
Surface proteomics on nanoparticles: a step to simplify the rapid prototyping of nanoparticles†
J.
Kuruvilla
a,
A. P.
Farinha
a,
N.
Bayat
b and
S.
Cristobal
 *ac
*ac
aDepartment of Clinical and Experimental Medicine, Cell Biology, Faculty of Medicine, Linköping University, Linköping, Sweden. E-mail: Susana.Cristobal@liu.se
bDepartment of Biochemistry and Biophysics, Arrhenius Laboratories, Stockholm University, Stockholm, Sweden
cIKERBASQUE, Basque Foundation for Science, Department of Physiology, Faculty of Medicine and Dentistry, University of the Basque Country, Leioa, Spain
First published on 21st October 2016
Abstract
Engineered nanoparticles for biomedical applications require increasing effectiveness in targeting specific cells while preserving non-target cells' safety. We developed a surface proteomics method for a rapid and systematic analysis of the interphase between the nanoparticle protein corona and the targeted cells that could implement the rapid prototyping of nanomedicines. Native nanoparticles entering in a protein-rich liquid medium quickly form a macromolecular structure called protein corona. This protein structure defines the physical interaction between nanoparticles and target cells. The surface proteins compose the first line of interaction between this macromolecular structure and the cell surface of a target cell. We demonstrated that SUSTU (SUrface proteomics, Safety, Targeting, Uptake) provides a qualitative and quantitative analysis from the protein corona surface. With SUSTU, the spatial dynamics of the protein corona surface can be studied. Data from SUSTU would ascertain the nanoparticle functionalized groups exposed at a destiny that could circumvent preliminary in vitro experiments. Therefore, this method could implement in the analysis of nanoparticle targeting and uptake capability and could be integrated into a rapid prototyping strategy which is a major challenge in nanomaterials science. Data are available via ProteomeXchange with the identifier PXD004636.
 J. Kuruvilla | Jacob Kuruvilla is a PhD student at Cristobal's lab at Linköping University, Sweden at the Department of Clinical and Experimental Medicine. He has finished his masters in molecular genetics and physiology from Linköping University, Sweden. His research projects are mainly focused on studying the effects and behavior of nanoparticles on humans and the environment by proteomics. He has also been using proteomics as a tool for monitoring and assessment of pollution in water bodies. |
 A. P. Farinha | Ana Paula Farinha received her PhD in molecular biology in 2010 at ITQB-Universidade Nova Lisboa, Portugal. She held a post-doctoral position in plant proteomics at IBET-Oeiras, Portugal. She was a postdoctoral researcher at Cristobal's lab at Linköping University, Sweden at the Department of Clinical and Experimental Medicine. Her main research interest is related with cellular responses to changing environmental conditions and more recently to nanoparticle impact in cells applying proteomics. |
 N. Bayat | Narges Bayat received her MS in nanobiotechnology engineering from the Royal Institute of Science and Technology, Sweden. She obtained her PhD in biochemistry at Cristobal's lab at Stockholm University, Department of Biochemistry and Biophysics in 2015. She is currently a postdoctoral researcher at the Children's Cancer Institute in Sydney, Australia. Her main research interest is nanomedicine for diagnostics and targeted drug delivery for cancer therapy. |
 S. Cristobal | Susana Cristobal received her PhD in biochemistry in 1997. She is a professor since 2012 at Linköping University, Sweden at the Department of Clinical and Experimental Medicine. Her fundamental research concept is based on integration of comprehensive and cost-efficient methodologies to evaluate the molecular and cellular response of nanomaterials, pollutants or small molecules on human health and the environment. She has been a pioneer in developing a proteomics-based method for marine pollution assessment. Her more recent interest is the development of a proteomics-based platform for rapid prototyping of nanodrug, nanotoxicology and metaproteomics in toxicity testing and environmental assessment. |
Conceptual insightsThe design and development of nanoparticles for nanomedicine is hampered by the lack of methods that could both evaluate the new material's potential value and provide information for further refinement. To simplify the functional characterization of nanoparticles we have focused on the ultimate line of interaction between the nanoparticle and the target cell, i.e. the surface of the protein corona. In this study we introduce SUSTU, a surface proteomics method applicable to the surface of nanoparticles after interaction with any biological media. We demonstrate that the application of this single analytical technique can deliver the minimal nanoparticle hallmarks for its functional characterization. Moreover, the comparison of SUSTU with classical quantitative analysis of the entire protein corona demonstrates that the surface of the protein corona differs from the entire protein corona. Considering the fact that by analysing only the surface of the protein corona the interaction between nanoparticles and destiny cells can be evaluated, this method could be integrated in a platform for rapid prototyping of nanoparticles, assisting nano-drug development and supporting the future development of precision medicine. |
Introduction
Nanotechnology has demonstrated the myriad of applications that nanoparticles (NPs) synthesized with specific properties can offer. The challenge of nanomaterials science is no longer to synthesize complex materials at the nano-size level, rather it is to develop methodologies for their functional characterization. In the context of NPs for nanomedicine, the main problem is that many novel formulations should be evaluated to confirm any improvement in their function compared to previous macro-sized compounds. The current testing methodologies for novel NPs are focused on NP synthesis and characterization combined with classical in vitro toxicology assays in a case-by-case basis.1–5 However, the initial NP functional evaluation should only ascertain or predict whether or not the NP administered in vivo can reach the action site. In addition, it would be required to confirm the cell's capability to uptake that NP, and whether the novel compound would accomplish the desired function at the target point. Any new method should also discern whether the NP could maintain its intrinsic particle properties during transit from the initial interaction with a biological fluid to the final destiny and moreover anticipate perilous effects for the target microenvironment or the surrounding site.Engineered NPs entering in a biological fluid are coated by biomolecules present in its vicinity forming a protein corona (PC).6–8 This protein-rich structure defines the physical interaction between NPs and target cells.9,10 This step transforms the nanostructure into a supramolecular structure that is biologically relevant for interactions and impact in cellular activities.11 In this work, we introduce a new method called SUSTU (SUrface proteomics, Safety, Targeting, and Uptake) that is based on the classical strategies to analyse the cell surface by proteomics. It is a proteomics-based method to analyse the most exposed proteins, accessible to biotinylated labelling, that are attached to a NP surface. This protein interphase could provide the key information required for a rapid prediction and evaluation of the potential value of a newly designed NP for nanomedicine or other targeted applications, where it is essential to predict the interaction between NPs and surfaces as well as cellular membranes.
