
Recent developments in self-oscillating polymeric systems as smart materials: from polymers to bulk hydrogels
Youn Soo
Kim†
,
Ryota
Tamate†
,
Aya Mizutani
Akimoto
and
Ryo
Yoshida
*
Department of Materials Engineering, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: ryo@cross.t.u-tokyo.ac.jp
First published on 14th November 2016
Abstract
As novel functional materials, we developed self-oscillating polymeric materials composed of synthetic polymers coupled with an oscillating chemical reaction, the so-called Belousov–Zhabotinsky (BZ) reaction. These materials are very different from traditional stimuli-responsive polymeric materials in terms of their autonomic, self-regulating, and rhythmically changing functions. In this review, we address recent advances in the field of self-oscillating smart materials from the microscopic scale (linear copolymers and block copolymers) to the macroscopic scale (cross-linked hydrogels). The unique features of self-oscillating materials are analogous to the diverse functions of living organisms. This overview has been prepared to promote the development of innovative self-oscillating materials for novel biomimetic material systems.
 Youn Soo Kim | Youn Soo Kim received her BS (2009) and MS (2011) degrees in the Dept. of Chemistry and Nanoscience at Ewha Womans University in Seoul, Korea. She Received her PhD degree from the University of Tokyo in 2015 under the direction of Professor Takuzo Aida. She is currently a JSPS Postdoctoral Fellow in prof. Yoshida Ryo's group at the Department of Materials Engineering, The University of Tokyo. Her research interests include the development of polymer-based smart soft materials. |
 Ryota Tamate | Ryota Tamate received his BS (2006) and MS (2008) degrees from Kyoto University. After working at Bridgestone Co. (2008–2013), he received a PhD degree in 2016 from The University of Tokyo under the supervision of Professor Ryo Yoshida. He is currently working as a postdoctoral researcher at The University of Tokyo with Professor R. Yoshida. Since 2014, he has been supported by a research fellowship for young scientists from the Japan Society for the Promotion of Science (JSPS). |
 Aya Mizutani Akimoto | Dr Aya Mizutani Akimoto received her PhD degree on Biomaterials from Keio University, Japan in 2010 under the direction of Professor Hideko Kanazawa and Professor Teruo Okano (Tokyo Women’s Medical University, Japan). She then joined the group of Professor Mizuo Maeda at RIKEN, Japan as a special postdoctoral researcher. In 2012, she began her academic career as a research associate of Material Engineering at The University of Tokyo, Japan, and was promoted in 2015 to Assistant professor. |
 Ryo Yoshida | Dr Ryo Yoshida is a full professor at The University of Tokyo. He received a PhD degree in chemical engineering in 1993 from Waseda University, Japan. He was a research associate at Tokyo Women’s Medical University in 1993, a researcher at the National Institute of Materials and Chemical Research from 1994–1997, and an assistant professor at University of Tukuba from 1997 to 2000. In 2001, he moved to The University of Tokyo as an associate professor and became a full professor in 2012. He won the Society of Polymer Science, Japan Wiley Award in 2009. |
1. Introduction
Living organisms which consist of cells and an extracellular matrix generate various motions and unique functions by molecular deformation, which are integrated to a macroscopic level through their hierarchical structures.1,2 In this respect, the most important substances are macromolecules such as nucleic acids, proteins, and polypeptides, which have the ability to adapt and change their conformations in response to the surrounding environment. Therefore, the ability to respond to external stimuli is a fundamental characteristic of living systems. Inspired by the stimuli-responsiveness of living matter, materials scientists have attempted to impart a stimuli-responsive nature to synthetic polymers. Upon variation of the surrounding environment, such as temperature, light, pH, electric or magnetic fields, the chemical environment, and ionic strength, stimuli-responsive polymers undergo changes in their conformation or chemical properties. These adaptive changes of stimuli-responsive polymers at the microscopic scale are accompanied by changes in their bulk properties such as shape, optical properties, surface characteristics, and solubility at the macroscopic scale. Because of their responsiveness, these polymers play important roles in a diverse range of applications, such as responsive interfaces,3 controlled drug delivery systems,4–6 and soft actuators.7–9However, in these systems, the response of the polymer is unidirectional; i.e., these polymers only produce an action, such as the expansion or collapse of the polymer chains, where thermodynamic forces drive the system towards a new equilibrium state. In consequence, a repetitive on–off switching of external stimuli is necessary to provoke bidirectional action of these polymers.
In contrast, there are many cyclic phenomena in physiological systems that undergo cyclic changes with temporal periodicity, the so-called “temporal structures.” These spontaneous and autonomous oscillations exist over a wide range of length scales, as typified by the cell cycle, pulsatile secretion of hormones, heart contractions, and circadian rhythms. If such self-oscillatory phenomena could be achieved by using a synthetic polymer system alone, the control of various non-unidirectional processes and the construction of new biomimetic materials with autonomic functions would be possible.
Toward this aim, we have utilized the Belousov–Zhabotinsky (BZ) reaction, which is a well-known oscillatory chemical reaction.10–12 The BZ reaction was first investigated as a model reaction for the tricarboxylic acid (TCA) cycle, an important metabolic cycle. During the overall BZ reaction, an organic substance, such as malonic acid, is oxidized by an oxidizing agent (bromate) in the presence of a metal catalyst under acidic conditions. Commonly used metal catalysts for this cyclic process include cerium ions, ferroin, or ruthenium tris(2,2′-bipyridine) (Ru(bpy)3). In the course of the reaction, while the substrate is present, the valence of the metal catalyst periodically changes between the oxidized and reduced states. Consequently, under homogeneously stirred conditions, cyclic changes in the color of the solution are observed, and these follow the changes in the redox state of the metal catalyst. In contrast, if the solution is unstirred, spatiotemporal patterns such as concentric or spiral waves, the so-called “chemical waves”, occur.
By coupling these chemical oscillatory reactions with adaptive synthetic polymers, we have developed a novel biomimetic energy transformation system: a “self-oscillating polymer” exhibiting spontaneous and periodic transitions of the polymer conformation under constant conditions without any on–off switching of external stimuli. The self-oscillating polymer is synthesized by random copolymerization of N-isopropylacrylamide (NIPAAm) and Ru(bpy)3, which has been modified with a vinyl group. The homopolymer of NIPAAm (poly(NIPAAm)) is thermosensitive and undergoes an abrupt coil-to-globule transition when heated to around 32 °C, which is the lower critical solution temperature (LCST).13 Therefore, because of the thermoresponsive NIPAAm component, poly(NIPAAm-co-Ru(bpy)3) also has an LCST. If the Ru(bpy)3 moiety of poly(NIPAAm-co-Ru(bpy)3) is oxidized from Ru(bpy)32+ to Ru(bpy)33+, the increased oxidation number of Ru(bpy)3 causes an increase in the hydrophilicity of the polymer, resulting in an increase in the LCST of the polymer. As a result, it is expected that the cyclic redox changes in the Ru(bpy)3 moiety will induce the hydration and dehydration of the polymer chains (Fig. 1; linear copolymer). When poly(NIPAAm-co-Ru(bpy)3) is dissolved in an aqueous solution containing the substrates, except for the metal catalyst, necessary for the BZ reaction (malonic acid (MA), sodium bromate (NaBrO3), and nitric acid (HNO3)), the BZ reaction is catalyzed by the Ru(bpy)3 units incorporated into the polymer chain. Synchronized with the periodic changes in the redox state of Ru(bpy)3, the hydrophilicity of the polymer changes. Consequently, poly(NIPAAm-co-Ru(bpy)3) exhibits cyclic and rhythmical changes in its solubility under constant and homogeneous conditions. The periodic changes in the polymer conformation can be easily visualized as cyclic changes in the optical transmittance of the polymer solution and also by changes in the color of the solution arising from the changes in the redox state of the Ru(bpy)3 chromophores (Fig. 2).14 In this system, the chemical energy of the BZ reaction can be transformed into rhythmic changes in the polymer conformation.
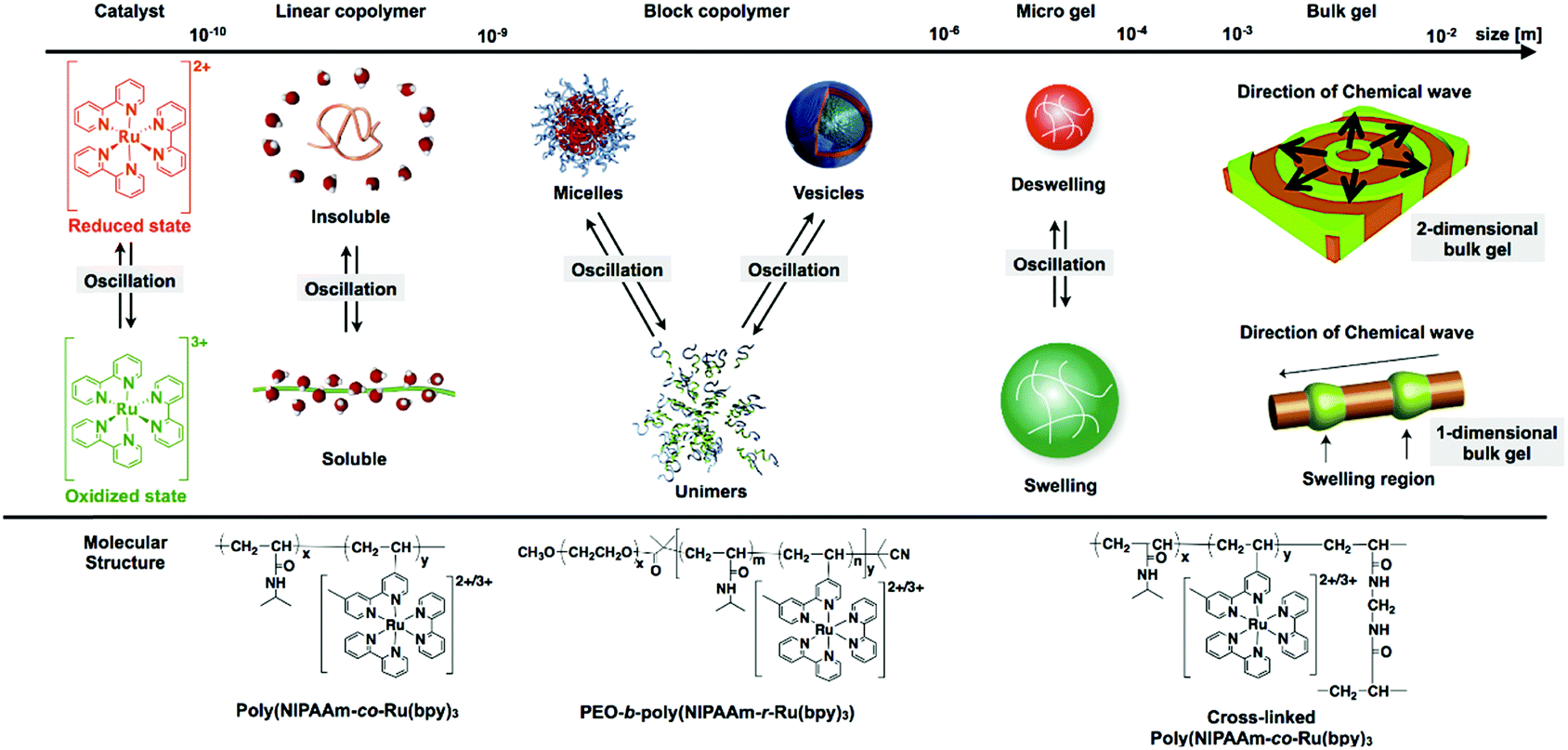 | ||
| Fig. 1 Self-oscillations at several scales, from the microscopic to the macroscopic. | ||
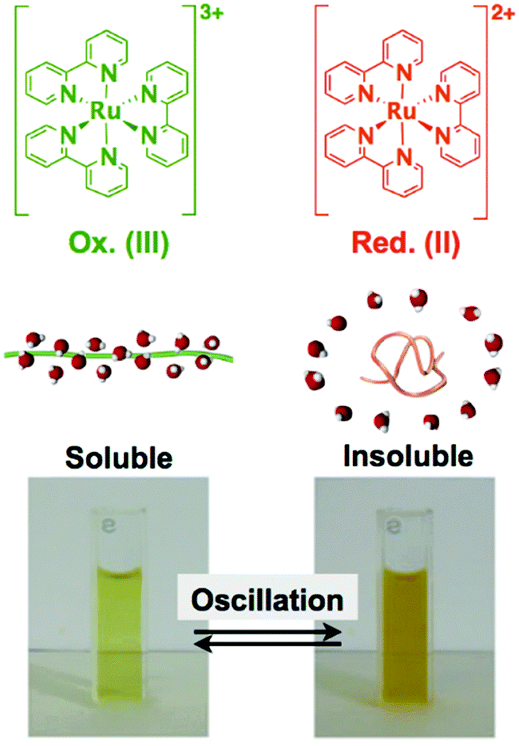 | ||
| Fig. 2 Soluble–insoluble periodic changes for a solution of poly(NIPAAm-co-Ru(bpy)3) synchronized with the redox changes of the Ru(bpy)3 complexes. Reproduced with permission from ref. 14. American Chemical Society 2002. | ||
Generally, when polymer chains are chemically cross-linked, conformational changes in the polymer chains are converted into macroscopic mechanical changes. By cross-linking the polymer chains, chemomechanical gels that can transform chemical energy into mechanical energy can been prepared. For example, a polymer gel containing immobilized enzymes can transform the chemical energy of the enzymatic reaction into mechanical energy expressed as changes in the volume of the polymer gel (i.e., a biochemomechanical gel).15
Based on this knowledge, the cross-linking of poly(NIPAAm-co-Ru(bpy)3) was used to prepare a “self-oscillating gel”.16 In the self-oscillating gel, the conformational changes of the poly(NIPAAm-co-Ru(bpy)3) gel are induced by the redox changes of the Ru(bpy)3 catalyst, which are amplified to macroscopic swelling/deswelling of the polymer network (Fig. 1; microgel and bulk gel). In the presence of the BZ reactants in a closed system, the polymer gel undergoes spontaneous swelling/deswelling oscillations without any external stimuli, significantly different behavior from that of conventional stimuli-responsive polymer gels.16 The self-oscillating gel has a built-in energy conversion circuit, generating periodic mechanical oscillations from the chemical energy of the BZ reaction. Since the first report in 1996,16 self-oscillating polymer materials have been studied systematically17–23 and their applications in biomimetic or intelligent materials have been explored, for example, as biomimetic soft actuators,24–27 mass transport systems,28–30 and functional fluids.31–36 Self-oscillatory phenomena can be realized on several length scales, from the microscopic to the macroscopic scale (Fig. 1). In the case of non-cross-linked poly(NIPAAm-co-Ru(bpy)3), the optical transmittance of the polymer solution exhibits self-oscillations because of the spontaneous cyclic coil–globule transitions of the polymer chains. Furthermore, when self-oscillating polymers are introduced into one segment of block copolymers, unique oscillation phenomena of the self-assembled structures, such as self-oscillating formation and break-up of micelles20,35,37 or vesicles38 at the nano to micron scale, can be observed. If the polymer is chemically cross-linked, as in polymer gels, the characteristics of the mechanical oscillations are size dependent. When the cross-linked polymer materials are much smaller than the wavelength of the chemical waves of the BZ reaction, redox changes in the material occur homogeneously and without pattern formation. Consequently, the self-oscillatory swelling and deswelling in the gel are isotropic. In contrast, when the size of the cross-linked polymer material is larger than the wavelength of the chemical waves, spatiotemporal patterns in the mechanical changes inside the material occur, and these are synchronized with the propagation of the chemical wave. By utilizing this peristaltic motion, new biomimetic transporting systems have been created by fabricating tubular self-oscillating gels, as discussed later.
