DOI:
10.1039/C6MH00434B
(Communication)
Mater. Horiz., 2017,
4, 88-97
Quaternisation-polymerized N-type polyelectrolytes: synthesis, characterisation and application in high-performance polymer solar cells†
Received
19th October 2016
, Accepted 25th November 2016
First published on 25th November 2016
Abstract
Perylene diimide (PDI) based semiconductors with high mobility are promising electron-transporting materials (ETMs), which are used to fabricate polymer solar cells (PSCs) using roll-to-roll (R2R) processing. However, PDI-based molecular semiconductors have a strong tendency to aggregate, which hinders their use as ETMs in the fabrication of high-performance thin-film devices. Additionally, multi-layer organic opto-electronic devices require that the materials for different layers should possess orthogonal solubility. Here, we develop an in situ polymerisation method to successfully prepare ion-containing PDI-based polyelectrolytes with good water/alcohol solubility, which can enable high-performance PSCs. The doping behaviour, self-assembling and charge-transporting properties of these polyelectrolytes can be fine-tuned by their anions, which allows the fabrication of high-quality and high-mobility electron-transporting thin films for PSCs. PSCs with these polyelectrolytes can maintain high power conversion efficiencies of over 8% when the thickness of the polyelectrolyte is up to 50 nm, which offers a remarkable processing window for the mass-fabrication of PSCs using R2R techniques. Our findings on the structure–property–performance relationships of these polyelectrolytes provide insights and guidelines for the design of high-performance n-type polyelectrolytes for organic opto-electronic devices.
Conceptual insights
Doping is an important strategy to improve the charge transport properties of both n-type and p-type polymer semiconductors. Compared to the widely reported p-type polymeric semiconductors, polymeric semiconductors with n-type doping properties have been scarcely studied. Herein, we report a series of novel perylene diimide (PDI)-based high-performance n-type polyelectrolytes for application in polymer solar cells (PSCs). The novel PDI-based polyelectrolytes are prepared via a simple quaternization polymerization/ion-exchanging process and have good water/alcohol solubility, which allows the interface engineering of high performance multilayer PSCs. More interestingly, it was found that the doping behavior, optoelectronic and morphological properties of the resulting polyelectrolytes could be simply tuned by using different contour ions. Furthermore, due to the unique doping interactions between the contour ions and the PDI units on their main chain, all the resulting polyelectrolytes show good electron-transporting properties and can be used as a thickness-insensitive electron-transporting layer to fabricate high-performance PSCs, which are more compatible for a roll-to-roll printing process and may pave the way for potential commercialization of the organic photovoltaics technology. All these results will also provide more insights and stimulations to further develop a new generation of self-doped n-type polymer semiconductors for optoelectronic devices.
|
Introduction
Organic-based semiconductors are widely applied in organic electronics (such as organic light-emitting diodes, organic field-effect transistors, organic solar cells and thermoelectric devices) due to their physical and opto-electrical properties.1–4 Compared to inorganic semiconductors, organic polymeric semiconductors can be deposited through various low-cost processing methods including spin-coating, printing, blade coating and roll-to-roll (R2R) processing, which offer great opportunities for large-area and high-throughput manufacturing.5,6 As the charge transport properties are vital to the performance of organic semiconductors, various methods including improving polymer backbone coplanarity, reducing grain boundaries and enhancing long-range orderings of polymers have been developed to enhance the charge transport properties of polymeric semiconductors.7–10 Doping is also an important strategy to improve the charge transport properties of both n-type and p-type organic semiconductors. For example, PEDOT:PSS (poly(3,4-ethylenedioxythiophene): poly(styrene sulfonate)), the most well-studied p-type semiconductor in organic electronics, has high ductility, good film-formation capability and high charge conductivity almost comparable to metals. It is well known that the high charge conductivity of PEDOT:PSS originates from p-type doping to the polythiophene conjugated backbone from its counter-sulfonated polystyrene.11,12 Other p-type semiconductors with high conductivity can also be achieved through a p-type doping strategy.13–16 In comparison to p-type polymeric semiconductors, polymeric semiconductors with n-type doping properties are much less well-studied.17–21 N-doped organic semiconductors have been realised previously by introducing an alkali metal (such as K) into an organic semiconductor.22,23 However, a strategy based on active alkali metals is not applicable to the fabrication of organic thin films using solution processing methods. Another successful doping strategy is to introduce electron-rich moieties into electron-deficient organic semiconductors to improve their charge conductivities.24–35 For example, perylene diimide (PDI) derivatives possess very planar structures, high electron affinity and high photochemical stability, characteristics that suggest their great potential as high-mobility charge-transporting materials in organic opto-electronic devices.7,24,36 Additionally, PDI derivatives can be further doped by different anions through electron transfer or charge transfer processes, leading to improved conductivity.24–27 However, the poor film-formation ability of PDI-based molecular semiconductors caused by strong aggregation between molecules hinders the fabrication of high-performance organic opto-electronic devices.24 Moreover, as most of the conjugated polymers are only soluble in non-polar solvents, it is necessary to develop PDI-based water/alcohol soluble polymers to realise the fabrication of multi-layer organic opto-electronic devices using orthogonal solvents.37,38
Herein, we design and synthesise a series of ion-containing n-type polyelectrolytes using in situ quaternisation polymerisation and an ion-exchanging process. The polyelectrolyte prepared through quaternisation polymerisation is endowed with good water solubility immediately. Moreover, the attached anions on the polyelectrolyte can be easily exchanged with other anions, which induces controllable doping and self-assembly in the deposited film. The results of the optical absorption and electron paramagnetic resonance study show that polyelectrolytes with F−, OH−, and CH3COO− as anions are strongly self-doped, and polyelectrolytes with Cl−, Br−, and I− possess weak self-doping properties. The space-charge limited current study shows that the charge mobilities of these polyelectrolytes correlate with their doping behaviour. Furthermore, the film morphology of these polyelectrolytes can be fine-tuned by the anions. In particular, PPDI-Ac with CH3COO− can self-assemble into nanowires in thin films, which contributes to its higher mobility than that of polyelectrolytes with other anions. These polyelectrolytes can be utilised as thickness-insensitive electron-transporting materials (ETMs) to fabricate high-performance PSCs. When the thickness of these polyelectrolytes is up to 50 nm, the power conversion efficiencies (PCEs) of PSCs with the polyelectrolytes can be over 8%, indicating that the polyelectrolytes are promising candidates as ETMs for fabricating large-area PSC devices via R2R processing.