Methods
NP characterization
The hydrodynamic size, polydispersity index (PDI) and zeta potential of TiO2-NPs rutile (10 nm) (4023, Nanograde LIc, Zurich, Switzerland), TiO2-NPs (30 nm) (4025, Nanograde LIc, Zurich, Switzerland) were characterized in suspension. The measurements were performed in triplicate at 500 mg l−1 in the exposure medium at 37 °C. The measurements were assessed with a Malvern Zetasizer Nano series V5.03 (PSS0012-16 Malvern Instruments, Worcestershire, UK) and the analysis program dispersion technology software, (Malvern Instruments). Before each experiment the NPs were suspended in appropriate media and sonicated using a sonicator bath (1510 Branson) at room temperature for 30 min at 40 kHz and vortexed vigorously to avoid NP aggregation. The surface area of the TiO2-NPs was measured at the Department of Materials and Environmental Chemistry at Stockholm University, Stockholm, Sweden. Nitrogen adsorption–desorption isotherms were recorded at the liquid nitrogen temperature of 77 K using a Micromeritics ASAP2020 volumetric adsorption analyser. The NP samples were treated under near-vacuum conditions (<10−5 Torr) at 200 °C for 10 h. The specific surface areas of the materials were calculated from the recorded data according to the BET method in the range of relative pressures of 0.05–0.15.Preparation of titanium dioxide nanoparticle protein corona (TiO2-NP-PC)
TiO2-NPs of 10 nm (anatase) and 30 nm (anatase/rutile) were purchased from Nanograde LIc (Zurich, Switzerland, #4023 and #4025, respectively). An initial suspension of 1 mg ml−1 nanoparticles was prepared in a glass vial with phosphate buffered saline (PBS) and 1 mM EDTA, and further dispersed by sonication for 30 min in a water bath sonicator (1510 Branson). Dispersed TiO2-NPs were introduced into a MCDB 131 cell culture medium (Gibco, Life Technologies, #10372019) supplemented with 10% heat inactivated fetal bovine serum (FBS) (Gibco, Life Technologies) in order to obtain a final concentration of 100 μg ml−1 in 12 ml. This NP-cell culture medium solution was prepared in 15 ml Falcon tubes and incubated at 37 °C for an hour in a Thermomixer (Eppendorf Thermomixer C) at 550 rpm. The TiO2-NP-PC complex was sedimented by centrifugation at 2700 × g for 5 min at 4 °C. The TiO2-NP-PC pellet was washed 3 times with PBS and 1 mM EDTA plus protease and phosphatase inhibitors (Pierce, #88668) to remove loosely bound proteins. The first wash was performed by re-suspending the TiO2-NP-PC pellet in 1 ml PBS solution using a vortex; thereafter 250 μl PBS solution was used. During the last wash, TiO2-NP-PC complexes were transferred to Eppendorf tubes for further analysis. For labelling assays, samples were re-suspended in PBS and prepared in triplicate plus an additional one to estimate the protein concentration. For this purpose, proteins adhering to TiO2-NPs were solubilized with 2% SDS and further diluted to 0.1% in order to be compatible with the Bradford assays. In the case of unlabelled TiO2-NP-PC, samples were prepared in duplicate and proteins were solubilized in 2% SDS and 100 mM DTT and submitted to Filter Aided Sample Preparation (FASP).12 The protein concentration was estimated by the Bradford (Bio-Rad) microassay using BSA as the standard.13 The selection of main parameters that would affect the formation of PC including NP size, type of media, concentration of serum, incubation time, and concentration of NPs in the medium was evaluated in preliminary experiments not included here as they have been extensively reported in the literature.14Labelling of TiO2-NP-PC with EZ-Link TM Sulfo-NHS-SS Biotin
A stock solution of 10 mM EZ-Link TM Sulfo-NHS-SS Biotin (Thermo Scientific) was prepared immediately before use by dissolving 2.2 mg in 360 μl ultrapure water. This type of biotin is water soluble and includes a cleavable disulphide bond allowing the biotin label to be removed in the presence of reducing agents like DTT. The TiO2-NP PC complex was re-suspended in 50 μl PBS and labelled with 0.4 mM Sulfo-NHS-SS Biotin for 3 h at 4 °C on a Thermomixer (Eppendorf) at 1400 rpm. Excess biotin was removed by washing 3 times with 0.5 ml of 50 mM ammonium bicarbonate buffer (ABC) and later re-suspended after centrifugation at 2700 × g for 5 min. After the last wash, the PC biotinylated surface was re-suspended in 50 μl of 50 mM ABC buffer and digested with 0.1 μg μl−1 trypsin (recombinant, proteomics grade, Roche # 03708985001) at an enzyme to protein ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20. This was performed overnight at 37 °C, using a rotator at the lowest speed. On the next day, half of the amount of trypsin was added to complete the digestion for an additional 3 h at 40 °C. The biotinylated tryptic peptides were centrifuged for 15 min at 16000 × g and 18 °C, and the supernatant, free of NPs and undigested proteins, was transferred into new Eppendorf tubes. The remaining pellet was washed twice with 50 μl of 50 mM ABC buffer and later the 2 supernatants were pooled. A final centrifugation step was performed to completely clarify the supernatant. Peptides were quantified by measuring the optical absorbance of tryptophan/tyrosine residues at 280 nm using the NanoDrop 2000 UV-Vis spectrophotometer (Thermo Scientific).15,16
:20. This was performed overnight at 37 °C, using a rotator at the lowest speed. On the next day, half of the amount of trypsin was added to complete the digestion for an additional 3 h at 40 °C. The biotinylated tryptic peptides were centrifuged for 15 min at 16000 × g and 18 °C, and the supernatant, free of NPs and undigested proteins, was transferred into new Eppendorf tubes. The remaining pellet was washed twice with 50 μl of 50 mM ABC buffer and later the 2 supernatants were pooled. A final centrifugation step was performed to completely clarify the supernatant. Peptides were quantified by measuring the optical absorbance of tryptophan/tyrosine residues at 280 nm using the NanoDrop 2000 UV-Vis spectrophotometer (Thermo Scientific).15,16
Sample preparation of the TiO2-NP-PC surface
All reagents (NeutrAvidin agarose resin, purification columns, PBS and DTT) except guanidine–HCl (Sigma) were from Thermo Scientific, upon purchasing the Pierce Cell Surface Protein Isolation Kit (# 89881). Reagents were brought to room temperature and NeutrAvidin agarose resin was added to pack the columns (100 μl of the 50% slurry was equivalent to 50 μl settled resin; this amount was suitable to purify approximately 50 μg of the biotinylated peptides). The columns were centrifuged at 500 × g for 1 min to remove the storage solution, washed 3 times with 200 μl binding buffer, phosphate buffered saline (PBS) and placed into new collection tubes to proceed for purification of biotinylated peptides. The biotinylated sample was incubated with the resin for 30 min at room temperature, followed by washing four times with 200 μl PBS. The biotinylated peptides were eluted by adding 50 μl 8 M guanidine–HCl pH 1.5 and 50 mM DTT after centrifuging at 500 × g for 1 min. This step was repeated 3 additional times and the fractions were combined. Desalting and concentration of the samples were achieved via reversed-phase extraction using C18 Top Tips (TT2C18.96, Glygen, Columbia, MD, USA) following the manufacturer's instructions. The peptide concentration was estimated by measuring the optical absorbance at 280 nm using a NanoDrop 2000 UV-Vis spectrophotometer (Thermo Scientific). The desalted peptides were concentrated to dryness in a Speedvac concentrator (Savant SPD 1010) and stored at −20 °C until further analysis by mass spectrometry (MS).Control sample preparation of TiO2-NP-PC
Protein samples were prepared according to Wiśniewski et al. (2009).12 This method uses a standard filter device to perform in-solution protein reduction, alkylation and digestion, enabling simultaneously the exchange of SDS by urea buffer. After solubilisation of the proteins adsorbed onto the TiO2-NPs with 2% SDS, 100 mM DTT and 100 mM Tris-HCl pH 8.0, the protein samples (30 μg) were alkylated and digested in a solution with 0.1 μg μl−1 trypsin (recombinant, proteomics grade, Roche # 03708985001) at an enzyme to protein ratio of 1:100 for 16 h at 37 °C. Digestion was performed in a 30 kDa Microcon centrifugal filter unit (Merck Millipore, # MRCPRT030). Peptides were recovered, acidified with formic acid (FA), desalted and concentrated via reversed phase extraction (C18 Top-Tips, TT2C18, Glygen, Columbia, MD, USA) using acetonitrile (CAN) (70%, v/v) with FA (0.1%, v/v) for elution. Before nano LC-MS/MS, the peptides were dried and recovered in 0.1% FA and the peptide concentration was adjusted to the required concentration.
Liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) analysis
Biotinylated and non-biotinylated peptides were reconstituted in 0.1% v/v FA and the peptide concentration was adjusted to the instrument required concentrations. Peptides were analysed via reversed-phase nano liquid chromatography (nLC) coupled online to a hybrid ion trap mass spectrometer (LTQ Orbitrap Velos Pro-ETD, Thermo Scientific). Samples were separated using an EASY nLC II system (Thermo Scientific) and tryptic peptides were injected into a pre-column (NS-MP-10 BioSphere C18, 5 μm particle size, 120 Å pore size, 100 μm inner diameter × 20 mm length; NanoSeparations, the Netherlands) and subsequently separated on the analytical column (NS-AC-10 BioSphere C18, 5 μm particle size, 120 Å pore size, 75 μm × 100 mm; NanoSeparations, the Netherlands). A linear gradient of 100% buffer A (water, 0.1% FA) and 2% buffer B (0.1% FA in 100% acetonitrile) to 100% buffer B was used for peptide separation over 90 min at a constant flow rate of 300 nl min−1. Biotinylated samples were run in triplicate and non-biotinylated ones in duplicate. Full scan MS spectra were recorded in the positive mode electrospray ionization with an ion spray voltage power frequency (pf) of 2.4 kilovolt (kV), a radio frequency (RF) lens voltage of 69 and a capillary temperature of 235 °C. This was acquired over an m/z of 380–2000 Da at a resolution of 30000 and the top 20 most intense multiply charged ions were selected for MS/MS under an isolation width of 2 amu, an activation time of 10 milliseconds and a Q value of 0.25. Collision induced dissociation (CID) was performed for fragmentation with a normalized collision energy of 30%. The selected masses were included in a dynamic exclusion list for 60 s with a repeat duration of 30 s and a threshold of more than 500 counts. Charge state filtering that disqualified singly charged peptides was activated and the predictive automatic gain control was enabled. The centroid mode was used for CID MS/MS.
Protein identification and data analysis
Raw MS files were searched against the Bovine UniProt database (23848 entries; 23901 total entries with contaminants) using Sequest as the search engine (Thermo Fisher Scientific, San Jose, CA, USA, version 1.4.0.288) in the Proteome Discoverer software (Thermo Scientific, version 1.4).17,18 The initial parent and fragment ion maximum mass deviation was set to 10 ppm and 0.6 Da, respectively. A maximum of 2 tryptic missed cleavages were allowed, protein N-terminal acetylation and oxidation of methionine as the variable modifications, and in the case of the biotinylated samples the parameters of oxidation of cysteine were established as a fixed modification. In the case of non-biotinylated samples, carbamidomethylation of cysteine was established as a fixed modification during searches, and protein N-terminal acetylation, oxidation of methionine and phosphorylation of serine, threonine and tyrosine as the variable modifications. Peptides with at least six amino acids were considered for identification and a false discovery rate (FDR) of 0.01 was set for both proteins and peptides in the search against the decoy database. The Magellan storage files (MSFs) were imported into Scaffold (version Scaffold_4.3.4, Proteome Software Inc., Portland, OR, USA) for further validation of protein identification and quantitation analysis based on extracted ion chromatograms and for the top 3 resulting ion peak areas. A second search was performed using the X! Tandem algorithm (minimal package manager for Go (GPM), version CYCLONE 2010.12.01.1).19 Oxidation of cysteine was specified as a fixed modification. Dehydration of the n-terminus, glu → pyro-Glu of the n-terminus, ammonia-loss of the n-terminus, gln → pyro-Glu of the n-terminus, oxidation of methionine, acetyl of the n-terminus and CAMthiopropanoyl of lysine were specified in X! Tandem as the variable modifications. Peptide identifications were accepted if they could be established at greater than 95% probability to achieve a FDR less than 1.0%. Peptide probabilities from X! Tandem were assigned by the Peptide Prophet algorithm20 with Scaffold delta-mass correction. Protein identifications were accepted if they could be established at greater than 99% probability to achieve an FDR less than 1.0% by the Protein Prophet algorithm21 and contained at least 2 identified peptides. Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium22via the PRIDE (proteomics identification) partner repository23 with the dataset identifier PXD004636. The scaffold output files were merged for comparison between PC and the surface PC. Only those proteins present in at least 2 out of 3 sample replicates were considered as reproducible for analysis. Relative protein abundance was estimated based on the top 3 most intense ion precursors and the normalized values (against the sum of all ion intensities in each replicate) were used to compute mean values based on all replicates, e.g., 3 distinct sample preparations of biotinylated TiO2-NP-PC 10 nm, representing each sample in turn, the merging of 3 independent runs in one single output file. For comparison between the entire PC and the PC surface, the top 3 precursor intensity normalized mean values were normalized a second time to the sum of intensities per protein corona and expressed in percentage of abundance.Results and discussion
SUSTU, methodological basis
This method is based on classical cell surface proteomics methodologies24 that could be applied to analyse the interacting protein surface of the nanoparticle protein corona (NP-PC). We have implemented a biotin labelling method for its application to the NP-PC surface and integrated into a label free quantitative proteomics pipeline. This method includes biotin-labelling of the PC surface, then performing in situ trypsin digestion, and finally specifically recovering tryptic peptides from the PC surface for analysis using label-free quantitative mass spectrometry (Fig. 1). Briefly, SUSTU consisted of the biotin labelling of the PC surface interface by adding Sulfo-NHS-SS biotin to NP-PC suspensions and incubating for 3 h at 4 °C. Then biotin-labelled proteins were digested using endopeptidase trypsin. The biotinylated tryptic peptides were captured on NeutrAvidin resin and eluted with DTT and guanidine allowing the cleavage of biotin from peptides before identification and quantification by label-free quantitative proteomics analysis. The mass spectrometric analysis was performed via reversed-phase nano liquid chromatography coupled online to a hybrid ion trap mass spectrometer (LTQ Orbitrap Velos Pro-ETD, Thermo Fisher Scientific Inc.) (Fig. 1).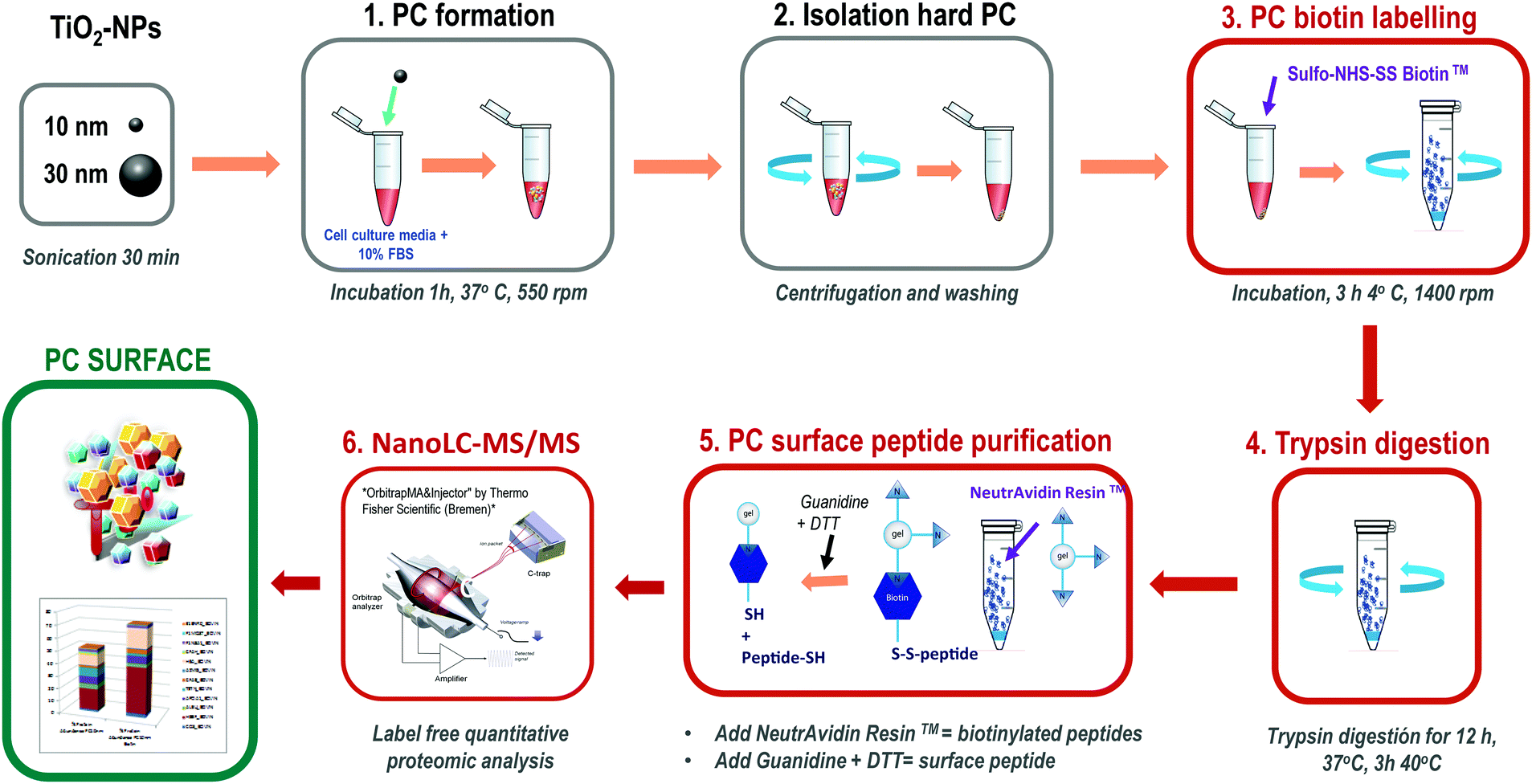 | ||
| Fig. 1 Outline of the surface proteomics workflow for analyzing the surface of the nanoparticle protein corona. | ||
The classical functional characterization of a novel engineered NP is based on combining extended characterization of the native or functionalized NP,5,25in vitro functional studies,4,26,27 and more recently a systematic analysis of the entire PC.28 In contrast to current methods,29–34 SUSTU is a surface proteomics-based approach focused on the characterization of the external protein surface adsorbed onto NPs in solutions, the PC surface, which is the accessible structure for protein–biomolecule interactions, and the subsequent impact in cellular processes.
The method rationale is based on the basic structure of the PC macromolecular assembly. In any biological environment, the bare NP surface will be hidden by layers of biomolecules, predominantly proteins.29,30 The relative protein abundance and the spatial distribution of proteins in this entity are very dynamic. The PC protein profile evolves as NPs cross biological environments with different protein content.35 The structural arrangements on the PC surface could determine the selection of specific cell surface receptors at the target cell.36 Therefore, we suggested that by focusing on a NP–cell interacting microenvironment that is dictated by the properties of this interface, the assessment of many variations of novel formulations could be achieved. Thus, the systematic characterization of the PC surface interface is a reductionist strategy to analyse the key components of a protein corona system that are essential to understand the NP targeting and uptake capability.
Characterization of the corona bio-interface of titanium dioxide nanoparticles (TiO2-NPs)
| NPs | Powder | Dispersed | |||||
|---|---|---|---|---|---|---|---|
| Crystal structure | Size (nm) | Specific surface area (m2 g−1) | Suspension | Hydrodynamic size (nm) | PDI | Z-potential (mV) | |
| TiO2 | Anatase | 10 | 163.94 | H2O | 858.5 ± 15.57 | 0.241 ± 0.05 | −9.62 ± 1.79 |
| Medium | 880.7 ± 20.90 | 0.396 ± 0.05 | −5.87 ± 1.86 | ||||
| TiO2 | Anatase/rutile | 30 | 50.36 | H2O | 243.0 ± 2.35 | 0.172 ± 0.05 | −10.20 ± 2.08 |
| Medium | 229.4 ± 2.02 | 0.195 ± 0.02 | −10.20 ± 1.50 | ||||
Similar values have been obtained with 30 nm TiO2-NPs dispersed in different cell culture media.4
Quantitative analysis of the NP-PC surface interface
We tested our method on TiO2-NPs, inorganic type NPs which were previously studied by our group.4,37–41 We prepared the NP-PC by incubating TiO2-NPs with a mean diameter of 10 and 30 nm, with cell culture media MCDB131 supplemented with 10% fetal bovine serum (FBS). The conditions selected for this comparative study were based on our preliminary experiments that confirmed the reproducibility of the NP-PC studied. The conditions that determine the formation of the PC were maintained constant in experiments for the analysis of the entire PC and the analysis of the PC surface including technical and experimental replicates (ESI,† S1 and S2). The protein samples corresponding to the entire PC were prepared according to the FASP method12 and the PC surface samples were prepared according to SUSTU (Fig. 1). We used nano-high-performance liquid chromatography coupled to a high-resolution mass spectrometer to analyse the peptides from proteins adsorbed onto NPs and for the surface proteins. The peptides were analysed using reverse phase nano liquid chromatography (nLC) coupled online to a hybrid ion trap mass spectrometer (LTQ Orbitrap Velos Pro-ETD, Thermo Scientific). Each of these peptide samples was separated using an EASY nLC II system (Thermo Scientific). The data analysis workflow includes MS raw data processing through Proteome Discoverer using a SEQUEST algorithm, and a second analysis with ScaffoldQ+ v4.2.1 using X! tandem algorithm,19 (peptide threshold: FDR 1.0% by the Peptide Prophet algorithm,20 protein threshold: FDR 1.0%; min 2 peptides). The relative protein abundance was estimated from the top 3 most intense precursor ions. We determined the protein composition of 4 sets of protein samples: 10 nm-sized TiO2-NPs entire PC (PC_10), 10 nm-sized TiO2-NPs surface PC (PC_10bio), 30 nm-sized TiO2-NPs entire PC (PC_30), and 30 nm-sized TiO2-NPs surface PC (PC_30bio).We provide a complete identification and quantitation of the proteins adsorbed onto the TiO2-NPs and analysed the proteins localized on the surface interface of the PC by comparing them to the entire corona. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository42 with the dataset identifier PXD004636 and 10.6019/PXD004636 (ESI,† S1 and S2).
This method firstly aims to simplify the analysis of PC profiles associated to targeting and uptake probabilities while offering accurate protein identification and quantitation via high-resolution mass spectrometry. The complexity of PC has been characterized using mass spectrometry-based methods in the last few years providing accurate information about the protein content adsorbed onto organic and inorganic NPs.28,33,43 An alternative method based on the assumption that the PC protein content can be correlated to its sulphur concentration has been developed. It combined electrospray ionization-tandem mass spectrometry (ESI-MS/MS) and total analysis of sulphur and the corresponding metal being present in the NPs, using inductively coupled plasma-mass spectrometry (ICP-MS).30 However, SUSTU is a targeted proteomic analysis focused exclusively on surface proteins, which are the molecular features responsible for targeting and uptake in cellular processes.44 The samples containing NP-PC with biotin-labelled surfaces could be directly digested into peptides and the biotinylated surface peptides were selectively recovered via affinity purification with the NeutrAvidin agarose resin. This procedure was tested with different sizes of TiO2-NPs and could provide a systematic characterization of the PC surface with equivalent parameters of accuracy and reproducibility (ESI,† S1 and S2). Therefore, SUSTU offers a simpler and faster method compared to previous techniques employed for the characterization of the entire PC. The quantitative analysis of NP-PC includes several additional steps of separation and purification of all tryptic digested peptides from the entire PC.28 However, SUSTU reduces the complexity of the final peptide sample for analysis.
Comparative analysis between the NP-PC and the PC surface interface
Secondly this method aims to quantify the distribution of proteins on the PC surface and evaluate if the PC surface could provide the hallmarks for the prediction of NP targeting and uptake. We compare the qualitative and quantitative differences between the entire NP-PC and the subset of labelled surface proteins. The definition of a PC surface using this method is based on the assumption that the surface proteins are the most exposed proteins from the supramolecular structure and therefore are accessible to form stable labelling with a biotin-reactive compound in solution.The number of distinct proteins on the PC surface was considerably reduced compared to the systematic identification of proteins from the entire PC (Fig. 2; ESI,† S1 and S2). These different features are sharper on the 10 nm- than on the 30 nm-sized NP-PCs. In the 10 nm-sized NPs the 6 most abundant proteins covered 80% of the PC surface compared to more than 20 in the entire PC. Similarly, in 30 nm-sized NPs we found that 80% of the PC surface was formed by half of the proteins that composed the entire corona (Fig. 2A and B; ESI,† S1 and S2). The relative protein abundance in the PC surface followed a different pattern than the entire PC. The ranking of the topmost abundant proteins from the PC and the PC surface differed in the NPs tested. We found that some of the PC high abundant proteins stayed away from the PC surface. Platelet factor 4 was completely buried in the PC and absent from the PC surface in the top 5 and top 10 most abundant proteins in the 10 nm-sized and 30 nm-sized TiO2-NP-PCs, respectively. Similarly, other proteins such as complement factor H, which could enhance the uptake of apoptotic cells by monocytes,45 were also hidden in the PC inner layers. Some types of apolipoproteins, molecules that could compromise targeting and uptake,46 were enriched in PC_10 and PC_30, whereas they are totally absent from the PC surface (Fig. 2A and B; ESI,† S1 and S2). Other studies have shown a correlation between size, NP surface and the number of layers of proteins composing the PC.30 Our results verified that the lack of homogeneous distribution of proteins is based on the PC relative protein abundance and that the corona is likely organized in layers or domains and specific proteins have higher probability to occupy the surface.