In this review, we highlight recent progress in the study of self-oscillating polymer materials. This review is divided into the following sections: first, we introduce the latest developments in self-oscillating non-cross-linked polymeric systems, from polymer design to their oscillating characteristics. These polymeric systems are further divided into three groups, which are discussed separately: polymer brushes, branched polymers, and block copolymers. Secondly, recent developments in cross-linked self-oscillating polymeric systems including gels, cross-linked polymersomes, and colloidosomes will be reviewed. In the section concerning self-oscillating gels, we discuss the specific functionalities and the improvement in the performance of autonomic gel actuators obtained by precisely designing the physical and chemical polymer structures. In the section concerning cross-linked polymersomes and colloidosomes, we introduce the fabrication methods for hollow, cell-like structures, formed by the self-assembly of block copolymers or microgels and post-crosslinking, as well as their specific shape and volume oscillations, which are similar to the dynamic behavior of living cell membranes.
2. Self-oscillating polymeric systems prepared without cross-linking
2.1. Self-oscillating polymer brushes
Functional surfaces that can control their wettability, permeability, and adhesiveness have attracted much attention because of their diverse potential applications. In particular, by incorporating stimuli-responsive polymers on material surfaces, the physical and chemical characteristics of the surfaces can change in response to external stimuli. Such stimuli-responsive surfaces can be used as smart surfaces with switchable properties, for example, in cell culture substrates, microfluidics, self-cleaning coatings, and filtration devices.39–46 To fabricate well-defined functional surfaces, surface modification techniques to graft synthetic polymers onto substrates have been intensively investigated. Among them, surface-initiated atom transfer radical polymerization (SI-ATRP) is a useful method to prepare well-defined, high-density polymer brush structures on substrates.47–50In contrast to functional surfaces modified by stimuli-responsive polymers, functional surfaces modified by self-oscillating polymers exhibit autonomous changes in their surface properties without requiring external stimuli, and novel applications such as autonomous mass transport systems on the micro- or nanoscale are expected. In the past, several active functional surfaces prepared by immobilizing motor proteins51 or utilizing Marangoni convection have been reported.52 In contrast with these materials, autonomous surfaces comprised of well-defined synthetic polymer brushes would widen the range of their uses and pave the way for more versatile applications. Building on this concept, “self-oscillating polymer brushes” were fabricated for the design of novel autonomous functional surfaces.
Self-oscillating polymer brushes have been prepared by grafting self-oscillating polymers onto a glass substrate using the SI-ATRP method.30,53 In the first step, a copolymer of NIPAAm and N-3-(aminopropyl)methacrylamide (NAPMAm) was grafted on the glass surface by SI-ATRP. Then, a succinimidyl ester of Ru(bpy)3 (Ru(bpy)3-NHS) was introduced into the polymer brush by coupling to the amino groups of NAPMAm. Characterization of the resultant poly(NIPAAm-r-NAPMAm-r-Ru(bpy)3NAPMAm) brush surface (Fig. 3(a)) was carried out using X-ray photoelectron spectroscopy (XPS) and atomic force microscopy (AFM). The molecular weight and layer thickness were determined using GPC, and ellipsometry measurements indicated that the dense polymer brushes had been successfully grafted onto the substrate. Based on this protocol, self-oscillating polymers were also modified on the inner surface of a glass capillary. Fig. 3(c) shows the fluorescence image of the modified glass capillary, indicating that the self-oscillating polymer brush had been successfully grafted onto the inner surface of the capillary. When the catalyst-free BZ solution flowed through the capillary, chemical wave propagation was clearly observed using a fluorescence microscope. The oscillating fluorescence intensity profiles during the BZ reaction at different locations through the capillary tube are compared in Fig. 3(c). These profiles indicate that the oscillating changes in the redox state of the Ru(bpy)3 complex conjugated to the grafted polymer brushes occurred with phase differences at each location.
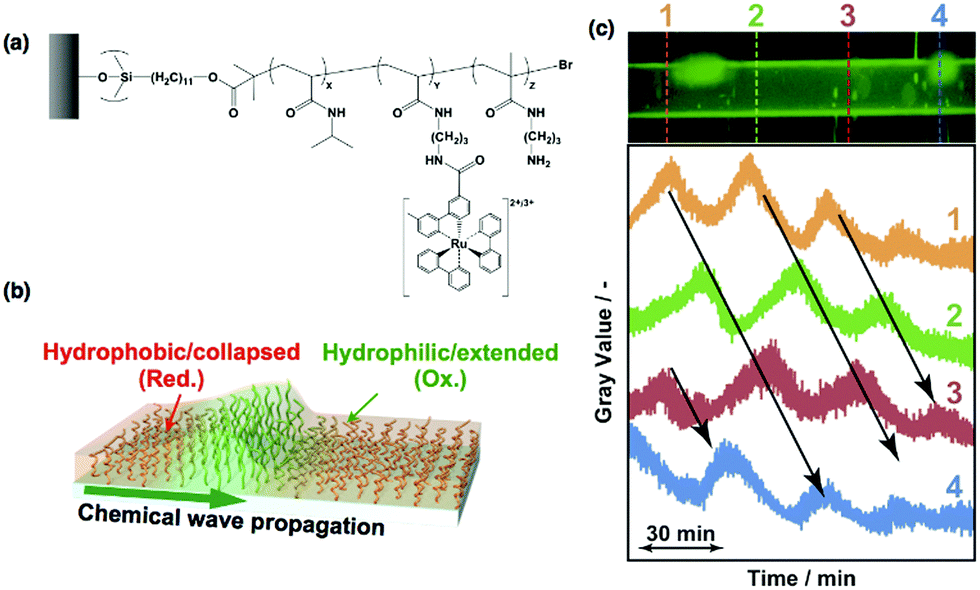 | ||
| Fig. 3 (a) Chemical structure of the self-oscillating polymer brush (poly(NIPAAm-co-NAPMAm-co-Ru(bpy)3NAPMAm)). (b) Illustration of the self-oscillating polymer brushes during the BZ reaction. (c) Top: A fluorescence image of a glass capillary modified with the self-oscillating polymer brushes that were grafted onto the inner surface of the capillary. Bottom: Oscillating profiles of fluorescence intensity along with the BZ reaction at different locations of the capillary. Reproduced with permission from ref. 30. Wiley 2013. | ||
To obtain more stable oscillations and control the self-oscillating behavior of the grafted polymer brushes, the relationship between the surface nanostructures and the oscillatory dynamics was investigated.53 The synthetic conditions were modulated by changing the concentrations of the monomer, the initiator, and Ru(bpy)3-NHS so that self-oscillating polymer brushes with various lengths of grafted polymers and different concentrations of immobilized Ru(bpy)3 were prepared. As a result, stable chemical wave propagation could be obtained by immobilizing an appropriate amount of Ru(bpy)3 on the grafted polymer, whereas inadequate or excessive amounts of Ru(bpy)3 resulted in no oscillation or oscillations for shorter durations. Furthermore, the actual hydration–dehydration behaviors of the polymer brush were studied using quartz crystal microbalance with dissipation (QCM-D) measurements. To carry out these measurements, we grafted the self-oscillating polymer brush onto the surface of a SiO2-coated QCM-D sensor. The temperature dependences of the change in frequency (Δf) and the dissipation energy (ΔD) for the polymer brush in the reduced state are shown in Fig. 4(a). Here, Δf is related to the change in the mass detected by the sensor, and ΔD represents energy dissipation due to the viscoelasticity of the polymer brushes. Upon heating, Δf increases because of the dehydration of the polymer brushes. On the other hand, ΔD decreases as a consequence of the conformational change from a stretched to a collapsed state. When the Ru(bpy)3 center of the polymer brush was oxidized by the addition of an oxidant (NaBrO3), the polymer brush swelled, as shown in Fig. 4(b). At constant temperature, the addition of NaBrO3 caused an increase in Δf and a decrease in ΔD, implying that the self-oscillating polymer brush was more swollen in the oxidized state than in the reduced state as a result of increased hydrophilicity.
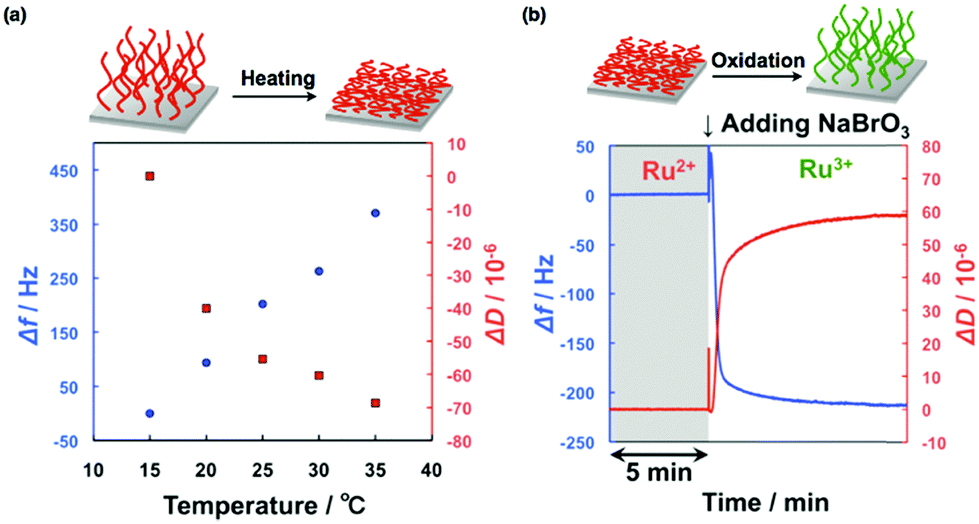 | ||
| Fig. 4 (a) The temperature dependence of the change in frequency (Δf) as well as the energy dissipation (ΔD) for the self-oscillating polymer brush in the reduced state. (b) Changes in Δf and ΔD in response to the oxidation of the self-oscillating polymer brush, induced by adding an oxidant (NaBrO3) at 25 °C. Reproduced with permission from ref. 53. American Chemical Society 2015. | ||
Thus far, the propagation wave in the self-oscillating polymer brush occurs in random directions. However, considering the potential applications in autonomous mass transport systems, e.g., ciliary-like actuators, the direction of the propagating wave must be controlled. According to previous reports concerning the control of the chemical wave direction in self-oscillating gels,54,55 heterogeneous structures are effective in the BZ reaction. Therefore, using sacrificial-anode atom transfer radical polymerization (saATRP), we have fabricated gradient self-oscillating polymer brushes that have a gradient of brush thicknesses (Fig. 5(a and b)).56 The gradient brush structures were experimentally confirmed by observing the spatial gradient of the immobilized Ru(bpy)3 chromophores by fluorescence microscopy (Fig. 5(a)), as well as the C/Si gradient values, which were measured using XPS. By using the resultant gradient self-oscillating polymer brush, the direction of the chemical wave was successfully regulated to move from areas of low to high Ru(bpy)3 concentration, as shown in Fig. 5(c). This can be explained by the characteristics of the BZ reaction, i.e., regions that have a lower Ru(bpy)3 concentration behave as a pacemaker for the oscillatory reaction. The introduction of the gradient structure into the self-oscillating polymer brush enhanced the functionality of the oscillatory phenomena, and this is an important step in developing applications in spatiotemporally controlled autonomous mass transport systems.
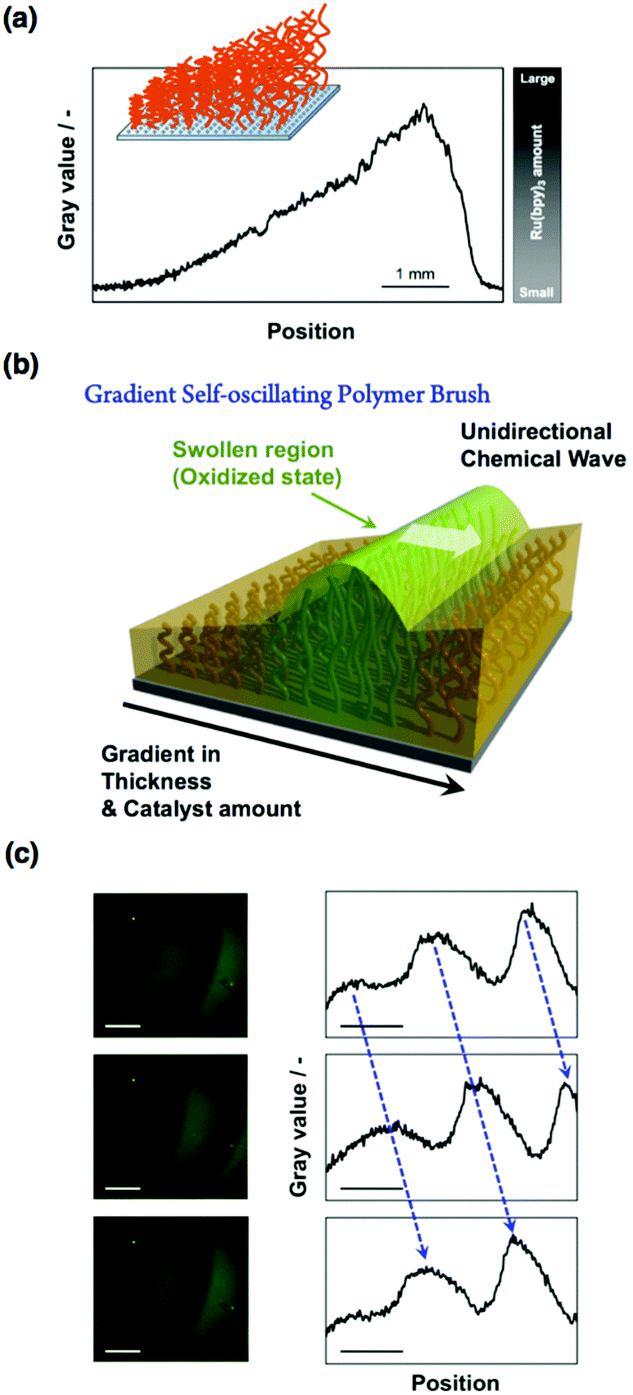 | ||
| Fig. 5 (a) Spatial gradient of the fluorescence intensity of the gradient self-oscillating polymer brush observed by cross-sectional analysis of fluorescence microscopy images. (b) Schematic illustration of the unidirectional propagation of the chemical wave of the gradient self-oscillating polymer brush. The direction of the chemical wave was from the area with a low concentration of Ru(bpy)3 to that with a high concentration of Ru(bpy)3. (c) Left: Time-course images of the gradient self-oscillating polymer brush with the propagation of the chemical wave, as observed by fluorescence microscopy. Scale bar: 500 μm. Time interval between each image: 10 s. Right: Chemical wave profiles expressed as fluorescence intensity by image analysis of the pictures shown on the left-hand side. Reproduced with permission from ref. 56. AAAS 2016. | ||
2.2. Self-oscillating branched polymers possessing dynamic metal–ligand coupling
Inspired by the recent advances in supramolecular chemistry, we have tried to integrate supramolecular interactions into self-oscillating polymers to achieve new types of self-oscillating materials. The supramolecular interactions used are non-covalent and are dynamic and reversible, such as hydrogen bonds and ionic interactions. Among these interactions, metal–ligand interactions are very significant because of the many possible combinations of metals and ligands. In particular, we have focused on metal–ligand interactions using ruthenium–terpyridine complexes57 because the binding of terpyridines to ruthenium ions is dependent on the redox state of the central ruthenium ion.58 When the ruthenium ion is in the reduced state (Ru2+), a bis-complex (Ru(terpy)2) is formed; in contrast, in the oxidized state (Ru3+), a mono-complex (Ru(terpy)) is formed (Fig. 6(a)). Therefore, if the ruthenium–terpyridine complex can act as a metal catalyst for the BZ reaction, oscillatory changes in the binding states of the complex are possible, generating a dynamic connection between supramolecular polymer networks.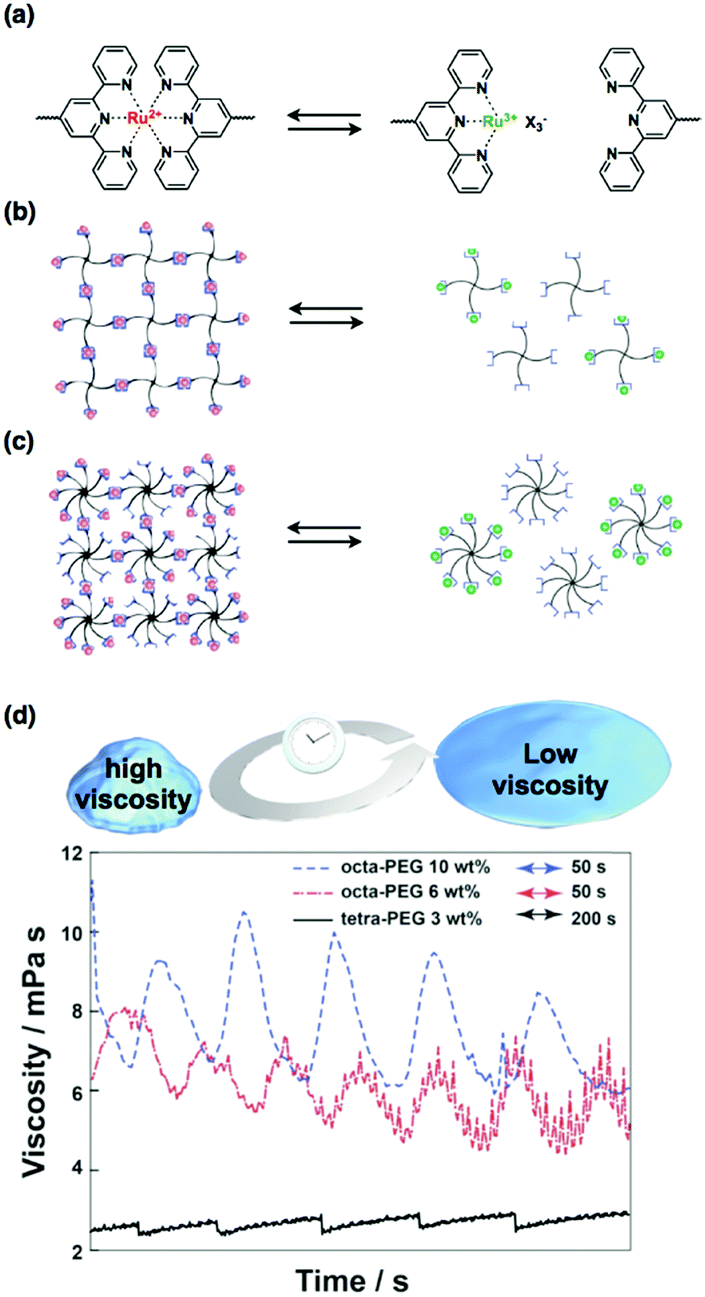 | ||
| Fig. 6 (a) Reversible metal–ligand complex formation between the bis-complex (Ru(terpy)2) in the reduced state (Ru2+) and the mono-complex (Ru(terpy)) in the oxidized state (Ru3+). (b) Reversible complex formation of terpyridine-terminated tetra-PEG and (c) that of octa-PEG. (d) The autonomous viscosity oscillations for the octa-PEG and tetra-PEG systems during the BZ reaction. Reproduced with permission from ref. 34. Royal Society of Chemistry 2014. | ||
Motivated by computational simulations that have demonstrated the applicability of the ruthenium–terpyridine complex as a dynamic junction point in the BZ reaction,59 we have experimentally shown that supramolecular interactions of ruthenium–terpyridine complexes can be coupled with the BZ reaction, allowing unprecedented oscillatory phenomena such as oscillations in solution viscosity that are synchronized with the cyclic formation and deformation of the metal–ligand complex.34,36 Consequently, linear-, tetra-, and octa-polyethylene glycols (PEGs) that have terpyridine ligands at their terminals were synthesized (Fig. 6(b and c)). When the ruthenium ions were in the reduced state, stable gels were obtained for the tetra- and octa-PEG, whereas no gelation occurred for the linear-PEG. This result suggests that three-dimensional polymer networks formed via ruthenium–terpyridine coordination were more easily formed for branched PEG structures because the number of crosslinking points necessary for gelation is lowered as the number of PEG branches increases. This hypothesis is supported by rheological measurements, which show that when the ruthenium ions are reduced, the sol-to-gel transition of the octa-PEG system is significantly quicker than that of the tetra-PEG systems. Finally, autonomous viscosity oscillations were observed for the polymer solutions with the BZ substrates (Fig. 6(d)). The largest oscillation amplitude was obtained for the octa-PEG system because the network structure, which is formed concomitantly with the complex, was efficiently constructed even during the BZ reaction. This system provides a new strategy to create novel self-oscillating materials by utilizing redox-responsive ruthenium–terpyridine complexes that act as both the catalyst for the BZ reaction and a supramolecular junction point.
2.3. Self-oscillating block copolymers
The self-assembly of block copolymers (BCPs) in bulk and solution has attracted significant attention for many decades.60 In the bulk, AB diblock copolymers having two immiscible segments show various microphase-separated structures that depend on the volume fraction of the A and B segments (fA and fB), the overall polymerization degree N, and the incompatibility of the A and B segments (i.e., the Flory–Huggins χ parameter). When χ and N are fixed, the microphase-separated structures change depending on fA, forming a body-centered-cubic sphere, a hexagonally packed cylinder, double gyroid, and lamellar structures. The phase diagram of AB diblock copolymers has been well studied, both theoretically61 and experimentally,62 and they correspond well. In addition, the self-assembly of BCPs in solution has been studied broadly, but, for BCPs, the situation is more complex because of the presence of additional components such as solvents. In particular, the self-assembly of amphiphilic diblock copolymers in aqueous solutions has been extensively investigated, for example, in the seminal work of Eisenberg et al., who investigated self-assembled structures of poly(styrene)-b-poly(acrylic acid) (PS-b-PAA) diblock copolymers in aqueous solutions. They demonstrated that diverse self-assembled morphologies can be obtained by varying the PS-b-PAA block length63 and the solution conditions, such as the presence of salts and cosolvents.64 Similarly, Jain and Bates investigated the effect of the block length of poly(butadiene)-b-poly(ethylene oxide) (PB-b-PEO) on the self-assembled structures formed in aqueous solution.65 In general, dilute aqueous solutions of BCPs form spherical micelles, cylindrical micelles, and vesicles (or polymersomes), depending on the block ratio between the hydrophilic and hydrophobic segments.66 In addition to these basic morphologies, many complex morphologies have been reported. Furthermore, beyond the AB diblock copolymers, multiblock copolymers that link more than two segments offer many distinct self-assembled morphologies.67Self-assembled BCP structures are either thermodynamically stable or metastable. In contrast, temporal or spatiotemporal structures, which are one of the unique characteristics of living systems, are dissipative structures in a dynamic state far from equilibrium.68 By coupling the self-assembly of BCPs with dissipative structures formed by the BZ reaction, a novel class of “self-oscillating BCPs” has been fabricated, where the self-assembled structures of BCPs, driven by the BZ reaction, autonomously oscillate.35 A diblock copolymer composed of poly(ethylene oxide) (PEO) as the hydrophilic segment and poly(NIPAAm-co-Ru(bpy)3) as the self-oscillating segment was synthesized by reversible addition-fragmentation transfer (RAFT) polymerization (Fig. 7(a)). When the temperature was increased, the PEO-b-poly(NIPAAm-co-Ru(bpy)3) block copolymer underwent a phase transition from unimers to micelles, driven by the change in the hydrophilicity of the self-oscillating segment. Importantly, in this system, the phase transition temperature is dependent on the redox states of the Ru(bpy)3 centers. Therefore, there is a bistable temperature region, where micelles are stable in the reduced state, whereas unimers are stable in the oxidized state, as shown in Fig. 7(b). Consequently, during the BZ reaction, autonomous structural oscillations between unimers and micelles (i.e., ‘self-oscillating micelles’) can be observed at the bistable temperature, and these oscillations follow the periodic changes in the redox state of Ru(bpy)3 (Fig. 7(c)).35 From this starting point, many self-oscillating BCPs with dynamic autonomous behavior have been developed. In this section, we review the recent development of self-oscillating BCP systems.
