
- A. J. J. Lennox, S. Fischer, M. Jurrat, S.-P. Luo, N. Rockstroh, H. Junge, R. Ludwig and M. Beller, Chem. – Eur. J., 2016, 22, 1233–1238 CrossRef CAS PubMed.
- A. Rosas-Hernández, P. G. Alsabeh, E. Barsch, H. Junge, R. Ludwig and M. Beller, Chem. Commun., 2016, 52, 8393–8396 RSC.
- M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed.
- N. Armaroli, G. Accorsi, F. Cardinali and A. Listorri, Top. Curr. Chem., 2007, 280, 69–115 CrossRef CAS.
- Y. Tamaki, K. Koike, T. Morimoto and O. Ishitani, J. Catal., 2013, 304, 22–28 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, quantum yield calculations and emission-quenching experiments procedure. See DOI: 10.1039/c6gc03527b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |