
PEG–peptide hydrogels reveal differential effects of matrix microenvironmental cues on melanoma drug sensitivity†
Emi Y.
Tokuda
ac,
Caitlin E.
Jones
ad and
Kristi S.
Anseth
*ab
aDepartment of Chemical and Biological Engineering, University of Colorado, Boulder, CO 80309, USA. E-mail: kristi.anseth@colorado.edu
bHoward Hughes Medical Institute and The BioFrontiers Institute, University of Colorado, Boulder, CO 80309, USA
cBen Towne Center for Childhood Cancer Research, Seattle Children's Research Institute, Seattle, WA 98101, USA
dDepartment of Biomedical Engineering, Ohio State University, Columbus, OH 43210, USA
First published on 5th December 2016
Abstract
Metastatic melanoma is highly drug resistant, though the exact mechanisms of this resistance are not completely understood. One method to study melanoma drug responsiveness in vitro is through the use of multicellular spheroids, which have been found to exhibit decreased drug sensitivity compared to traditional 2D culture on various substrates. Because it is unclear whether dimensionality, cell–matrix interactions, and/or cell–cell contacts may influence melanoma drug responsiveness, we utilized a synthetic PEG-based hydrogel to compare the responses of cells cultured on top of or encapsulated within matrices with the same adhesive ligand density, polymer density, and material properties. We found that depending on the stage of progression at which the melanoma cells were derived, the cells responded differently to PLX4032 treatment, a commercially available melanoma drug. In particular, early stage WM35 cells were insensitive to dimensionality (i.e., 2D versus 3D culture), while metastatic A375 cells exhibited decreased responsiveness in 3D compared to 2D. To further understand the role of the microenvironment in early stage melanoma cells, we tested single WM35 cells and multicellular WM35 spheroids in 3D. The results revealed that the spheroids were similarly sensitive to PLX4032 treatment compared to single cell encapsulations. Collectively, this study implicates the role that 3D microenvironments (i.e., dimensionality) may play in observed melanoma drug responsiveness, and the potential lack of influence of cell–matrix interactions over cell–cell contacts in early stages of melanoma resistance to PLX4032-induced apoptosis.
Insight, innovation, integrationUsing a tunable, fully synthetic, cytocompatible hydrogel scaffold, we compared melanoma cells seeded on top (2D) or encapsulated as a single cell suspension (3D) to assess the role of cell–matrix interactions and dimensionality in apoptotic responses to drug treatment. Current in vitro methods to evaluate drug responsiveness include culture on traditional plastic (2D) and multicellular spheroids embedded within protein matrices (3D). While 3D spheroid models have shown decreased drug sensitivity in cells compared to 2D, it is difficult to determine whether dimensionality, cell–matrix interactions, cell–cell contacts, and matrix elasticity are the most important microenvironmental factors affecting drug responsiveness. Here, we systematically control and elucidate which microenvironmental factors may be important when screening for drug effects in vitro. |
Introduction
Traditional two-dimensional (2D) culture of cells on tissue culture-treated polystyrene (TCPS) has allowed invaluable characterization and improved the quantitative understanding of basic cell signaling and function. However, these 2D surfaces are often aphysiologically stiff (six orders of magnitude stiffer than most soft tissues), can unnaturally polarize cells, and do not necessitate any matrix remodeling for proliferation and migration as occurs in vivo.1,2 As a result, three-dimensional (3D) culture platforms have evolved to present a more in vivo-like microenvironment or recapitulate organ or tissue function.3–7 For example, a pioneering study by Petersen et al. illustrated a fundamental difference in breast epithelial cells, which proliferated indefinitely as a monolayer culture on TCPS but formed in vivo-like multicellular acinar structures when embedded in Matrigel.8 Since this landmark publication, 3D matrix environments have become more common in the study of cancer biology and drug responsiveness, as key parameters of matrix signaling can be quite important in predicting in vivo outcomes from in vitro screens.9–11In cancer research, 3D models often utilize multicellular spheroids, where cells are either aggregated or allowed to proliferate when embedded within hydrogel microenvironments, which are typically composed of collagen or Matrigel.12–14 Numerous studies have reported altered or increased resistance to drug treatment in these multicellular aggregates compared to traditional 2D culture on TCPS.9,14–18 Researchers have hypothesized that a 3D environment better recapitulates the native environment that cancer cells might experience, where cell–matrix and cell–cell interactions can promote survival.5,19,20 As a result, the use of 3D models has advanced to become a more standard method to better evaluate and predict drug candidate efficacy before studying their effects in animal models.21
While experiments using 3D spheroids have shown differential responses to the same drug treatment compared to cells in monolayer culture, numerous differences exist between cell aggregates and cells on hard plastic surfaces.22 For instance, on TCPS, cells are unnaturally polarized, are exposed to a sink of nutrients or drugs without any diffusion length scale, and cell–matrix interactions are generally extensive.1,23 This is in stark contrast to 3D spheroids, where spatial positioning of the cells can matter, cell–cell interactions are numerous, and the elasticity and chemistry of the microenvironment is dramatically different than TCPS.24,25
With this in mind, we sought to explore the role of the matrix microenvironment and its dimensionality, in a more controlled manner, on melanoma apoptotic responses to clinically available drugs. We utilized fully synthetic PEG-based hydrogels in order to simplify the culture system compared to naturally derived 3D systems, such as collagen.26 These PEG–peptide hydrogels were formed via the thiol–ene “photo-click” reaction through step-growth network formation27 between norbornene-functionalized multi-arm PEG and cysteine containing peptides.28 The thiol–ene reaction is cytocompatible and therefore allows for culture as both a 2D and 3D culture platform with wide tunability of bulk properties.29,30 Furthermore, the chemistry allows systematic control and manipulation of the adhesive ligand density, as well as susceptibility to degradation by cell-secreted proteases, through the incorporation of cysteine-containing peptide sequences or thiolated proteins.
Previously,31 we reported that early stage radial growth phase melanoma cells (WM35) were sensitive to substrate elasticity in 2D, and this in turn, affected their drug responsiveness to PLX4032 (clinically, Zelboraf or vemurafenib). Metastatic A375 cells, however, appeared less dependent on cell–matrix interactions and substrate elasticity, by exhibiting minimal changes in drug responsiveness as a function of the stiffness of the local environment. The WM35 and A375 cells were chosen because of previously established sensitivity to PLX403231 and they represent two different stages of melanoma progression originally established from patient samples. To make more direct comparisons between 2D and 3D environments, WM35 or A375 cells were seeded on top of and also encapsulated within hydrogels of the same composition as a single cell suspension. Viability was then challenged with PLX4032 for 48 hours and metabolic activity and apoptosis were measured. Finally, studies comparing single cell encapsulations with embedded spheroids were performed. Collectively, experiments were designed to better understand what aspects of the spheroid “tumor-like” environment contribute to its more “drug resistant” phenotype that is often studied and described in the literature.
Methods
Reagents and materials
Unless otherwise noted, all cell culture reagents were purchased from Life Technologies (Carlsbad, CA, USA) and all chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA). PEG was purchased from JenKem (Plano, TX, USA) and functionalized with norbornene as previously described.28 All peptides were purchased from American Peptide Company (Sunnyvale, CA, USA). The photoinitiator lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) was synthesized as previously described.32 PLX4032 was purchased from Chemietek (Indianapolis, IN, USA) and made into a stock of 10 mM in DMSO.Cell culture
Radial growth phase human melanoma cells (WM35) and metastatic A375 cells were a generous gift from Professor Natalie Ahn (University of Colorado, Department of Chemistry and Biochemistry). Cells were cultured in RPMI 1640 without phenol red in 10% FBS. Experiments were performed with 1% FBS.Hydrogel preparation for 2D and single cell encapsulations
Hydrogels were prepared similarly as previously described.31 To allow for cell-mediated degradation of the network, matrix metalloproteinase-degradable peptides (KCGPQG↓IWGQCK, down arrow denotes the sight of cleavage, MW 1304.6 g mol−1) were used to crosslink the hydrogel, while the fibronectin-derived adhesion sequence RGD (CRGDS, MW 536.6 g mol−1) was covalently tethered to aid in cell adhesion and spreading. Macromer solutions were prepared with 3 mM four-arm 20 kDa norbornene-functionalized PEG, 3.18 mM MMP crosslinker, 1 mM RGD, and 1.7 mM LAP. Two-dimensional hydrogels were prepared by placing thiolated 12 mm circular coverslips on top of a 12 μL drop of macromer solution, polymerized for 3 minutes with UV light (XX Series) centered around 365 nm light, ∼5 mW cm−2. Coverslips were first cleaned by passing them through a flame and then functionalized with 0.55% (v/v) (3-mercaptopropyl)trimethoxysilane in 95% ethanol (pH ∼ 5.5) to result in thiol-functionalized coverslips. The 2D culture gels were prepared the day before seeding; on the day of seeding, gels were sterilized for one hour at room temperature in 5% isopropanol in PBS under a UV light, rinsed twice with PBS, and then seeded at 1 × 105 cells per cm2. The cells were encapsulated at 2 × 106 cells per mL as previously described (Fig. 1).33 Briefly, the cells were suspended in PBS to achieve the desired cell concentration and added to the macromer solution described above. 2D hydrogels were seeded the same day as 3D encapsulations and both conditions used the same cell solution stock. To characterize the modulus of the hydrogels, 30 μL gels were formed in the cut end of a 1 mL syringe and allowed to swell overnight. The moduli of all hydrogels were measured using an Ares DHR3 (TA Instruments, New Castle, DE, USA) shear rheometer to quantify the shear elastic modulus and then converted to the Young's modulus, where E = 2G(1 + ν) assuming a Poisson's ratio (ν) of 0.5. The hydrogels were found to have a Young's modulus of approximately 0.4 kPa.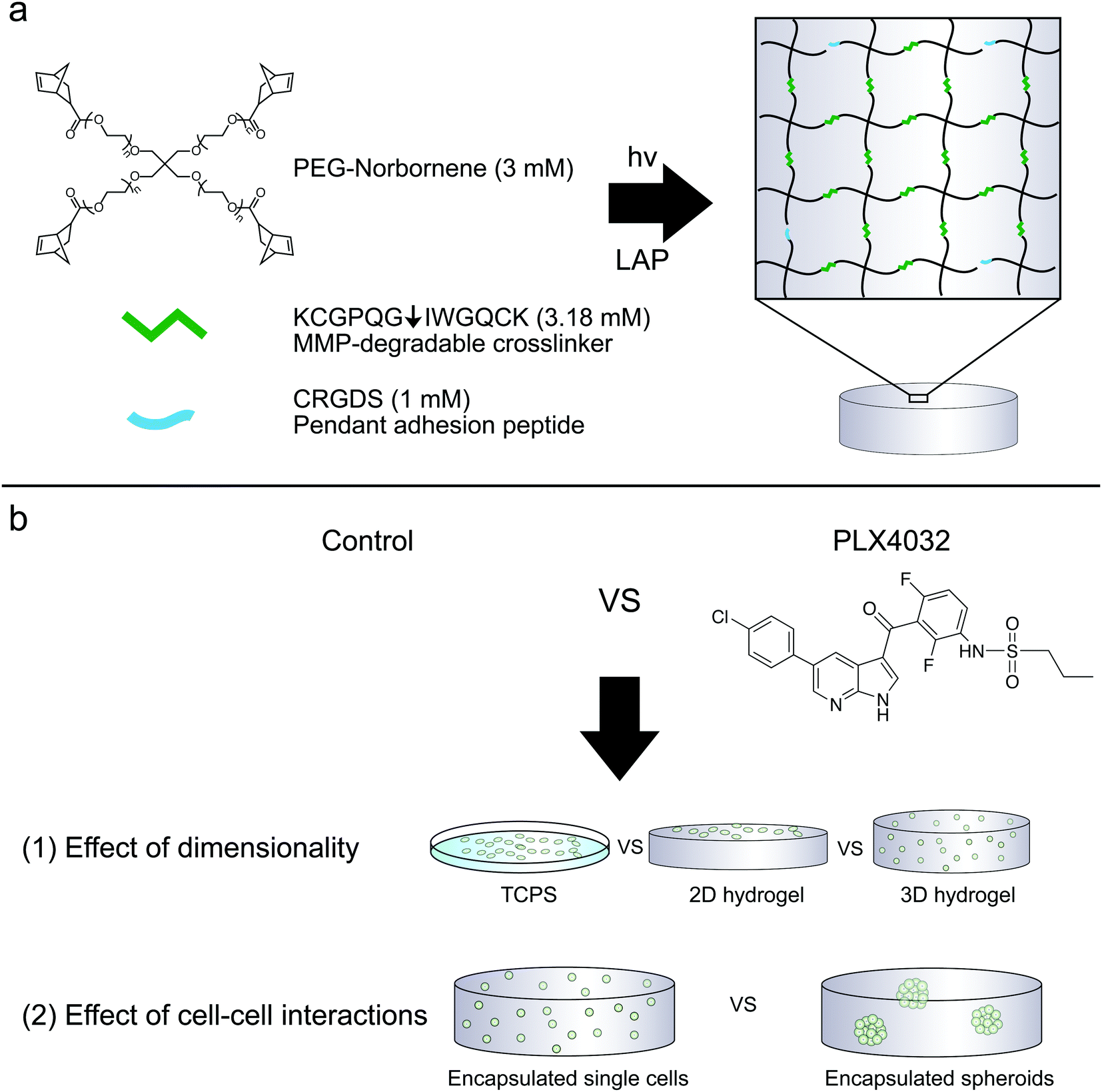 | ||
| Fig. 1 (a) Components of PEG–peptide hydrogels utilized for these studies. Four-arm norbornene-functionalized PEG (-ene) was crosslinked with an MMP-degradable peptide (KCGPQG↓IWGQCK) containing cysteines (-thiol). The PEG was used at a concentration of 3 mM (12 mM ene), and reacted 47% off stoichiometry (6.36 mM thiol, equivalently 3.18 mM crosslinker peptide) to form soft (0.4 kPa) hydrogels. The cysteines on either side of the MMP crosslinker or the pendant adhesion peptide CRGDS can react with norbornene to form a step-growth thiol–ene network. Integrins can engage the RGDS peptide, and combined with the proteolytically cleavable linker, aid in spreading and cell survival. (b) Cells can either be encapsulated or seeded on top of the PEG–peptide hydrogels. (1) Here, WM35 and A375 were cultured on TCPS, on top of a hydrogel matrix or encapsulated within the same formulation and tested for sensitivity to PLX4032 treatment. (2) The WM35 cells were then tested as single cells or multicellular spheroids against drug treatment to better understand what component of spheroid culture may contribute to its more drug resistant nature. | ||
Spheroid formation and encapsulation
Spheroids were formed via the liquid overlay method.14,34 Briefly, 1.5% Noble agar (Thermo Fisher Scientific, Waltham, MA, USA) dissolved in diH2O was heated in a microwave until liquefied. Then, 50 μL of the solution was pipetted into the bottom of a 96-well plate and allowed to solidify. 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells in 200 μL, as a single cell suspension, were added on top of the agar and allowed to cluster for 2 days in 10% FBS growth media. Spheroids were transferred using a 1 mL pipette to minimize their dissociation, and three spheroids were collected in a 1.7 mL Eppendorf tube and spun down for 5 minutes at 3000 rpm. As much media as possible were aspirated from the spheroids, which were then re-suspended in the macromer solution defined above and polymerized. For measuring metabolic activity and apoptosis, samples were prepared in a 1 mL syringe with the end cut off and made into 30 μL gels. 15 μL of the macromer solution was polymerized at the bottom of the syringe for 30 seconds, then a solution containing 3 spheroids was added on top and polymerized for 3 minutes. Single cell encapsulations were prepared with 2.5 × 106 cells per mL to yield ∼75000 cells in both the spheroid (3 spheroids per hydrogel) and single cell samples for comparisons. Both single cell and spheroid encapsulations were performed the same day with the same passage number of cells. Imaged samples were visualized using the Live/Dead Viability/Cytotoxicity Kit (Life Technologies) containing calcein AM which is metabolized by live cells and becomes fluorescent and ethidium homodimer-1 to indicate plasma membrane permeability.
000 cells in 200 μL, as a single cell suspension, were added on top of the agar and allowed to cluster for 2 days in 10% FBS growth media. Spheroids were transferred using a 1 mL pipette to minimize their dissociation, and three spheroids were collected in a 1.7 mL Eppendorf tube and spun down for 5 minutes at 3000 rpm. As much media as possible were aspirated from the spheroids, which were then re-suspended in the macromer solution defined above and polymerized. For measuring metabolic activity and apoptosis, samples were prepared in a 1 mL syringe with the end cut off and made into 30 μL gels. 15 μL of the macromer solution was polymerized at the bottom of the syringe for 30 seconds, then a solution containing 3 spheroids was added on top and polymerized for 3 minutes. Single cell encapsulations were prepared with 2.5 × 106 cells per mL to yield ∼75000 cells in both the spheroid (3 spheroids per hydrogel) and single cell samples for comparisons. Both single cell and spheroid encapsulations were performed the same day with the same passage number of cells. Imaged samples were visualized using the Live/Dead Viability/Cytotoxicity Kit (Life Technologies) containing calcein AM which is metabolized by live cells and becomes fluorescent and ethidium homodimer-1 to indicate plasma membrane permeability.
Metabolic activity, apoptosis, and DNA content
After seeding cells on 2D gel surfaces or encapsulating them in 3D gel samples, the cells were allowed to adhere to the materials and recover overnight. The medium was then changed to introduce drug treatment (1 μM PLX4032) or as a negative control (DMSO). The 2D gel samples were moved to clean plates before adding fresh media. Cells were treated for 48 hours. PrestoBlue (Life Technologies) was used to measure the metabolic activity; the samples were allowed to incubate for 1 hour and then read on a plate reader (Biotek Synergy H1, Winooski, VT, USA). To measure the metabolic activity, a well scan was performed in order to measure the fluorescence across the entire well in a grid, and the values within the grid were averaged to result in a single value for a well/sample.33 To collect apoptosis and DNA samples, media from each untreated and treated well were saved to capture any potential floating cells, and 2 mg mL−1 collagenase (Type 1, Worthington Biochemicals, Lakewood, NJ, USA) was used to dissolve the gels and recover the embedded cells (3D gels required ∼1 hour to degrade completely). The samples were spun down for 4 minutes at 1100 rpm, rinsed with 2 mL PBS, and then centrifuged again. The cell pellet was lysed using the lysis buffer provided by the EnzCheck Caspase 3 Kit #2 (Life Technologies). The cell lysate was then exposed to one freeze–thaw cycle to ensure complete lysis, spun down at 7000 rpm for 7 minutes, and then the company protocol was followed to measure caspase 3 activity. The DNA content from the same lysate was quantified using the Quant-iT PicoGreen assay kit (Life Technologies); 10 μL of the sample lysate was used for DNA content analysis. Both caspase 3 and DNA content were measured on a Biotek Synergy H1 plate reader. Fig. 3a, b, 5b and Fig. S1a, b, S2 (ESI†) show caspase 3 activity normalized to the DNA content to take into account any potential differences in cell numbers between conditions.Statistical analysis
All experiments were performed at least 3 times with a minimum of 2 technical replicates. Graphs were produced in Graphpad Prism 5. Statistical analysis was performed in Graphpad Prism 5 by a two-way ANOVA and Bonferonni post-tests for p < 0.05 (Fig. 3a and b, 5 and Fig. S1a and b, S2, ESI†); one-way ANOVA with Tukey post-tests for p < 0.05 (Fig. 2b, 3c and d and Fig. S1c and d, ESI†).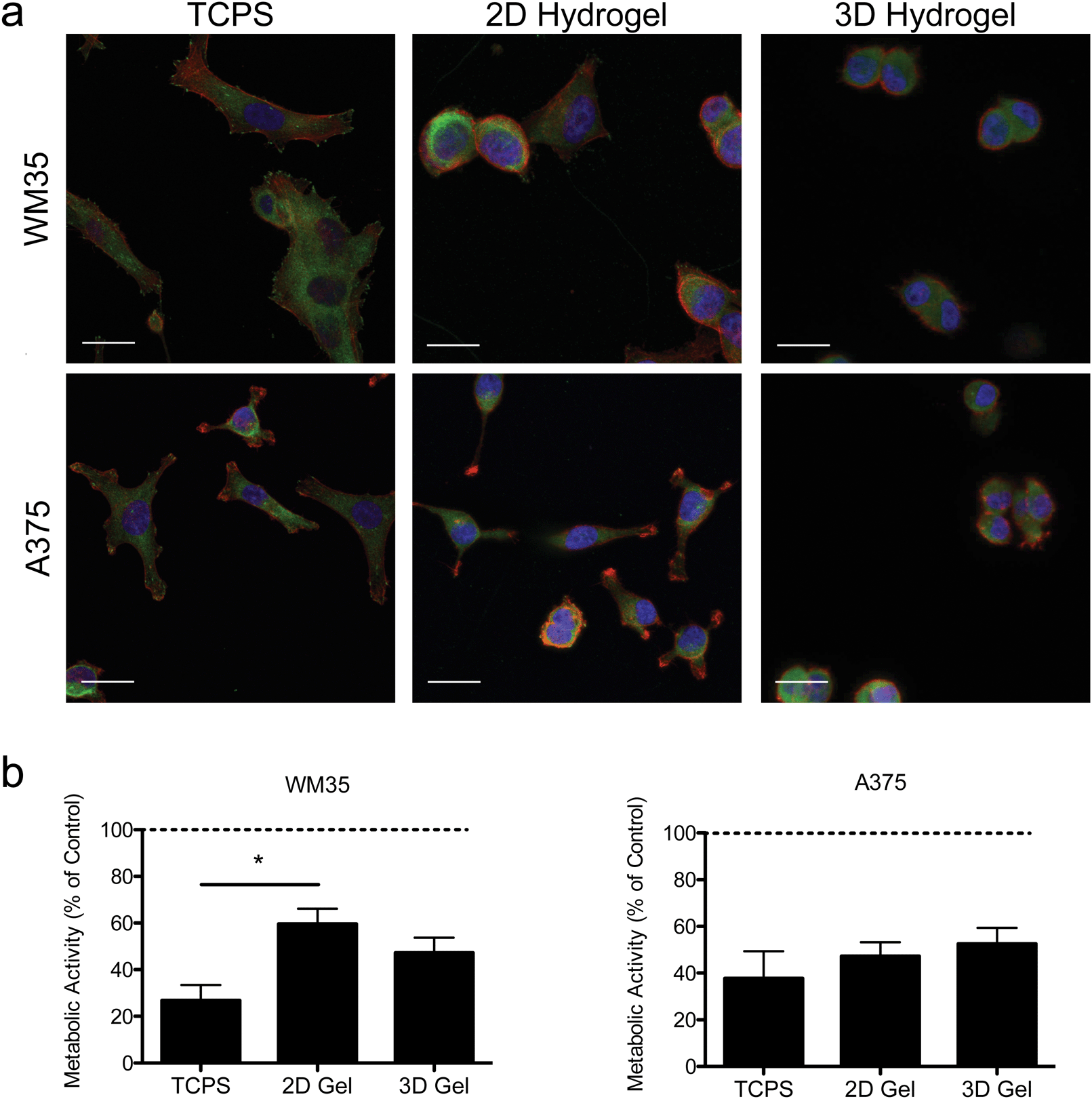 | ||
| Fig. 2 Morphology and activity in 2D and 3D microenvironments. (a) Immunostaining for focal adhesions was performed using two melanoma cell lines (WM35 and A375) cultured on TCPS, gel surfaces (2D), or encapsulated in hydrogels (3D) after 24 hours of culture. Images of cells seeded on TCPS show cell spreading and more focal adhesions, while cells on the soft gels appear slightly more rounded. When encapsulated within the hydrogels, both cell types become very rounded, presumably due to the longer time scale for remodeling and degradation of the surrounding matrix. Paxillin (green), f-actin (red), nucleus (blue), scale bar, 10 μm. (b) Metabolic activity as measured by PrestoBlue of WM35 and A375 cells after 48 hours of treatment. WM35 cells exhibited a statistically significant increase in metabolic activity on hydrogel surfaces compared to TCPS. No statistically significant differences were observed in metabolic activity between the culture conditions in the A375 cells. The dashed line represents values of the control samples. *p < 0.05. | ||
Results
Morphology and metabolic activity of 2D versus 3D cell culture
Hydrogels were synthesized from 3 mM four-arm PEG-norbornene (MW: 20000) crosslinked with 3.18 mM MMP-degradable peptides (KCGPQG↓IWGQCK), and 1 mM pendant CRGDS (fibronectin-derived adhesive sequence), in the presence of 1.7 mM LAP (photoinitiator lithium phenyl-2,4,6-trimethylbenzoylphosphinate) and ∼5 mW cm−2 light centered around 365 nm and resulted in a final Young's modulus of approximately 0.4 kPa (Fig. 1). This formulation was selected based on previous work showing that 1 mM RGDS promoted cell attachment and spreading while the MMP-crosslinker allowed for cell-mediated degradation and motility of both encapsulated primary and cancer cells.35–38 To study the effect of dimensionality (i.e., 2D versus 3D) on cell function and drug responsiveness, two different human melanoma cell lines were selected: an early stage radial growth phase (RGP) cell line (WM35), and a metastatic line (A375). Both WM35 and A375 cells were seeded on the surface of the hydrogels (2D) at a density of 1 × 105 cells per cm2, as well as encapsulated within the same hydrogel formulation at a density of 2 × 106 cells per mL (3D). These cell numbers resulted in similar final cell densities under both 2D and 3D conditions, assuming a 50 μm slice through a 3D hydrogel is then projected in the XY plane (i.e., 2D). Overall, cell morphology was assessed via immunostaining for focal adhesion formation (Fig. 2a), and cells cultured on TCPS were included as a reference for comparison. On TCPS, the WM35 cells were spread, exhibited clearly defined f-actin stress fibers, and formed distinct focal adhesions as indicated by punctate paxillin staining (green), a focal adhesion component. When cultured on top of the relatively soft hydrogels, the cells became more rounded and focal adhesions appeared smaller; encapsulated cells were mostly rounded with only cytoplasmic paxillin staining patterns. The metastatic A375 cells were not as large as the WM35 cells on TCPS and formed small lamellipodia-like structures with smaller focal adhesions. When cultured on the surface of the hydrogels, some of the cells maintained small lamellipodia, but others were more rounded. A375 cells encapsulated in the hydrogel matrices were typically rounded, though a few began to form short, wide protrusions, indicative of local matrix degradation.
To study the influence of PLX4032 treatment on cultured melanoma cells, the metabolic activity was measured after 48 hours of exposure to 1 μM of PLX4032, and the results are reported as a percent of activity relative to the control conditions (i.e., no PLX4032 treatment, Fig. 2b). While both cell types exhibited an overall decrease in absolute metabolic activity in the presence of PLX4032, the WM35 cells revealed a more sensitive response to drug treatment when cultured on TCPS compared to either 2D or 3D hydrogel conditions; the reduced response in the 2D hydrogel conditions was statistically significant compared to TCPS. There were no statistically significant differences in the metabolic activity as a function of dimensionality in the A375 cells (i.e., 2D versus 3D culture). In all A375 drug-treated samples, the metabolic activity was between 40% and 60% compared to the untreated control conditions. While the metabolic activity provides a general screen of cell health, PLX4032 has been shown to be both cytostatic and cytotoxic, so we next measured apoptosis in order to better understand whether there were any differential effects of PLX4032 as a function of the culture environment.
Apoptosis as a function of inhibitor treatment in 2D versus 3D
Apoptosis in both the radial growth phase (WM35s) and the metastatic (A375s) melanoma cells were measured based on caspase 3 activity after 48 hours of drug treatment. Caspase 3 activity was normalized to the TCPS control (i.e., the TCPS control had a value of 1), and cells were seeded on TCPS and on top of or encapsulated within hydrogels. The WM35 cells were previously shown to respond to PLX4032 exposure in a manner that was dependent on the underlying substrate elasticity.31 Here, WM35s exhibited sensitivity to PLX4032 under all culture conditions tested (Fig. 3a). In both 2D and 3D hydrogel samples, PLX4032 treatment caused an increase in apoptosis (∼4.3- and 4.1-fold increase compared to respective controls) compared to a 2.4-fold increase on TCPS (Fig. 3c). Despite the lower absolute values of caspase 3 activity in both 2D and 3D samples compared to TCPS, the fold changes in caspase 3 were much higher under the hydrogel conditions; though not statistically significant, the basal levels of caspase 3 were lower on top of or within hydrogels compared to TCPS.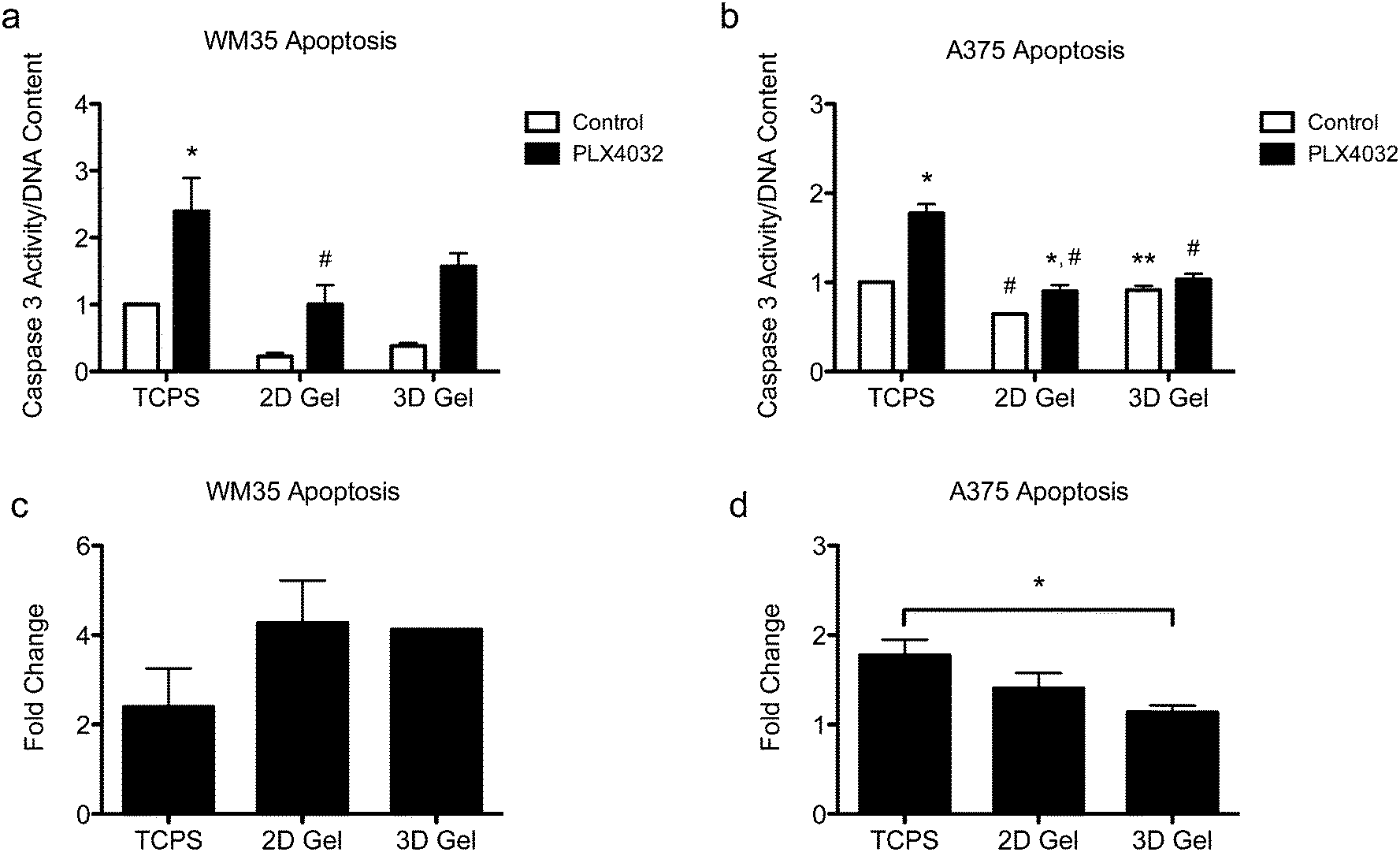 | ||
| Fig. 3 Apoptosis of WM35 and A375 cells after 48 hours of treatment with PLX4032 in 2D and 3D microenvironments. (a) WM35 cells exhibited sensitivity to PLX4032 treatment, as measured by increased caspase 3 activity, for all culture microenvironments, TCPS and 2D and 3D hydrogel cultures. The 2D hydrogel PLX4032-treated sample was statistically significantly different than the TCPS PLX4032 sample. (b) A375 cells exhibited overall less sensitivity to PLX4032 treatment as observed by the smaller increases in caspase 3 activity with PLX4032 treatment. The 3D control sample was significantly higher than the 2D hydrogel control sample, and the 2D hydrogel control was statistically lower than that of the TCPS control. (c and d) Fold change in caspase 3 activity with PLX4032 treatment relative to the respective control without treatment. *p < 0.05 compared to respective control, #p < 0.05 compared to corresponding conditions in TCPS, **p < 0.05 between A375 2D and 3D control samples. | ||
Apoptosis was also assessed in the metastatic A375 cells (Fig. 3b), and overall, the differential changes in caspase 3 activity were much lower (∼1.8-, 1.4-, and 1.1-fold increase over the controls in TCPS, 2D and 3D gels, respectively, Fig. 3d) compared to the radial growth phase WM35s (up to an ∼4.3-fold increase). For the A375 cells cultured on TCPS and 2D gels, PLX4032 induced a significant increase in apoptosis compared to the control, though the basal level of caspase 3 activity on the 2D hydrogel control was statistically significantly lower than the TCPS control (Fig. 3b). Additionally, the level of apoptosis caused by PLX4032 treatment for A375s cultured on 2D gel surfaces or embedded in 3D gels was significantly lower than that under the TCPS PLX4032 conditions. In fact, the 3D hydrogels did not exhibit a statistically significant increase in apoptosis in the presence of PLX4032, and instead when comparing the fold change with respect to the controls, A375s embedded in 3D hydrogels exhibited a statistically significantly lower level of apoptosis compared to TCPS (Fig. 3d).
Apoptosis was also evaluated for both cell types in a stiffer hydrogel formulation (∼4 kPa) (Fig. S1, ESI†), as matrix elasticity can be important for cell function and subsequent signaling.39–42 WM35s remained sensitive to PLX4032 in the stiff hydrogels (∼4.2 and 4.8-fold increase in caspase 3 over corresponding controls in 2D gels and 3D, respectively) though there were no significant differences between the PLX4032-treated samples in 2D and 3D gels (Fig. S1a and c, ESI†). The A375s exhibited approximately a 1.4-fold increase in caspase 3 activity when compared to respective controls in both 2D gels and 3D (Fig. S1b and d, ESI†). Again, the maximum change in caspase 3 activity was only ∼1.8 on TCPS in the A375s compared to ∼4.8 in the WM35 cells in 3D. Cell death was also measured in soft hydrogels with either RGDS, derived from fibronectin, or a P15 collagen I peptide mimic. Collagen I is one of the most abundant proteins in the dermis and spheroid studies often embed cells within a collagen matrix; therefore, we asked whether the adhesive ligand might change the apoptotic response to PLX4032. Both WM35 and A375 cells responded similarly to PLX4032 regardless of the adhesive peptide used (Fig. S2, ESI†).
Single cells versus spheroids
Since we observed increased levels of apoptosis in radial growth phase WM35 cells cultured in 3D compared to 2D hydrogels or TCPS, as well as the large volume of literature comparing aggregates to single cells, we next asked whether multicellular spheroids embedded in hydrogels might confer protection from PLX4032-induced apoptosis compared to single cells. In order to study spheroids, we utilized the liquid overlay method to form spheroids.14,34 Briefly, 200 μL of a WM35 single-cell suspension at a concentration of 1.25 × 105 cells per mL (∼25000 cells) was pipetted on top of an agar-coated 96-well plate and cultured for 2 days (Fig. 4a). The resulting spheroids were then collected, and sizes were observed to be approximately 1000 μm in diameter. A typical phase contrast image showed that the melanoma spheroids were generally rounded and had clearly defined edges (Fig. 4b). Three spheroids were encapsulated within the soft hydrogel formulation used in the 2D versus 3D studies (0.4 kPa) and compared to WM35s encapsulated as single cells. The WM35 cells were selected for this study based on their stronger and differential response to drug treatment despite dimensionality.
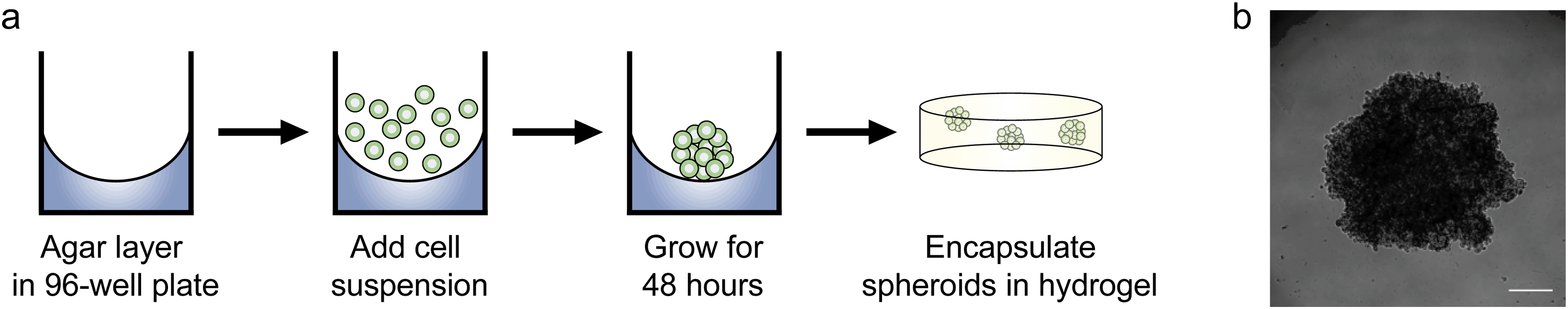 | ||
| Fig. 4 Procedure for forming spheroids. (a) A 50 μL layer of 1.5% Noble agar was plated in a 96-well plate and allowed to solidify at room temperature. 200 μL of WM35 single cell suspension (1.25 × 105 cells per mL) was pipetted on top of the agar layer and left to grow for approximately 48 hours in normal growth media. After 2 days, the spheroids were transferred using a 1 mL pipette, and then encapsulated within PEG–peptide hydrogels. (b) A representative melanoma spheroid after 48 hours of culture on top of agar. Spheroids were approximately 1 mm in diameter. Scale bar, 250 μm. | ||
Drug responsiveness of single cells and spheroids
To further assess the effects of PLX4032 treatment on WM35 single cells versus spheroids embedded in hydrogel matrices, metabolic activity, apoptosis, and DNA content were quantified (Fig. 5). The metabolic activity was measured after 48 hours of culture with or without PLX4032, and in both cases, it caused a decrease in the average metabolic activity (approximately 43% and 32% decrease in single cells and spheroids, respectively) (Fig. 5a). When PLX4032 treatment is assessed, drug treatment may be less effective in spheroids due to lack of availability of PLX4032 to the interior cells, and therefore, on a per cell basis, single cells exhibit a bigger decrease in metabolic activity. The level of cell death via apoptosis was also assessed, where caspase 3 activity increased with PLX4032 treatment (Fig. 5b). With drug treatment, caspase 3 increased by approximately 1.7 and 2.6-fold in single cells and spheroids, respectively (Fig. S4, ESI†), though the fold changes were not statistically significantly different. The DNA content of single cells and spheroids was also measured. The DNA content of spheroid controls was significantly higher (1.8 times) than that of single cell encapsulations (Fig. 5c) implying increased proliferation in the spheroid cells. Furthermore, with drug treatment, the DNA content significantly decreased in both culture conditions (∼72% and 53% lower compared to controls in single cells and spheroids, respectively), and the DNA measured in PLX4032 spheroids was significantly higher (approximately 1.8 times higher) than that under the single cell PLX4032 conditions.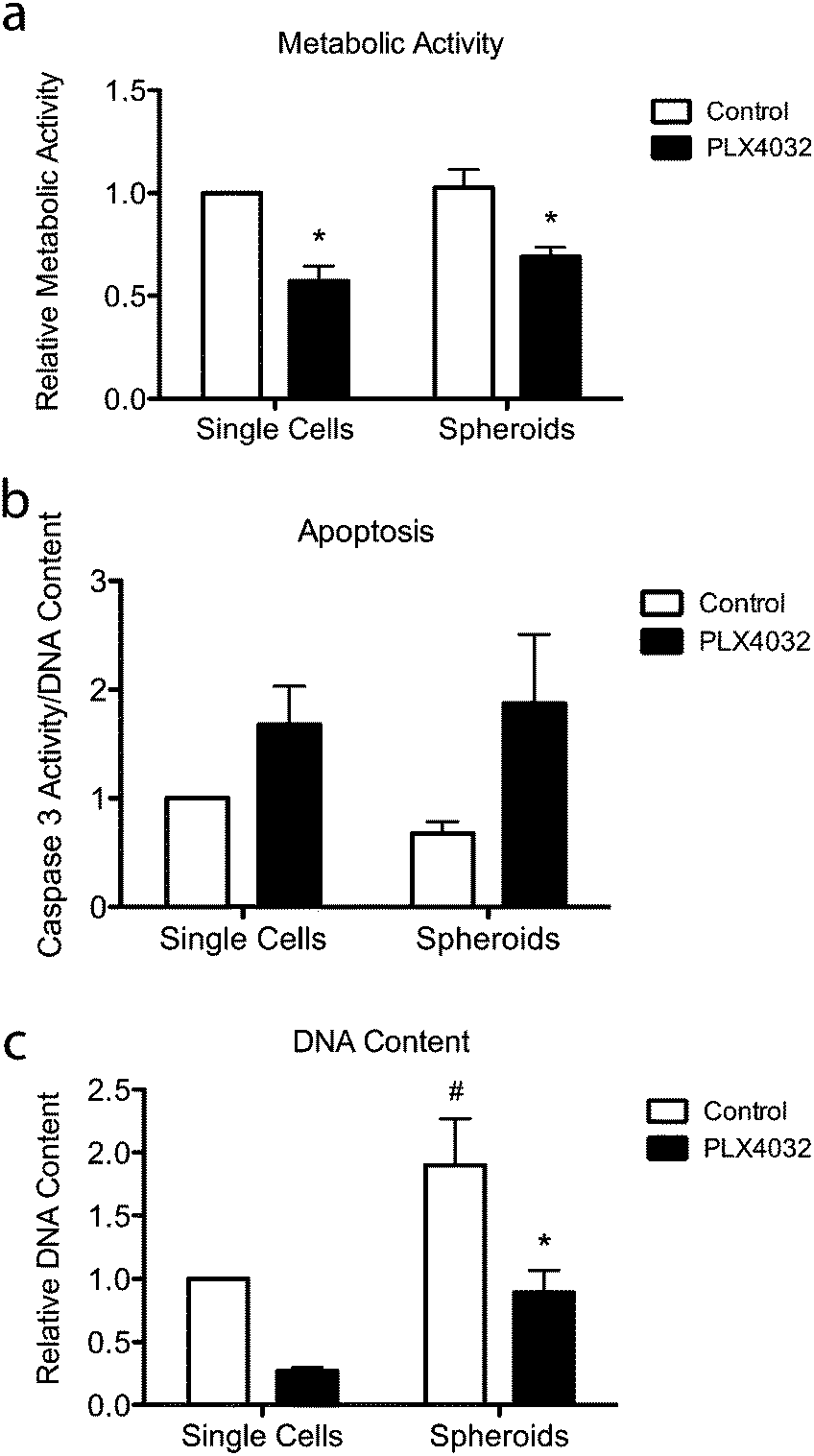 | ||
| Fig. 5 Overall responsiveness of WM35 cells to PLX4032 treatment for 48 hours. (a) Metabolic activity was measured and found to decrease with PLX4032 treatment for all conditions. (b) Apoptosis was measured via caspase 3 activity and found to increase with PLX4032 treatment in both culture conditions. (c) DNA content was measured to quantify changes in cell number in the single cell encapsulations versus spheroid culture. PLX4032 caused a significant decrease in cell number in both culture conditions (∼72% and 53% lower in single cells and spheroids, respectively). *p < 0.05 compared to respective control, #p < 0.05 compared to the corresponding conditions in single cell encapsulation samples. | ||
After successfully forming spheroids, temporal changes in cell aggregates were examined via confocal microscopy (Fig. 6) using calcein and ethidium homodimer to visualize both overall morphology and live and dead cells. Live/dead images show both single cells and spheroids encapsulated within hydrogels with and without PLX4032 after 48 hours of treatment. In the single cell control, cells were rounded and many appeared to proliferate significantly over the relatively short time course of this experiment (as indicated by numerous pairs of cells in close proximity to one another). In the PLX4032-treated sample, there were fewer “paired” cells and many had small protrusions. Interestingly, in the spheroids under control conditions, the aggregates had well defined borders with a few loosely dissociated cells near the perimeter. However, PLX4032-treatment led to what appeared to be a more invasive morphology, as characterized by an elongated cell shape and perimeter cells moving large distances away from the aggregate (up to ∼200 μm in 48 hours). Brightfield images of the spheroids confirm the compact shape of the control spheroid and the potential invasiveness of the drug-treated spheroid (Fig. S3, ESI†).
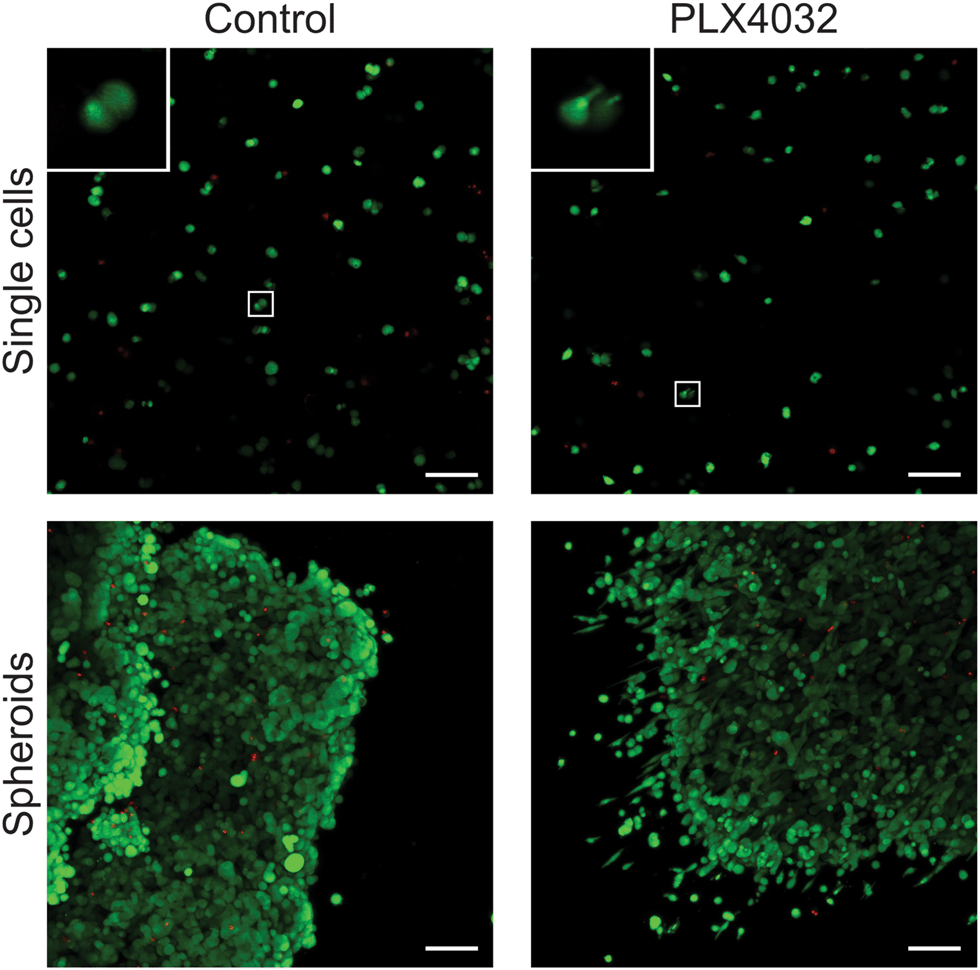 | ||
| Fig. 6 Overall morphology of WM35 single cells and spheroids encapsulated within PEG hydrogels. Live/Dead staining of single cells or spheroids within 0.4 kPa PEG–peptide gels in the presence or absence of PLX4032. Single cell encapsulations exhibited a very rounded morphology in the control conditions, whereas many single cells had small, thin protrusions in the presence of PLX4032. Inset image shows magnification of cells exhibiting rounded or elongated morphology. Spheroids cultured under control conditions formed relatively well-defined edges with some cells loosely dissociated from the edge of the cluster. In contrast, spheroids treated with PLX4032 displayed many cells with an invasive morphology appearing to migrate away from the spheroid. Scale bar, 100 μm. | ||
Discussion
Experiments were designed to determine whether the dimensionality of a cell's microenvironment (i.e., 2D versus 3D culture) might influence the apoptotic response of melanoma cells to PLX4032. Previous reports have compared cells cultured on TCPS to spheroids and their sensitivity to different types of cancer drugs.14,15,17,18 These results have demonstrated that aggregated cells can respond differently, often in a less sensitive manner, when cultured as a multicellular structure embedded within collagen or seeded on top of Matrigel compared to traditional 2D TCPS culture.9,43 While these studies have highlighted the role that the tumor microenvironment may play in a cell's drug response, several variables are changing when single cells are plated on TCPS versus those that are aggregated into spheroids.22 For example, spheroids introduce cell–cell contacts, a heterogeneous 3D cellular architecture with respect to diffusional distances, as well as a 3D matrix platform that may have a change in the modulus or chemistry of the surrounding environment (e.g., adhesion ligands, sequestered growth factors1,16,26). Therefore, advances in biomaterial scaffolds that mimic aspects of the tumor ECM, but also allow tunability in their properties, were designed to better understand how specific properties of a cell's microenvironment might create a more drug resistant response as measured by activity and survival.Specifically, we investigated whether radial growth phase (WM35) or metastatic (A375) melanoma cells, when cultured as single cells, would respond differently to PLX4032 treatment when cultured on TCPS, a soft 2D hydrogel environment, or embedded in the same hydrogel system (3D). Both metabolic activity and apoptotic responses were evaluated with PLX4032 treatment. Then, the possible role of cell–cell versus cell–matrix contacts in melanoma drug responsiveness was inferred by comparing single cell studies to spheroid cultures. Overall, the metastatic A375 cells were less responsive to drug treatment regardless of culture conditions, perhaps not unexpectedly. In contrast, the early stage radial growth phase cells were found to be more sensitive when cultured on 2D hydrogels and in 3D culture compared to TCPS, and when aggregated into multicellular structures, may become slightly more responsive to PLX4032.