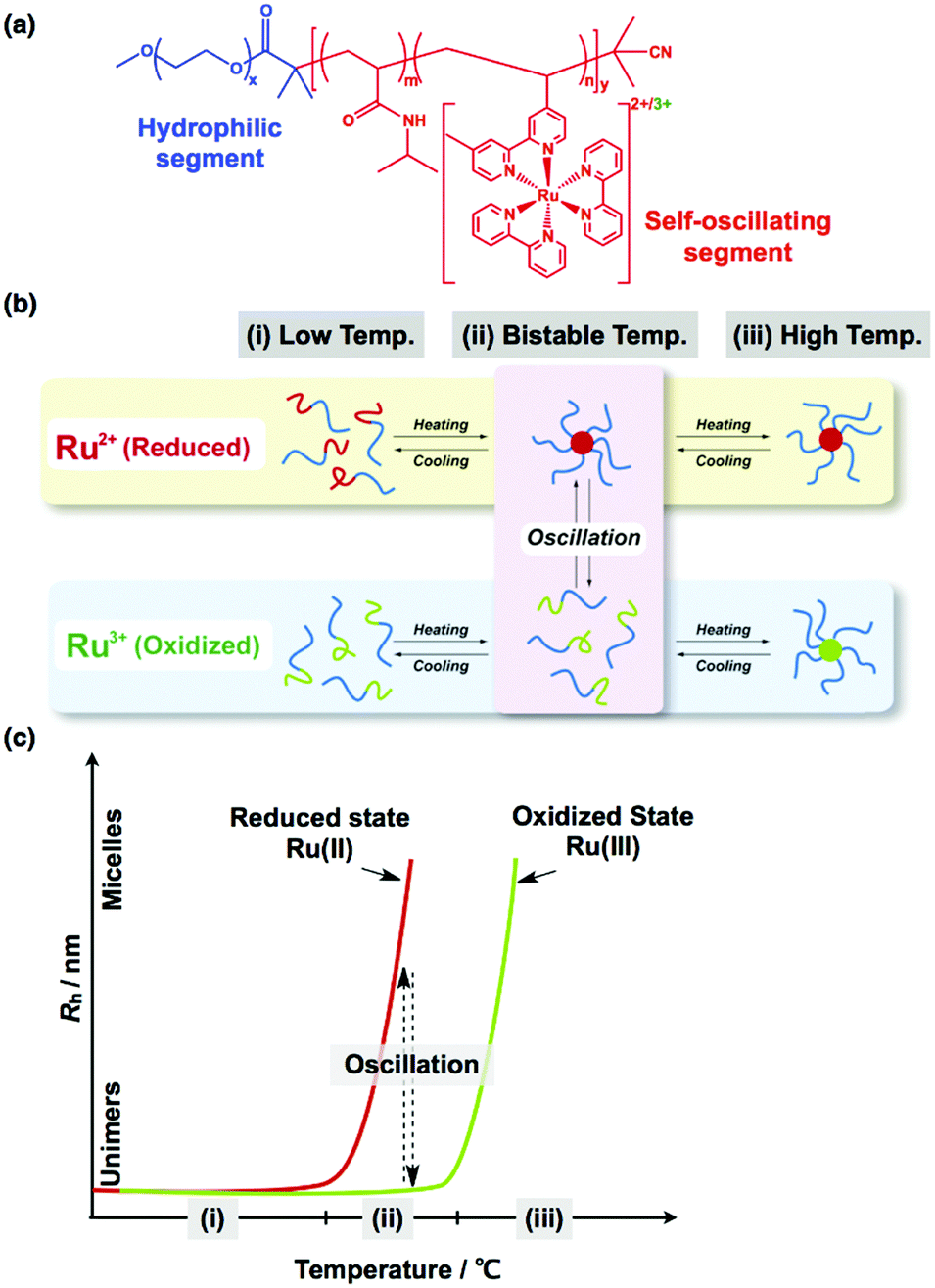 | ||
| Fig. 7 (a) Chemical structure of the self-oscillating diblock copolymer (PEO-b-poly(NIPAAm-co-Ru(bpy)3)) composed of poly(ethylene oxide) (PEO) as a hydrophilic segment and poly(NIPAAm-co-Ru(bpy)3) as a self-oscillating segment. (b) Conceptual illustration of self-oscillating block copolymers showing autonomous oscillations between unimers and micelles. (c) Temperature dependence of the hydrodynamic radius (Rh) of the diblock copolymer in the reduced and oxidized states. Reproduced with permission from ref. 35. Royal Society of Chemistry 2013. | ||
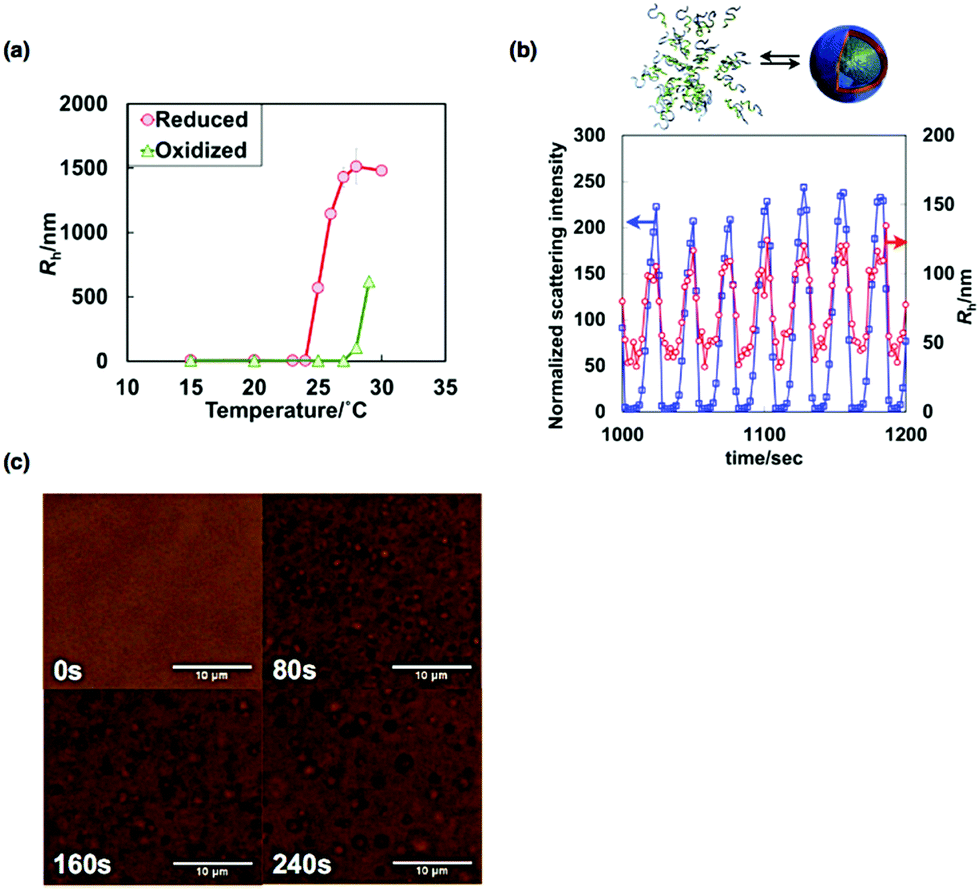 | ||
| Fig. 8 (a) Temperature dependences of Rh of the diblock copolymer solution in both the reduced and oxidized states. (b) Time variation of the normalized scattering intensities (blue) and Rh values (red) during the BZ reaction. (c) Optical microscopic images of the association process of vesicles during the BZ reaction. Reproduced with permission from ref. 38. Wiley 2014. | ||
The self-oscillatory behavior driven by periodic redox changes during the BZ reaction was observed via time-resolved DLS measurements. Fig. 8(b) shows the time variation of the normalized scattering intensities and the Rh values of a dilute aqueous solution of the diblock copolymer (0.1 wt%) during the BZ reaction. As shown, oscillations in both the scattering intensity and the Rh values were confirmed. These results indicate that autonomous structural changes in the self-assembly of the BCPs occurred. However, the Rh values during the self-oscillations were relatively small compared to those at equilibrium. The main reason for this observation is that the oscillatory periods were too short to allow the formation of large, equilibrated vesicles.
With an increase in polymer concentration, large vesicles form more easily. As a result, for the 1.5 wt% diblock copolymer solution, autonomous vesicle assembly and disassembly were successfully observed using time-lapse optical microscopy (Fig. 8(c)). In the reduced state, vesicles underwent Brownian motion and gradually grew in size. In contrast, vesicles suddenly disappeared on the change from the reduced state to the oxidized state. Consequently, the autonomous association/dissociation of vesicles with micrometer dimensions was observed directly. In common with vesicles in living cells, vesicle fusion was also confirmed during the association process. Considering the dynamic nature of biological vesicles, such as nuclear envelopes, synaptic vesicles and cell membranes, self-oscillating vesicles are a novel type of biomimetic vesicle with dynamic behavior.
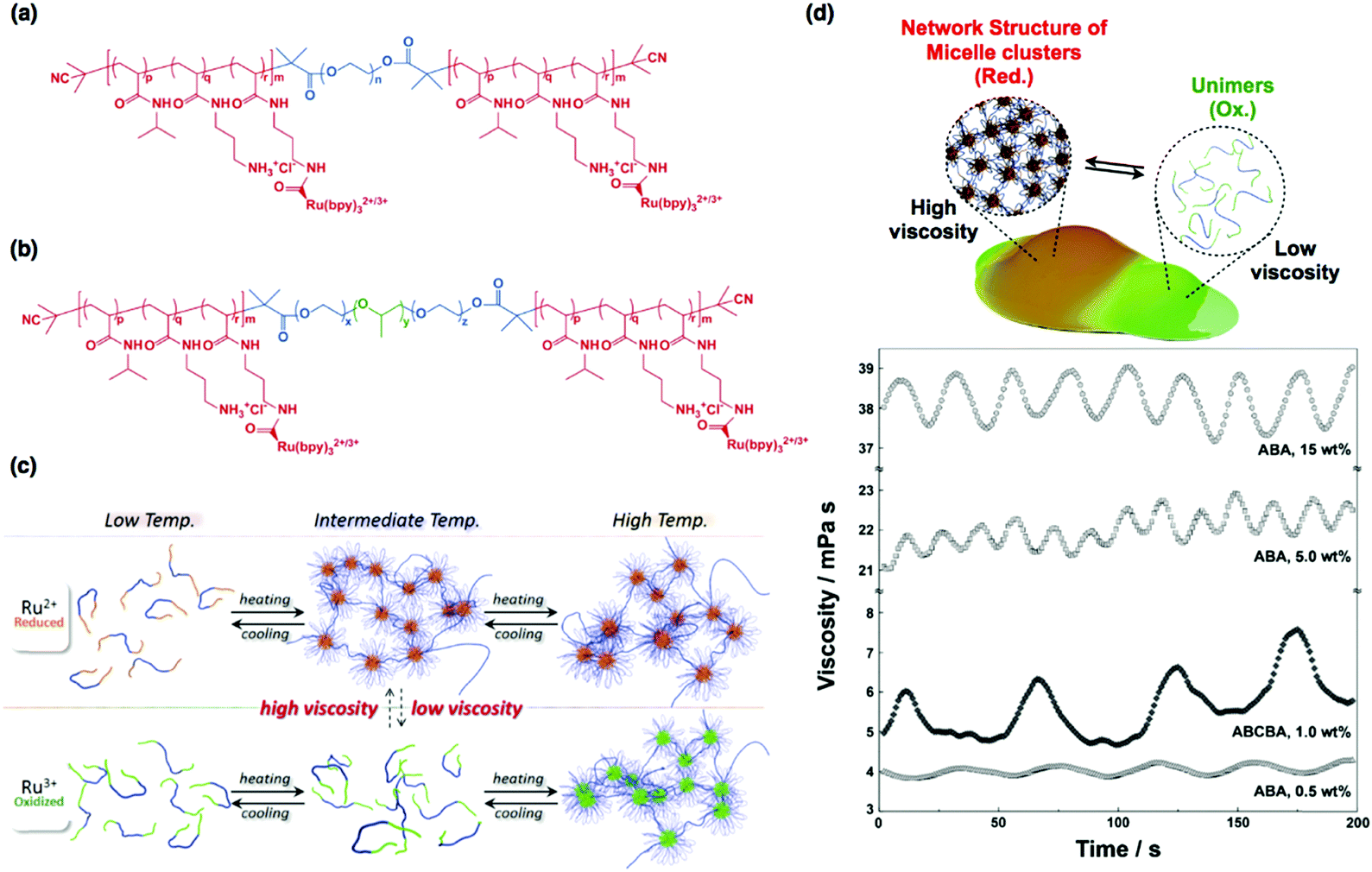 | ||
| Fig. 9 (a) Chemical structures of the ABA triblock copolymer and (b) ABCBA pentablock copolymer. (c) Conceptual illustration of self-oscillating multiblock copolymers. (d) Viscosity oscillations of ABA and ABCBA multiblock copolymers with different concentrations. Reproduced with permission from ref. 69. Nature Publishing Group 2015. | ||
Fig. 9(d) shows the time-evolution of the viscosity of self-oscillating ABA triblock copolymers in a catalyst-free BZ solution at various polymer concentrations. Oscillations in the viscosity are clear, and the amplitude of oscillations increases with increasing polymer concentration. As expected, the amplitude of the viscosity oscillations for the ABA triblock copolymer was much greater than that for AB diblock copolymers,37 implying that the micellar network formed by the ABA triblock copolymers affects the macroscopic physical properties. To further amplify viscosity oscillations, ABCBA pentablock copolymers were synthesized, where the C-segment is hydrophobic poly(propylene oxide) (Fig. 9(b)). Due to the presence of the hydrophobic C-segment, it was anticipated that the micellar structures would be maintained, even in the oxidized state. As a result, two-component micellar networks would be formed in the reduced state via the additional micellar core formed of A-segments. As shown in Fig. 9(d), although the polymer concentration was relatively low (1.0 wt%), the oscillation amplitude of the ABCBA pentablock copolymers was comparable to that of the 15 wt% ABA triblock copolymer solution. However, it was not possible to induce larger oscillation amplitudes by using higher pentablock copolymer concentrations because of the low solubility of the pentablock copolymer in water. Despite this, the results strongly suggest that the introduction of a hydrophobic C-segment can accelerate the assembly and disassembly of the micellar network. Therefore, by optimizing the polymer by controlling the block ratio and molecular weight and by careful monomer selection, sol-to-gel and gel-to-sol oscillations can be realized via the formation and deformation of an elastic micellar network. Such sol-to-gel and gel-to-sol conversions through hierarchical structural changes are characteristic of an amoeba in motion.69 Therefore, this strategy could pave the way to fabricate novel self-propelled autonomous micromachines.
3. Self-oscillating polymeric systems with cross-linking
3.1. Self-oscillating hydrogels
For the design of a self-oscillating hydrogel, a cross-linked copolymer of NIPAAm and Ru(bpy)3 was prepared, where Ru(bpy)3 was covalently linked to the NIPAAm polymer chains (Fig. 10(a)). Similar to a linear polymer of poly(NIPAAm-co-Ru(bpy)3), the poly(NIPAAm-co-Ru(bpy)3) gel has a volume phase transition temperature (VPTT) due to the thermosensitive NIPAAm component. The oxidation of Ru(bpy)3 increased not only the swelling degree of the gel but also the VPTT (Fig. 10(b)). This result may be construed as an increase in the hydrophilicity of the polymer network, which originates from the oxidation of the Ru(bpy)3 moiety. Consequently, the gel exhibits swelling/deswelling changes that are synchronized with the redox changes of the Ru(bpy)3 moiety. If the gel is submerged in a solution containing all of the substrates for the BZ reaction except for the catalyst (malonic acid (MA), sodium bromate (NaBrO3), and nitric acid (HNO3)), the substrates penetrate into the polymer network, and the BZ reaction takes place in the gel. Accordingly, autonomous and periodic mechanical oscillations (swelling/deswelling) of the gel occur over the course of the BZ reaction (Fig. 10(c)).16,73 Consequently, these self-oscillating gels have their own cyclic reaction network, yielding mechanical energy from the chemical energy of the BZ reaction.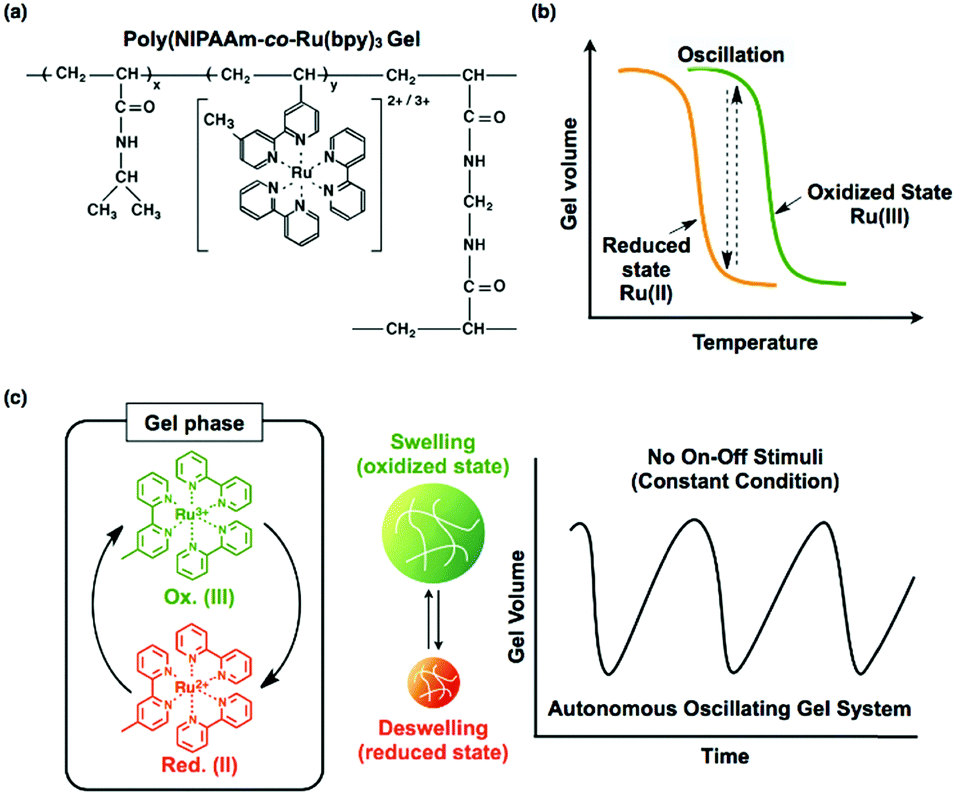 | ||
| Fig. 10 (a) Chemical structure of the poly(NIPAAm-co-Ru(bpy)3) gel. (b) Temperature dependence of the volume of the poly(NIPAAm-co-Ru(bpy)3) gel, where Ru(bpy)3 is either in the reduced or oxidized state. (c) Concept of mechanical self-oscillation of the poly(NIPAAm-co-Ru(bpy)3) gel coupled with the BZ reaction. | ||
As mentioned previously, if the gel is smaller than the wavelength of the BZ chemical wave (typically several millimeters), the gel color should oscillate between orange and green, synchronized with the oscillating redox state of the chromophore. Furthermore, the mechanical swelling/deswelling oscillations of the gel are isotropic, as in the beating of a heart muscle cell, and have the same period as the redox oscillation. The mechanical oscillations (where the gel swells and deswells in the oxidized and reduced states, respectively) coincide well with the chemical oscillations, and there is no phase difference.