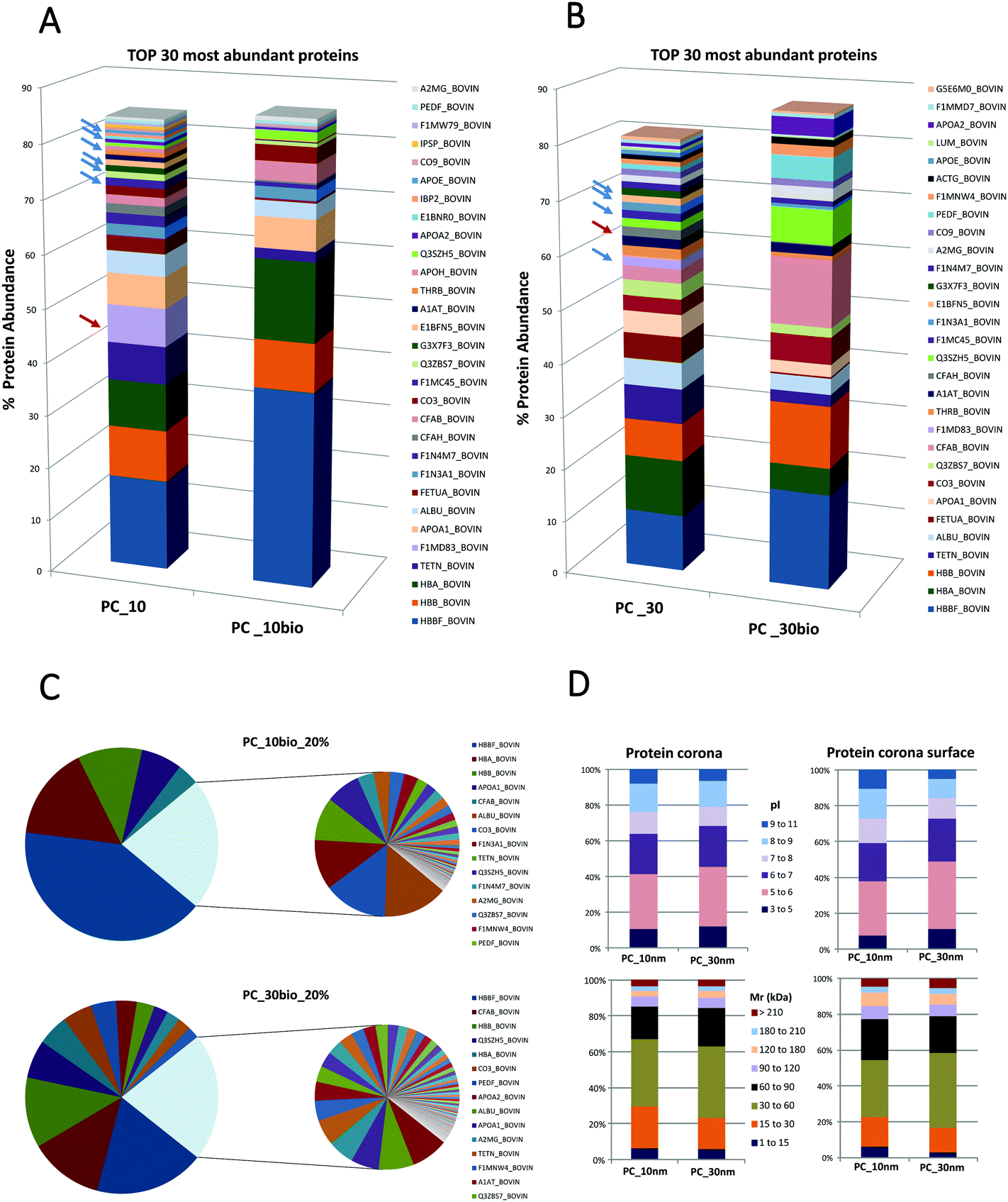 | ||
| Fig. 2 Differential distribution of proteins (% protein abundance) between the surface and the entire nanoparticle protein corona (NP-PC). (A) Composition of protein corona (PC) formed around 10 nm-sized TiO2-NPs. Differential protein distribution of the top 30 most abundant proteins on the surface (PC_10bio), compared to the entire PC (PC_10) formed around 10 nm-sized TiO2-NPs based on quantitative mass spectrometry analysis. (B) Composition of the PC formed around 30 nm-sized TiO2-NPs. Differential protein distribution of the top 30 most abundant proteins on the surface (PC_30bio), compared to the entire PC (PC_30) formed around 30 nm-sized TiO2-NPs based on quantitative mass spectrometry analysis. (C) The low abundant proteins have been merged in a sector that corresponds to 20% and the high abundant proteins contribute up to 80% of the total content. These two fractions have been extended in two individual pie charts for clarification. PC_10bio_20% corresponded to the 10 nm-sized TiO2-NPs and PC_30bio_20% corresponded to the 30 nm-sized TiO2-NPs. (D) Differential protein distribution of the PC and PC surface has been classified according to their calculated molecular mass (Mr) and their calculated isoelectric points (pI). | ||
The PC surface low abundant proteins composed approximately 20% of the total surface protein. This low abundant protein profile was particle- and size-dependent (Fig. 2C). The differential distribution of proteins on the PC structure was not based on protein properties such as their calculated molecular mass or their calculated isoelectric points (Fig. 2D). This analysis of the PC surface from TiO2-NPs is an example to illustrate how SUSTU could identify protein enrichment or depletion in the PC surface. It has been proposed that the relative abundance of PC could influence the enhanced or decreased cellular uptake of NPs by cancer cells.44 Thus the key surface features should be exposed if they are crucial for targeting of a specific cell type or are connected to receptor recognition and receptor-mediated uptake.46
The data provided by SUSTU indicate that the number of proteins found on the labeled surface was lower than that in the entire PC, and that the low abundant proteins were NP-specific. These are key features on the surface PC that could modulate the NP's destiny. It has been accepted that the initial requirement to predict the NP's targeting and uptake was a deep knowledge of the NP-PC's composition and dynamics.6,30,31,33 In that line, SUSTU is a surface proteomics method applied for the first time to identify an accessible NP-PC surface and would help understand the key protein components that could mediate NP's targeting and uptake.
SUSTU, a shortcut strategy for rapid prototyping of NPs
Our solution for simplifying the characterization of newly synthesized NPs is the implementation of one single analytical technique that would facilitate the prediction and supported rational guess about their targeting capabilities. This study provides the initial data that support a methodological shift from holistic approaches to atomistic approaches for the initial evaluation of novel NPs. This method could provide minimal data for the initial characterization of four key properties of a new material: targeting, uptake, function and safety. These four key features by themselves could assist and direct a more rapid and safe design of novel NPs by integrating the concept of rapid prototyping into the field of materials science (Fig. 3A and B). The rapid prototyping classically includes preliminary design, testing and refinement.47 By integrating new methods such as SUSTU into a platform for rapid prototyping of novel NPs, we could accelerate the process from the NP synthesis to its functional validation for nanoscience or nanomedicine.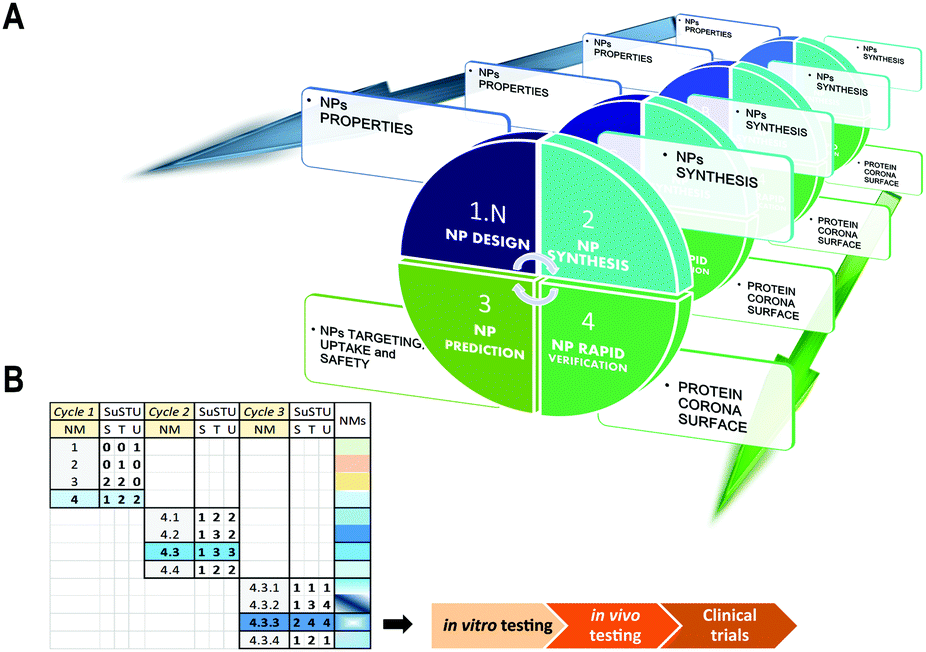 | ||
| Fig. 3 Graphical representation of a concept of rapid prototyping for nanomaterials with future applications in nanomedicines. (A) Scheme of the four proposed phases and deliverables including: NP design, NP synthesis, NP prediction, and NP verification through SUSTU. (B) Table illustrating an example of the application of the 4-step cycle (described in a) through several repetitions until the nanomaterial obtained could be selected for further analysis: in vitro testing, in vivo testing, and finally clinical trials. | ||
Conclusions
In conclusion, we have shown that SUSTU can provide a quantitative distribution and qualitative composition of the surface of the NP-PC that is the active interphase between NPs and target cells. We demonstrate that the distribution of protein on the surface differs from the distribution along the entire PC. Therefore the characterization of the surface of the PC cannot be deciphered by quantitative analysis of the entire PC under the assumption of a homogenous distribution of protein throughout the entire PC. We anticipate that the surface proteomic analysis of the PC could deliver the minimal hallmarks of any NPs that might simplify their functional characterization. We expect that SUSTU will introduce the concept of rapid prototyping in the future development of nanoparticles for biomedical applications. This method could be used as an analytical tool to assist nano-drug design and development and thus to support the future of precision medicine. Precision medicine, patient oriented healthcare from diagnosis to optimal therapies, is based on integrating a patient's genetic profile with a patient specific molecular and cellular disease phenotype. In this global initiative, methods that support the rapid prototyping of nanomaterials for nanomedicine could provide the inflexion point for developing finely tuned novel therapies.Abbreviations
| NP | Nanoparticle |
| PC | Protein corona |
| NP-PC | Nanoparticle protein corona |
| SUSTU | Surface proteomics for nanoparticle safety, targeting and uptake |
| TiO2-NPs | Titanium dioxide nanoparticles |
| MS | Mass spectrometry |
| ESI-MS/MS | Electrospray ionization-tandem mass spectrometry |
| ICP-MS | Inductively coupled plasma-mass spectrometry |
| FBS | Fetal bovine serum |
| PBS | Phosphate buffered saline |
| PDI | Polydispersity index |
| FASP | Filter aided sample preparation |
| FDR | False discovery rate |
| CID | Collision induced dissociation |
| ABC | Ammonium bicarbonate buffer |
| Mr | Calculated molecular mass |
| pI | Calculated isoelectric points |
Acknowledgements
This work was supported by grants from the Swedish Research Council-Natural Science, VR-NT (SC), Carl Trygger Foundation (SC), Oscar and Lilli Lamms Minne Foundation (SC), Magnus Bergvall Foundation (SC), Ångpanneförening Research foundation (SC), VINNOVA (SC), support from the County Council of Östergötland, Sweden (SC), IKERBASQUE, Basque Country Foundation for Science (SC), and Linköping University, Sweden (SC). We would like to thank Prof. Begona Ochoa from the Basque Country University Medical School for her initial contribution to our protein corona studies.References
- J. P. Almeida, A. Y. Lin, E. R. Figueroa, A. E. Foster and R. A. Drezek, Small, 2015, 11, 1453–1459 CrossRef CAS PubMed.
- B. M. Barth, E. I Altinoğlu, S. S. Shanmugavelandy, J. M. Kaiser, D. Crespo-Gonzalez, N. A. DiVittore, C. McGovern, T. M. Goff, N. R. Keasey, J. H. Adair, T. P. Loughran Jr., D. F. Claxton and M. Kester, ACS Nano, 2011, 5, 5325–5337 CrossRef CAS PubMed.
- N. Bayat, V. R. Lopes, M. Sanchez-Dominguez, R. Lakshmanan, G. K. Rajarao and S. Cristobal, Environ. Sci.: Nano, 2015, 2, 380–394 RSC.
- N. Bayat, V. R. Lopes, J. Scholermann, L. D. Jensen and S. Cristobal, Biomaterials, 2015, 63, 1–13 CrossRef CAS PubMed.
- R. Liu, W. Jiang, C. D. Walkey, W. C. Chan and Y. Cohen, Nanoscale, 2015, 7, 9664–9675 RSC.
- T. Cedervall, I. Lynch, S. Lindman, T. Bergg å rd, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS PubMed.
- D. Dell'Orco, M. Lundqvist, C. Oslakovic, T. Cedervall and S. Linse, PLoS One, 2010, 5, e10949 Search PubMed.
- A. Lesniak, F. Fenaroli, M. P. Monopoli, C. Aberg, K. A. Dawson and A. Salvati, ACS Nano, 2012, 6, 5845–5857 CrossRef CAS PubMed.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. B. Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed.
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi, C. Reinhardt, K. Landfester, H. Schild, M. Maskos, S. K. Knauer and R. H. Stauber, Nat. Nanotechnol., 2013, 8, 772–781 CrossRef CAS PubMed.
- J. R. Wisniewski, A. Zougman, N. Nagaraj and M. Mann, Nat. Methods, 2009, 6, 359–362 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- S. C. Gill and P. H. von Hippel, Anal. Biochem., 1989, 182, 319–326 CrossRef CAS PubMed.
- H. Edelhoch, Biochemistry, 1967, 6, 1948–1954 CrossRef CAS PubMed.
- J. K. Eng, A. L. Mccormack and J. R. Yates, J. Am. Soc. Mass Spectrom., 1994, 5, 976–989 CrossRef CAS PubMed.