Results and discussion
Materials design, synthesis and characterisation
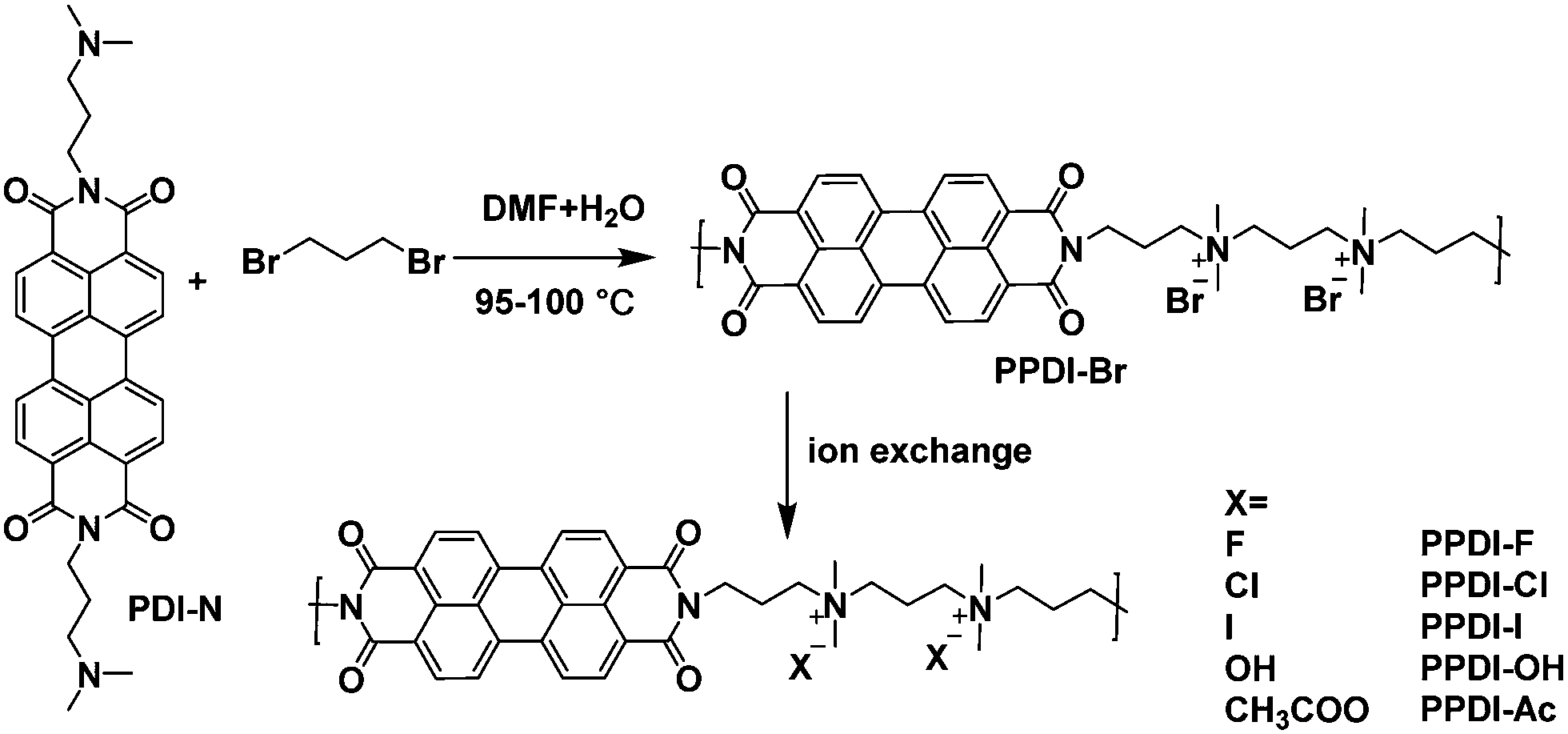
The synthetic routes of the polyelectrolytes in this study are shown in Scheme 1, and the synthetic process can be found in the ESI.† The first precursor polyelectrolyte, PPDI-Br, was prepared through a quaternisation reaction from 1,3-dibromopropane and an amine functionalised monomer PDI-N36 in a mixture of N,N-dimethylformamide (DMF) and H2O at 95–100 °C for 72 h. The resulting PPDI-Br was purified via Soxhlet extraction with acetone and ethanol to remove the low molecular weight fraction, and the remaining product was extracted by water, followed by precipitation into tetrahydrofuran (THF) and drying under a vacuum. The polymerisation process is different from Pa-catalyst coupling polymerisation,39 which requires much longer synthesis and purification steps. Another advantage of such an in situ polymerisation method is that the resulting polyelectrolytes do not contain any metal catalyst residue, and no further procedure is needed to remove deleterious catalyst residues. It should be noted that the synthesis and purification of polyelectrolytes using this polymerisation strategy are very economical, which are especially advantageous for high-throughput industrial production. The molecular weight of PPDI-Br was estimated using gel permeation chromatography (GPC) with water as the eluent. The weight-average molecular weight (Mw) and the number-average molecular weight (Mn) of PPDI-Br are about 14.2k and 9.96k, and thus the polydispersity is calculated to be 1.42. Other polyelectrolytes (PPDI-I, PPDI-F, PPDI-Cl, PPDI-OH and PPDI-Ac) with different counterions (I−, F−, Cl−, OH−, and CH3COO−) were prepared via ion-exchanging processes using anion exchange resins and the details are shown in the ESI.† Analysis of polyelectrolyte powders using X-ray photoelectric spectroscopy (XPS) (shown in Fig. S1, ESI†) confirmed greater than 99% bromide exchange, indicating the successful preparation of the designed polyelectrolytes with different anions. The resulting PPDI-Br is readily soluble in water and dimethyl sulfoxide (DMSO), but is slightly soluble in alcohol solvents and is insoluble in non-polar solvents. Interestingly, the solubility of polyelectrolytes with the same main chain can be fine-tuned by their attached anions. PPDI-I showed similar alcohol solubility to PPDI-Br, and PPDI-x with F−, Cl−, OH−, and CH3COO− dissolved in methanol with concentrations over 8 mg mL−1. The excellent water solubility of these polyelectrolytes can be attributed to their alternative conjugated–non-conjugated (conjugated PDI aromatic units–quaternary ammonium salts) structures, which give the main chains great flexibility. The different solubilities of these polyelectrolytes in methanol are determined by the non-conjugated part-quaternary ammonium salts, which have different association constants in methanol.40 Tetrabutylammonium halide (TBAX) with Br− and I− showed larger association constants in methanol,40 indicating that Br− and I− are prone to bind with ammonium cations in methanol, but do not separate into free anions and cations, which results in their lower solubility in methanol. These polyelectrolytes show the orthogonal solubility of most of the conjugated polymers, which are soluble in non-polar solvents (toluene, chlorobenzene and chloroform, etc.). Therefore, these polyelectrolytes are suitable for the fabrication of multi-layer thin films because the interface mixing during the processing of multi-layer thin films can be effectively avoided. In sum, these polyelectrolytes provide an opportunity to fabricate multi-layer organic opto-electronic devices using low-temperature solution processing techniques.37,38
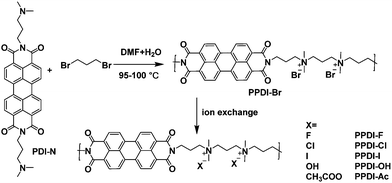 |
| | Scheme 1 Synthetic routes of the designed polyelectrolytes. | |
Optical properties
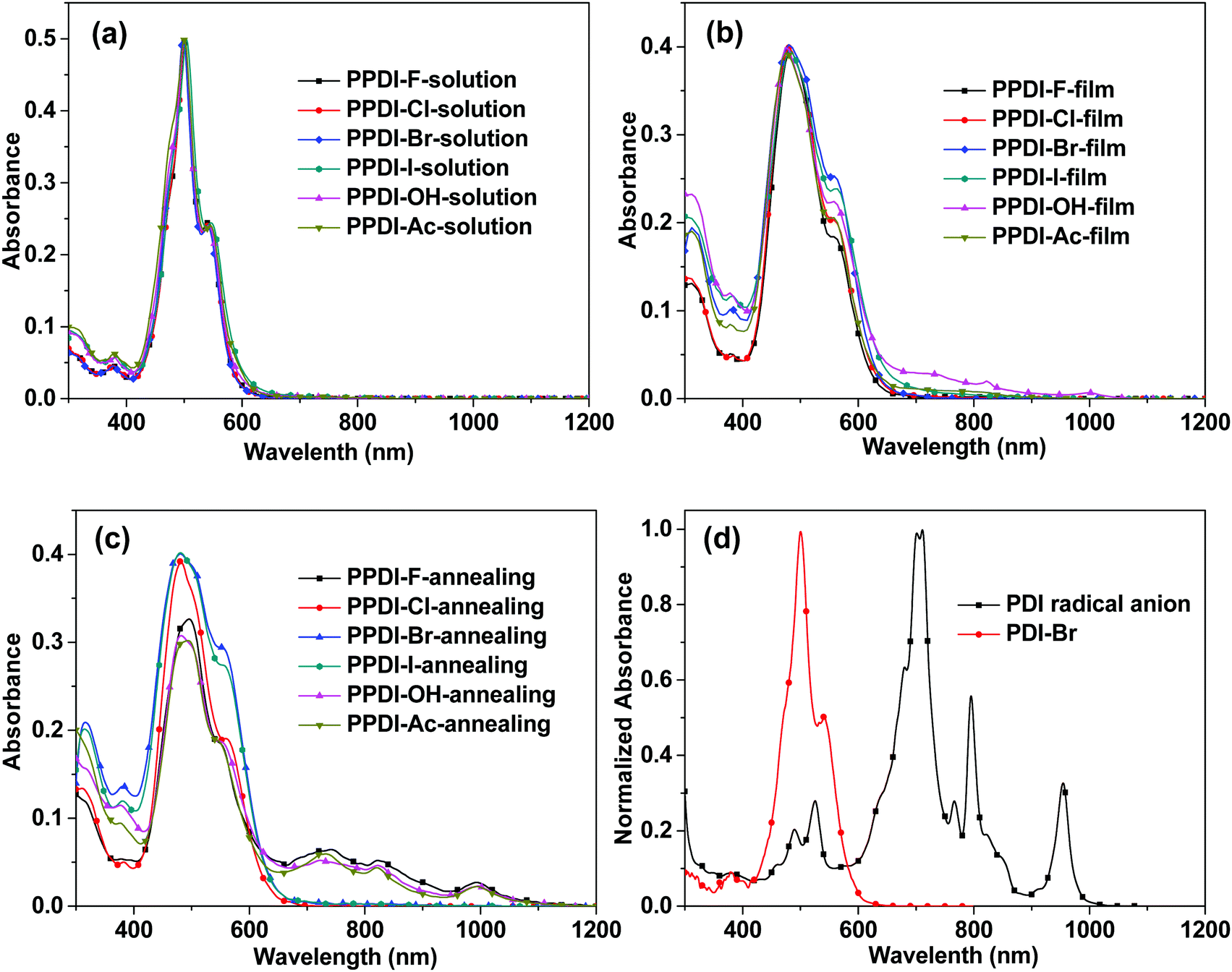
Fig. 1a–c show the UV-vis-NIR absorption spectra of the polyelectrolytes in solution and as films. In solution, all the polyelectrolytes with different counterions show the same absorption spectra, but in solid thin-film states differences emerge. In a diluted water solution, all the polyelectrolytes show an absorption peak at 542 nm and a blue-shifted sharp absorption peak at 502 nm, which can be attributed to the H-aggregation of PDI moieties.41 The H-aggregation in PDI derivatives leads to a face-to-face self-assembly of PDI moieties, which facilitates charge transport in the π–π stacking direction.42,43 In the film state, all the polyelectrolytes deliver broadened absorption spectra, with blunt peaks at 475 nm and 562 nm. In addition, PPDI-OH shows slight absorption between 700 and 1100 nm in the film state, with three small peaks, and these characteristics of PPDI-OH films become more pronounced after an annealing process at 120 °C for 15 min. However, the absorption spectra of the PPDI-Br, PPDI-Cl and PPDI-I films after annealing exhibit no changes excepting slight increases in their shoulder peaks at 562 nm. In the annealed film of polyelectrolytes with F−, OH− and CH3COO− as counterions, the strongest peaks at about 475 nm all weaken compared to their drying films, and the most pronounced change is the emergence of absorption bands ranging from 650 to 1100 nm with peaks at 740 nm, 830 nm and 1010 nm in the annealing films, which are typical of PDI radical anions.24,26 To examine the characteristics of PDI radical anions more closely, we synthesised a PDI radical anion (details are shown in the ESI†) and examined its absorption spectrum in DMF solution (shown in Fig. 1d). Three sharp absorption peaks appeared at 720 nm, 800 nm and 960 nm, which match well with those of the reported PDI radical anion in THF prepared using an electrochemical method.24 The absorption characteristics of the polyelectrolytes with F−, OH− and CH3COO− clearly indicate that strong electron transfer occurs between F−, OH−, and CH3COO− to the PDI aromatic units, leading to the formation of PDI radical anions in the film state. It can also be concluded from the absorption spectra of the annealed films of PPDI-Cl, PPDI-Br and PPDI-I that electron transfer from anions (Cl−, Br− and I−) to PDI aromatic units is hard to achieve. The electron transfer from these anions to PDI units can be described by the following reaction: PDI + X− → X˙ + PDI˙−, where PDI˙− is denoted as the PDI radical anion.27 For the polyelectrolytes with F−, OH−, and CH3COO−, this reaction will proceed more easily, while for the polyelectrolytes with Cl−, Br− and I−, the reaction is much more difficult to achieve. The differences in electron transfer from anions to PDI aromatic units can be explained by the Lewis basicities of anions, which are often used to evaluate the electron-donating ability of the anions.44 The Lewis basicities of these anions can be determined by gas-phase proton affinities.44 The gas-phase proton affinities of F−, OH− and CH3COO− are reported to be 1554, 1635 and 1459 kJ mol−1, respectively.44 Cl−, Br− and I− show lower gas-phase proton affinities of 1395, 1354, and 1315 kJ mol−1, respectively. A comparison of the Lewis basicities clearly indicates that F−, OH− and CH3COO− are stronger electron-donating species than Cl−, Br− and I−,44,45 and electron transfer would occur much more easily from F−, OH− and CH3COO− to PDI aromatic units, which coincides very well with the radical anion characteristics in the absorption spectra.