In contrast, when the gel size is larger than the wavelength of the chemical wave of the BZ reaction, chemical wave propagation occurs within the gel at a constant rate. Typically, the spatiotemporal patterns of the chemical waves differ depending on the gel shapes.55 For example, Fig. 11(d) shows the chemical wave pattern formed within a one-dimensional cylindrical gel.16 As shown in the time-course images of the cylindrical gel in Fig. 11(d), the green and orange parts of the gel correspond to the oxidized and reduced states of Ru(bpy)3, respectively. During the BZ reactions, chemical waves propagate at a constant rate in the direction of the long axis of the gel. If we consider that the green (Ru(III)) and orange (Ru(II)) regions are the swollen and shrunken parts, respectively, these partially swollen and shrunken parts proceed with the chemical wave, similar to the peristaltic motion of the intestines and worms.
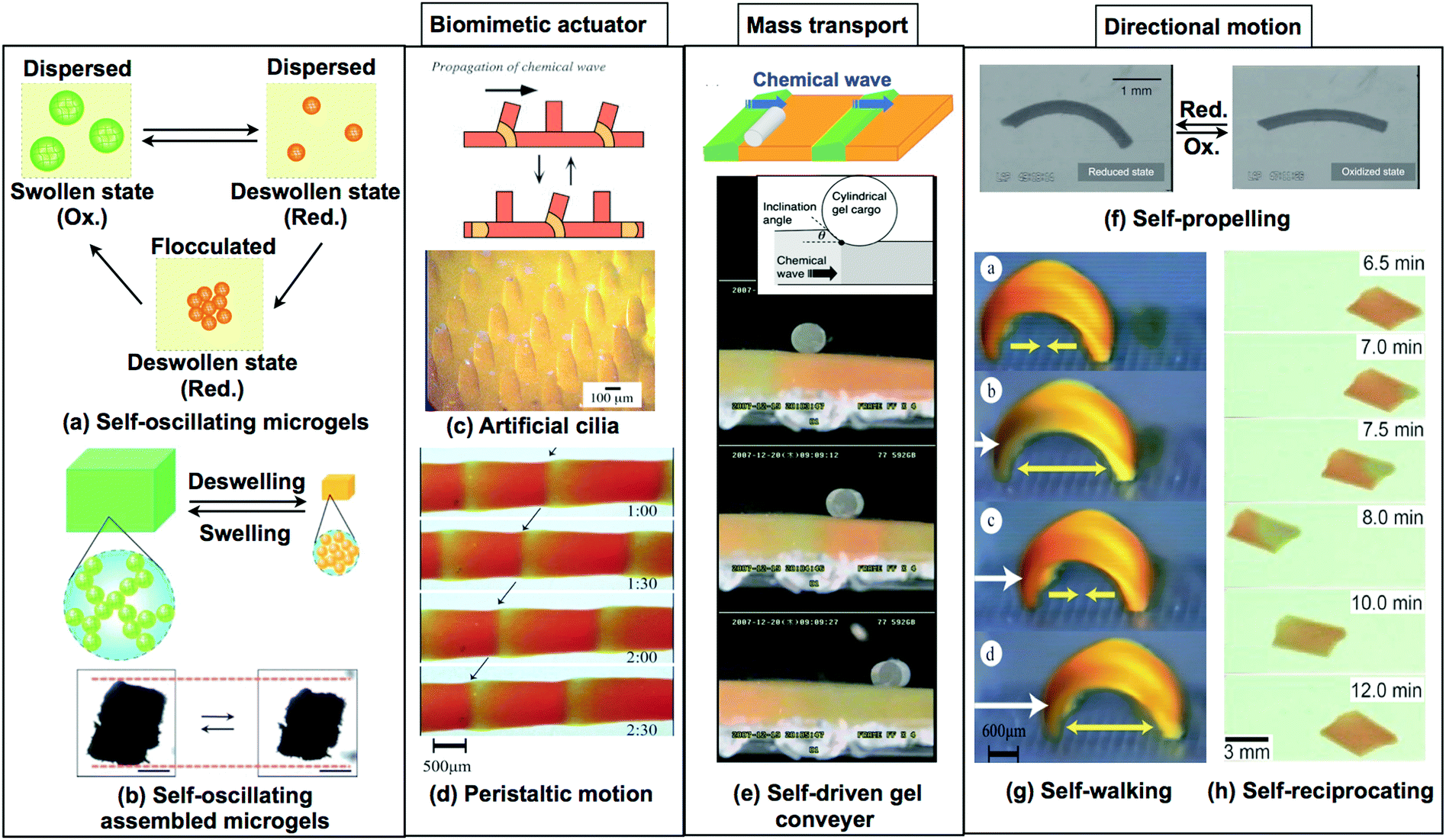 | ||
| Fig. 11 Recent developments in self-oscillating gels as functional materials. (a) Reproduced with permission from ref. 33. American Chemical Society 2009. (b) Reproduced with permission from ref. 76. Royal Society of Chemistry 2012. (c) Reproduced with permission from ref. 87. Wiley 2015. (d) Reproduced with permission from ref. 90. Wiley 2016. (e) Reproduced with permission from ref. 79. Royal Society of Chemistry 2011. (f) Reprinted with permission from ref. 25. Wiley 2007. (g) Reprinted with permission from ref. 80. American Chemical Society 2014. (h) Reprinted with permission from ref. 24. Elsevier 2002. | ||
Recently, autonomous viscosity oscillation in microgel dispersions has been achieved by using self-oscillating microgels.31–33,74,75 These sub-micron sized oscillatory microgels are fabricated using surfactant-free aqueous precipitation polymerization. When the BZ reaction was carried out in the microgels, which were dispersed in an aqueous solution containing all substrates for the BZ reaction except for the catalyst, viscosity oscillations in the dispersion took place in two different ways: a simple pulsatile waveform and a more complicated waveform with two peaks per period. Viscosity is associated with the effective volume fraction of microgels and the type of dispersion (either flocculated or dispersed). Therefore, it has been suggested that the two types of waveforms originate from the differences in the oscillatory modes of the microgels, i.e., swelling/deswelling or dispersing/flocculating oscillations (Fig. 11(a)). The microgels exhibited swelling/deswelling oscillations at a low temperature (20 °C), whereas flocculating–dispersing oscillations were observed at high temperatures (23 °C). These results indicate that temperature can be used to control the type of viscosity oscillation in the microgel dispersion during the BZ reaction.
By taking advantage of the unique characteristics of the self-oscillating microgels, self-oscillating macrogels were fabricated by assembling macrogels from previously prepared microgels. The macrogels showed considerably enhanced mechanical oscillations with larger amplitudes of swelling and deswelling (Fig. 11(b)).76 To prepare the macrogels, microgels were assembled and chemically cross-linked with each other using the reaction between the amino groups at the microgel surface and glutaric dialdehyde. Large-amplitude mechanical oscillations of the macrogel were achieved using the dispersing–flocculating oscillation of the component microgels. The microgels were movable even in the macrogel owing to the limited crosslinking.
By utilizing the peristaltic motion of the macro-sized self-oscillating gels, as shown in Fig. 11(d and e), a novel functional surface for autonomous mass transport was obtained.28,29,77 When a cylindrical poly(acrylamide) (PAAm) gel was placed on the self-oscillating gel surface, it rolled in one direction and so could be transported on the gel surface by the propagating chemical wave, as shown in Fig. 11(e). In addition, the factors determining the mass transport of self-oscillating gels were investigated. A phase diagram of transportable conditions was suggested, defined by the inclination angle θ between the gel surface and the cylindrical PAAm gel and the velocity of the chemical wave front. It was found that the cylindrical PAAm gel was transportable only if the inclination angle was larger than approximately 0.05 rad. The mass transportability did not depend on the velocity of the chemical wave but on the inclination angle and diameter of the cylindrical PAAm gel.
In particular, large-amplitude peristaltic motion was crucial to generate the rotational movement of the cylindrical gel, allowing cylindrical gel transport. Using a model describing the mass transport phenomena based on the Hertz contact theory, the relationship between the transportability and the peristaltic motion of the self-oscillating gel was investigated. In addition, the physically processed gel surface was fabricated for use as a versatile, self-driven gel conveyor that is capable of the unidirectional transport of microparticles, such as gel beads.28 The self-oscillating gel with a grooved surface was prepared by using a PDMS template, and the influence of the surface design on transportability was investigated. It was found that when the groove distance was shorter than the wavelength of the chemical wave, the direction of the peristaltic motion could be restricted to the direction along the grooves, thereby allowing several gel beads to be transported in parallel.
The design concepts of novel biomimetic actuators by using self-oscillating gels have been extensively explored. For example, a self-oscillating gel plate with a micro-sized structural array was fabricated using a micro-molding technique, and this gel exhibited ciliary motion of the microactuator array with spontaneous oscillations (Fig. 11(c)).24,78
Furthermore, control over the motion of self-oscillating gel actuators, which is essential in robotic or microfluidic applications, was also attempted (Fig. 11(f–h)).25,79,80 For example, a self-propelled gel and a self-walking gel were obtained by forming a self-oscillating gel with a hierarchical, heterogeneous structure. A cylindrical self-oscillating gel with a gradient in crosslinking density was fabricated by directionally initiated photopolymerization. This process imparts a gradient in the crosslink density to the gel interior. Due to the heterogeneous structure of the gel, the prepared gel shows rhythmic bending and stretching mechanical oscillations. The propagation of the chemical waves in the bent sample, caused by the BZ reaction, enables the gels to undergo self-propelled motion in the direction of lower crosslinking density (Fig. 11(f)).79 Based on these results, computational simulations of this heterogeneous self-oscillating gel were carried out. These simulations demonstrate the contribution of the bending/stretching oscillating motion to the direction of locomotion of the self-oscillating gel, as well as the influence of the cross-linking density and responsiveness of the gel to the oscillatory behavior. By taking advantage of this asymmetrical bending motion, a new biomimetic self-walking gel actuator was developed (Fig. 11(g)).25 To convert the bending/stretching mechanical oscillations into unidirectional movement, a ratchet mechanism was used. On the ratchet base, repetitive and autonomous bending/stretching motions allowed the gel to move forward, whereas the gel was prevented from sliding back by the teeth of the ratchet. Fig. 11(g) shows the “self-walking” motion of the self-oscillating gel, which acts like a looper.
In addition, a reciprocating motion, similar to that of water-walking insects, has been achieved by utilizing a self-oscillating gel sheet floating in an aqueous solution (Fig. 11(h)).80 The key to the periodic reciprocal movement of the gel is that the contact angle between the liquid/gel interface oscillates, becoming small when Ru(bpy)3 is oxidized and large when reduced. Accordingly, when the chemical wave propagates through the gel, which contains both partially oxidized and reduced regions, the gel accelerates away from the oxidized region. During the slow reduction, the gel reverses its motion and returns to its original position.