As part of our characterization, we first assessed overall morphology of cells seeded on TCPS and on top of or encapsulated within hydrogels (Fig. 2a). Not surprisingly, both cell types were spread and formed focal adhesions on TCPS, indicating a high level of cell–substrate interactions. On hydrogel substrates, cells were able to attach, but did not spread to the same degree, possibly due to the softness of the underlying substrate.39,44,45 Because the modulus of skin is not well established, we chose two moduli for this set of studies, 0.4 and 4 kPa, which encompass a range of physiologically relevant tissues. The moduli capture soft tissues such as brain, healthy breast, and liver (0.4 kPa), and malignant breast tissue, smooth muscle and fat (4 kPa).46–48 Upon encapsulation, the melanoma cells were generally rounded, in part because they are completely surrounded and confined by the hydrogel network, which necessitates that the cells degrade the local matrix to allow spreading. Perhaps not enough time had elapsed to allow the cells to spread (e.g., degrade the local matrix) and more elongated morphologies would be observed at longer culture times.38 Future work might track cells in real time and treat with PLX4032 at different stages of spreading or motility.
Next, cellular responses due to PLX4032 treatment were assessed. The metabolic activity showed similar responses to PLX4032 treatment in the A375 cells for each of the culture conditions (Fig. 2b). The WM35 cells had a significantly lower metabolic activity compared to the 2D hydrogels. PLX4032, however, can induce apoptosis and prevent proliferation,49,50 and so the differences in metabolic activity reflect both the cytotoxic and cytostatic effects of drug treatment on melanoma cells. To better understand how drug treatment affected melanoma cells, the apoptotic response to PLX4032 was evaluated. We hypothesized that dimensionality would lead to differential cell responses, and the results showed an increase in apoptosis in WM35 cells cultured on top of or encapsulated within gels compared to TCPS (Fig. 3a). In the case of the radial growth phase WM35 cells, 3D culture by itself was not sufficient to protect the cells from apoptosis compared to traditional monolayer culture. Instead, a soft environment, whether cells were on or encapsulated within a hydrogel, induced an increased apoptotic response. We previously showed a correlation between smaller focal adhesions and increased apoptosis in the WM35 cells,31 and perhaps even in a 3D environment, WM35s are unable to form mature focal adhesions and therefore receive decreased survival signaling from the local environment.
The metastatic A375 cells were generally less responsive to PLX4032, regardless of the microenvironment, as evidenced by the fold changes in caspase 3 activity (approximately 1.8- to 1.1-fold increases) (Fig. 3b and d), compared to the ∼4.3-fold increase observed for the WM35 cells (Fig. 3c). Interestingly, the A375 cells exhibited decreased levels in caspase 3 induction from TCPS to soft 2D hydrogels to 3D culture (Fig. 3d); there was a statistically significant difference between the fold changes in caspase 3 activity between TCPS and 3D cultures. The A375 cells also appeared to have a slightly lower overall apoptotic response to PLX4032, but the basal (control conditions) level of caspase 3 in 3D environments was higher than that on 2D hydrogels. While the materials and encapsulation conditions have been used for numerous types of primary cells,30,51,52 perhaps the process of encapsulation is slightly harsher on the melanoma cells compared to their seeding in 2D, and this may account for the elevation in caspase 3 at this relatively early time point. These data suggest that 3D cell culture may help to protect the A375 cells from PLX4032 treatment. We hypothesize that for the metastatic A375 cells, 3D culture is sufficient to cause a lower apoptotic response, and this potentially contributes to more drug resistant spheroid studies. Drug resistance in melanoma is a complex biological problem53 involving both intrinsic and acquired drug resistance.54 Recent examples have found that metastatic melanoma can overcome BRAF inhibition by reactivation of the ERK signaling pathway by acquiring EGFR expression55 or BRAF inhibition results in melanoma senescence and may explain the transient efficacy of PLX4032.56 In the A375 cells in this study, it is possible that inhibition-induced activation of another pathway is aiding the cells' survival, which in turn, could be aided by cell–matrix interactions. The cells encapsulated in 3D had less of a response to PLX4032 which could be due to mechanotransduction of a 3D environment, where integrins and cadherins57 could be engaged on all surfaces of a cell, compared to only on the basal side in a 2D environment.25,58 More studies, however, are needed to fully understand the intracellular signaling involved in the A375 cells that may be directly contributing to the decreased sensitivity in 3D.
Though we observed differences between TCPS and 2D hydrogel cultures, we must note that there are several differences between the two culture conditions. TCPS does not inherently contain binding moieties, is orders of magnitude stiffer than soft tissues, and nonspecifically adsorbs serum proteins. The 2D hydrogel platform used in these studies is nearly six orders of magnitude softer than TCPS, can allow the diffusion of nutrients to both sides of the cells, and generally resists nonspecific protein adsorption. The 2D hydrogels maintain a polarized binding environment similar to TCPS in that cells can only bind on one of their surfaces; however, a direct comparison of TCPS to any hydrogel system is often difficult due to the multiple variables that can change.
We also investigated the possible role of the adhesive ligand in conferring survival. In most published research work 3D spheroids are embedded within commercially available collagen gels. This motivated us to examine differences between an RGDS adhesive peptide derived from fibronectin, to a P15 sequence, derived from collagen I.59 However, the results indicated no significant change in how WM35 or A375 cells responded to PLX4032 treatment as a function of these different integrin-binding epitopes (Fig. S2, ESI†). We had hypothesized that a collagen mimic might increase susceptibility to drug treatment,60 but perhaps these particular melanoma cell lines are less sensitive to specific cell–ECM interactions in 3D compared to other cell types, such as breast cancer cells.61,62 Additionally, another collagen I mimic peptide could be tested, such as the GFOGER or DGEA.59,63
Because the WM35 cells appeared to have increased susceptibility to drug treatment on both 2D hydrogels and 3D culture conditions as a single cell suspension, we focused on these radial growth phase cells and asked whether aggregation into spheroids might confer a protective effect from apoptosis. We also analyzed viability metrics for drug responsiveness in single cells versus spheroids. Beyond metabolic activity and apoptosis, the DNA content of the control spheroids was significantly higher than that of single cell encapsulations (Fig. 5c), which may point to increased proliferation rates of cells (and the effect of high cell–cell contacts) within the spheroid. We hypothesize that cells in the outer shell of the spheroid are likely proliferating more than those in the potentially nutrient-deprived core.64 In the studies performed here, the spheroids were approximately 1 mm in diameter. As such, with spheroid culture conditions, there is an inherent heterogeneity among the cells sampled when measuring apoptosis or DNA content. A spheroid contains a fraction of cells that are in contact with the ECM, some that are nutrient deprived at the core, and others that have numerous cell–cell contacts.65,66 When assessing metrics for the spheroid population, it is difficult to determine what components are contributing to this signaling – cell–cell contacts or cell–matrix interactions of the outer ring of cells in the spheroid. Here, caspase 3 activity, DNA content, and metabolic activity (Fig. 5) were all measured based on the average of the collective population. For metabolic activity (Fig. 5a) and live/dead staining (Fig. 6), the experiments may likely only measure the outer shell of cells that come into contact with PLX4032, as cells at the periphery can consume most of the probe molecules and imaging in the 3D interior of large aggregates is difficult.
With this in mind, we hypothesize that the observed changes in apoptosis in spheroids (Fig. 5b) could be due to competing issues. First, the inner mass of cells may be protected from exposure to drug treatment by the outer cells,67 and cell–cell contacts in spheroids may promote survival signaling;68 however, there is most likely a core of cells undergoing cell death due to nutrient deprivation. Further experiments are needed to spatially identify where apoptotic cells reside in the aggregates, and this may necessitate new methods to form smaller aggregates of more uniform size.69 In addition, understanding the growth of these spheroids in real time (e.g., proliferation markers, tracking cell numbers) would aid in understanding why the DNA content within spheroids is higher than that of single cells, even though both encapsulation conditions started with approximately similar numbers of cells. By measuring the proliferation and subsequent growth of the spheroids over time, one might better understand why the spheroids respond differently to drug treatment compared to single cells. Collectively, the 2D hydrogel and encapsulated single cells vs. spheroid studies suggest that perhaps cell–matrix interactions can be more important than cell–cell contact in the context of soft hydrogel culture scaffolds by increasing susceptibility to drug treatment.
These measures of drug responsiveness, however, are bulk measurements of spheroid activity, which do not capture heterogeneity on a single cell level. Despite similar responses in metabolic activity and apoptosis, inherent cellular heterogeneity due to cell–microenvironment interactions in spheroids may require more studies that focus on single or subpopulations of cells. Interestingly, we observed a more elongated morphology with PLX4032 treatment of 3D single cells and in cells around and near the periphery of drug-treated spheroids (Fig. 6), which might have been otherwise missed by solely studying the population in bulk. Cells embedded in these molecularly crosslinked gels must locally remodel their environment to allow spreading and motility. As such, this significant change in morphology in 3D most likely correlates with increased MMP activity, which allows degradation of the surrounding network (Fig. 6). We have previously observed that PLX4032 treatment correlates with increased single cell migration and MMP activity,70 and as a result, the cells that had an elongated morphology and were separate from the spheroid most likely migrated from the peripheral edge of the mass of cells. It is also worth noting that the invasive phenotype seen in PLX4032-treated spheroid samples was using a RGP melanoma cell line, whereas previous outgrowth from spheroids has been observed in metastatic cell lines.15,71 Two previous studies have observed increased metastatic melanoma invasion with MEK or BRAF treatment via transwell assays or spheroid outgrowth.72,73 Here, we show a change in morphology associated with migration in an early stage melanoma cell line, which could have important implications for melanoma treatment with the clinically available PLX4032 drug.