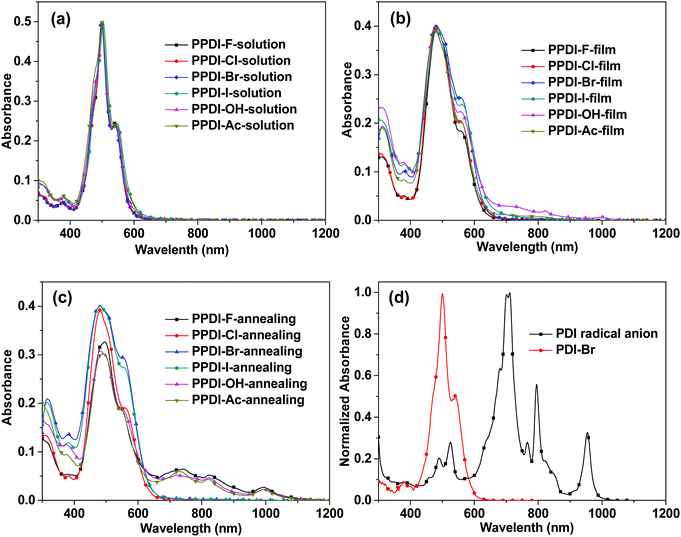 |
| | Fig. 1 (a–c) The UV-vis-NIR absorption spectra of the designed polyelectrolytes in solution and film state with/without annealing; (d) the UV-vis-NIR absorption spectra of PDI-Br and PDI anions. | |
Electron paramagnetic resonance study
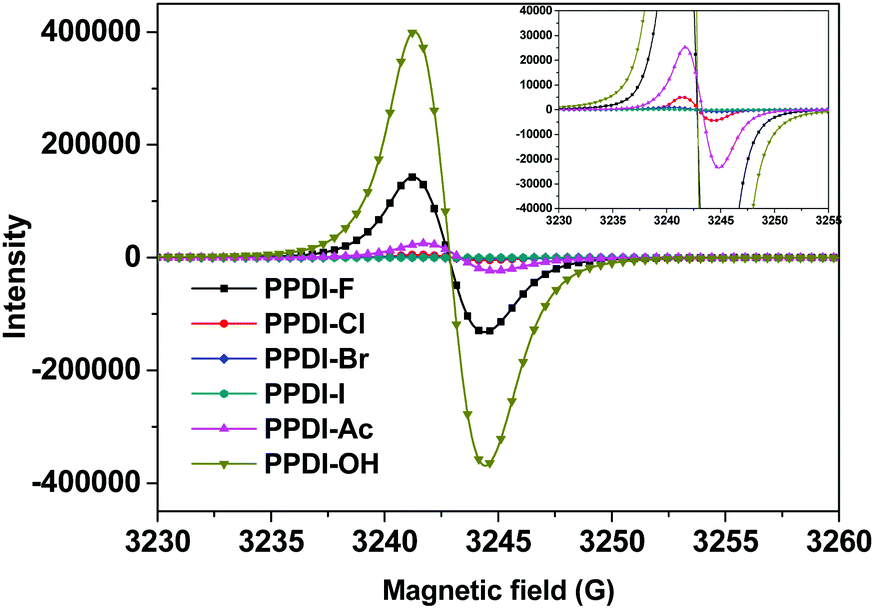
The different electron transfer intensities of these anions from the PDI aromatic units were also verified using electron paramagnetic resonance (EPR) spectroscopy, which can provide direct evidence for the formation of radical anions.31 Equal amounts of the polyelectrolytes in the solid state were prepared and measured under the same conditions, and the results are shown in Fig. 2. Clearly, polyelectrolytes with OH− and F− show much higher signal intensities than other polyelectrolytes, indicating that electron transfer occurs easily from OH− and F− to PDI aromatic units. In particular, PPDI-OH with OH− shows the highest EPR signal intensity, which is also consistent with its strongest absorption characteristic. The strongest electron transfer phenomena of OH− to PDI can be attributed to the strong electron-donating ability of OH−, which is also evidenced by its highest Lewis basicity among these anions. For the polyelectrolytes with CH3COO−, the signal intensity is lower than those of PPDI-F and PPDI-OH, implying that moderate electron transfer occurs from CH3COO− to PDI aromatic units. PPDI-Cl delivers a much weaker EPR signal than PPDI-Ac, indicating that the electron transfer process between Cl− and PDI aromatic units is relatively weaker. For PPDI-Br and PPDI-I, the EPR signals are even weaker. The relatively high EPR signal intensities of polyelectrolytes with OH−, CH3COO− and F− agree well with the absorption characteristics of the PDI radical anions. The trend in the EPR signal intensities of these polyelectrolytes coincides quite well with their absorption characteristics and the gas-phase proton affinity values of their anions. It can be concluded from the experimental and theoretical data that OH−, CH3COO− and F− in these polyelectrolytes can easily self-dope PDI aromatic units and form radical anions in films, which will potentially improve the conductivity of the polyelectrolytes.24,25
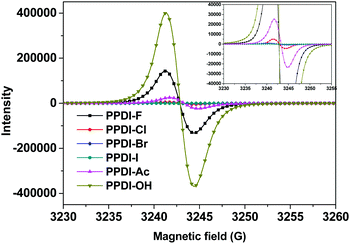 |
| | Fig. 2 EPR spectra of the designed polyelectrolytes in the solid state. | |
Electrochemical properties
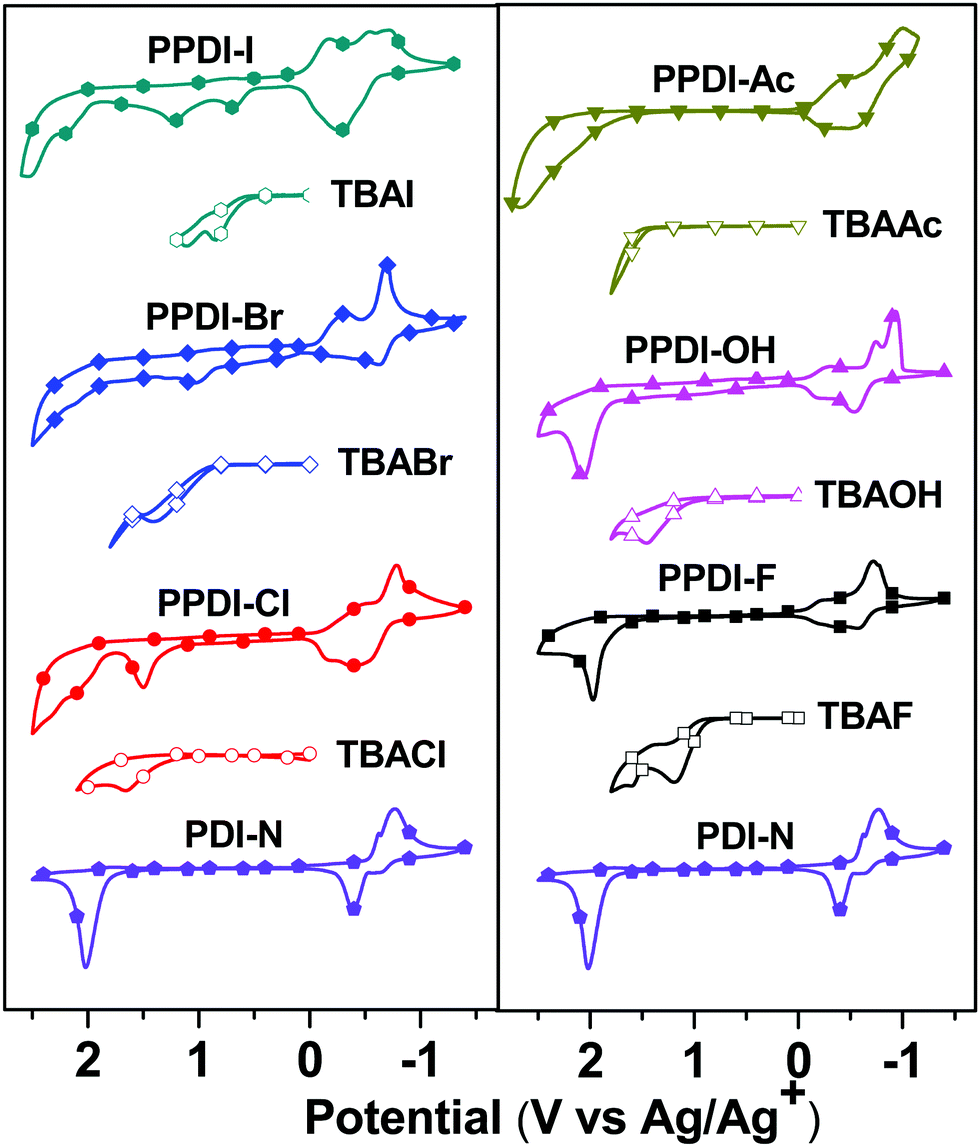
The electrochemical behaviour of the polyelectrolytes was investigated via cyclic voltammetry (CV) analysis and the results are shown in Fig. 3. The ferrocene/ferrocenium (Fc/Fc+) reference was used as an internal standard that delivered a potential onset of 0.35 V with respect to the Ag/Ag+ electrode. For comparison, the CV curves of PDI-N and small molecular TBAX (X = F, Cl, Br, I, OH or Ac) were also recorded. PDI-N delivers a first reduction onset of −0.52 V (vs. Ag/Ag+), indicating the lowest unoccupied molecular orbital (LUMO) energy levels of −3.93 eV (the calculation of the energy level46 can be found in the ESI†). The first reduction onsets of PPDI-F, PPDI-Cl, PPDI-Br, PPDI-I, PPDI-OH and PPDI-Ac are 0.12, 0.21, 0.24, 0.09, 0.14 and 0.23 V, respectively; thus the LUMO levels of these polyelectrolytes are calculated to be −4.33, −4.24, −4.21, −4.36, −4.31 and −4.22 eV, respectively. The much lowered LUMO level of the polyelectrolytes with respect to PDI-N may be caused by electrostatic and anion-induced polarisation due to the anion–π interaction between the anions and the electron-deficient PDI aromatic units.47–49 For the oxidation process, the TBAX electrolytes exhibit different oxidation onsets of anions (0.92 V for F−, 1.25 V for Cl−, 0.85 V for Br−, 0.52 V for I−, 1.08 V for OH− and 1.42 V for CH3COO−vs. Ag/Ag+), which are all before the oxidation of PDI aromatic units (1.72 V vs. Ag/Ag+), indicating that the anions are potential electron donors to PDI aromatic units. For the polyelectrolytes with Cl−, Br− and I−, the oxidation onsets of the anions are clearly observed, and are the same as those of their corresponding small molecular salts-TBAX (X = Cl−, Br− and I−). The oxidation onsets of the anions in PPDI-Ac, PPDI-OH and PPDI-F cannot be observed clearly, which may be explained by the strong interaction between the stronger electron donors (CH3COO−, OH− and F−) and the electron-deficient PDI aromatic units, in which electron transfer occurs more easily.30,49,50 The oxidation potential of the PDI aromatic units in these polyelectrolytes is around 1.7 V, indicating the deep highest occupied molecular orbital (HOMO) energy levels of around −6.15 eV. The deep HOMO levels of these polyelectrolytes indicate that they are endowed with good hole-blocking properties and are suitable ETMs for various donor materials in PSCs.36,51
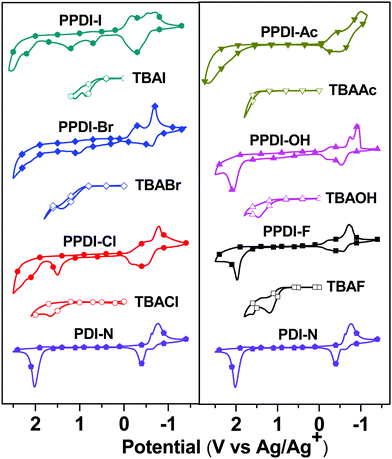 |
| | Fig. 3 The cyclic voltammetry curves of the designed polyelectrolytes. | |
Morphology study