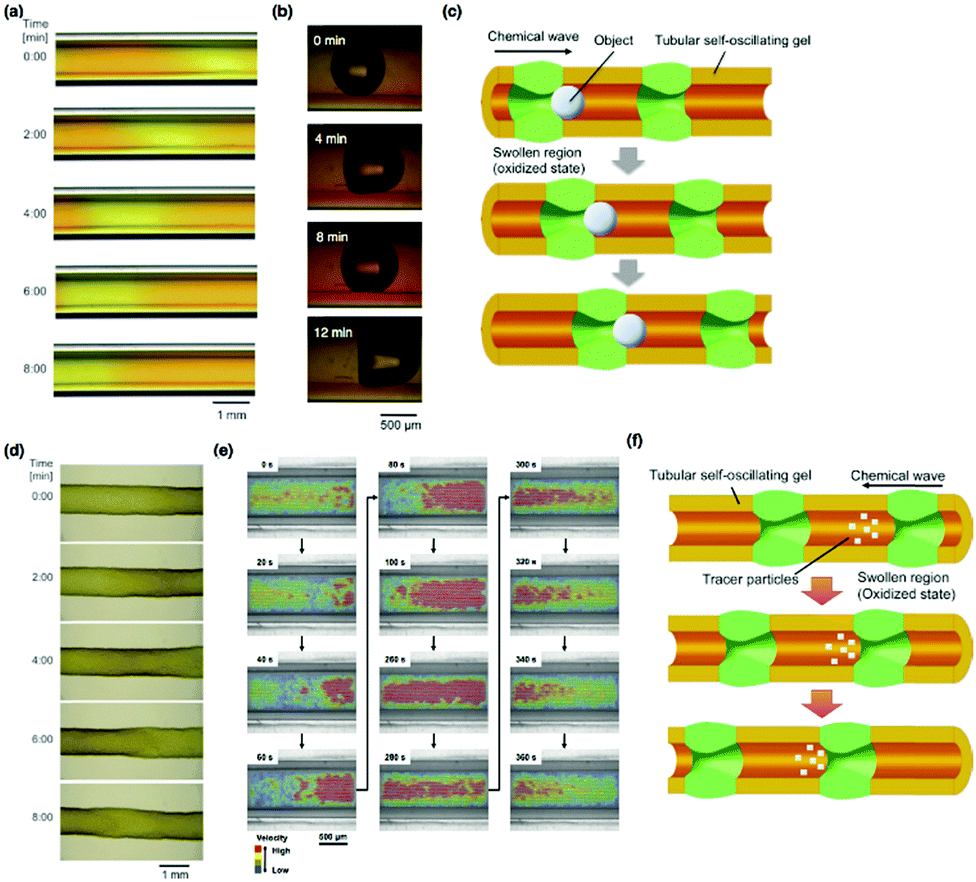 | ||
| Fig. 12 (a) Time-course images of the tubular self-oscillating gel adhered to the inner wall of a glass capillary. (b) The autonomous transport of a CO2 bubble in the self-oscillating tubular gel via peristaltic pumping motion and (c) a schematic illustration of the process. (d) Time-course images of the large peristaltic motion of the free poly(NIPAAm-co-Ru(bpy)3-co-AMPS) tubular gel. Reproduced with permission from ref. 26. Wiley 2012. (e) Distribution of the velocity vector in the tubular self-oscillating gel during the autonomous peristaltic motion. (f) Schematic illustration of autonomous pulsatile flow via the peristaltic motion of a tubular self-oscillating gel. Reproduced with permission from ref. 27. American Chemical Society 2014. | ||
Further, by dissolving the glass capillary using hydrofluoric acid, a free tubular gel that can be freely swollen without mechanical restriction was obtained. To improve both the mechanical strength and swelling degree of the gel, 2-acrylamide-2-methylpropanesulfonate (AMPS) was copolymerized into the polymer network so that a poly(NIPAAm-co-Ru(bpy)3-co-AMPS) tubular gel was obtained. When the free-swelling poly(NIPAAm-co-Ru(bpy)3-co-AMPS) tubular gel was immersed in an aqueous solution containing all the substrates of the BZ reaction except for the catalyst, peristaltic motion with an observably large deformation of the outer diameter of the tubular gel occurred (Fig. 12(d)).
Besides the construction of tubular self-oscillating gels, the analysis of an autonomous fluidic behavior inside the tubular gel was investigated by using latex beads as tracer particles (Fig. 12(e and f)).27 From the comparison between the velocities of the tracer particles and the thickness changes in the gel layer with time, it has been suggested that the velocities of the tracer particles change periodically in an oscillatory manner that is synchronized with the swelling/deswelling mechanical oscillation of the gel layer. The velocity increases during the propagation of the chemical wave and slows down after the wave has passed. The flow velocity of the tracer particle was the highest during the swelling of the tubular gel layer. Fig. 12(e) shows the distribution of the flow velocity vectors inside the tubular gel, where each color represents the relative magnitude of the velocity (red, higher; blue, lower). These results indicate that the velocity of the fluid increased rapidly in the swollen region of the chemical wave. In contrast, the velocity decreased after the wave had passed the swollen region. A pause in the flow or backward flow was observed transiently before the flow velocity increased, as a result of the negative hydrodynamic pressure caused by swelling of the gel. Therefore, this tubular, self-oscillating gel could be used as an autonomous micro-pumping actuator.
We have demonstrated such a design concept by replacing NIPAAm with N,N′-ethylmethylacrylamide (EMAAm).82 Poly(EMAAm) is a thermosensitive polymer exhibiting an LCST at around 56 °C. The VPTT of poly(EMAAm-co-Ru(bpy)3) shifts to higher temperatures in both the reduced and oxidized states. Therefore, a notable swelling/deswelling mechanical oscillation in the poly(EMAAm-co-Ru(bpy)3) gel can be observed even at body temperature (37 °C).
Recently, a copolymer of NIPAAm and NAPMAm has been used to broaden the temperature window of self-oscillating gels.83 At first, a copolymer of NIPAAm and NAPMAm was prepared, and then, post-conjugation of Ru(bpy)3 was carried out by a chemical reaction between the succinimidyl group of the Ru(bpy)3 ester (Ru(bpy)3-NHS) and the amino group of NAPMAm. The resulting polymer is poly(NIPAAm-co-NAPMAm-co-Ru(bpy)3NAPMAm). The amount of Ru(bpy)3 introduced in this two-step method is higher than that in poly(NIPAAm-co-Ru(bpy)3), which is synthesized by the copolymerization of NIPAAm and a vinyl monomer of Ru(bpy)3 in one-step. Moreover, properties such as the hydrophilicity and the LCST of poly(NIPAAm-co-NAPMAm-co-Ru(bpy)3NAPMAm) can be tuned accurately by changing the composition of NIPAAm, NAPMAm, and Ru(bpy)3. In particular, if we left unreacted amino groups in the polymer chain by adding a lower amount of Ru(bpy)3-NHS in the second step, the remaining amino groups could alter the hydrophilicity of the polymer chain. In the case of poly(NIPAAm-co-NAPMAm-co-Ru(bpy)3NAPMAm), the difference in LCSTs between the reduced and oxidized states is about 50 °C, as shown in Fig. 13(b). In contrast, in the case of poly(NIPAAm-co-Ru(bpy)3), the difference in LCSTs between the reduced and oxidized states is about 8 °C, as shown in Fig. 13(d). The larger difference in LCST can be interpreted as a consequence of the greater amount of Ru(bpy)3 in the polymer, as well as an increase in the total hydrophilicity due to the presence of residual amino groups. As the difference in LCST becomes large, aqueous solutions of poly(NIPAAm-co-NAPMAm-co-Ru(bpy)3NAPMAm) show self-oscillation over a wider temperature range, from 15 to 37 °C.
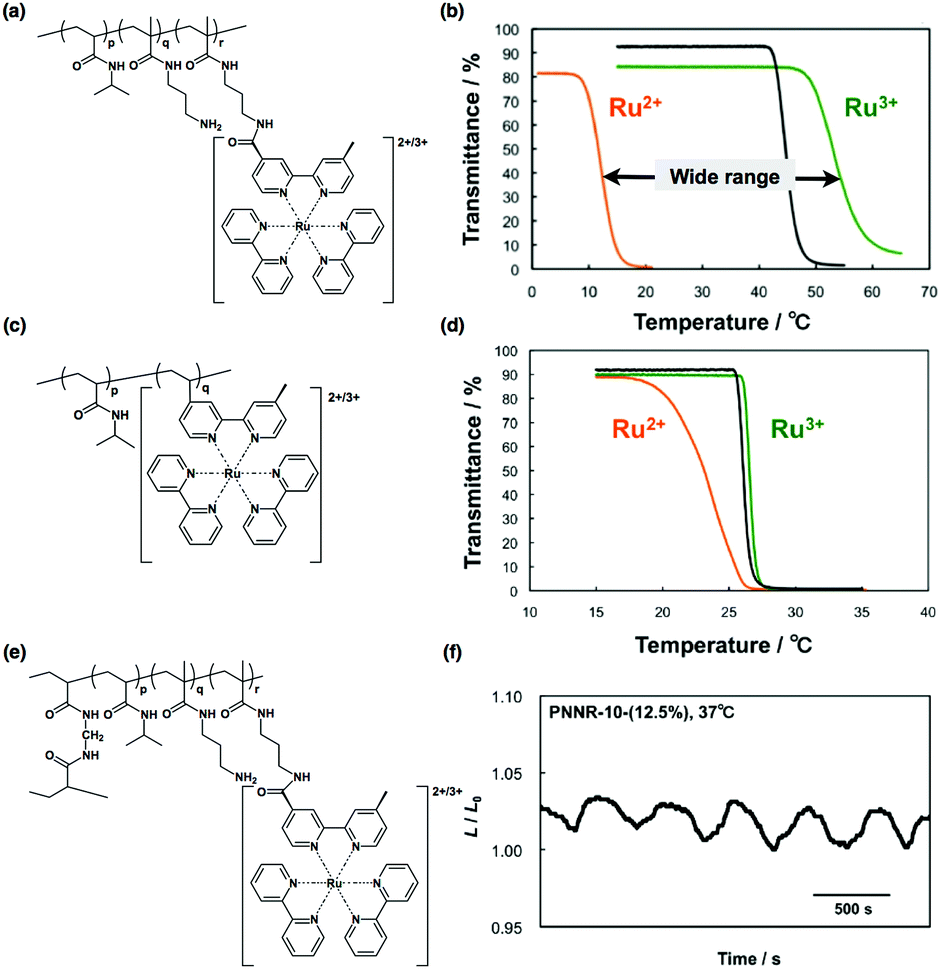 | ||
| Fig. 13 (a) Chemical structure of poly(NIPAAm-co-NAPMAm-co-Ru(bpy)3NAPMAm) synthesized by post-modification of Ru(bpy)3-NHS. (b) Temperature dependence of the optical transmittance for (a) in the oxidized (green line) and reduced (orange line) states and poly(NIPAAm-co-NAPMAm) (black line). (c) Chemical structure of the conventional self-oscillating polymer (poly(NIPAAm-co-Ru(bpy)3)). (d) Temperature dependence of optical transmittance for (c) in the oxidized (green line) and reduced (orange line) states, and poly(NIPAAm) (black line). (e) Chemical structure of cross-linked poly(NIPAAm-co-NAPMAm-co-Ru(bpy)3NAPMAm) gel and (f) its swelling/deswelling oscillating profile at 37 °C. Reproduced with permission from ref. 83. Royal Society of Chemistry 2015. | ||
An additional advantage of the post-conjugation method is that Ru(bpy)3 can be applied to the self-oscillating gel. Consequently, the poly(NIPAAm-co-NAPMAm-co-Ru(bpy)3NAPMAm) gel (Fig. 13(e)) was prepared using this method, and swelling/deswelling mechanical oscillations of the poly(NIPAAm-co-NAPMAm-co-Ru(bpy)3NAPMAm) gel were observed at 37 °C (Fig. 13(f)). At 37 °C, the period of oscillation became shorter owing to the increased temperature, although the amplitude (ca. 3%) was comparable to that of the poly(NIPAAm-co-Ru(bpy)3) gels.
3.2. Cross-linked polymersomes showing self-beating motion
Microscopic structural changes are characteristic of living cells. In particular, oscillatory or pulsatile dynamic behaviors are often observed during the process of morphogenesis,84 cytokinesis,85 and migration.86 The unique oscillatory motion of vesicle membranes similar to living cell membranes based on BCP self-assembly have also been demonstrated by covalently cross-linking the membranes of BCP vesicles (i.e., polymersomes).87 Therefore, we prepared cross-linkable self-oscillating diblock copolymers containing double bonds in the self-oscillating segment (Fig. 14(a)). As described in Section 2.3.1, the diblock copolymer formed vesicles at high temperatures. The cross-linked diblock copolymer vesicles were obtained by UV irradiation of the aqueous solution of the diblock copolymer with photo-initiators at a temperature above the unimer-to-vesicle phase transition temperature. After the cross-linking reaction, the cross-linked vesicles did not dissolve as unimers, even at a low temperature, because of the chemical cross-linking of the membrane. Instead, they were swollen due to the hydration of the membrane. Consequently, both the swelling and deswelling of vesicles are temperature dependent. Furthermore, the temperature dependence of the swelling ratio was also dependent on the redox state of the Ru(bpy)3 incorporated into the vesicle membranes (Fig. 14(b)). Thus, we anticipated that autonomous volume changes would occur following the changes in the redox state of Ru(bpy)3 during the BZ reaction.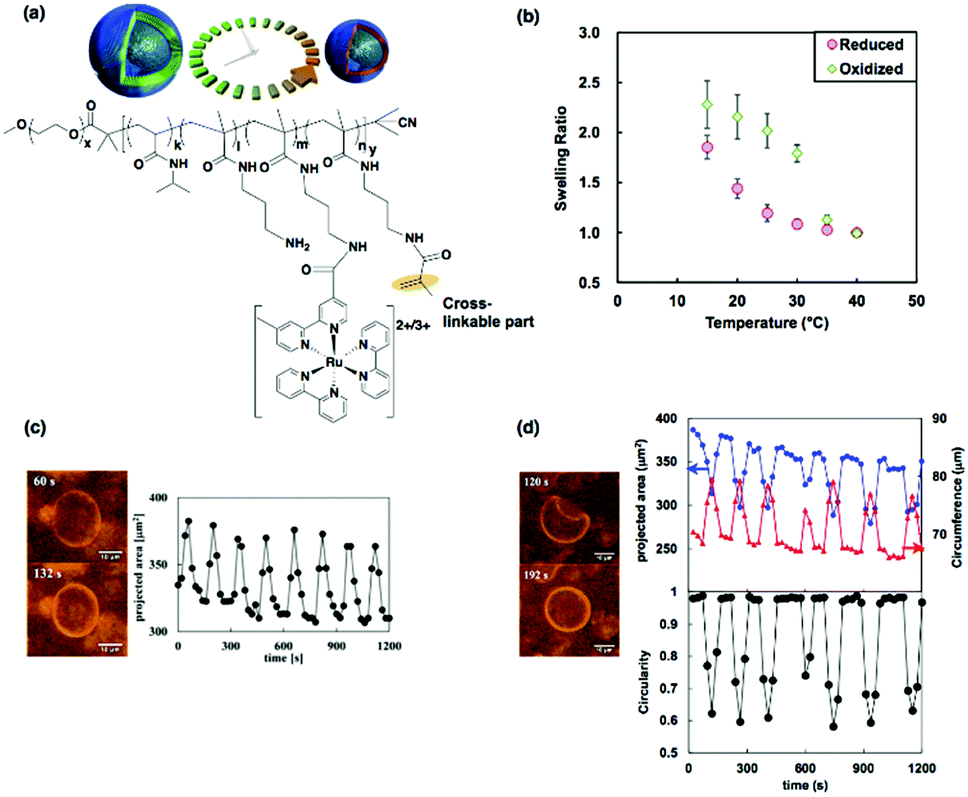 | ||
| Fig. 14 (a) Schematic illustration of the self-oscillations of cross-linked vesicles and chemical structure of the cross-linkable self-oscillating diblock copolymer. (b) Temperature dependences of the equilibrium swelling ratio of the cross-linked vesicles in the reduced and oxidized states. (c) Optical images of the swollen (upper) and deswollen (lower) states of the vesicle and time-variation of the projected area during the volume oscillation. (d) Optical images of the buckled (upper) and unbuckled (lower) states of the vesicle and time-variation of the projected area (upper, blue), circumference (upper, red) and circularity (lower) during the shape oscillation. Reproduced with permission from ref. 87. Wiley 2015. | ||
Microscopic observation of the dynamic behavior of the cross-linked vesicles during the BZ reaction was carried out. The autonomous volume oscillations of cross-linked vesicles are shown in Fig. 14(c). The oscillation was quantified by the change in the projected areas of the vesicles with time. Due to the hollow structure and small sizes of the vesicles, the amplitude of the volume oscillation was larger than that of a conventional self-oscillating gel.16 In addition to the volume oscillations, unique oscillations in the shapes of some cross-linked vesicles were also observed. The images on the left of Fig. 14(d) show the two states observed during the shape oscillation, i.e., the buckled and the unbuckled states. The oscillation profiles of the circumference and the projected area were in the reverse phase (Fig. 14(d), the upper graph). The circularities (a measure of closeness to a circle) of the vesicles were calculated from the circumferences and the projected areas, and the circularity varied in time in a pulsing manner (lower graph in Fig. 14(d)).