Despite some of the complexities in understanding the heterogeneity that exists within spheroids and how that might affect cell responses, spheroids may be the most appropriate in vitro models of apoptotic responses to drug treatment. Spheroids can bridge the gap between traditional 2D TCPS culture and animal models,11,20 and they recapitulate aspects of the natural heterogeneity of a tumor that contains a protected core of cells and an outer ring that may be exposed to a drug.74 Even so, single cell encapsulations and soft 2D hydrogel cell culture platforms still have importance in enabling researchers to deconvolute the potential effects of matrix signaling, which is likely important to cancer biology and drug responses.
For example, single cells embedded in 3D hydrogels are useful for studying cell migration,75 as 3D environments necessitate matrix remodeling, which occurs in vivo.76,77 When combined with drug treatment, these hydrogel systems can encompass protease activity and functional motility in response to drug treatment, and are not limited to metabolic activity and apoptosis. While aspects of this could be studied on 2D substrates, cells are less likely to secrete proteases, as there is no barrier to proliferation or migration on planar substrates.2 We have also learned important facets of cell behavior from 2D studies; for instance, 2D stiffness studies have elucidated the role in matrix elasticity in mesenchymal stem cell differentiation,40 mammary epithelial cell transformation,39 and myofibroblast activation associated with aortic calcification.78 In addition, 2D studies on soft substrates and single cell encapsulations complement spheroids by allowing for the control of cell density and allowing one to focus on cell–matrix interactions and signaling analysis, as each cell would theoretically experience the same type of environment. Conversely, monolayer and dispersed cell encapsulation studies may not be as physiologically relevant because they lack the cell–cell contacts inherent to an in vivo tumor. Overall, single cell encapsulations can work in concert with spheroid studies by simplifying the local environment and help with the interpretation of the results from spheroid experiments.
Another advantage in developing more physiologically relevant culture systems is their use as better predictive models for drug screening and development and understanding cancer biology. Recent opinion articles79,80 have put forth the argument for integrating more complex culture systems to better inform our understanding of cell function and recapitulate more tissue- or organ-like behavior. Three-dimensional hydrogel culture platforms provide a tunable and versatile network that has the ability to bridge traditional 2D cell culture and in vivo models. Added complexity can arise from developing co-culture models or changing the extracellular matrix with which the cells interact with. The aim of this study was to further understand and quantify how particular components of cell culture platforms may affect the results the researchers observe.
In summary, we aimed to design experiments to reduce the complexity and better control some of the variables between 2D and 3D culture conditions. Rather than using TCPS culture as the sole 2D comparison, we exploited hydrogel substrates for cell seeding. For the 3D comparison, we used both a single cell suspension and spheroids that could be encapsulated within the same hydrogel formulation used as the 2D hydrogel substrate. In doing so, we were able to maintain the same modulus, use the same material chemistry for both 2D and 3D studies, and control cell–matrix interactions (single cell experiments) or allow cell–cell contacts (spheroid experiments). Collectively, these results contribute to the broader characterization of the role of dimensionality on cells, and particularly melanoma apoptotic responses to PLX4032 treatment.
Conclusions
Experiments were designed to test whether dimensionality alters PLX4032-induced apoptosis in an early stage radial growth phase (WM35) and a metastatic (A375) melanoma cell line. The metastatic A375 cells were not as sensitive to PLX4032 treatment as the WM35 cells and exhibited decreased apoptosis in 3D compared to TCPS or 2D hydrogel culture conditions. Interestingly, the WM35 cells appeared to be more sensitive to PLX4032 when cultured on (2D) or in (3D) soft hydrogels, exhibiting higher levels of apoptosis compared to cells cultured on TCPS (4.3- and 4.1-fold increase on 2D and 3D hydrogel samples). Due to the increased sensitivity of the WM35 cells cultured in hydrogel matrices, especially compared to TCPS, we then asked whether the addition of cell–cell contacts through spheroid formation might confer anti-apoptotic protection from PLX4032. The WM35 spheroids exhibited similar levels of apoptosis in response to PLX4032 and potentially increased proliferation. Overall, 3D culture in itself is not sufficient to confer significantly decreased apoptotic responses to pharmacological inhibition in RGP WM35s; however, it may protect metastatic cells. Many of the reported results support the paradigm that spheroid culture results in more “drug resistant” responses in early stage melanoma cells. We hypothesize that 3D culture alone is not adequate to protect WM35 cells, but perhaps cell–cell contacts or spheroid heterogeneity may play a more significant role in contributing to altered responses that might be missed by bulk measurements of drug responsiveness.Acknowledgements
All cell lines were a generous gift from Dr Natalie Ahn (Department of Chemistry and Biochemistry, University of Colorado Boulder). The authors would like to thank Dr Chun Yang for her assistance with the schematics and table of contents figure; Dr William Wan and Dr Jennifer Leight (Department of Biomedical Engineering, Ohio State University) for helpful discussions and aid with statistical analysis; Dr Kelly Mabry and Kelly Shekiro for technical discussions and feedback on early versions of this manuscript; and Dr MK Tarrant-Conacher for aid with the spheroid assays. This work was funded in part by the National Institutes of Health (R01CA132633 and EB018505) and The Howard Hughes Medical Institute.References
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS PubMed.
- P. Friedl and K. Wolf, Nat. Rev. Cancer, 2003, 3, 362–374 CrossRef CAS PubMed.
- H. Hall, D. Farson and M. Bissell, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 4672 CrossRef CAS.
- A. Freeman and R. Hoffman, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 2694 CrossRef CAS.
- T. Jacks and R. Weinberg, Cell, 2002, 111, 923–925 CrossRef CAS PubMed.
- D. Huh, B. D. Matthews, A. Mammoto, M. Montoya-Zavala, H. Y. Hsin and D. E. Ingber, Science, 2010, 328, 1662–1668 CrossRef CAS PubMed.
- S. R. Khetani and S. N. Bhatia, Nat. Biotechnol., 2008, 26, 120–126 CrossRef CAS PubMed.
- O. W. Petersen, L. Ronnov-Jessen, A. R. Howlett and M. J. Bissell, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 9064–9068 CrossRef CAS.
- C. Fischbach, R. Chen, T. Matsumoto, T. Schmelzle, J. Brugge, P. Polverini and D. Mooney, Nat. Methods, 2007, 4, 855–860 CrossRef CAS PubMed.
- T. W. Ridky, J. M. Chow, D. J. Wong and P. A. Khavari, Nat. Med., 2010, 16, 1450–1455 CrossRef CAS PubMed.
- C. Caicedo-Carvajal, Q. Liu, A. Goy, A. Pecora and K. S. Suh, Transl. Med. Ser., 2012, 1, 1–8 Search PubMed.
- C. S. Szot, C. F. Buchanan, J. W. Freeman and M. N. Rylander, Biomaterials, 2011, 32, 7905–7912 CrossRef CAS PubMed.
- V. Weaver, O. Petersen, F. Wang, C. A. Larabell, P. Briand, C. Damsky and M. Bissell, J. Cell Biol., 1997, 137, 231–245 CrossRef CAS PubMed.
- K. Smalley, N. Haass, P. Brafford, M. Lioni, K. Flaherty and M. Herlyn, Mol. Cancer Ther., 2006, 5, 1136–1144 CrossRef CAS PubMed.
- N. Haass, K. Sproesser, T. Nguyen, R. Contractor, C. Medina, K. Nathanson, M. Herlyn and K. Smalley, Clin. Cancer Res., 2008, 14, 230–239 CrossRef CAS PubMed.
- B. Weigelt and M. J. Bissell, Semin. Cancer Biol., 2008, 18, 311–321 CrossRef CAS PubMed.
- B. Weigelt, A. T. Lo, C. C. Park, J. W. Gray and M. J. Bissell, Breast Cancer Res. Treat., 2010, 122, 35–43 CrossRef CAS PubMed.
- D. Loessner, K. S. Stok, M. P. Lutolf, D. W. Hutmacher, J. A. Clements and S. C. Rizzi, Biomaterials, 2010, 31, 8494–8506 CrossRef CAS PubMed.
- K. Smalley, M. Lioni and M. Herlyn, In Vitro Cell. Dev. Biol.: Anim., 2006, 42, 242–247 CrossRef CAS PubMed.
- K. Yamada and E. Cukierman, Cell, 2007, 130, 601–610 CrossRef CAS PubMed.
- L. Griffith and M. Swartz, Nat. Rev. Mol. Cell Biol., 2006, 7, 211–224 CrossRef CAS PubMed.
- J. B. Kim, Semin. Cancer Biol., 2005, 15, 365–377 CrossRef PubMed.
- C. M. Nelson and M. J. Bissell, Annu. Rev. Cell Dev. Biol., 2006, 22, 287–309 CrossRef CAS PubMed.
- M. J. Bissell, A. Rizki and I. S. Mian, Curr. Opin. Cell Biol., 2003, 15, 753 CrossRef CAS PubMed.
- M. G. Rubashkin, G. Ou and V. M. Weaver, Biochemistry, 2014, 53, 2078–2090 CrossRef CAS PubMed.
- M. Cushing and K. Anseth, Science, 2007, 316, 1133–1134 CrossRef CAS PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- B. D. Fairbanks, M. P. Schwartz, A. E. Halevi, C. R. Nuttelman, C. N. Bowman and K. S. Anseth, Adv. Mater., 2009, 21, 5005–5010 CrossRef CAS PubMed.
- S. T. Gould, N. J. Darling and K. S. Anseth, Acta Biomater., 2012, 8, 3201–3209 CrossRef CAS PubMed.