Most PDI-based small molecules have a strong tendency to aggregate in thin films, resulting in various nanostructures and morphologies.52–56 Considering that our designed polyelectrolytes have a very planar conjugated moiety and very soft connecting chains, it is very interesting to determine the morphologies of these polyelectrolytes in the deposited films.57,58 The morphologies of these polyelectrolytes were studied through atom force microscopy (AFM) by spin-coating fresh methanol solutions of the polyelectrolytes onto indium tin oxide (ITO) substrates. Since the water solutions of these polyelectrolytes are hard to spread out on the top of the active layer and get good films,59 we prefer to use methanol as the processing solvent for these polyelectrolytes. The AFM topographies are shown in Fig. 4 (the morphologies of PPDI-Br and PPDI-I were not determined because of their lower solubility in methanol). It is clear that the polyelectrolytes have very different morphologies on ITO substrates. The surface of PPDI-F is quite rough with many small aggregates, and the root mean roughness (RMS) is 3.50 nm, which may cause adverse effects with respect to contact with the active layer.60 Unlike PPDI-F, PPDI-Cl delivers a quite smooth surface with an RMS of about 0.78 nm, which would increase the contact between PPDI-Cl and other layers in organic electronics. For PPDI-OH, very small aggregates were formed on the surface, leading to an increased RMS of 2.12 nm. To our surprise, PPDI-Ac formed nanowires, which is obviously different from the other polyelectrolytes with anions of F−, Cl− and OH−. The nanowires embedded in the polyelectrolyte film can be several microns in length. It should be noted that the RMS of the PPDI-Ac film is also maintained at 2.10 nm, which is conducive to the contact between the polyelectrolyte film and other layers in organic electronics.59 To further investigate the effect of anions on the morphologies of these polyelectrolytes, we added different amounts (10% and 50% by weight) of PPDI-Ac, which can form nanowires, to PPDI-Cl, which can form a smooth film without aggregates, and studied the morphologies of the resulting films using AFM. It can be seen that only a few small nano-aggregates embedded in the polyelectrolytes for PPDI-Cl0.9Ac0.1 when PPDI-Ac was only 10% in the polyelectrolyte film. When the amount of PPDI-Ac is increased to 50%, the PPDI-Cl0.5Ac0.5 film shows a different morphology with nanowires dispersed in the film, but the density of the nanowires is lower than that of the pure PPDI-Ac film. This comparison of the morphologies clearly shows that PPDI-Ac possess the capability of self-assembling into nanowires. We also conducted a slow drying process to deposit the PPDI-Ac film (by drop-casting fresh methanol solution of PPDI-Ac onto ITO substrates and letting the methanol evaporate slowly) and investigated the nanostructures of PPDI-Ac using optical microscopy (the results are shown in Fig. S3, ESI†). Well-defined one-dimensional nanowires of PPDI-Ac formed, up to more than ten microns in length with diameters of dozens of nanometres. The unique self-assembly properties of PPDI-Ac might be attributed to its anion, CH3COO−, which possesses a hydrophobic methyl group at the end of the molecule. During the deposition of the thin films, the large-sized CH3COO− might induce ordered stacking of PDI aromatic units.55,61 To sum up, our results demonstrate that the nanostructures of these polyelectrolytes can be well-tuned by the choice of anions. In particular, it can be expected that a PPDI-Ac film with nanowires will exhibit better charge-transporting properties.62
 |
| | Fig. 4 Tapping mode AFM topography of the spin-coated films of the designed polyelectrolytes. | |
Space-charge limited current study
The electron mobility of the polyelectrolytes was determined using a space-charge limited current (SCLC) method using electron-only devices with an ITO/Al/polyelectrolyte/Al structure.
The results are shown in Fig. 5 and the relevant data are summarised in Table 1. The mobility of PC71BM was also determined for comparison, and a mobility of 5.93 × 10−4 cm2 V−1 s−1 was obtained. The electron mobilities of polyelectrolytes with stronger self-doping properties (PPDI-F, PPDI-OH and PPDI-Ac) are 4.3 × 10−4, 3.34 × 10−4 and 4.76 × 10−4 cm2 V−1 s−1, respectively, whereas PPDI-Cl, with weak self-doping, delivered an obviously lower mobility of 0.66 × 10−4 cm2 V−1 s−1, indicating that electron transfer from stronger electron donors (F−, OH− and CH3COO−) to PDI aromatic units can substantially improve the mobility of the polyelectrolytes. Among the three strongly self-doped polyelectrolytes, PPDI-Ac, which possesses the weakest doping behaviour, surprisingly delivered the highest mobility. This phenomenon can be explained by the film morphology of PPDI-Ac, which exhibited self-assembled nanowires in the deposited film. The nanowires formed in the deposited films possess ordered molecular packing, which will contribute to the charge-transporting properties of the material, and thus lead to higher mobility.62 The lower mobility of PPDI-Cl is also consistent with its weak doping behaviour. The results on the mobility of the polyelectrolytes are in agreement with the recently reported results that the mobilities of organic semiconductors are much higher than those of pristine-type semiconductors.63,64 The increased mobilities of the strongly doped polyelectrolytes may be attributed to the increased carrier density in the deposited thin films. To study the effect of doping concentration on the strongly doped polyelectrolytes, the charge transport properties of PPDI-Cl0.9Ac0.1 and PPDI-Cl0.5Ac0.5 in which CH3COO− and Cl− coexisted in the film were studied. It was found that PPDI-Cl0.9Ac0.1 delivered a slightly higher mobility of 0.85 × 10−4 cm2 V−1 s−1 than pristine PPDI-Cl, and that PPDI-Cl0.5Ac0.5 showed a higher mobility of 2.31 × 10−4 cm2 V−1 s−1, which is much closer to that of PPDI-Ac, indicating that increasing the doping concentration of CH3COO− in the polyelectrolyte film can directly improve the mobility of the polyelectrolyte.