- J. R. Yates, J. K. Eng, K. R. Clauser and A. L. Burlingame, J. Am. Soc. Mass Spectrom., 1996, 7, 1089–1098 CrossRef CAS PubMed.
- T. Muth, M. Vaudel, H. Barsnes, L. Martens and A. Sickmann, Proteomics, 2010, 10, 1522–1524 CrossRef CAS PubMed.
- A. Keller, A. I. Nesvizhskii, E. Kolker and R. Aebersold, Anal. Chem., 2002, 74, 5383–5392 CrossRef CAS PubMed.
- A. I. Nesvizhskii, A. Keller, E. Kolker and R. Aebersold, Anal. Chem., 2003, 75, 4646–4658 CrossRef CAS PubMed.
- J. A. Vizcaino, E. W. Deutsch, R. Wang, A. Csordas, F. Reisinger, D. Rios, J. A. Dianes, Z. Sun, T. Farrah, N. Bandeira, P. A. Binz, I. Xenarios, M. Eisenacher, G. Mayer, L. Gatto, A. Campos, R. J. Chalkley, H. J. Kraus, J. P. Albar, S. Martinez-Bartolome, R. Apweiler, G. S. Omenn, L. Martens, A. R. Jones and H. Hermjakob, Nat. Biotechnol., 2014, 32, 223–226 CrossRef CAS PubMed.
- J. A. Vizcaino, R. G. Cote, A. Csordas, J. A. Dianes, A. Fabregat, J. M. Foster, J. Griss, E. Alpi, M. Birim, J. Contell, G. O'Kelly, A. Schoenegger, D. Ovelleiro, Y. Perez-Riverol, F. Reisinger, D. Rios, R. Wang and H. Hermjakob, Nucleic Acids Res., 2013, 41, D1063–1069 CrossRef CAS PubMed.
- S. Elschenbroich, Y. Kim, J. A. Medin and T. Kislinger, Expert Rev. Proteomics, 2010, 7, 141–154 CrossRef CAS PubMed.
- D. H. Tsai, F. W. DelRio, J. M. Pettibone, P. A. Lin, J. Tan, M. R. Zachariah and V. A. Hackley, Langmuir, 2013, 29, 11267–11274 CrossRef CAS PubMed.
- V. R. Lopes, N. Bayat, M. Sanchez-Dominguez, R. Lakshmanan, G. K. Rajarao and S. Cristobal, Environ. Sci.: Nano, 2015, 2, 380–394 RSC.
- L. Momtazi, S. Bagherifam, G. Singh, A. Hofgaard, M. Hakkarainen, W. R. Glomm, N. Roos, G. M. Maelandsmo, G. Griffiths and B. Nystrom, J. Colloid Interface Sci., 2014, 433, 76–85 CrossRef CAS PubMed.
- X. Cai, R. Ramalingam, H. S. Wong, J. Cheng, P. Ajuh, S. H. Cheng and Y. W. Lam, Nanomedicine, 2013, 9, 583–593 CAS.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggard, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- N. Fernandez-Iglesias and J. Bettmer, Nanoscale, 2015, 7, 14324–14331 RSC.
- P. Kamath, A. Fernandez, F. Giralt and R. Rallo, Curr. Top. Med. Chem., 2015, 15, 1930–1937 CrossRef CAS PubMed.
- P. M. Kelly, C. Aberg, E. Polo, A. O'Connell, J. Cookman, J. Fallon, Z. Krpetic and K. A. Dawson, Nat. Nanotechnol., 2015, 10, 472–479 CrossRef CAS PubMed.
- M. P. Monopoli, A. S. Pitek, I. Lynch and K. A. Dawson, Methods Mol. Biol., 2013, 1025, 137–155 CAS.
- L. F. Pease, J. I. Feldblyum, S. H. Depaoli Lacaerda, Y. Liu, A. R. Hight Walker, R. Anumolu, P. B. Yim, M. L. Clarke, H. G. Kang and J. Hwang, ACS Nano, 2010, 4, 6982–6988 CrossRef CAS PubMed.
- F. Darabi Sahneh, C. Scoglio and J. Riviere, PLoS One, 2013, 8, e64690 Search PubMed.
- C. C. Fleischer and C. K. Payne, J. Phys. Chem. B, 2014, 118, 14017–14026 CrossRef CAS PubMed.
- K. Rajapakse, D. Drobne, D. Kastelec, K. Kogej, D. Makovec, C. Gallampois, H. Amelina, G. Danielsson, L. Fanedl, R. Marinsek-Logar and S. Cristobal, Nanotoxicology, 2016, 10, 542–556 CrossRef CAS PubMed.
- V. R. Lopes, V. Loitto, J. N. Audinot, N. Bayat, A. C. Gutleb and S. Cristobal, J. Nanobiotechnol., 2016, 14, 22 CrossRef PubMed.
- S. Garcia-Sanchez, I. Bernales and S. Cristobal, BMC Genomics, 2015, 16, 341 CrossRef PubMed.
- N. Bayat, K. Rajapakse, R. Marinsek-Logar, D. Drobne and S. Cristobal, Nanotoxicology, 2014, 8, 363–373 CrossRef CAS PubMed.
- S. Tedesco, N. Bayat, G. Danielsson, X. Buque, P. Aspichueta, O. Fresnedo and S. Cristobal, OMICS, 2015, 5, 44–57 Search PubMed.
- T. Ternent, A. Csordas, D. Qi, G. Gomez-Baena, R. J. Beynon, A. R. Jones, H. Hermjakob and J. A. Vizcaino, Proteomics, 2014, 14, 2233–2241 CrossRef CAS PubMed.
- H. Zhang, K. E. Burnum, M. L. Luna, B. O. Petritis, J. S. Kim, W. J. Qian, R. J. Moore, A. Heredia-Langner, B. J. Webb-Robertson, B. D. Thrall, D. G. Camp 2nd, R. D. Smith, J. G. Pounds and T. Liu, Proteomics, 2011, 11, 4569–4577 CrossRef CAS PubMed.
- S. Ritz, S. Schottler, N. Kotman, G. Baier, A. Musyanovych, J. Kuharev, K. Landfester, H. Schild, O. Jahn, S. Tenzer and V. Mailander, Biomacromolecules, 2015, 16, 1311–1321 CrossRef CAS PubMed.
- Y. H. Kang, B. C. Urban, R. B. Sim and U. Kishore, Immunobiology, 2012, 217, 455–464 CrossRef CAS PubMed.
- V. Bitsikas, I. R. Correa Jr. and B. J. Nichols, eLife, 2014, 3, e03970 Search PubMed.
- I. Lim, R. Walkup and M. W. Vannier, Biomed. Instrum. Technol., 1992, 26, 209–214 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nh00162a |
| This journal is © The Royal Society of Chemistry 2017 |