- K. A. Kyburz and K. S. Anseth, Acta Biomater., 2013, 9, 6381–6392 CrossRef CAS PubMed.
- E. Y. Tokuda, J. L. Leight and K. S. Anseth, Biomaterials, 2014, 35, 4310–4318 CrossRef CAS PubMed.
- B. Fairbanks, M. Schwartz, C. Bowman and K. Anseth, Biomaterials, 2009, 30, 6702–6707 CrossRef CAS PubMed.
- J. L. Leight, D. L. Alge, A. J. Maier and K. S. Anseth, Biomaterials, 2013, 34, 7344–7352 CrossRef CAS PubMed.
- G. M. Argast, C. H. Croy, K. L. Couts, Z. Zhang, E. Litman, D. C. Chan and N. G. Ahn, Oncogene, 2009, 28, 2697–2709 CrossRef CAS PubMed.
- E. Ruoslahti, Cell, 1986, 44, 517–518 CrossRef CAS PubMed.
- S. B. Anderson, C.-C. Lin, D. V. Kuntzler and K. S. Anseth, Biomaterials, 2011, 32, 3564–3574 CrossRef CAS PubMed.
- M. P. Schwartz, B. D. Fairbanks, R. E. Rogers, R. Rangarajan, M. H. Zaman and K. S. Anseth, Integr. Biol., 2010, 2, 32–40 RSC.
- M. Lutolf, J. Lauer-Fields, H. Schmoekel, A. Metters, F. Weber, G. Fields and J. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5413 CrossRef CAS PubMed.
- M. J. Paszek, N. Zahir, K. R. Johnson, J. N. Lakins, G. I. Rozenberg, A. Gefen, C. A. Reinhart-King, S. S. Margulies, M. Dembo, D. Boettiger, D. A. Hammer and V. M. Weaver, Cancer Cell, 2005, 8, 241–254 CrossRef CAS PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- C. Yang, M. W. Tibbitt, L. Basta and K. S. Anseth, Nat. Mater., 2014, 13, 645–652 CrossRef CAS PubMed.
- P. A. Janmey, R. G. Wells, R. K. Assoian and C. A. McCulloch, Differentiation, 2013, 86, 112–120 CrossRef CAS PubMed.
- M. Pickl and C. H. Ries, Oncogene, 2008, 28, 461–468 CrossRef PubMed.
- F. J. Byfield, Q. Wen, I. Levental, K. Nordstrom, P. E. Arratia, R. T. Miller and P. A. Janmey, Biophys. J., 2009, 96, 5095–5102 CrossRef CAS PubMed.
- P. A. Janmey, J. P. Winer, M. E. Murray and Q. Wen, Cell Motil. Cytoskeleton, 2009, 66, 597–605 CrossRef PubMed.
- I. Levental, K. R. Levental, E. A. Klein, R. Assoian, R. T. Miller, R. G. Wells and P. A. Janmey, J. Phys.: Condens. Matter, 2010, 22, 194120 CrossRef CAS PubMed.
- D. T. Butcher, T. Alliston and V. M. Weaver, Nat. Rev. Cancer, 2009, 9, 108–122 CrossRef CAS PubMed.
- D. E. Discher, D. J. Mooney and P. W. Zandstra, Science, 2009, 324, 1673–1677 CrossRef CAS PubMed.
- J. T. Lee, L. Li, P. A. Brafford, M. van den Eijnden, M. B. Halloran, K. Sproesser, N. K. Haass, K. S. M. Smalley, J. Tsai, G. Bollag and M. Herlyn, Pigm. Cell Melanoma Res., 2010, 23, 820–827 CrossRef CAS PubMed.
- F. M. Kaplan, Y. Shao, M. M. Mayberry and A. E. Aplin, Oncogene, 2010, 30, 366–371 CrossRef PubMed.
- B. V. Sridhar, N. R. Doyle, M. A. Randolph and K. S. Anseth, J. Biomed. Mater. Res., Part A, 2014, 102, 4464–4472 Search PubMed.
- S. T. Gould, E. E. Matherly, J. N. Smith, D. D. Heistad and K. S. Anseth, Biomaterials, 2014, 35, 3596–3606 CrossRef CAS PubMed.
- D. Grossman and D. C. Altieri, Cancer Metastasis Rev., 2001, 20, 3–11 CrossRef CAS PubMed.
- A. D. Bucheit and M. A. Davies, Biochem. Pharmacol., 2013, 87, 381–389 CrossRef PubMed.
- C. Sun, L. Wang, S. Huang, G. J. J. E. Heynen, A. Prahallad, C. Robert, J. Haanen, C. Blank, J. Wesseling, S. M. Willems, D. Zecchin, S. Hobor, P. K. Bajpe, C. Lieftink, C. Mateus, S. Vagner, W. Grernrum, I. Hofland, A. Schlicker, L. F. A. Wessels, R. L. Beijersbergen, A. Bardelli, F. Di Nicolantonio, A. M. M. Eggermont and R. Bernards, Nature, 2014, 508, 118–122 CrossRef CAS PubMed.
- S. Haferkamp, A. Borst, C. Adam, T. M. Becker, S. Motschenbacher, S. Windhövel, A. L. Hufnagel, R. Houben and S. Meierjohann, J. Invest. Dermatol., 2013, 133, 1601–1609 CrossRef CAS PubMed.
- P. A. Janmey and C. A. McCulloch, Annu. Rev. Biomed. Eng., 2007, 9, 1–34 CrossRef CAS PubMed.
- F. Spill, D. S. Reynolds, R. D. Kamm and M. H. Zaman, Curr. Opin. Biotechnol., 2016, 40, 41–48 CrossRef CAS PubMed.
- K. M. Hennessy, B. E. Pollot, W. C. Clem, M. C. Phipps, A. A. Sawyer, B. K. Culpepper and S. L. Bellis, Biomaterials, 2009, 30, 1898–1909 CrossRef CAS PubMed.
- Y. Shao and A. E. Aplin, Cancer Res., 2010, 70, 6670–6681 CrossRef CAS PubMed.
- P. A. Kenny, G. Y. Lee, C. A. Myers, R. M. Neve, J. R. Semeiks, P. T. Spellman, K. Lorenz, E. H. Lee, M. H. Barcellos-Hoff, O. W. Petersen, J. W. Gray and M. J. Bissell, Mol. Oncol., 2007, 1, 84–96 CrossRef CAS PubMed.
- V. Weaver, S. Lelievre, J. Lakins, M. Chrenek, J. Jones, Z. Werb and M. Bissell, Cancer Cell, 2002, 2, 205–216 CrossRef CAS PubMed.
- M. Mehta, C. M. Madl, S. Lee, G. N. Duda and D. J. Mooney, J. Biomed. Mater. Res., Part A, 2015, 103, 3516–3525 CrossRef CAS PubMed.
- W. Mueller-Klieser, Crit. Rev. Oncol. Hematol., 2010, 36, 123–139 CrossRef.
- R. Sutherland, Science, 1988, 240, 177–184 CAS.
- D. Loessner, J. A. Flegg, H. M. Byrne, J. A. Clements and D. W. Hutmacher, Integr. Biol., 2013, 5, 597–605 RSC.
- L. Kunz-Schughart, Cell Biol. Int., 1999, 23, 157–161 CrossRef CAS PubMed.
- R. C. Bates, A. Buret, D. F. van Helden, M. A. Horton and G. F. Burns, J. Cell Biol., 1994, 126, 403–425 CrossRef.
- A. B. Bernard, C.-C. Lin and K. S. Anseth, Tissue Eng., Part C, 2012, 18, 583–592 CrossRef CAS PubMed.
- J. L. Leight, E. Y. Tokuda, C. E. Jones, A. J. Lin and K. S. Anseth, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5366–5371 CrossRef CAS PubMed.
- K. H. T. Paraiso, Y. Xiang, V. W. Rebecca, E. V. Abel, Y. A. Chen, A. C. Munko, E. Wood, I. V. Fedorenko, V. K. Sondak, A. R. A. Anderson, A. Ribas, M. D. Palma, K. L. Nathanson, J. M. Koomen, J. L. Messina and K. S. M. Smalley, Cancer Res., 2011, 71, 2750–2760 CrossRef CAS PubMed.
- R. Halaban, W. Zhang, A. Bacchiocchi, E. Cheng, F. Parisi, S. Ariyan, M. Krauthammer, J. P. McCusker, Y. Kluger and M. Sznol, Pigm. Cell Melanoma Res., 2010, 23, 190–200 CrossRef CAS PubMed.
- A. Vultur, J. Villanueva, C. Krepler, G. Rajan, Q. Chen, M. Xiao, L. Li, P. A. Gimotty, M. Wilson, J. Hayden, F. Keeney, K. L. Nathanson and M. Herlyn, Oncogene, 2014, 33, 1850–1861 CrossRef CAS PubMed.
- J. Friedrich, R. Ebner and L. A. Kunz-Schughart, Int. J. Radiat. Biol., 2007, 83, 849–871 CrossRef CAS PubMed.
- K. Wolf, I. Mazo, H. Leung, K. Engelke, U. Von Andrian, E. Deryugina, A. Strongin, E. Bröcker and P. Friedl, J. Cell Biol., 2003, 160, 267 CrossRef CAS PubMed.
- P. Friedl and K. Wolf, J. Cell Biol., 2010, 188, 11–19 CrossRef CAS PubMed.
- C. Gaggioli and E. Sahai, Pigm. Cell Res., 2007, 20, 161–172 CrossRef CAS PubMed.
- A. M. Kloxin, J. A. Benton and K. S. Anseth, Biomaterials, 2010, 31, 1–8 CrossRef CAS PubMed.
- C. S. Chen, Trends Cell Biol., 2016, 26, 798–800 CrossRef PubMed.
- P. Horvath, N. Aulner, M. Bickle, A. M. Davies, E. D. Nery, D. Ebner, M. C. Montoya, P. Östling, V. Pietiäinen, L. S. Price, S. L. Shorte, G. Turcatti, C. von Schantz and N. O. Carragher, Nat. Rev. Drug Discovery, 2016, 15, 751–769 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ib00229c |
| This journal is © The Royal Society of Chemistry 2017 |