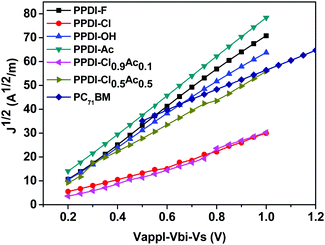 |
| | Fig. 5
J–V curves of electron-only devices with an ITO/Al/polyelectrolyte/Al structure. | |
Table 1 Electron mobilities of the designed polyelectrolytes
| |
PPDI-F |
PPDI-Cl |
PPDI-OH |
PPDI-Ac |
PPDI-Cl0.9Ac0.1 |
PPDI-Cl0.5Ac0.5 |
PC71BM |
| Mobility (×10−4 cm2 V−1 s−1) |
4.30 |
0.66 |
3.34 |
4.76 |
0.85 |
2.31 |
5.93 |
Metal/metal oxide electrode modification
It has been reported that a polymer or small molecules with polar side chains can effectively alter the work function (WF) of metal/metal oxide electrodes, leading to a more suitable energy level alignment with the organic semiconductor.65–68 We investigated the modification capability of our designed polyelectrolytes with ITO and Ag, and the results of the WF change are listed in Table 2. The polyelectrolytes effectively reduced the WF of both Ag and ITO electrodes. The WFs of the ITO modified by our polyelectrolytes were all close to 4.0 eV, which is approximately 0.7 eV lower than that of pristine ITO (4.72 eV). Similar to ITO, the WFs of the Ag electrode were also efficiently reduced to about 4.0 eV by the polyelectrolytes. These results show that quaternary ammonium salts with different anions in these polyelectrolytes can induce interfacial dipoles on the surface of metal/metal oxide electrodes, which reduce the WF and lead to a better ohmic contact at the cathode.6–69 The much lowered WF of Ag and ITO will be more suitable for collecting electrons in conventional and inverted polymer solar cells66–68 and other organic electronic devices.70
Table 2 The work function (WF) of ITO and Ag with/without different polyelectrolytes
| |
None |
PPDI-F |
PPDI-Cl |
PPDI-OH |
PPDI-Ac |
| WF of ITO |
4.65 |
3.95 |
4.05 |
4.00 |
4.07 |
| WF of Ag |
4.69 |
3.98 |
4.04 |
3.99 |
3.96 |
Photovoltaic performance
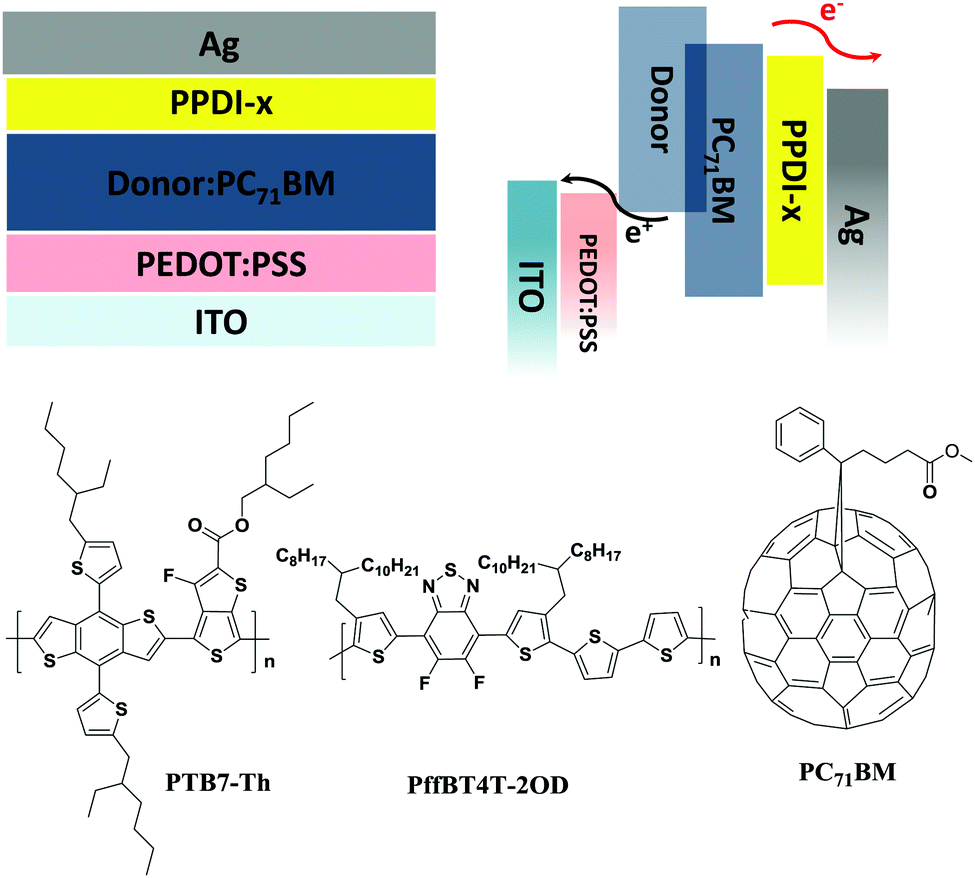
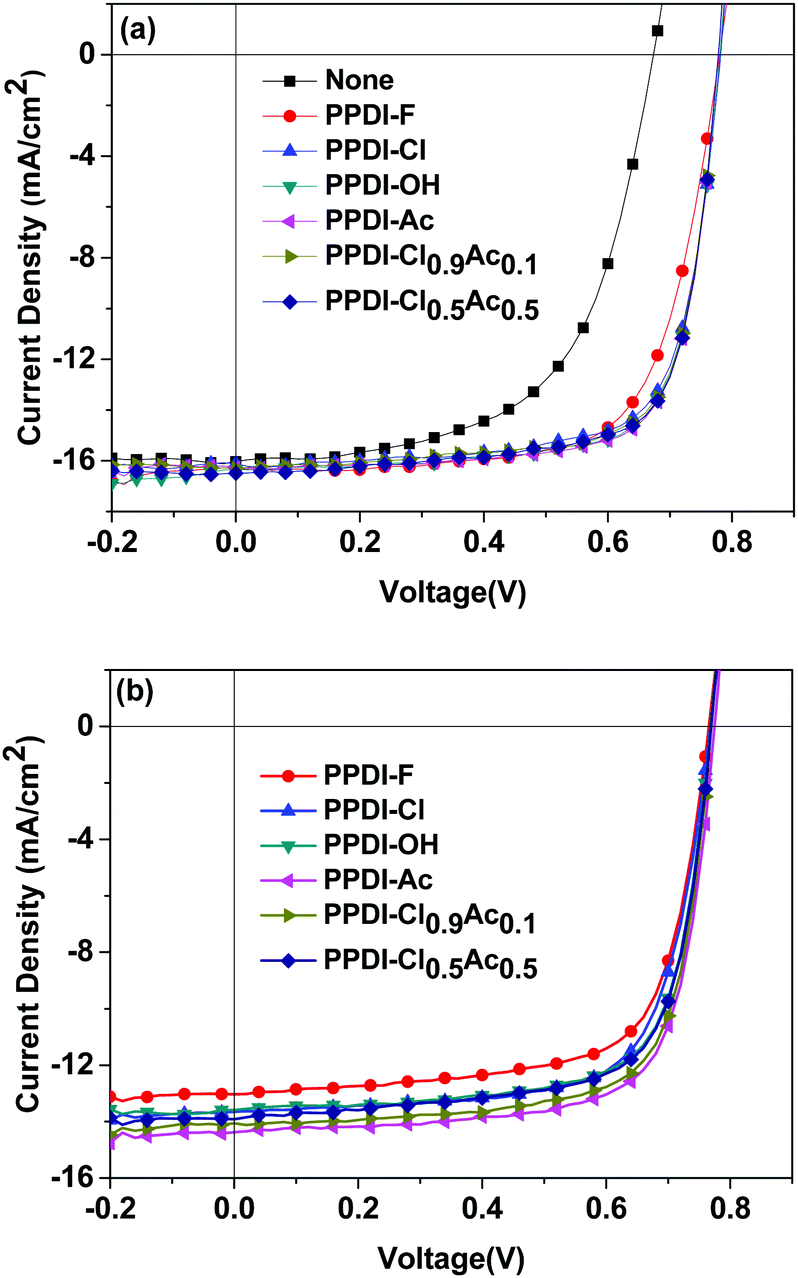
As discussed above, n-type polyelectrolytes possess multiple functions including doping-induced high electron mobility and electrode modification ability, so it can be predicted that a better performance might be achieved when these polyelectrolytes are used as ETMs in organic electronic devices. The performance of PSCs with widely used thin ETMs (such as PFN) can be very high.71 However, the performance decreases quickly as the thickness of the ETMs increases.72,73 Additionally, very thin ETMs are not compatible with the requirements of R2R processing of PSCs.18,72 We applied these polyelectrolytes as ETMs on polymer solar cells with varied thicknesses and fabricated devices with configurations of ITO/PEDOT:PSS/active layer/polyelectrolyte/Ag (shown in Fig. 6). Poly[[2,6′-4,8-di(5-ethylhexylthienyl)benzo[1,2-b;3,3-b]dithiophene][3-fluoro-2[(2-ethylhexyl)carbonyl]thieno[3,4-b]-thiophenediyl]] (PTB7-Th) and [6,6]-phenyl-C71-butyric acid methyl ester (PC71BM) (shown in Fig. 6) were used as the donor and the acceptor, respectively. The current–voltage (J–V) characteristics of the devices are shown in Fig. 7, and the detailed data are summarised in Table 3. PSC devices without ETMs were also fabricated for comparison, and a low PCE of 6.32% was obtained. The effect of the thickness of the polyelectrolytes on the device performance was studied by inserting 20 nm and 50 nm polyelectrolytes between the active layer and the Ag electrode. The devices with 20 nm PPDI-Cl, PPDI-OH and PPDI-Ac delivered average PCEs of 9.03%, 9.27% and 9.33%, which are higher than that of the PPDI-F devices. Devices based on the self-doped polyelectrolyte PPDI-F showed a lower PCE of 8.85%, which can be attributed to the rough surface of the PPDI-F film causing poor contact with the Ag electrode and the resulting relatively poor performance of the PPDI-F based devices.60 The FFs of devices with PPDI-Cl, PPDI-OH and PPDI-Ac as ETMs were all greater than 70%, reaching 72.46%, 73.13% and 73.31%, respectively, indicating that PPDI-Cl, PPDI-OH, and PPDI-Ac are excellent ETMs for PSCs. Additionally, the Rs obtained in devices with PPDI-Cl was slightly larger than that in the PPDI-Ac based devices (3.61 Ω cm2vs. 2.53 Ω cm2), due to the relatively lower mobility of PPDI-Cl. When PPDI-Ac was added to PPDI-Cl, the Rs in devices with PPDI-Cl0.9Ac0.1 and PPDI-Cl0.5Ac0.5 was reduced to 2.75 and 2.55 Ω cm2, respectively, with efficiencies of 9.38% and 9.39%, respectively. Similar trends became more pronounced in the PSC devices when the thicknesses of the ETMs were increased to 50 nm. The devices with PPDI-Ac maintained a high PCE of about 8%, and the devices with PPDI-Cl had a relatively lower average PCE of 7.34%. PPDI-Cl0.9Ac0.1 and PPDI-Cl0.5Ac0.5 based devices in which PPDI-Ac and PPDI-Cl coexisted in the ETMs gave efficiencies of 7.91% and 7.98%, which are almost the same as those obtained from PPDI-Ac based devices. These results clearly show that PPDI-Ac can enhance the electron-transporting/collecting properties of PPDI-Cl, indicating that strongly doped polyelectrolytes are more suitable to act as ETMs in PSCs.
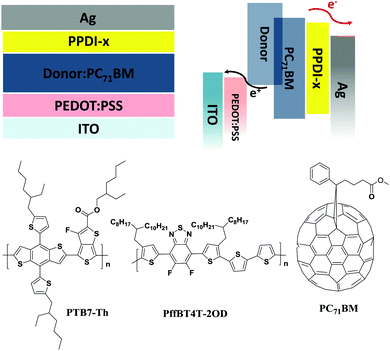 |
| | Fig. 6 The device structure and active layer materials of the PSCs. | |
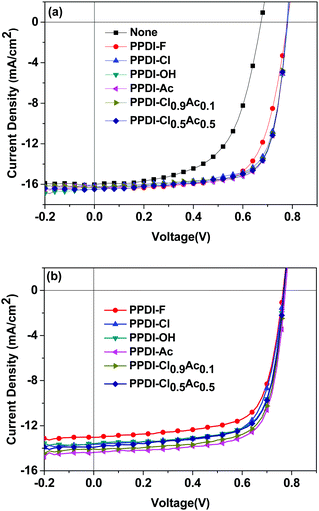 |
| | Fig. 7 The J–V characteristics of the PTB7-Th:PC71BM based PSC devices with 20 nm (a) and 50 nm (b) polyelectrolytes as the ETMs. | |
Table 3 Device parameters of PSC devices based on PTB7-Th/PC71BM and PffBT4T-2OD:PC71BM with 20 nm and 50 nm polyelectrolytes as the ETMs
| ETM |
Thickness (nm) |
V
oc (V) |
J
sc (mA cm−2) |
FF (%) |
PCE (%) |
Best PCE (%) |
R
s
(Ω cm2) |
|
Defined from the J–V curves at V = Voc.
|
| None |
|
0.673 ± 0.002 |
15.80 ± 0.19 |
59.42 ± 0.15 |
6.32 ± 0.07 |
6.40 |
6.90 |
|
|
| PPDI-F |
20 |
0.782 ± 0.001 |
16.26 ± 0.14 |
68.47 ± 0.94 |
8.71 ± 0.14 |
8.85 |
5.44 |
| 50 |
0.765 ± 0.001 |
12.96 ± 0.12 |
69.40 ± 0.25 |
6.88 ± 0.03 |
6.91 |
5.32 |
|
|
| PPDI-Cl |
20 |
0.781 ± 0.001 |
15.98 ± 0.04 |
72.46 ± 1.30 |
9.03 ± 0.19 |
9.17 |
3.61 |
| 50 |
0.768 ± 0.001 |
13.73 ± 0.07 |
69.66 ± 0.74 |
7.34 ± 0.04 |
7.37 |
5.26 |
|
|
| PPDI-OH |
20 |
0.781 ± 0.001 |
16.23 ± 0.16 |
73.13 ± 0.60 |
9.27 ± 0.16 |
9.38 |
3.02 |
|
|
50 |
0.768 ± 0.001 |
13.48 ± 0.11 |
71.89 ± 0.08 |
7.44 ± 0.07 |
7.51 |
4.12 |
|
|
| PPDI-Ac |
20 |
0.782 ± 0.001 |
16.28 ± 0.04 |
73.31 ± 0.68 |
9.33 ± 0.11 |
9.45 |
2.53 |
| 50 |
0.770 ± 0.004 |
14.08 ± 0.30 |
72.89 ± 0.73 |
7.90 ± 0.13 |
8.04 |
4.06 |
|
|
| PPDI-Cl0.9Ac0.1 |
20 |
0.782 ± 0.001 |
16.14 ± 0.28 |
73.22 ± 0.05 |
9.23 ± 0.16 |
9.38 |
2.75 |
| 50 |
0.781 ± 0.001 |
13.97 ± 0.06 |
71.62 ± 1.26 |
7.82 ± 0.12 |
7.91 |
4.40 |
|
|
| PPDI-Cl0.5Ac0.5 |
20 |
0.782 ± 0.001 |
16.19 ± 0.18 |
73.15 ± 0.17 |
9.26 ± 0.10 |
9.39 |
2.55 |
| 50 |
0.782 ± 0.001 |
14.01 ± 0.11 |
72.35 ± 0.03 |
7.92 ± 0.06 |
7.98 |
4.05 |
|
|
| PffBT4T-2OD:PC71BM with PPDI-Ac as ETM |
20 |
0.760 ± 0.001 |
17.27 ± 0.42 |
71.90 ± 1.01 |
9.40 ± 0.16 |
9.63 |
4.04 |
| 50 |
0.760 ± 0.001 |
16.00 ± 0.33 |
70.10 ± 1.50 |
8.44 ± 0.06 |
8.53 |
4.82 |
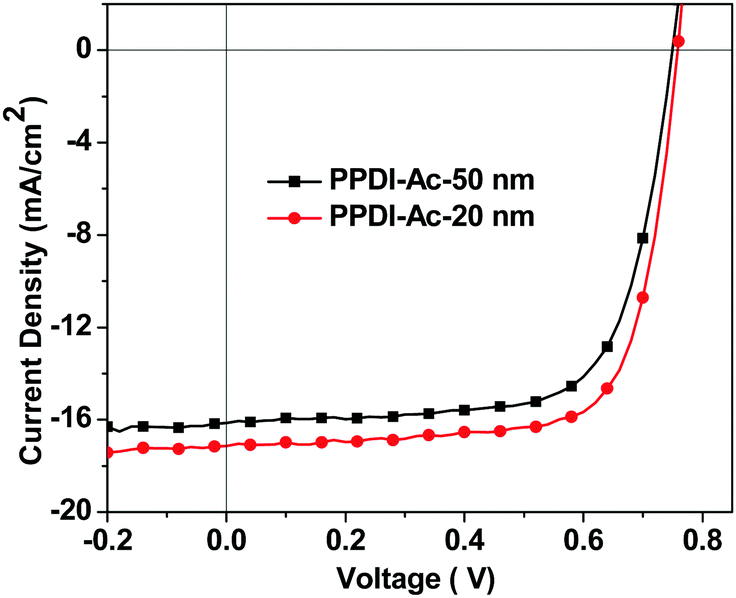
Another active material, PffBT4T-2OD74 (shown in Fig. 6), which can enable high-performance PSCs with active layer thicknesses of over 300 nm, was also used as a donor polymer to fabricate PSCs. The J–V characteristics are shown in Fig. 8 and the data are summarised in Table 3. It can be seen that a high PCE of 9.6% was obtained for the PSC devices when 20 nm PPDI-Ac was used as the ETM. When the thickness of PPDI-Ac was increased to 50 nm, a PCE of 8.53% was maintained in the PSC devices, and the obtained FF (70.1%) was almost the same as that in devices with 20 nm PPDI-Ac (71.9%), indicating that PPDI-Ac can be an excellent thick ETM in PSCs, and is a promising candidate for thickness-insensitive ETMs for R2R processing of PSCs.18,36,75 The major loss in the photovoltaic devices with thicker electron transport materials is the reduction of current density, which is evidenced by the EQE study (shown in Fig. S5, ESI†). The EQE curve in devices with 50 nm polyelectrolytes exhibit a decrease in the wavelength range from 450 nm to 750 nm, which partly coincide with the absorption spectrum of PPDI-Ac (from 450 nm to 1100 nm), indicating that that the thick ETL with a broad absorption spectrum might compete with the active layer in the absorbance of incident photons.18
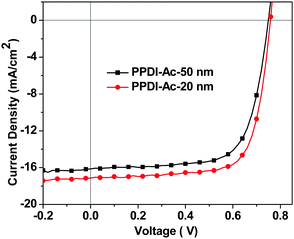 |
| | Fig. 8 The J–V characteristics of the PffBT4T-2OD:PC71BM based PSC devices with 20 nm and 50 nm PPDI-Ac as the ETMs. | |
Interface contact study
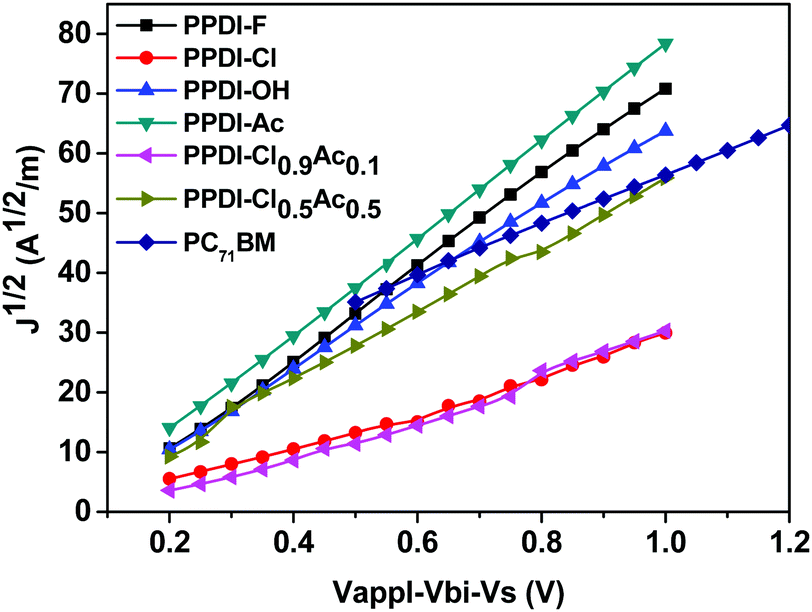
Given that anions may n-dope and improve the conductivity/mobility of fullerene derivatives,30,31,33,76 the interface contacts between PC71BM and our polyelectrolytes were further investigated. Electron-only devices with an ITO/Al/PC71BM/polyelectrolyte/Al structure were fabricated and tested, and the results are shown in Fig. S6 and Table S1 (ESI†). As expected, the electron mobilities of the polyelectrolyte/PC71BM devices were all much higher than those of the pure PC71BM film, reaching 1.83 × 10−3 cm2 V−1 s−1, 1.60 × 10−3 cm2 V−1 s−1, 1.17 × 10−3 cm2 V−1 s−1, and 2.32 × 10−3 cm2 V−1 s−1 for PPDI-F/PC71BM, PPDI-Cl/PC71BM, PPDI-OH/PC71BM and PPDI-Ac/PC71BM, respectively, indicating that these polyelectrolytes can also dope PC71BM at their interface.30,31,33,76 The Jsc obtained in the photovoltaic devices with these 20 nm polyelectrolytes were slightly higher than those of the devices without ETMs, indicating that interface doping between the PC71BM and the polyelectrolytes can improve the interface contact in the photovoltaic devices, and can thus improve the electron collection and current density of the PSCs.77 The improved mobility in the bilayer electron-only devices is also consistent with the decreased Rs in the photovoltaic devices, indicating that the insertion of these polyelectrolytes can substantially improve the electron collection in the photovoltaic devices. When the thickness of the polyelectrolytes was increased to 50 nm, the mobilities of polyelectrolytes/PC71BM exhibited a minor decrease compared to those of devices with 20 nm polyelectrolytes, but was still located in the region of 10−3 cm2 V−1 s−1, reaching 1.26 × 10−3 cm2 V−1 s−1, 1.26 × 10−3 cm2 V−1 s−1, 1.22 × 10−3 cm2 V−1 s−1, and 1.87 × 10−3 cm2 V−1 s−1 for PPDI-F/PC71BM, PPDI-Cl/PC71BM, PPDI-OH/PC71BM and PPDI-Ac/PC71BM, respectively. Of particular interest are the charge transport properties of polyelectrolyte/PC71BM based bilayer electron-only devices, the only devices in which CH3COO− and Cl− coexisted in the film. It can be seen that the bilayer electron-only devices with strongly self-doped PPDI-Ac show a higher mobility than the devices with PPDI-Cl, which showed weak doping behaviours. The mobility of the 20 nm PPDI-Cl0.5Ac0.5 based device (2.09 × 10−3 cm2 V−1 s−1) is higher than that of the PPDI-Cl0.9Ac0.1 based device (1.61 × 10−3 cm2 V−1 s−1), indicating that increasing the doping concentration of CH3COO− improves the electron-transporting properties of PPDI-Cl. Similar results were also obtained for the electron-only devices with 50 nm PPDI-Cl0.5Ac0.5 and PPDI-Cl0.9Ac0.1. These results support the above conclusion that the strongly self-doped polyelectrolytes possess high charge-transporting properties and are good candidates for fabricating thickness-insensitive ETMs in PSCs.
Conclusions
In conclusion, we have designed and synthesised a series of ion-containing n-type polyelectrolytes with different anions via a simple quaternisation polymerisation and ion-exchanging process. The water/alcohol solubility of these polyelectrolytes can be tuned by the choice of anion, which is advantageous for the fabrication of multi-layer organic opto-electronic devices. The doping behaviour and charge-transporting properties of these polyelectrolytes correlate with their attached anions, and higher mobility can be obtained from the strongly self-doped polyelectrolytes. One unique finding is that the film morphologies of these polyelectrolytes can be fine-tuned by the anions. These polyelectrolytes also possess functions including lowering the WF of electrodes and doping PC71BM at the interface. Polymer solar cells with these polyelectrolytes as ETMs of varied thicknesses were studied. The results show that the performance can maintain a high value with almost an unchanged FF of over 70% when the thickness of the polyelectrolyte is up to 50 nm, implying a great opportunity for large-area processing of PSCs. We have disclosed the structure–property–performance relationships of these polyelectrolytes and propose that the strategy we used can provide inspiration for the design of novel high-performance n-type polyelectrolytes for organic opto-electronic devices.
Acknowledgements
This work was financially supported by the Ministry of Science and Technology (No. 2014CB643501), the Natural Science Foundation of China (No. 21520102006, 21634004, 51521002 and 51361165301) and the Guangdong Natural Science Foundation (Grant No. S2012030006232).
Notes and references
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2011, 112, 2208–2267 CrossRef PubMed.
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS.
- K. Wang, C. Liu, T. Meng, C. Yi and X. Gong, Chem. Soc. Rev., 2016, 45, 2937–2975 RSC.
- A. C. Arias, J. D. MacKenzie, I. McCulloch, J. Rivnay and A. Salleo, Chem. Rev., 2010, 110, 3–24 CrossRef CAS PubMed.
- T. R. Andersen, H. F. Dam, M. Hösel, M. Helgesen, J. E. Carlé, T. T. Larsen-Olsen, S. A. Gevorgyan, J. W. Andreasen, J. Adams, N. Li, F. Machui, G. D. Spyropoulos, T. Ameri, N. Lemaître, M. Legros, A. Scheel, D. Gaiser, K. Kreul, S. Berny, O. R. Lozman, S. Nordman, M. Välimäki, M. Vilkman, R. R. Søndergaard, M. Jørgensen, C. J. Brabec and F. C. Krebs, Energy Environ. Sci., 2014, 7, 3284–3290 Search PubMed.
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed.
- R. J. Kline, M. D. McGehee, E. N. Kadnikova, J. Liu and J. M. J. Fréchet, Adv. Mater., 2003, 15, 1519–1522 CrossRef CAS.
- C. Luo, A. K. K. Kyaw, L. A. Perez, S. Patel, M. Wang, B. Grimm, G. C. Bazan, E. J. Kramer and A. J. Heeger, Nano Lett., 2014, 14, 2764–2771 CrossRef CAS PubMed.
- G. Giri, E. Verploegen, S. C. B. Mannsfeld, S. Atahan-Evrenk, D. H. Kim, S. Y. Lee, H. A. Becerril, A. Aspuru-Guzik, M. F. Toney and Z. Bao, Nature, 2011, 480, 504–508 CrossRef CAS PubMed.
- L. Groenendaal, F. Jonas, D. Freitag, H. Pielartzik and J. R. Reynolds, Adv. Mater., 2000, 12, 481–494 CrossRef CAS.
- F. Jonas, W. Krafft and B. Muys, Macromol. Symp., 1995, 100, 169–173 CrossRef.
- W. H. Nguyen, C. D. Bailie, E. L. Unger and M. D. McGehee, J. Am. Chem. Soc., 2014, 136, 10996–11001 CrossRef CAS PubMed.
- G. C. Welch and G. C. Bazan, J. Am. Chem. Soc., 2011, 133, 4632–4644 CrossRef CAS PubMed.
- B. H. Lee, J.-H. Lee, S. Y. Jeong, S. B. Park, S. H. Lee and K. Lee, Adv. Energy Mater., 2014, 5, 1401653 CrossRef.
- H. Zhou, Y. Zhang, C.-K. Mai, S. D. Collins, T.-Q. Nguyen, G. C. Bazan and A. J. Heeger, Adv. Mater., 2014, 26, 780–785 CrossRef CAS PubMed.
- C.-K. Mai, R. A. Schlitz, G. M. Su, D. Spitzer, X. Wang, S. L. Fronk, D. G. Cahill, M. L. Chabinyc and G. C. Bazan, J. Am. Chem. Soc., 2014, 136, 13478–13481 CrossRef CAS PubMed.
- Z. Wu, C. Sun, S. Dong, X.-F. Jiang, S. Wu, H. Wu, H.-L. Yip, F. Huang and Y. Cao, J. Am. Chem. Soc., 2016, 138, 2004–2013 CrossRef CAS PubMed.
- N. Cho, H.-L. Yip, J. A. Davies, P. D. Kazarinoff, D. F. Zeigler, M. M. Durban, Y. Segawa, K. M. O’Malley, C. K. Luscombe and A. K.-Y. Jen, Adv. Energy Mater., 2011, 1, 1148–1153 CrossRef CAS.
- K. Shi, F. Zhang, C.-A. Di, T.-W. Yan, Y. Zou, X. Zhou, D. Zhu, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2015, 137, 6979–6982 CrossRef CAS PubMed.
- R. A. Schlitz, F. G. Brunetti, A. M. Glaudell, P. L. Miller, M. A. Brady, C. J. Takacs, C. J. Hawker and M. L. Chabinyc, Adv. Mater., 2014, 26, 2825–2830 CrossRef CAS PubMed.
- R. Yamachika, M. Grobis, A. Wachowiak and M. F. Crommie, Science, 2004, 304, 281–284 CrossRef CAS PubMed.
- H. Araki, A. A. Zakhidov, K. Tada, K. Yakushi and K. Yoshino, Synth. Met., 1996, 77, 291–297 CrossRef CAS.
- T. H. Reilly, A. W. Hains, H.-Y. Chen and B. A. Gregg, Adv. Energy Mater., 2012, 2, 455–460 CrossRef CAS.
- B. A. Gregg and R. A. Cormier, J. Am. Chem. Soc., 2001, 123, 7959–7960 CrossRef CAS PubMed.
- B. Russ, M. J. Robb, F. G. Brunetti, P. L. Miller, E. E. Perry, S. N. Patel, V. Ho, W. B. Chang, J. J. Urban, M. L. Chabinyc, C. J. Hawker and R. A. Segalman, Adv. Mater., 2014, 26, 3473–3477 CrossRef CAS PubMed.
- F. S. Goodson, D. K. Panda, S. Ray, A. Mitra, S. Guha and S. Saha, Org. Biomol. Chem., 2013, 11, 4797–4803 CAS.
- A. Nollau, M. Pfeiffer, T. Fritz and K. Leo, J. Appl. Phys., 2000, 87, 4340–4343 CrossRef CAS.
- L. Chen, Y. Tan, X. Liu and Y. Chen, Nano Energy, 2016, 27, 492–498 CrossRef CAS.
- C.-Z. Li, C.-C. Chueh, F. Ding, H.-L. Yip, P.-W. Liang, X. Li and A. K.-Y. Jen, Adv. Mater., 2013, 25, 4425–4430 CrossRef CAS PubMed.
- C.-Z. Li, C.-C. Chueh, H.-L. Yip, F. Ding, X. Li and A. K.-Y. Jen, Adv. Mater., 2013, 25, 2457–2461 CrossRef CAS PubMed.
- C.-C. Chueh, C.-Z. Li and A. K.-Y. Jen, Energy Environ. Sci., 2015, 8, 1160–1189 CAS.
- W. Chen, W. Jiao, D. Li, X. Sun, X. Guo, M. Lei, Q. Wang and Y. Li, Chem. Mater., 2016, 28, 1227–1235 CrossRef CAS.
- P. Wei, J. Ha. Oh, G. Dong and Z. Bao, J. Am. Chem. Soc., 2010, 132, 8852–8853 CrossRef CAS PubMed.
- H. Li, C. Risko, J. H. Seo, C. Campbell, G. Wu, J.-L. Brédas and G. C. Bazan, J. Am. Chem. Soc., 2011, 133, 12410–12413 CrossRef CAS PubMed.
- Z.-G. Zhang, B. Qi, Z. Jin, D. Chi, Z. Qi, Y. Li and J. Wang, Energy Environ. Sci., 2014, 7, 1966–1973 CAS.
- Z. Hu, K. Zhang, F. Huang and Y. Cao, Chem. Commun., 2015, 51, 5572–5585 RSC.
- C. Duan, K. Zhang, C. Zhong, F. Huang and Y. Cao, Chem. Soc. Rev., 2013, 42, 9071–9104 RSC.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed.
- T.-T. Wang and C.-C. Kuo, J. Chin. Inst. Chem. Eng., 2003, 34, 187–199 CAS.
- E. E. Neuteboom, S. C. J. Meskers, E. W. Meijer and R. A. J. Janssen, Macromol. Chem. Phys., 2004, 205, 217–222 CrossRef CAS.
- M. C. R. Delgado, E.-G. Kim, D. A. da Silva Filho and J.-L. Bredas, J. Am. Chem. Soc., 2010, 132, 3375–3387 CrossRef PubMed.
- C. Huang, S. Barlow and S. R. Marder, J. Org. Chem., 2011, 76, 2386–2407 CrossRef CAS PubMed.
-
J. E. Huheey, E. A. Keiter and R. L. Keiter, Inorganic Chemistry Principles of Structure and Reactivity, Harper Collins, New York, 4th edn, 1993, pp. 332–333 Search PubMed.
- S. Guha, F. S. Goodson, L. J. Corson and S. Saha, J. Am. Chem. Soc., 2012, 134, 13679–13691 CrossRef CAS PubMed.
- Y. Li, Y. Cao, J. Gao, D. Wang, G. Yu and A. J. Heeger, Synth. Met., 1999, 99, 243–248 CrossRef CAS.
- B. L. Schottel, H. T. Chifotides and K. R. Dunbar, Chem. Soc. Rev., 2008, 37, 68–83 RSC.
- Y. Jiao, K. Liu, G. Wang, Y. Wang and X. Zhang, Chem. Sci., 2015, 6, 3975–3980 RSC.
- A. Robertazzi, F. Krull, E.-W. Knapp and P. Gamez, CrystEngComm, 2011, 13, 3293–3300 RSC.
- C. Duan, W. Cai, B. B. Y. Hsu, C. Zhong, K. Zhang, C. Liu, Z. Hu, F. Huang, G. C. Bazan, A. J. Heeger and Y. Cao, Energy Environ. Sci., 2013, 6, 3022–3034 CAS.
- S.-H. Liao, Y.-L. Li, T.-H. Jen, Y.-S. Cheng and S.-A. Chen, J. Am. Chem. Soc., 2012, 134, 14271–14274 CrossRef CAS PubMed.
- Z. Xie, B. Xiao, Z. He, W. Zhang, X. Wu, H. Wu, F. Würthner, C. Wang, F. Xie, L. Liu, Y. Ma, W.-Y. Wong and Y. Cao, Mater. Horiz., 2015, 2, 514–518 RSC.
- Z. Xie, V. Stepanenko, B. Fimmel and F. Würthner, Mater. Horiz., 2014, 1, 355–359 RSC.
- T. E. Kaiser, H. Wang, V. Stepanenko and F. Würthner, Angew. Chem., Int. Ed., 2007, 46, 5541–5544 CrossRef CAS PubMed.
- Y. Che, A. Datar, K. Balakrishnan and L. Zang, J. Am. Chem. Soc., 2007, 129, 7234–7235 CrossRef CAS PubMed.
- Y. Huang, B. Quan, Z. Wei, G. Liu and L. Sun, J. Phys. Chem. C, 2009, 113, 3929–3933 CAS.
- E. E. Neuteboom, R. A. J. Janssen and E. W. Meijer, Synth. Met., 2001, 121, 1283–1284 CrossRef CAS.
- W. Wang, L.-S. Li, G. Helms, H.-H. Zhou and A. D. Q. Li, J. Am. Chem. Soc., 2003, 125, 1120–1121 CrossRef CAS PubMed.
- Z. Hu, S. Dong, Q. Xue, R. Xu, H.-L. Yip, F. Huang and Y. Cao, Org. Electron., 2015, 27, 46–52 CrossRef CAS.
- J.-S. Yeo, M. Kang, Y.-S. Jung, R. Kang, S.-H. Lee, Y.-J. Heo, S.-H. Jin, D.-Y. Kim and S.-I. Na, Nano Energy, 2016, 21, 26–38 CrossRef CAS.
- Y. Guan, Y. Zakrevskyy, J. Stumpe, M. Antonietti and C. F. J. Faul, Chem. Commun., 2003, 894–895 RSC.
- S. G. Hahm, Y. Rho, J. Jung, S. H. Kim, T. Sajoto, F. S. Kim, S. Barlow, C. E. Park, S. A. Jenekhe, S. R. Marder and M. Ree, Adv. Funct. Mater., 2013, 23, 2060–2071 CrossRef CAS.
- B. H. Lee, G. C. Bazan and A. J. Heeger, Adv. Mater., 2015, 28, 57–62 CrossRef PubMed.
- J. Kim, D. Khim, K.-J. Baeg, W.-T. Park, S.-H. Lee, M. Kang, Y.-Y. Noh and D.-Y. Kim, Adv. Funct. Mater., 2016, 26, 7886–7894 CrossRef CAS.
- F. Liu, Z. A. Page, V. V. Duzhko, T. P. Russell and T. Emrick, Adv. Mater., 2013, 25, 6868–6873 CrossRef CAS PubMed.
- Z. A. Page, Y. Liu, V. V. Duzhko, T. P. Russell and T. Emrick, Science, 2014, 346, 441–444 CrossRef CAS PubMed.
- T. Yang, M. Wang, C. Duan, X. Hu, L. Huang, J. Peng, F. Huang and X. Gong, Energy Environ. Sci., 2012, 5, 8208–8214 CAS.
- K. Zhang, Z. Hu, R. Xu, X.-F. Jiang, H.-L. Yip, F. Huang and Y. Cao, Adv. Mater., 2015, 27, 3607–3613 CrossRef CAS PubMed.
- S. Braun, W. R. Salaneck and M. Fahlman, Adv. Mater., 2009, 21, 1450–1472 CrossRef CAS.
- Y. Zhou, C. Fuentes-Hernandez, J. Shim, J. Meyer, A. J. Giordano, H. Li, P. Winget, T. Papadopoulos, H. Cheun, J. Kim, M. Fenoll, A. Dindar, W. Haske, E. Najafabadi, T. M. Khan, H. Sojoudi, S. Barlow, S. Graham, J.-L. Bredas, S. R. Marder, A. Kahn and B. Kippelen, Science, 2012, 336, 327–332 CrossRef CAS PubMed.
- Z. He, B. Xiao, F. Liu, H. Wu, Y. Yang, S. Xiao, C. Wang, T. P. Russell and Y. Cao, Nat. Photonics, 2015, 9, 174–179 CrossRef CAS.
- S. J. Liu, K. Zhang, J. Lu, J. Zhang, H.-L. Yip, F. Huang and Y. Cao, J. Am. Chem. Soc., 2013, 135, 15326–15329 CrossRef CAS PubMed.
- X. Liu, R. Xu, C. Duan, F. Huang and Y. Cao, J. Mater. Chem. C, 2016, 4, 4288–4295 RSC.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293–5301 CrossRef CAS PubMed.
- Y. Liu, Z. A. Page, T. P. Russell and T. Emrick, Angew. Chem., Int. Ed., 2015, 54, 11485–11489 CrossRef CAS PubMed.
- C.-Y. Chang, W.-K. Huang and Y.-C. Chan, Chem. Mater., 2016, 28, 6305–6312 CrossRef CAS.
- K. Zhang, C. Zhong, S. Liu, C. Mu, Z. Li, H. Yan, F. Huang and Y. Cao, ACS Appl. Mater. Interfaces, 2014, 6, 10429–10435 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, X-ray photoelectric spectroscopy, thermogravimetric, optical microscopy results of the polyelectrolytes, electron-only devices, and external quantum efficiency of the devices. See DOI: 10.1039/c6mh00434b |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 * and
Yong
Cao
* and
Yong
Cao