The mechanism of the shape oscillations with buckling and unbuckling deformations can be explained as follows: when Ru(bpy)3 in the vesicles is oxidized, membrane hydration occurs, causing tangential stress and increasing the surface area of the vesicles. If the tangential stress is larger than the buckling threshold, buckling deformation occurs, increasing the surface area and reducing the stress. In contrast, when Ru(bpy)3 is reduced, tangential stress is applied in the opposite direction, reducing the surface area. Consequently, recovery from the buckled state to the unbuckled state takes place. As a result, periodic shape changes, including buckling and unbuckling deformations, are driven by the cyclic changes in the redox state of Ru(bpy)3 over the course of the BZ reaction. Self-oscillating cross-linked vesicles showing dynamic structural changes provide an artificial cell model and might be useful to understand the dynamic nature of biological membranes.
3.3. Self-oscillating colloidosomes
In this section, we highlight another strategy to fabricate a dynamic artificial cell model that shows oscillatory behavior. Recently, an increasing number of researchers have investigated the fabrication of hollow microstructures based on templating emulsions stabilized by colloidal particles. Velev et al. first reported the fabrication of hollow structures made from colloidal particles from oil-in-water emulsions.88 Later, Dinsmore et al. introduced the term ‘colloidosomes’ by analogy with liposomes and polymersomes.89 Based on this strategy using the self-assembly of colloidal particles onto water/oil emulsions, which act as hollow particle templates, we have fabricated “self-oscillating colloidosomes” that exhibit unprecedented and complex shape oscillations.90Fig. 15(a) shows a schematic illustration of the fabrication of the self-oscillating colloidosomes. Thermoresponsive poly(NIPAAm-co-NAPMAm) microgels were used to stabilize water-in-1-octanol emulsions. In addition, the aqueous phase contained poly(NIPAAm-r-N-acryloxysuccinimide) as a cross-linker. After the formation of the water-in-oil emulsions, the microgels at the liquid–liquid interface were covalently fixed by cross-linkers through a coupling reaction between the amino groups of the microgel and the succinimide ester groups of the cross-linker. Then, succinimide esters of Ru(bpy)3 were reacted with the residual amino groups of the microgels. Finally, the resulting colloidosomes were transferred to water. Fluorescence from Ru(bpy)32+ in the membrane was observed, clearly indicating that the Ru(bpy)3 catalysts had been successfully introduced into the surfaces of the colloidosomes (Fig. 15(b)).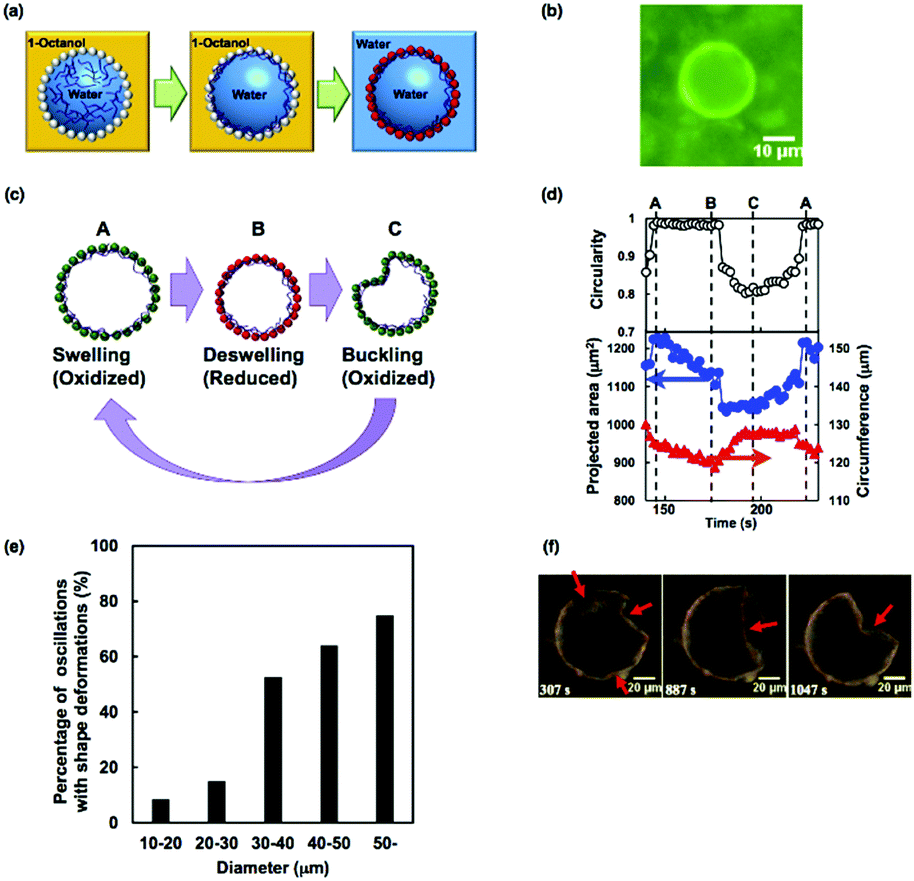 | ||
| Fig. 15 (a) Schematic illustration of the preparation of self-oscillating colloidosomes. (b) Fluorescent image of the colloidosome in water. (c) Schematic illustration of the shape oscillation of the colloidosome. (d) Circularity (upper), projected area (blue, lower) and circumference (red, lower) of the colloidosome during one cycle of the shape oscillations. The times marked A, B, and C correspond to those shown in (c). (e) Occurrence ratio of shape oscillations with buckling for each diameter range of colloidosomes. (f) The complex oscillatory behavior is accompanied by multiple buckling deformations and the movement of buckling points. Reproduced with permission from ref. 90. Wiley 2016. | ||
The autonomous oscillatory behavior of the obtained colloidosomes was closely investigated. During the BZ reaction, the shapes of the colloidosomes undergo buckling deformations, similar to the cross-linked BCP vesicles described in Section 3.2. However, compared to the shape oscillations of the cross-linked BCP vesicles, where oscillations occurred only between the buckled and unbuckled states, the oscillatory profiles of colloidosomes had more complex waveforms, attributed to the existence of three stages of oscillations, i.e., (i) unbuckled and swollen, (ii) unbuckled and deswelled, and (iii) buckled (Fig. 15(c)). This three-stage behavior can be explained by the fast relaxation kinetics of colloidosomes due to their porous structure.91 As a result, relaxation from the buckled state to the unbuckled swollen state occurred in the oxidized state, resulting in complex oscillatory waveforms (Fig. 15(d)).
In addition, it was found that the diameter of the colloidosomes influences their oscillatory behavior. Fig. 15(e) shows the ratio of shape oscillations with the buckling deformations for each range of colloidosome diameters. The shape oscillations were more frequent for the larger diameter colloidosomes. The ratio of the stretching energy (Es) and the bending energy (Eb) of the spherical shells scales as Eb/Es ∼ (h/R)2, where h is the thickness and R is the diameter of the shell.92 In this study, because h was equal to the diameter of the microgels and almost the same for all colloidosomes, the ratio Eb/Es decreased as the colloidosome diameter increased. In consequence, the bending deformations were easier in colloidosomes with larger diameters. Furthermore, other complex oscillatory changes to the colloidosome shape, such as multiple buckling deformations and movement of buckling points, were observed for large colloidosomes (Fig. 15(e)). Interestingly, complex oscillatory shape deformation of cell membranes have been observed in cells with large diameters, for example, fibroblast cells.93
4. Summary and outlook
Self-oscillating polymeric materials are a breakthrough and have emerged as an independent field of intelligent functional material research. In this review, we have introduced the fundamentals, as well as the latest developments in the design of self-oscillating materials. Since the first synthetic concept conjugating a chemical transducer (i.e., a metal catalyst such as Ru(bpy)3) to adaptive polymers was demonstrated, the diversity of polymeric structures has broadened significantly, from linear copolymers to polymer brushes and branched polymers, and, more recently, block copolymers. Each of these self-oscillating systems shows distinctive behavior, such as the autonomous and unidirectional ciliary motion of polymer brushes, the assembly/disassembly of block copolymers, and the association/dissociation of branched polymers. Recent approaches combining self-oscillatory systems with concepts of supramolecular chemistry have yielded unexpected practical applications; for example, the autonomous and cyclic formation/fragmentation of vesicles can be used for the autonomous encapsulation and release of specific agents in the same manner as synaptic vesicles. In addition, we have highlighted recent studies of cross-linked self-oscillating polymeric systems with their behaviors and functions, including self-walking gels, self-propelled gels, self-reciprocating gels, intestine-like tubular gels, mass transportable gels, and multi-deformable cross-linked polymersomes and colloidosomes. Furthermore, we have introduced several approaches to enhance the mechanical performance of these self-oscillating gels, including the preparation of porous macrogels composed of cross-linked microgels and thermally stable gels exhibiting higher VPTTs. These self-oscillating cross-linked polymeric systems could offer versatile practical applications; self-walking motions and peristaltic pumping could be used for autonomous mass transport systems conveying various scale objects. Viscosity oscillations of self-oscillating microgels could be applied as novel autonomous rheological modifiers.To date, studies of self-oscillating polymeric materials at the microscopic and macroscopic scales have progressed independently. In this regard, the hierarchical integration of self-oscillating polymeric units used as microscale building blocks may be an innovative method for the design of novel self-oscillating materials. By coupling building blocks, the concerted motion of each block can yield interesting macroscopic changes. Such hierarchical organization maximizes the advantages of self-oscillating materials by enhancing their intrinsic performance and offers a greater diversity in function.
Although a rational model for self-oscillating materials based on chemomechanics has been proposed, there is still no exact explanation at the molecular level that clarifies the driving forces and decisive parameters causing these autonomous oscillating behaviors. In this regard, it is necessary to understand how materials can, in effect, convert chemical energy into various mechanical actions, such as buckling, swelling/deswelling, and disintegration/reconstruction, by using theoretical and computational investigations. In-depth studies of mechanisms to account for these underlying scientific phenomena may provide guidelines to create tailor-made structural oscillations at the microscopic scale, as well as new types of autonomous mechanical functions at the macroscopic scale.
Investigating other oscillatory chemical reactions for the fabrication of self-oscillating polymeric materials is also a fascinating challenge. Although there are a few reports regarding self-oscillating systems using pH oscillating reactions, they need inflow and outflow of reactants (i.e., a continuous stirred-tank reactor).94–96 We have also reported self-oscillating gels by using calcium oscillations in a batch reactor, but the oscillation had much less stability and smaller amplitude than that with the BZ reaction.97 Very recently, however, several novel oscillatory reactions have been reported using biomolecules such as DNA, RNA and enzymes.98–100 Utilizing these reactions would pave the way to realize unprecedented biocompatible self-oscillating polymeric systems.
We hope that the experimental efforts described and the concepts presented in this review will promote, inspire, and motivate further studies to establish new design strategies for self-oscillating polymeric materials and develop applications for these autonomous functional materials.
Acknowledgements
This work was supported in part by Grants-in-Aid for Scientific Research (No. 15H02198 to R. Y.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan and Research Fellowship of the Japan Society for the Promotion of Science for Young Scientists (No. 15F15044 to Y. S. K. and No. 14J02019 to R. T.).References
- H. E. Huxley, Clin. Orthop. Relat. Res., 2002, 164, S6–S17 CrossRef.
- G. H. Pollack, APBME 2003 – IEEE EMBS Asian-Pacific Conference on Biomedical Engineering 2003, 2003, pp. 14–18 Search PubMed.
- W. Senaratne, L. Andruzzi and C. K. Ober, Biomacromolecules, 2005, 6, 2427–2448 CrossRef CAS PubMed.
- S. J. Jhaveri, M. R. Hynd, N. Dowell-mesfin, J. N. Turner, W. Shain and C. K. Ober, Biomacromolecules, 2009, 10, 174–183 CrossRef CAS PubMed.
- A. S. Hoffman, J. Controlled Release, 2008, 132, 153–163 CrossRef CAS PubMed.
- C. L. Bayer and N. A. Peppas, J. Controlled Release, 2008, 132, 216–221 CrossRef CAS PubMed.
- Y. Osada, H. Okuzaki and H. Hori, Nature, 1992, 355, 242–244 CrossRef CAS.
- R. Yoshida, K. Uchida, Y. Kaneko, K. Sakai, A. Kikuchi, Y. Sakurai and T. Okano, Nature, 1995, 374, 240–242 CrossRef CAS.
- Y. S. Kim, M. Liu, Y. Ishida, Y. Ebina, M. Osada, T. Sasaki, T. Hikima, M. Takata and T. Aida, Nat. Mater., 2015, 14, 1002–1007 CrossRef CAS PubMed.
- R. J. Field, E. Koros and R. M. Noyes, J. Am. Chem. Soc., 1972, 94, 8649–8664 CrossRef CAS.
- R. J. Field and R. M. Noyes, J. Chem. Phys., 1974, 60, 1877–1884 CrossRef CAS.
- I. R. Epstein and J. A. Pojman, An introduction to nonlinear chemical dynamics: Oscillations, Waves, Patterns, and Chaos, Oxford University Press, New York, 1998 Search PubMed.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- R. Yoshida, T. Sakai, S. Ito and T. Yamaguchi, J. Am. Chem. Soc., 2002, 124, 8095–8098 CrossRef CAS PubMed.
- E. Kokufuta and Y. Aman, Polym. Gels Networks, 1997, 5, 439–454 CrossRef CAS.
- R. Yoshida, T. Takahashi, T. Yamaguchi and H. Ichijo, J. Am. Chem. Soc., 1996, 118, 5134–5135 CrossRef CAS.
- R. Yoshida, T. Sakai, Y. Hara, S. Maeda, S. Hashimoto, D. Suzuki and Y. Murase, J. Controlled Release, 2009, 140, 186–193 CrossRef CAS PubMed.
- R. Yoshida, Adv. Mater., 2010, 22, 3463–3483 CrossRef CAS PubMed.
- R. Yoshida, Bull. Chem. Soc. Jpn., 2008, 81, 676–688 CrossRef CAS.
- T. Ueki and R. Yoshida, Phys. Chem. Chem. Phys., 2014, 16, 10388–10397 RSC.
- R. Yoshida, Conf. Proc. IEEE Eng. Med. Biol. Soc., 2013, 318–321 Search PubMed.
- R. Tamate, A. Mizutani Akimoto and R. Yoshida, Chem. Rec., 2016, 16, 1852–1867 CrossRef CAS PubMed.
- R. Yoshida and T. Ueki, NPG Asia Mater., 2014, 6, e107 CrossRef CAS.
- O. Tabata, H. Hirasawa, S. Aoki, R. Yoshida and E. Kokufuta, Sens. Actuators, A, 2002, 95, 234–238 CrossRef CAS.
- S. Maeda, Y. Hara, T. Sakai, R. Yoshida and S. Hashimoto, Adv. Mater., 2007, 19, 3480–3484 CrossRef CAS.
- Y. Shiraki and R. Yoshida, Angew. Chem., Int. Ed., 2012, 51, 6112–6116 CrossRef CAS PubMed.
- Y. Shiraki, A. M. Akimoto, T. Miyata and R. Yoshida, Chem. Mater., 2014, 26, 5441–5443 CrossRef CAS.
- R. Yoshida and Y. Murase, Colloids Surf., B, 2012, 99, 60–66 CrossRef CAS PubMed.
- Y. Murase, S. Maeda, S. Hashimoto and R. Yoshida, Langmuir, 2009, 25, 483–489 CrossRef CAS PubMed.
- T. Masuda, M. Hidaka, Y. Murase, A. M. Akimoto, K. Nagase, T. Okano and R. Yoshida, Angew. Chem., Int. Ed., 2013, 52, 7468–7471 CrossRef CAS PubMed.
- T. Sakai and R. Yoshida, Langmuir, 2004, 20, 1036–1038 CrossRef CAS PubMed.
- H. Taniguchi, D. Suzuki and R. Yoshida, J. Phys. Chem. B, 2010, 114, 2405–2410 CrossRef CAS PubMed.
- D. Suzuki, H. Taniguchi and R. Yoshida, J. Am. Chem. Soc., 2009, 131, 12058–12059 CrossRef CAS PubMed.
- T. Ueki, Y. Takasaki, K. Bundo, T. Ueno, T. Sakai, Y. Akagi and R. Yoshida, Soft Matter, 2014, 10, 1349–1355 RSC.
- T. Ueki, M. Shibayama and R. Yoshida, Chem. Commun., 2013, 49, 6947–6949 RSC.
- T. Ueno, K. Bundo, Y. Akagi, T. Sakai and R. Yoshida, Soft Matter, 2010, 6, 6072–6074 RSC.
- T. Ueki, M. Onoda, R. Tamate, M. Shibayama and R. Yoshida, Chaos, 2015, 25, 064605 CrossRef PubMed.
- R. Tamate, T. Ueki, M. Shibayama and R. Yoshida, Angew. Chem., Int. Ed., 2014, 53, 11248–11252 CrossRef CAS PubMed.
- T. Okano, N. Yamada, H. Sakai and Y. Sakurai, J. Biomed. Mater. Res., 1993, 27, 1243–1251 CrossRef CAS PubMed.
- A. Mizutani, A. Kikuchi, M. Yamato, H. Kanazawa and T. Okano, Biomaterials, 2008, 29, 2073–2081 CrossRef CAS PubMed.
- H. Takahashi, M. Nakayama, M. Yamato and T. Okano, Biomacromolecules, 2010, 11, 1991–1999 CrossRef CAS PubMed.
- K. Nagase, J. Kobayashi, A. Kikuchi, Y. Akiyama, H. Kanazawa and T. Okano, Langmuir, 2011, 27, 10830–10839 CrossRef CAS PubMed.
- N. Idota, A. Kikuchi, J. Kobayashi, K. Sakai and T. Okano, Adv. Mater., 2005, 17, 2723–2727 CrossRef CAS.
- L. Ionov, S. Minko, M. Stamm, J. F. Gohy, R. Jéróme and A. Scholl, J. Am. Chem. Soc., 2003, 125, 8302–8306 CrossRef CAS PubMed.
- I. Lokuge, X. Wang and P. W. Bohn, Langmuir, 2007, 23, 305–311 CrossRef CAS PubMed.
- J. A. Howarter and J. P. Youngblood, Macromol. Rapid Commun., 2008, 29, 455–466 CrossRef CAS.
- D. Xiao and M. J. Wirth, Macromolecules, 2002, 35, 2919–2925 CrossRef CAS.
- B. Zhao and W. J. Brittain, Prog. Polym. Sci., 2000, 25, 677–710 CrossRef CAS.
- H. Tu, C. E. Heitzman and P. V. Braun, Langmuir, 2004, 20, 8313–8320 CrossRef CAS PubMed.
- D. Bontempo, N. Tirelli, K. Feldman, G. Masci, V. Crescenzi and J. A. Hubbell, Adv. Mater., 2002, 14, 1239–1241 CrossRef CAS.
- T. Nitta and H. Hess, Nano Lett., 2005, 5, 1337–1342 CrossRef CAS PubMed.
- Y. Sumino, N. Magome, T. Hamada and K. Yoshikawa, Phys. Rev. Lett., 2005, 94, 068301 CrossRef PubMed.
- T. Masuda, A. M. Akimoto, K. Nagase, T. Okano and R. Yoshida, Chem. Mater., 2015, 27, 7395–7402 CrossRef CAS.
- S. Tateyama, Y. Shibuta and R. Yoshida, J. Phys. Chem. B, 2008, 112, 1777–1782 CrossRef CAS PubMed.
- P. Yuan, O. Kuksenok, D. E. Gross, A. C. Balazs, J. S. Moore and R. G. Nuzzo, Soft Matter, 2013, 1231–1243 RSC.
- T. Masuda, A. M. Akimoto, K. Nagase, T. Okano and R. Yoshida, Sci. Adv., 2016, 2, e1600902 Search PubMed.
- U. S. Schubert, H. Hofmeier and G. R. Newkome, Modern Terpyridine Chemistry, Wiley-VCH, Weinheim, 2006 Search PubMed.
- B. G. G. Lohmeijer and U. S. Schubert, Angew. Chem., Int. Ed., 2002, 41, 3825–3829 CrossRef CAS.
- V. V. Yashin, O. Kuksenok and A. C. Balazs, J. Phys. Chem. B, 2010, 114, 6316–6322 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- M. W. Matsen and F. S. Bates, Macromolecules, 1996, 29, 1091–1098 CrossRef CAS.
- A. K. Khandpur, S. Foerster, F. S. Bates, I. W. Hamley, A. J. Ryan, W. Bras, K. Almdal and K. Mortensen, Macromolecules, 1995, 28, 8796–8806 CrossRef CAS.
- N. S. Cameron, M. K. Corbierre and A. Eisenberg, Can. J. Chem., 1999, 77, 1311–1326 CrossRef CAS.
- P. Lim Soo and A. Eisenberg, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 923–938 CrossRef.
- S. Jain and F. S. Bates, Science, 2003, 300, 460–464 CrossRef CAS PubMed.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- F. S. Bates, M. A. Hillmyer, T. P. Lodge, C. M. Bates, K. T. Delaney and G. H. Fredrickson, Science, 2012, 336, 434–440 CrossRef CAS PubMed.
- M. C. Cross and P. C. Hohenberg, Rev. Mod. Phys., 1993, 65, 851–1112 CrossRef CAS.
- M. Onoda, T. Ueki, M. Shibayama and R. Yoshida, Sci. Rep., 2015, 5, 15792 CrossRef PubMed.
- C. Li, Y. Tang, S. P. Armes, C. J. Morris, S. F. Rose, A. W. Lloyd and A. L. Lewis, Biomacromolecules, 2005, 6, 994–999 CrossRef CAS PubMed.
- T. G. O’Lenick, N. Jin, J. W. Woodcock and B. Zhao, J. Phys. Chem. B, 2011, 115, 2870–2881 CrossRef PubMed.
- C. Tsitsilianis, Soft Matter, 2010, 6, 2372–2388 RSC.
- R. Yoshida, T. Takahashi, T. Yamaguchi and H. Ichijo, Adv. Mater., 1997, 9, 175–178 CrossRef CAS.
- D. Suzuki, T. Sakai and R. Yoshida, Angew. Chem., Int. Ed., 2008, 47, 917–920 CrossRef CAS PubMed.
- D. Suzuki and R. Yoshida, Macromolecules, 2008, 41, 5830–5838 CrossRef CAS.
- D. Suzuki, T. Kobayashi, R. Yoshida and T. Hirai, Soft Matter, 2012, 8, 11447–11449 RSC.
- Y. Murase, R. Takeshima and R. Yoshida, Macromol. Biosci., 2011, 11, 1713–1721 CrossRef CAS PubMed.
- O. Tabata, H. Kojima, T. Kasatani, Y. Isono and R. Yoshida, Proceedings of the International Conference on MEMS 2003, 2003, pp. 12–15 Search PubMed.
- O. Kuksenok, V. V. Yashin, M. Kinoshita, T. Sakai, R. Yoshida and A. C. Balazs, J. Mater. Chem., 2011, 21, 8360–8371 RSC.
- S. Nakata, M. Yoshii, S. Suzuki and R. Yoshida, Langmuir, 2014, 30, 517–521 CrossRef CAS PubMed.
- P. Ruoff, Physica D, 1995, 84, 204–211 CrossRef CAS.
- M. Hidaka and R. Yoshida, J. Controlled Release, 2011, 150, 171–176 CrossRef CAS PubMed.
- T. Masuda, A. Terasaki, A. M. Akimoto, K. Nagase, T. Okano and R. Yoshida, RSC Adv., 2015, 5, 5781–5787 RSC.
- A. C. Martin, M. Kaschube and E. F. Wieschaus, Nature, 2009, 457, 495–499 CrossRef CAS PubMed.
- J. Sedzinski, M. Biro, A. Oswald, J. Y. Tinevez, G. Salbreux and E. Paluch, Nature, 2011, 476, 462–466 CrossRef CAS PubMed.
- G. Giannone, B. J. Dubin-Thaler, H. G. Döbereiner, N. Kieffer, A. R. Bresnick and M. P. Sheetz, Cell, 2004, 116, 431–443 CrossRef CAS PubMed.
- R. Tamate, T. Ueki and R. Yoshida, Adv. Mater., 2015, 27, 837–842 CrossRef CAS PubMed.
- O. D. Velev, K. Furusawa and K. Nagayama, Langmuir, 1996, 12, 2374–2384 CrossRef CAS.
- A. D. Dinsmore, M. F. Hsu, M. G. Nikolaides, M. Marquez, A. R. Bausch and D. A. Weitz, Science, 2002, 298, 1006–1009 CrossRef CAS PubMed.
- R. Tamate, T. Ueki and R. Yoshida, Angew. Chem., Int. Ed., 2016, 55, 5179–5183 CrossRef CAS PubMed.
- I. Tokarev and S. Minko, Adv. Mater., 2010, 22, 3446–3462 CrossRef CAS PubMed.
- L. D. Laudau and E. M. Lifshitz, Theory of Elasticity, Butterworth-Heinemann, Oxford, 1986 Search PubMed.
- G. Salbreux, J. F. Joanny, J. Prost and P. Pullarkat, Phys. Biol., 2007, 4, 268–284 CrossRef CAS PubMed.
- R. Yoshida, H. Ichijo, T. Hakuta and T. Yamaguchi, Macromol. Rapid Commun., 1995, 16, 305–310 CrossRef CAS.
- I. Lagzi, D. Wang, B. Kowalczyk and B. A. Grzybowski, Langmuir, 2010, 26, 13770–13772 CrossRef CAS PubMed.
- G. Wang, B. Tang, Y. Liu, Q. Gao, Z. Wang and X. Zhang, Chem. Sci., 2016, 7, 1151–1155 RSC.
- R. Yoshida and Y. Uesusuki, Biomacromolecules, 2005, 6, 2923–2926 CrossRef CAS PubMed.
- J. Kim and E. Winfree, Mol. Syst. Biol., 2011, 7, 465 CrossRef PubMed.
- K. Montagne, R. Plasson, Y. Sakai, T. Fujii and Y. Rondelez, Mol. Syst. Biol., 2011, 7, 466 CrossRef PubMed.
- S. N. Semenov, A. S. Y. Wong, R. M. van der Made, S. G. J. Postma, J. Groen, H. W. H. van Roekel, T. F. A. de Greef and W. T. S. Huck, Nat. Chem., 2015, 7, 160–165 CrossRef CAS PubMed.
Footnote |
| † These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |