
Interactions of the innate immune system with carbon nanotubes
Kirsten M.
Pondman
ab,
Carolina
Salvador-Morales
c,
Basudev
Paudyal
ad,
Robert B.
Sim
 e and
Uday
Kishore
*a
e and
Uday
Kishore
*a
aDepartment of Life Sciences, College of Health and Life Sciences, Heinz Wolff Building, Brunel University London, Uxbridge UB8 3PH, UK. E-mail: uday.kishore@brunel.ac.uk; ukishore@hotmail.com
bNeuro Imaging, MIRA Institute, University of Twente, Enschede, The Netherlands
cBioengineering Department and Krasnow Institute for Advanced Study, George Mason University, 4400 University Drive MS 1G5, Fairfax, VA 22030, USA
dFaculty of Science, Engineering and Computing, Penrhyn Road, Kingston upon Thames, Surrey, KT1 2EE, UK
eDepartment of Pharmacology, University of Oxford, Oxford, UK
First published on 3rd April 2017
Abstract
The therapeutic application of nanomaterials requires that they are biocompatible and can reach the desired target. The innate immune system is likely to be the first defence machinery that would recognise the nanomaterials as ‘non-self’. A number of studies have addressed the issue of how carbon nanotubes (CNTs) interact with phagocytic cells and their surface receptors that can impact on their intracellular processing and subsequent immune response. In addition, soluble innate immune factors also get involved in the recognition and clearance of CNTs. The interaction of CNTs with the complement system, the most potent and versatile innate immune mechanism, has shed interesting light on how complement activation on the surface of CNTs can modulate their phagocytosis and effector cytokine response. The charge or altered molecular pattern on the surface of CNTs due to functionalization and derivatization can also dictate the level of complement activation and subsequent inflammatory response. It is becoming evident that complement deposition may facilitate phagocytic uptake of CNTs through receptor routes that leads to dampening of pro-inflammatory response by complement–receptor bearing macrophages and B cells. Thus, recombinant complement regulators decorated on the CNT surface can constructively influence the therapeutic strategies involving CNTs and other nanoparticles.
 Kirsten M. Pondman | Kirsten Pondman obtained a BSc and MSc in biomedical engineering at the University of Twente. During an internship at Oxford University, she became interested in nanomedicine, especially targeted drug delivery and interactions with the immune system. The main focus in her PhD was the application of nanoparticles in medicine; she worked both at developing particles that could be used for magnetic drug delivery and on interactions of various nanoparticles with different components of the (innate) immune system. Furthermore she developed ways to either suppress or stimulate the immune system using nanoparticles. |
 Carolina Salvador-Morales | Carolina Salvador Morales received her Doctor in Philosophy (DPhil) in Chemistry in 2007 at Oxford University. She was a postdoctoral fellow at Brigham and Women's Hospital, Harvard Medical School (2007–2009) and a postdoctoral associate in the laboratory of Prof. Robert Langer at MIT during 2007–2011. She holds BSc in Physics and MS in Biochemistry from Universidad Nacional Autonoma de Mexico and Trinity College, Dublin, respectively. She joined the Department of Bioengineering at George Mason University as Assistant Professor in 2011. She was promoted to Associate Professorship in 2017. Her research interests are in Colloidal Chemistry and Nanomedicine. |
 Basudev Paudyal | Basudev Paudyal received his BSc (Hons) in Microbiology in 2012 at University of the West of England, Bristol. He later joined MSc (Cancer Biology) at the Kingston University in 2013. In 2014, he joined PhD in Kingston University. His research interests are focused on the immune interaction of different nanoparticles. |
 Robert B. Sim | Robert B. Sim is an Honorary Professor at University of Leicester, Department of Infection, Immunity and Inflammation. He studied Biochemistry at the University of Edinburgh, and received his DPhil in Immunochemistry from the University of Oxford, MRC Immunochemistry Unit, Department of Biochemistry. He was an MRC/INSERM Research Fellow at Centre d'Etudes Nucleaires in Grenoble, then returned to Oxford as a group leader in the MRC Immunochemistry Unit. His research interests are focused on the structure and functions of complement and other innate immune system proteins, and their interactions with microorganisms, altered host cells and macromolecules, and with synthetic materials. |
 Uday Kishore | Uday Kishore is a teacher and a scientist researching in immunology. After his PhD from the University of Delhi, India, he spent a year at the Salk Institute for Biological Studies, San Diego, California as a NASA Fellow. His post-doctoral training and senior fellowship took place at the University of Oxford, first at the Medical Research Council (MRC) Immunochemistry Unit, and then Weatherall Institute of Molecular Medicine, John Radcliffe Hospital. In addition to being the founder director of the Centre for Infection, Immunity and Disease mechanisms at Brunel University London (2009–2015), Dr Kishore has been the recipient of MRC Investigator award, European Commission Young Scientist Prize, and Mother Teresa Excellence award. He holds adjunct and professorial positions at a number of universities, research institutions and teaching hospitals worldwide. His current interest includes devising drug delivery strategies in allergy and cancer through nanoparticles that can exploit the best of what the innate immunity has to offer. |
Introduction
CNT characteristics and applications
Since Iijima described their synthesis in 19911 carbon nanotube (CNT)-based nanotechnology has rapidly emerged as a platform for a variety of uses, including many biomedical applications.2 CNTs can be described as seamless cylindrical tubes, composed of graphene, with the carbon atoms hybridised in hexagonal sp2 arrangements in honeycomb lattices. Each atom is joined to three other neighbour atoms just like in graphene. Based on the rolling orientation of graphene sheet, CNTs are classified as arm chair, zigzag and chiral nanotubes.3 Depending on the number of concentric carbon tubes, they can be divided into single-walled (SWNT), double-walled (DWNT) and multi-walled (MWNT) carbon nanotubes (Fig. 1a and c). A principal characteristic is their high aspect ratio, resulting from their small diameter (1–3 nm for SWNT up to 2–100 nm for MWNTs) and extended length (up to 500 μm), which arise from their method of synthesis.4 CNTs are grown on a substrate (Fig. 1b), either with or without (metallic) catalyst particles; the preparation methods include arc discharge, laser ablation and chemical vapour deposition.5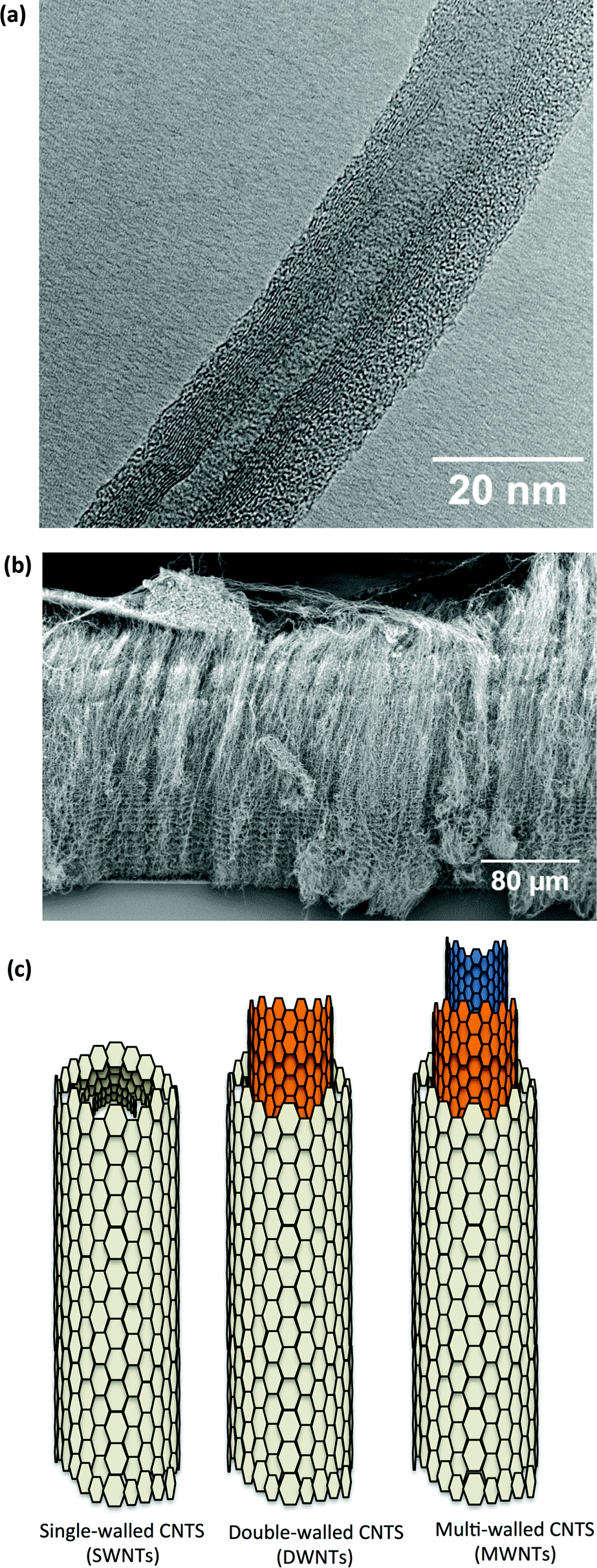 | ||
| Fig. 1 (a) TEM micrograph of MWNTs, clearly showing the high number of concentric carbon sidewalls and a 5 nm inner tube diameter. The outer walls of the MWNT are undamaged. (b) SEM micrograph of “as grown” MWNTs on a surface, also known as nanotube forest grown by chemical vapour deposition. These MWNTs are approximately 300 μm in length and 50 nm in diameter. On the top, remainder of the catalyst layer can be seen. (c) Sketch of the structure of carbon nanotubes showing different number of concentric carbon sidewalls. | ||
Biomedical applications of CNTs include drug delivery,6–11 immunoassays12 and scaffold.13,14 In combination with magnetic filling or particles, CNTs can be used as an MRI contrast agent,15–20 and in hyperthermia treatment.21,22 As their most promising application, CNTs as drug delivery platforms have been reviewed extensively elsewhere.6,23–25 The large surface area of CNTs offers a substantially higher drug loading capacity, compared to other nanoparticles while the dimensions of CNTs allow for entry in the smallest capillaries.26 Compatible with the requirement for targeted drug delivery, CNTs are able to cross the cell and nuclear membrane.27–30 Drugs can be either entrapped inside the CNTs,31 or absorbed or attached on the surface.9,10,32 Using these methods, CNTs have been shown to be versatile carriers for a range of drugs,6,11,23,25,32–37 genes,38 proteins39 and peptides.28 The drugs can, in principle, be delivered to specific targets (e.g. tumours) by attaching target-specific molecules (e.g. antibodies).40,41
In order to be used for a variety of applications, CNTs have to be individually dispersed in physiological buffers. Owing to their hydrophobicity, strong π–π interactions and length, CNTs are prone to rope and cluster formation; therefore, functionalizing or coating CNTs is essential.42 Non-covalent modifications of the CNT surfaces include pre-coating with proteins,43 surfactants,44 synthetic polymers45 and nucleic acids.46 Covalent functionalization involves introducing new functional groups on the external walls, usually beginning with the oxidation of the walls creating defects and carboxyl groups.47 The biocompatibility of CNTs can only be achieved by the covalent or non-covalent functionalization of their surface.34,48–51
In all biomedical applications, permanent or transient contact between CNTs and blood, cells or tissues is unavoidable, and, hence, an encounter with the immune system. These interactions may lead to severe inflammatory responses and tissue damage,52 which is likely to interfere with the tissue targeting or intended destination of the CNTs. It is, therefore, essential to study and understand the interactions between CNTs and all components of the immune system. In this review, we focus on the interactions of CNTs with the innate immune system, the body's first line of defence, which is likely to have the most profound influence on host–CNT interaction.
The innate immune system
The immune system is responsible for protection against micro-organisms (bacteria, fungi, viruses and parasites). In addition, altered or damaged cells and tissues are cleared via the cellular and molecular immune components. Recognition of these altered self or non-self materials (e.g. synthetic nanoparticles) is mediated by specific proteins, which bind to their targets and trigger downstream effector functions with the goal of eliminating the imminent danger to homeostasis. The human immune system consists of a complex conglomeration of interacting proteins and cells. In order to enhance short and long term efficiency of the clearance mechanisms, the immune system operates via two wings: innate immunity (rapid and broad in specificity) and adaptive immunity (slow and highly specific). The innate immune system involves proteins, always present in the blood, body fluids and tissues, while in adaptive immunity new recognition proteins (e.g. antibodies) are generated specifically towards a newly presented threat.A major part of the innate immune response involves opsonisation of the target (e.g., a pathogen) by proteins of the complement system, as well as migration and activation of phagocytic cells, mainly macrophages and immature dendritic cells (DCs) to the site of infection. The complement system consists of more than 40 soluble and cell surface proteins, working together via three activation pathways in order to recognise and opsonise foreign and altered-self components (Fig. 2).53 The recognition proteins of the complement system work through “multiple low-affinity binding”. A single binding between the recognition protein with its target, which can be a molecular motif such as a charge cluster, single neutral sugar, vicinal hydroxyl groups or a single acetyl group, is not strong enough to hold the target and the complement recognition protein together. Therefore, the recognition proteins have a multimeric structure with multiple contact/binding sites. The complement cascade is only activated when multiple bonds are formed allowing for a strong interaction.
 | ||
| Fig. 2 The three complement pathways: classical, lectin and alternative pathways, have different recognition strategies. The classical pathway is activated by recognition and binding of antigen–antibody complexes binding via C1q, which in turn activates C1r and C1s. C1, cleaves inactive C4 and C2 in sequence to form active C3 convertase, C4b2a. The lectin pathway is initiated by binding of mannose or carbohydrates moieties with MBL or ficolins activating membrane associated serine proteases (MASPs), which cleave C4 and C2 to form common C3 convertase (C4b2a). The alternative pathway is spontaneously activated by spontaneous lysis of C3 to C3b. Factor D cleaves factor B and forms C3 convertase (C3bBb) which in turns cleaves C3 leading to amplification of C3b formation and C5 convertase (C3bBbC3b). C3 convertase is stabilized by properdin while Factor H acts as an antagonist. Factor H inactivates C3b to form inactive iC3b. C5 convertase cleaves C5 forming C5b which combines with C6, C7, C8 and C9 to form membrane attack complex (MAC) leading to cell lysis. C3b acts as an opsonin and facilitates rapid macrophage mediated clearance of foreign particles. C4a, C3a and C5a act as anaphylatoxins and enhance the inflammation. | ||
The complement classical pathway is initiated by C1q (Fig. 3), a charge pattern recognition protein (460 kDa), consisting of 18 homologous polypeptide chains (6A, 6B and 6C chains) each consisting of a short N-terminal region, followed by a collagen like region with repeating Gly-X-Yaa triplets, and a heterotrimeric globular head (gC1q) domain composed of C-terminal region of A, B and C chains (ghA, ghB and ghC, respectively) (Fig. 3). The gC1q domain binds to charge clusters or hydrophobic patches on the targets which can be altered or non-self ligands.54 In the lectin pathway, the recognition proteins include mannan-binding lectin (MBL) that mainly binds to vicinal diols on sugars such as mannose, fucose or glucosamine; collectin 11 (CL-11) that binds to more complex glycan motifs; or one of the three ficolins (L-, H- and M-ficolin) that bind to acetyl groups and possibly other motifs.55 After C1q is bound to its targets, proteases C1r and C1s are activated (or for MBL, CL-11 and ficolins, MBL-associated serine proteases, MASP-1, 2 and 3, are activated). This then activates complement proteins C4 and C2 forming a C3 convertase (C4b2a), which cleaves C3 to form C3b that then binds to the target surface. C3b and its breakdown products, iC3b and C3dg, interact with C3 receptors on phagocytic and other cells. C3b is also a binding site for C5, which is activated by the same protease which cleaves C3, and then forms a complex with C6, C7, C8 and C9 (C5–9), called membrane attack complex (MAC), which disrupts the lipid bilayer of cells (Fig. 2).55
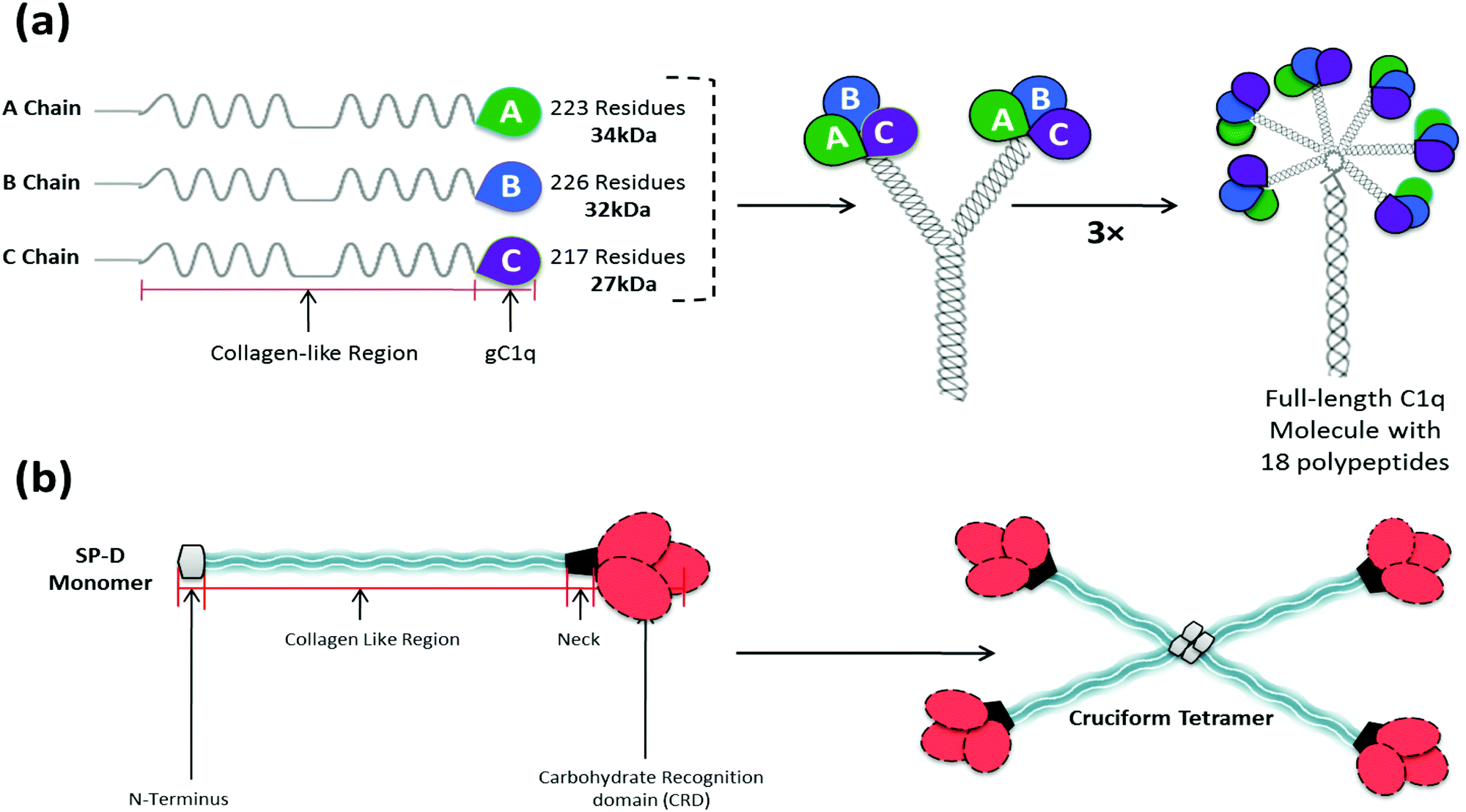 | ||
| Fig. 3 Overall structure of C1q and surfactant protein SP-D. (a) C1q is a charge pattern recognition protein (460 kDa), consisting of 18 homologous polypeptide chains (6A, 6B and 6C chains) with by a collagen like region (N terminal) with repeating Gly-X-Yaa triplets, and a globular head domain (C terminal), which bind to charge clusters or hydrophobic patches on targets. Each trimeric subunit has three globular head domains, called ghA, ghB, ghC. (b) SP-D has an N-terminal triple-helical collagen region, followed by a trimerising α-helical coiled-coil neck region, and C-terminal homotrimeric carbohydrate recognition domain (CRD). This primary subunit structure can associate further to yield a cruciform tetrameric supramolecule. | ||
The activation of the complement alternative pathway involves a constant slow hydrolysis of C3 in solution, which forms C3(H2O), and alters the shape of the protein. This conformational change allows the formation of a complex between factor B and C3(H2O), which allows factor D to cleave the bound factor B into Ba, which is removed, while Bb remains bound. C3(H2O)Bb is a protease which cleaves more C3 to form C3b, which can bind to target surfaces, and form a complex with factor B, which is converted to C3bBb, by factor D, as above. This leads to coating of the target particle with C3bBb, which is a homologue of the classical pathway C3 convertase, C4b2a. C3bBb, the alternative pathway C3 convertase, can be further stabilized by properdin (factor P) to C3bBbP. This complex is an enzyme able to generate more C3b to bind to targets. This is an amplification mechanism in order to increase turnover of C3 and coating of targets with C3b. To avoid consuming all available C3, the amplification mechanism needs to be balanced by down regulators: factor H binds to C3b inhibiting C3 convertase formation, and together with factor I it cleaves C3b to iC3b, which is unable to form C3bBb.55–57
After the complement proteins have tagged a particle (opsonisation), interaction with cell bound receptors (e.g. red blood cells through CR1/CR35, a receptor for C3b) and phagocytosis follows (via CR3 and CR4, which are receptors for iC3b). These complement–receptor interactions also promote uptake of complement-activating targets by DCs. Once immature DCs ingest an antigen, they undergo a directed activation and maturation towards becoming a potent antigen presenting cell (APC), after which they migrate towards lymph nodes. This makes DCs the main link between the innate and adaptive immune system, as they provide signals for T lymphocytes with the specific receptors for the presented epitopes to become activated.58
Interactions of CNTs with human plasma proteins
CNTs not only interact with components of the immune system but also show highly specific interactions with other soluble plasma proteins.59 The bound proteins form a corona, which plays an important role in determining the effective size, surface charge, physicochemical properties and aggregation state of the nanoparticles.60 In addition, it changes the recognition patterns, possibly presenting novel peptide or glycan motifs to the immune system, and can therefore alter the nature of interaction with the complement system, cells and ultimately immune response and bio-distribution.61–64The composition of the protein corona changes with time, depending on the binding affinities and stoichiometry of the nanoparticle and proteins.65 Affinities can be affected by surface properties such as available functional groups, but also the surface area and curvature. In general, a nanoparticle will be first covered by the most abundant plasma proteins (e.g. albumin and fibrinogen). These proteins are then replaced by proteins with higher affinity towards the particle surface, a process called the “Vroman effect”.66–68 The initial coating on the CNT can influence the binding of proteins, as some proteins have affinities towards charge, hydrophilicity, nucleic acids or carboxyl groups.66
Oxidation of CNTs offers a more negatively charged surface, which binds more protein.69,70 Shannahan et al. have performed an extensive proteomics analysis to identify the proteins in the corona of SWNTs (1 nm) and MWNTs (20–30 nm) unmodified, PVP (Polyvinylpyrrolidone) coated, or oxidised.70 All CNT coronas contained 14 common proteins: serum albumin, titin, apolipoprotein-A-I, apolipoprotein A-II, α1-anti-proteinase, α2-HS-glycoprotein, α-S1-casein and keratin. A much larger variety of proteins was found to bind only onto specific types of CNTs. A similar binding profile was found by Salvador-Morales et al., but it was reported that more albumin bound to chemically modified MWNTs, suggesting that the plasma could enter the larger diameter MWNTs by capillary forces; these entrapped proteins are likely to be difficult to wash out.71,72 Cai et al. showed that larger diameter CNTs were also able to bind a wide range of proteins on their surfaces, although increasing the diameter of CNTs above 20 nm did not have any additional effect.73
Complement absorption and activation
Certain components of the corona, opsonins, which include IgG and complement, can enhance uptake of the nanoparticles by macrophages and other cells of the reticulo-endothelial system.48,64 The importance of complement activation by nanoparticles used in drug targeting was highlighted by a study on liposome-encapsulated-doxorubicin. After hypersensitivity reaction was reported in clinical application of these particles, it was found that these side effects were due to complement activation.74Previous studies have shown that non-functionalized CNTs, when placed in contact with human serum, activate complement via the classical and, to a lesser extent, via the alternative pathway.71,75 However, the mode of binding of the recognition proteins to the CNTs has not fully been characterised and questions remain whether complement proteins bind directly to the CNTs or bind via other deposited (serum) proteins that can act as adaptors. Complement proteins C1q and MBL, as well as C-reactive protein, an acute phase protein which itself mediates binding of C1q (an “adaptor” for C1q), are known to recognise repetitive structures or charge patterns, which are not found on pristine CNTs but commonly found on the surface of functionalized CNTs.76 Ling et al. presented evidence that C1q “crystallizes” on pristine and functionalized CNTs, but is not bound in a way that allows it to activate the next step of the complement cascade.77 Other serum proteins would thus have to form a stable layer on the CNTs for indirect C1 binding and subsequent complement activation. Others,71,75,78,79 however, observed direct high affinity binding of C1q to CNTs by hydrophobic interactions, and concluded that direct binding of C1 would allow complement activation. Binding of C1q onto CNTs is not ionic or calcium-ion-dependent and is of high affinity since denaturation of C1q is required to remove the C1q from the CNTs.80
Recombinant forms of individual globular head regions of C1q A, B and C chains can be bound to pristine, oxidised and carboxymethyl (CMC) cellulose coated MWNTs,78,79 confirming that the binding of C1q to CNTs takes place via the gC1q domain, which is the principal ligand-recognition domain of C1q.81 Binding of C1 is followed by activation of C4 and C2, but activation may not go beyond that due to the lack of suitable covalent binding (OH, NH2 or SH) sites for C4b or C3b.82 However, it has been shown that C3 and C5 turnover did occur with pristine and various proteins coated CNTs.79 Therefore, it is likely that C3b binds and the MAC is subsequently formed. These interactions of C3b and C4b are most likely via direct hydrophobic interactions with the surface of the CNTs.80 Similarly, another key component and up-regulator of the complement alternative pathway, properdin, which has previously been shown to bind to apoptotic T cells83 and DNA exposed on apoptotic and necrotic cells,84 has recently been reported to show pattern recognition properties and binds to both CMC-CNTs and Ox-CNTs and enhances their uptake by macrophages in a complement-independent manner. Furthermore, CNTs bound properdin still retains its C3 and C5 convertase stabilisation properties and activates the alternative pathway. However, recombinant thrombospondin repeats (TSR) 4 and 5, the modules of human properdin that can bind C3b, act as a potent inhibitor of the alternative pathway and also inhibit the rapid macrophage mediated clearance of CNTs, raising the possibility of therapeutic use in a range of diseases, including tumour.85 CNTs, opsonised with human properdin or TSR4+5, triggered a robust pro-inflammatory response by macrophages, suggesting that local synthesis of complement proteins can alter the immune clearance of nanoparticles considerably, even when there is no complement activation involved.
Differential innate immune recognition of CNTs based on functionalization
Several studies have shown that functionalization, and therefore, alteration in the surface properties of the CNTs, can change the extent of complement activation.49,52,69,78–80,86,87 Pre-coating CNTs will increase the dispersion state, making more surface area available for complement proteins to recognise and deposit themselves. RNA and BSA, used as dispersing agents, do not uniformly coat the CNT surface, therefore, binding sides on the CNT surface are made more available and complement activation might increase compared to clustered pristine CNTs. RNA itself can interact with C1q providing an additional binding site for complement.80 Poly-ε-caprolactam (Nylon-6) and CMC-cellulose have been shown to reduce the level of complement activation via the classical pathway most efficiently, but fail to eliminate opsonisation.52,79Until a decade ago, PEGylation (poly ethylene glycol coating) was considered to provide a shielding surface on nanoparticles, but in 2002, it was shown that PEGylated polystyrene microspheres could activate complement, depending on the configuration of the PEG on the surface.88 The effects of PEGylation on SWNTs as well as MWNTs have been extensively studied by the Moghimi group. They showed that although PEG can reduce complement activation via both classical and alternative pathway, levels of both C4d (cleavage product of C4) and MAC significantly increased. They concluded that complement activation was likely to occur through the lectin pathway. For MWNTs, complement activation was independent of the molecular mass of PEG chains and the effect was not caused by uncoated regions of the CNTs. The surface domains of the PEG derivatives may thus act as templates for the lectin pathway activating molecules (L-ficolin and MASP-2).86,89,90
Complement activation can be influenced by coating specific humoral factors onto the outer walls of CNTs. For instance, binding of factor H, a down-regulator of the alternative pathway, lowers the activation of the alternative pathway.52 In contrast to full length C1q, the recombinant globular heads of C1q were shown to reduce complement activation.79 This phenomenon is likely to be caused by globular heads competing out the binding of whole C1q to CNTs, thereby diminishing complement activation. A similar technique to avoid recognition by the complement system is used by pathogenic bacteria, which have specific binding motifs on their surface to bind factor H, thus inhibiting alternative pathway activation.91
Innate immune receptors, phagocytosis and immune response
The cells of the innate immune system, including macrophages and DCs, have pattern recognition receptors (PRRs) that recognise and bind pathogens via pathogen-associated molecular patterns (PAMPs). These include toll-like receptors (TLRs), scavenger receptors, complement receptors, integrins, and lectin-like receptors, which are potentially capable of recognising nanoparticles. Once a particle is bound to receptors, the particle will be attached to the cell and taken on its path, but the particle can also be phagocytosed and ultimately, if possible, digested internally and cleared from the system.The most important complement-derived opsonin is C3b and its breakdown product iC3b. Multiple copies of C3b can bind onto the surface of the nanoparticle. C3b interacts with complement receptor 1 (CR1 or CD35) which is abundant on red blood cells. Once C3b has bound, it is gradually broken down into iC3b, which has lower affinity towards CR1, but high affinity towards CR3 and CR4, which are commonly found on phagocytic cells. Therefore, the nanoparticles will be transferred from red blood cells towards phagocytic cells, especially during the passage of the red blood cells through the liver where macrophages are present in high numbers. The iC3b will be further broken down into C3d, which can interact with CR2 (CD21) on the surface of B-lymphocytes, and therefore, interact with the adaptive immune system.
Opsonised CNTs absorb or bind onto the surface of red blood cells (Pondman et al., unpublished), indicating that C3b is bound in a conformation that allows interaction with CR1. PEGylation, which down-regulates complement activation, was shown to reduce uptake of CNTs by monocytes, spleen and liver phagocytes that correlated with increasing molecular weight and PEG coating density.92 Uptake of CNTs by macrophages (U937 cell line), blood monocytes and B cells (Raji cell line) is more efficient in the presence of serum; while complement inactivated (heat inactivated) serum does not enhance the phagocytosis of CNTs, indicating an important additive effect of complement.78,79 Most interestingly, Jurkat T cells, which are known to express complement receptors feebly on their surface, were able to take up CNTs poorly and serum treatment did not increase uptake.78 Complement adsorption on the surface of MWNTs was shown to reduce the expression of pro-inflammatory cytokines (TNF-α, IL-6 and IL-1β) and increase expression of anti-inflammatory cytokines (IL-10 and TGF-β) in monocytes and macrophages.79 This indicates that complement might signal the cells to silently remove the CNTs by phagocytosis, but do not give out stress signals to their microenvironment. Even when only the initial complement proteins C1q and MBL are bound on the surface of the CNT, receptor interactions are possible with calreticulin, a receptor molecule for C1q and MBL, working together with CD91 acting as a C1q receptor.93 These bindings are less efficient as the density of deposited C1q and MBL is far lower than C3b and for adhesion, hundreds of receptor–ligand pairs are needed. As was shown recently, pre-coating the CNTs with the recombinant globular head modules of human C1q and its full length counterpart87 can increase the phagocytosis by macrophages. Similar properties have been shown with properdin coated CNTs85 while factor H87 and small fragment of properdin molecule (TSR4+5)85 proved to be an inhibitor of phagocytosis. Recently, Meng et al. showed that after phagocytosis of ox-MWNTs, macrophages produced macrophage inflammatory proteins (MIP-1α and MIP-2) to recruit other macrophages to the site. They also confirmed that low levels of pro-inflammatory cytokines were produced.94
Non-complement dependent uptake of CNTs
The method of entry of CNTs into cells is a highly debated subject in the literature, complement dependent phagocytosis being one of the several proposed mechanisms. Covalent and non-covalent modification of CNT walls can alter its interaction with immune cells. In the case of the former, that phenomenon can be caused by the chemical nature of the coating. For example, macrophages are known to interact more strongly with positively charged particles due to the presence of negatively charged sialic acid on their surface.95 However, altered uptake and interactions can also be a direct effect of the higher dispersibility and, therefore, biocompatibility of the functionalized CNTs. In general, hydrophilic or acidic polymer coated MWNTs are more efficiently internalized by macrophages than hydrophobic polymer coated MWNTs.96 Direct penetration or “needling” through the plasma membrane is another described phenomenon.97,98 Others state that absorption of albumin or other serum proteins is essential to trigger scavenger receptor-mediated uptake.99 Kam et al. found that very short SWNTS (e.g., 50–200 nm) enter cells (e.g., HeLa and H60 cell lines) through clathrin-dependent endocytosis.100 However, Pantarotto et al. showed that slightly longer SWNTs (300–1000 nm) behave like cell penetrating peptides while entering human (3T6) and murine (3T3) fibroblasts.28After uptake by the cells, the chemically modified CNTs can be found in the cytoplasm, endosomes,11,28,98,101,102 and in some cases, inside the nucleus.28,97,102 These variations can be due to different functionalization.28,97 Exocytosis has not been reported often and the time course for the process varies between simultaneous with endocytosis103 and until after 5 h of incubation.78,79,101
Cytokine, inflammation and immune responses
In their bio-persistence as well as high aspect ratio, CNTs show similarities to asbestos, and therefore, an incomplete uptake and frustrated phagocytosis with the related inflammation and granuloma formation is a risk that has to be analysed.98,104,105 Frustrated phagocytosis was analysed by Brown et al. with a variety of elongated CNTs.106 In their study, only individually dispersed long straight CNTs led to frustrated phagocytosis using PBMCs (peripheral blood mononuclear cells) and THP-1 cells, (a monocyte-derived cell line), which correlated with superoxide anion and TNF-α release. The presence of CNTs interferes with the function of the macrophages as was shown by an inhibition of the ability of THP-1 cells to phagocytose E. coli. Clustered CNTs and nanofibers did not induce apoptotic or necrotic effects.106Exposure to long MWNTs resulted in a significant and dose-dependent release of IL-1β, TNF-α, IL-6 and IL-8 from THP-1, but not from mesothelial cells (Met5a).104 More interestingly, when cell medium from the THP-1 cells treated with long CNTs was added to Met5a cells, they too showed an increased cytokine production, indicating the essential role of macrophages in the immune response towards CNTs. Liu et al. showed that immune response with pluronic F127 coated MWNTs in RAW (a murine macrophage cell line) and MCF-7 (a breast cancer cell line) cells was length-dependent.105 RAW cells showed higher internalisation, resulting in higher toxicity due to CNTs than MCF-7. Long MWNTs (3–8 μm) were more toxic than short (<1.5 μm), but short MWNTs gave more TNF-α release than long MWNTs, which could lead to a stronger inflammatory response. However, complement deposited CNTs dampened the pro-inflammatory cytokines such as IL-1β and TNF-α despite higher internalization of these CNTs by human macrophage cell lines.79
Besides cytokine response, indications of inflammasome formation by CNTs have been reported.98,107,108 Many carbon nanomaterials (carbon black, short, long and tangled CNTs, long needle-like MWNTs, and asbestos) induced IL-1β secretion (indicator of inflammasome formation), but only long needle-like CNTs induced IL-1α secretion in LPS-primed macrophages.98 DWNTs can synergize with TLR4 antagonists; when K+ efflux is hindered, IL-1β secretion could be eliminated, indicating that phagocytosis was required for inflammasome activation. After phagocytosis, NF-kβ (nuclear factor kappa-light-chain-enhancer of activated B cells) and NLRP3 (nucleotide-binding oligomerization domain (NOD)-like receptors family, pyrin domain containing 3) inflammasome are activated.107,108
Various CNTs can modulate DC maturation.109 Short, purified SWNTs by an oxidation method and with free-endotoxin content induced no maturation of DC cultures and did not lead to the secretion of IL-6, TNF-α, or IL-1β following their uptake. In comparison, incubation of DCs with LPS and CNTs induced IL-1β secretion, which was dose and NLRP3-dependent, indicating that LPS contamination causes this effect.110 Dumortier et al. showed that PEG1500-SWNTs are taken up by B- and T-cells without affecting viability of the cells or causing damage, inhibiting or stimulating their function.50 Although they found no IL-2 and IFN-α secretion (reflecting T cell activation), PEGylated SWNTs did induce IL-6 and TNF-α secretion in peritoneal macrophages in vitro, which the authors attributed to the formation of CNT aggregates.
Lung innate immunity and CNTs
Most likely triggered by their asbestos like appearance,111 pulmonary toxicity of CNTs is one of the most discussed aspects in the toxicology field. Disagreement started from the very first studies published by Lam112 and Warheit113 who independently concluded that CNTs were highly toxic and non-toxic to the lungs, respectively. First, they both showed that CNTs induced granulomas, but only Lam showed subsequent fibrogenesis. This effect can be explained by the fact that granuloma formation is mediated by the accumulation of alveolar macrophages at sites of CNT deposition, which become activated by the phagocytosis of the nanoparticles. The activated macrophages produce growth factors that stimulate the proliferation of fibroblasts, the collagen producing cells driving the fibrogenesis.114 Whereas Lam found a dose- and time-dependent interstitial inflammation, Warheit did not see any inflammation and fibrosis; in addition, the granuloma formation was not dose-dependent. Warheit concluded that the toxicity of the CNTs was caused by aggregation of the CNTs due to the administration method (instillation), which also caused airway blocking. Shvedova et al. confirmed the results of Lam et al. in mice and also showed dose-dependent functional respiratory deficiencies.115 Subsequently, Mangum et al. found no inflammation in SWNT exposed (oropharyngeal aspiration) rats, although they did find a few focal interstitial fibrotic lesions at locations with clusters of macrophages containing micron sized aggregates of SWNTs in the alveolar region. In addition, they reported, in bronchoalveolar lavage fluid (BALF), macrophages linked together with bridges of parallel bundles of SWNTs. They stated that this bridge formation is not similar to frustrated or incomplete phagocytosis seen in asbestos and other long fibres.114 The origin in the variations of effects reported possibly owes it to a wide variation in the nanoparticles (single, double or multi-walled) with variable diameter and length, coating, aggregation states, contamination with other materials, and administration method and route.116 By comparing well-dispersed SWNTs with aggregated SWNTs, Shvedova et al. found that poorly dispersed SWNTs formed clumps of 5 to 20 μm in the lungs, which triggered granuloma formation, whereas highly dispersed SWNTs that did not form any clumps and were found free in the tissue, gave rise to interstitial fibrosis but no granulomatous lesions.117–119 This was confirmed by a study where well dispersed MWNTs were found in every cell and cell layer of the lung parenchyma, with signs of interstitial fibrosis of the alveolar wall but with very limited granuloma formation.120The lung innate immune defence is governed mainly by surfactant proteins A and D (SP-A and SP-D), together with lung leukocytes and the epithelial cells lining the alveolar surface. Like MBL, SP-A and SP-D are members of the collectin (collagenous lectins) family. SP-A and SP-D have a multimeric structure similar to C1q and MBL (Fig. 3). Among other roles, SP-A and SP-D bind to invading particles (commonly via vicinal diols) in a Ca2+-dependent manner and promote their binding to receptors on alveolar macrophages.121 The concentrations of SP-A and SP-D are very low, and therefore, binding of these pulmonary surfactant proteins to CNTs can cause significant depletion of the proteins and damage to the pulmonary innate immune defence mechanisms.72
Selective Ca2+-dependent binding of BALF SP-A and SP-D to the oxygen containing functional groups on the surface of CNTs was confirmed in a study by Salvador-Morales by using acid treated (oxidized) MWNTs, which could be coated entirely with SP-A.122 Oxidized DWNTs bound SP-A and SP-D more efficiently than non-oxidized DWNTs and purified DWNTs. Similar results were found for MWNTs and ox-MWNTs by Marchetti et al.123
SP-A-coated MWNTs were able to enter the cytoplasm and the nucleus of alveolar macrophages. Interestingly, the high nitric oxide secretion evoked by pristine MWNTs and BSA-coated MWNTs was not observed by SP-A-coated MWNTs, indicating a possible method to avoid an inflammatory response towards CNTs.122 Allowing SWNTs to obtain a lung surfactant corona, consisting of SP-A, B and D, enhanced the in vitro uptake of SWNT by RAW cells (murine macrophages).124 Gasser et al. showed that pre-coating MWNTs (pristine, oxidised and aminated) with pulmonary surfactant proteins induced clusters of coated MWNTs intracellulary in monocyte-derived macrophages, while more stable suspensions are obtained with coated MWNTs.125 It has been argued that both SP-A and phosphatidylserine might represent an “eat me” signal towards macrophages. A similar effect was seen by coating CNTs with SP-D.126 Coating the MWNTs with SP-D slightly increased apoptosis while necrosis slightly decreased.125 Interestingly, a decrease in TNF-α release was found, which might be attributed to the phosphatidylserine present in the surfactant.
Coating of CMC-MWNTs and Ox-MWNTs with recombinant SP-D (rhSP-D) increased the phagocytosis by macrophages 2-fold, at the same time enhanced the pro-inflammatory cytokine storm provoked by MWNTs (reduction of IL-12 and TGF-β; increased IL-1β, IL-6 and TNF-α production).126 Interestingly, SP-D coated CNTs enhanced the complement activation and SP-D coated and complement deposited CNTs showed the dramatic reduction of pro-inflammatory cytokine production compared to SP-D coated CNTs.126
Conclusions
A number of studies have addressed how CNTs interact with the innate immune system including complement proteins, macrophages, dendritic cells, cell surface pattern recognition receptors and soluble factors. The results obtained thus far paint a varied and heterogeneous picture: primarily owing to the diverse range of CNTs and in vitro and in vivo model systems used for investigation. However, it is evident that the previously suggested pro-inflammatory response to CNTs needs to be viewed in the context of complement. The recognition subcomponents of the three pathways of the complement system are fully capable of binding via patterns presented as an array over the surface of pristine or chemically-modified CNTs. This recognition can lead to complement deposition, thus enhancing phagocytosis by the immune cells bearing complement receptors such as macrophages and B cells. Complement deposition on the CNTs appears to skew the pro-inflammatory response towards an anti-inflammatory one, suggesting beneficial effects of complement. It is unclear how complement deposition enhances anti-inflammatory immune response although a link between heightened IL-10 levels and suppressed TNF-α and IL-1β is evident in recent studies. It is worth examining how altered pattern can affect CNT engagement with TLRs with or without complement deposition. Clearly, the ability of CNTs to induce pro-inflammatory and anti-inflammatory immune response requires a comprehensive in vivo assessment to prove their feasibility usage as therapeutic vehicles in the long term. Thus, their potency to deposit complement on the surface is going to acquire importance. There are a number of conflicting data in the literature that have arisen out of variability of the CNTs introduced by surface coating, shapes, sizes, dispersion, and surface charge. With advancement in the production of CNTs, such issues can be resolved. It is also important to note that the ability of CNTs to activate complement has been reported consistently by a range of research groups. Thus, any clinical trials involving CNTs, or for that matter, any nanoparticle, needs to include quality control involving innate immune aspects (Table 1).| Pristine | Covalently functionalized CNTs | Non covalently functionalized CNTs | PEG–CNTs | |
|---|---|---|---|---|
| Complement activation | Less activation of both classical and alternative pathway than functionalised-CNTs.52,71 | Activates both classical and alternative pathway71 | Activates both classical and alternative pathway79 | Activates all three complement pathway62,86,89 |
| Endocytosis (highly dependent on size, diameter, surface modification and ability to activate complement pathways) | — | Reduced uptake by macrophages, B-cells and T-cells compared to functionalized CNTs. Uptake highly dependent on level of complement activation.78 | High uptake compared to Ox-CNTs and PEG–CNTs.78 | PEGylation decreases the uptake by macrophages and increases circulation time in vivo92 |
| Cytokine response | — | Anionic functionalization decreases the production of pro-inflammatory cytokines/growth factors than cationic functionalization127 | CMC-CNTs are pro-inflammatory to immune cells.87 | PEGylated CNTs decreases the production of pro-inflammatory cytokines |
| Cytotoxicity (depends on impurities, functionalization, shape and size of CNTs) | Considered more toxic than any other form of CNTs128 diameter- and rigidity dependent toxicity and carcinogenicity: thin and rigid nanotubes the most toxic and carcinogenic129 | Based on the functional group. Oxides group present on CNTs are less toxic than amine group.38,130 | Toxicity dependent on dispersant131 | PEGylated SWNTs exhibits less cytotoxic potency132 non toxicity of PEGylated CNTs on T cells.133 |
Acknowledgements
We thank Valarmathy Murugaiah for drawing Fig. 3.References
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- F. Liang and B. Chen, Curr. Med. Chem., 2010, 17, 10–24 CrossRef CAS PubMed.
- A. Aqel, K. M. M. A. El-Nour, R. A. A. Ammar and A. Al-Warthan, Arabian J. Chem., 2012, 5, 1–23 CrossRef CAS.
- M. Keidar, J. Phys. D, 2007, 40, 2388 CrossRef CAS.
- J. Prasek, J. Drbohlavova, J. Chomoucka, J. Hubalek, O. Jasek, V. Adam and R. Kizek, J. Mater. Chem., 2011, 21, 15872–15884 RSC.
- A. Bianco, K. Kostarelos and M. Prato, Curr. Opin. Chem. Biol., 2005, 9, 674–679 CrossRef CAS PubMed.
- C. Biale, V. Mussi, U. Valbusa, S. Visentin, G. Viscardi, N. Barbero, N. Pedemonte and L. Galietta, in 2009 9th IEEE Conference on Nanotechnology, IEEE NANO 2009, ed. Anonymous, 2009, pp. 644–646.
- J. G. Li, W. X. Li, J. Y. Xu, X. Q. Cai, R. L. Liu, Y. J. Li, Q. F. Zhao and Q. N. Li, Environ. Toxicol., 2007, 22, 415–421 CrossRef CAS PubMed.
- H. Ali-Boucetta, K. T. Al-Jamal, K. H. Muller, S. Li, A. E. Porter, A. Eddaoudi, M. Prato, A. Bianco and K. Kostarelos, Small, 2011, 7, 3230–3238 CrossRef CAS PubMed.
- E. Heister, V. Neves, C. Tilmaciu, K. Lipert, V. S. Beltrán, H. M. Coley, S. R. Silva and J. McFadden, Carbon, 2009, 47, 2152–2160 CrossRef CAS.
- W. Wu, S. Wieckowski, G. Pastorin, M. Benincasa, C. Klumpp, J. P. Briand, R. Gennaro, M. Prato and A. Bianco, Angew. Chem., Int. Ed., 2005, 44, 6358–6362 CrossRef CAS PubMed.
- M. Yang, S. Sun, Y. Kostov and A. Rasooly, Lab Chip, 2010, 10, 1011–1017 RSC.
- N. Aoki, T. Akasaka, F. Watari and A. Yokoyama, Dent. Mater. J., 2007, 26, 178–185 CrossRef CAS.
- X. Shi, B. Sitharaman, Q. P. Pham, F. Liang, K. Wu, W. Edward Billups, L. J. Wilson and A. G. Mikos, Biomaterials, 2007, 28, 4078–4090 CrossRef CAS PubMed.
- J. H. Choi, F. T. Nguyen, P. W. Barone, D. A. Heller, A. E. Moll, D. Patel, S. A. Boppart and M. S. Strano, Nano Lett., 2007, 7, 861–867 CrossRef CAS PubMed.
- O. Vittorio, S. L. Duce, A. Pietrabissa and A. Cuschieri, Nanotechnology, 2011, 22, 095706 CrossRef PubMed . Epub 2011 Jan 27.
- P. K. Avti, Y. Talukdar, M. V. Sirotkin, K. R. Shroyer and B. Sitharaman, J. Biomed. Mater. Res., Part B, 2013, 101, 1039–1049 CrossRef PubMed.
- E. Fidiani, P. M. F. J. Costa, A. U. B. Wolter, D. Maier, B. Buechner and S. Hampel, J. Phys. Chem. C, 2013, 117, 16725–16733 CAS.
- J. T. Wang, L. Cabana, M. Bourgognon, H. Kafa, A. Protti, K. Venner, A. M. Shah, J. K. Sosabowski, S. J. Mather, A. Roig, X. Ke, G. Van Tendeloo, R. T. M. de Rosales, G. Tobias and K. T. Al-Jamal, Adv. Funct. Mater., 2014, 24, 1880–1894 CrossRef CAS PubMed.
- B. Chen, H. Zhang, C. Zhai, N. Du, C. Sun, J. Xue, D. Yang, H. Huang, B. Zhang, Q. Xie and Y. Wu, J. Mater. Chem., 2010, 20, 9895–9902 RSC.
- R. Klingeler, S. Hampel and B. Buchner, Int. J. Hyperthermia, 2008, 24, 496–505 CrossRef CAS PubMed.
- I. Moench, A. Meye and A. Leonhardt, Nanotechnologies for the Life Sciences, Wiley-VCH, 2007, DOI:10.1002/9783527610419.ntls0068.
- M. Foldvari and M. Bagonluri, Nanomedicine, 2008, 4, 173–182 CrossRef CAS PubMed.
- Z. Liu, S. Tabakman, K. Welsher and H. Dai, Nano Res., 2009, 2, 85–120 CrossRef CAS PubMed.
- M. Foldvari and M. Bagonluri, Nanomedicine, 2008, 4, 183–200 CrossRef CAS PubMed.
- N. A. Monteiro-Riviere and A. O. Inman, Carbon, 2006, 44, 1070–1078 CrossRef CAS.
- M. VanHandel, D. Alizadeh, L. Zhang, B. Kateb, M. Bronikowski, H. Manohara and B. Badie, J. Neuroimmunol., 2009, 208, 3–9 CrossRef CAS PubMed.
- D. Pantarotto, J. P. Briand, M. Prato and A. Bianco, Chem. Commun., 2004, 16–17 RSC.
- N. A. Monteiro-Riviere, R. J. Nemanich, A. O. Inman, Y. Y. Wang and J. E. Riviere, Toxicol. Lett., 2005, 155, 377–384 CrossRef CAS PubMed.
- A. E. Porter, M. Gass, K. Muller, J. N. Skepper, P. A. Midgley and M. Welland, Nat. Nanotechnol., 2007, 2, 713–717 CrossRef CAS PubMed.
- S. Hampel, D. Kunze, D. Haase, K. Kramer, M. Rauschenbach, M. Ritschel, A. Leonhardt, J. Thomas, S. Oswald, V. Hoffmann and B. Buchner, Nanomedicine, 2008, 3, 175–182 CrossRef CAS PubMed.
- Z. Liu, X. Sun, N. Nakayama-Ratchford and H. Dai, ACS Nano, 2007, 1, 50–56 CrossRef CAS PubMed.
- Z. Liu, W. Cai, L. He, N. Nakayama, K. Chen, X. Sun, X. Chen and H. Dai, Nat. Nanotechnol., 2007, 2, 47–52 CrossRef CAS PubMed.
- A. Bianco, K. Kostarelos, C. D. Partidos and M. Prato, Chem. Commun., 2005, 571–577 RSC.
- L. Lacerda, A. Bianco, M. Prato and K. Kostarelos, Adv. Drug Delivery Rev., 2006, 58, 1460–1470 CrossRef CAS PubMed.
- N. Sinha and J. T. Yeow, IEEE Trans Nanobioscience, 2005, 4, 180–195 CrossRef PubMed.
- T. L. Moore, S. W. Grimes, R. L. Lewis and F. Alexis, Mol. Pharmaceutics, 2014, 11, 276–282 CrossRef CAS PubMed.
- D. Pantarotto, R. Singh, D. McCarthy, M. Erhardt, J. P. Briand, M. Prato, K. Kostarelos and A. Bianco, Angew. Chem., Int. Ed., 2004, 43, 5242–5246 CrossRef CAS PubMed.
- R. A. Graff, T. M. Swanson and M. S. Strano, Chem. Mater., 2008, 20, 1824–1829 CrossRef CAS.
- S. K. Smart, A. I. Cassady, G. Q. Lu and D. J. Martin, Carbon, 2006, 44, 1034 CrossRef CAS.
- M. R. McDevitt, D. Chattopadhyay, B. J. Kappel, J. S. Jaggi, S. R. Schiffman, C. Antczak, J. T. Njardarson, R. Brentjens and D. A. Scheinberg, J. Nucl. Med., 2007, 48, 1180–1189 CrossRef CAS PubMed.
- R. Bandyopadhyaya, E. Nativ-Roth, O. Regev and R. Yerushalmi-Rozen, Nano Lett., 2002, 2, 25–28 CrossRef CAS.
- C. Bertulli, H. J. Beeson, T. Hasan and Y. Y. Huang, Nanotechnology, 2013, 24, 265102 CrossRef PubMed.
- M. F. Islam, E. Rojas, D. M. Bergey, A. T. Johnson and A. G. Yodh, Nano Lett., 2003, 3, 269–273 CrossRef CAS.
- N. Hadidi, F. Kobarfard, N. Nafissi-Varcheh and R. Aboofazeli, Int. J. Nanomed., 2011, 6, 737–746 CAS.
- M. Zheng, A. Jagota, E. D. Semke, B. A. Diner, R. S. McLean, S. R. Lustig, R. E. Richardson and N. G. Tassi, Nat. Mater., 2003, 2, 338–342 CrossRef CAS PubMed.
- Y. Li, X. Zhang, J. Luo, W. Huang, J. Cheng, Z. Luo, T. Li, F. Liu, G. Xu, X. Ke, L. Li and H. J. Geise, Nanotechnology, 2004, 15, 1645 CrossRef CAS.
- K. Bhattacharya, F. T. Andon, R. El-Sayed and B. Fadeel, Adv. Drug Delivery Rev., 2013, 65, 2087–2097 CrossRef CAS PubMed.
- V. A. Basiuk, C. Salvador-Morales, E. V. Basiuk, R. M. J. Jacobs, M. Ward, B. T. Chu, R. B. Sim and M. L. H. Green, J. Mater. Chem., 2006, 16, 4420–4426 RSC.
- H. Dumortier, S. Lacotte, G. Pastorin, R. Marega, W. Wu, D. Bonifazi, J. P. Briand, M. Prato, S. Muller and A. Bianco, Nano Lett., 2006, 6, 1522–1528 CrossRef CAS PubMed.
- M. Shim, N. W. Shi Kam, R. J. Chen, Y. Li and H. Dai, Nano Lett., 2002, 2, 285–288 CrossRef CAS.
- C. Salvador-Morales, E. V. Basiuk, V. A. Basiuk, M. L. Green and R. B. Sim, J. Nanosci. Nanotechnol., 2008, 8, 2347–2356 CrossRef CAS PubMed.
- Y. H. Kang, L. A. Tan, M. V. Carroll, M. E. Gentle and R. B. Sim, Adv. Exp. Med. Biol., 2009, 653, 117–128 CrossRef CAS PubMed.
- U. Kishore and K. B. Reid, Immunopharmacology, 1999, 42, 15–21 CrossRef CAS PubMed.
- M. V. Carroll and R. B. Sim, Adv. Drug Delivery Rev., 2011, 63, 965–975 CrossRef CAS PubMed.
- L. Kouser, M. Abdul-Aziz, A. Nayak, C. M. Stover, R. B. Sim and U. Kishore, Front. Immunol., 2013, 4, 93 Search PubMed.
- U. Kishore and R. B. Sim, Immunobiology, 2012, 217, 162–168 CrossRef CAS PubMed.
- J. Leleux and K. Roy, Adv. Healthcare Mater., 2013, 2, 72–94 CrossRef CAS PubMed.
- C. Salvador-Morales, E. Flahaut, E. Sim, J. Sloan, M. L. Green and R. B. Sim, Mol. Immunol., 2006, 43, 193–201 CrossRef CAS PubMed.
- C. Gräfe, A. Weidner, M. V. D. Lühe, C. Bergemann, F. H. Schacher, J. H. Clement and S. Dutz, Int. J. Biochem. Cell Biol., 2016, 75, 196–202 CrossRef PubMed.
- G. Maiorano, S. Sabella, B. Sorce, V. Brunetti, M. A. Malvindi, R. Cingolani and P. P. Pompa, ACS Nano, 2010, 4, 7481–7491 CrossRef CAS PubMed.
- C. Sacchetti, K. Motamedchaboki, A. Magrini, G. Palmieri, M. Mattei, S. Bernardini, N. Rosato, N. Bottini and M. Bottini, ACS Nano, 2013, 7, 1974–1989 CrossRef CAS PubMed.
- A. Gustafsson, E. Lindstedt, L. S. Elfsmark and A. Bucht, J. Immunotoxicol., 2011, 8, 111–121 CrossRef CAS PubMed.
- Y. K. Lee, E. J. Choi, T. J. Webster, S. H. Kim and D. Khang, Int. J. Nanomed., 2014, 10, 97–113 Search PubMed.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggard, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- P. Aggarwal, J. B. Hall, C. B. McLeland, M. A. Dobrovolskaia and S. E. McNeil, Adv. Drug Delivery Rev., 2009, 61, 428–437 CrossRef CAS PubMed.
- M. Rahman, S. Laurent, N. Tawil, L. Yahi and M. Mahmoudi, in Protein-nanoparticle interactions, ed. Anonymous, Springer, 2013, pp. 21–44 Search PubMed.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS PubMed.
- M. Rybak-Smith, K. M. Pondman, E. Flahaut, C. Salvador-Morales and R. B. Sim, in Carbon Nanotubes for Biomedical Applications, ed. R. Klingeler and B. R. Sim, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 183–210 Search PubMed.
- J. H. Shannahan, J. M. Brown, R. Chen, P. C. Ke, X. Lai, S. Mitra and F. A. Witzmann, Small, 2013, 9, 2171–2181 CrossRef CAS PubMed.
- C. Salvador-Morales, E. Flahaut, E. Sim, J. Sloan, M. L. Green and R. B. Sim, Mol. Immunol., 2006, 43, 193–201 CrossRef CAS PubMed.
- C. Salvador-Morales, P. Townsend, E. Flahaut, C. Vénien-Bryan, A. Vlandas, M. L. H. Green and R. B. Sim, Carbon, 2007, 45, 607–617 CrossRef CAS.
- X. Cai, R. Ramalingam, H. S. Wong, J. Cheng, P. Ajuh, S. H. Cheng and Y. W. Lam, Nanomedicine, 2013, 9, 583–593 CrossRef CAS PubMed.
- A. Chanan-Khan, J. Szebeni, S. Savay, L. Liebes, N. M. Rafique, C. R. Alving and F. M. Muggia, Ann. Oncol., 2003, 14, 1430–1437 CrossRef CAS PubMed.
- C. Salvador-Morales, M. L. H. Green and R. B. Sim, in Chemistry of carbon nanotubes, ed. E. V Basiuk and V. A. Basiuk, American Scientific Publishers, 2007 Search PubMed.
- S. M. Moghimi, A. J. Andersen, D. Ahmadvand, P. P. Wibroe, T. L. Andresen and A. C. Hunter, Adv. Drug Delivery Rev., 2011, 63, 1000–1007 CrossRef CAS PubMed.
- W. L. Ling, A. Biro, I. Bally, P. Tacnet, A. Deniaud, E. Doris, P. Frachet, G. Schoehn, E. Pebay-Peyroula and G. J. Arlaud, ACS Nano, 2011, 5, 730–737 CrossRef CAS PubMed.
- K. M. Pondman, A. G. Tsolaki, B. Paudyal, M. H. Shamji, A. Switzer, A. A. Pathan, S. M. Abozaid, B. Ten Haken, G. Stenbeck, R. B. Sim and U. Kishore, J. Biomed. Nanotechnol., 2016, 12, 197–216 CrossRef CAS PubMed.
- K. M. Pondman, M. Sobik, A. Nayak, A. G. Tsolaki, A. Jakel, E. Flahaut, S. Hampel, B. Ten Haken, R. B. Sim and U. Kishore, Nanomedicine, 2014, 10, 1287–1299 CrossRef CAS PubMed.
- M. J. Rybak-Smith and R. B. Sim, Adv. Drug Delivery Rev., 2011, 63, 1031–1041 CrossRef CAS PubMed.
- U. Kishore, R. Ghai, T. J. Greenhough, A. K. Shrive, D. M. Bonifati, M. G. Gadjeva, P. Waters, M. S. Kojouharova, T. Chakraborty and A. Agrawal, Immunol. Lett., 2004, 95, 113–128 CrossRef CAS PubMed.
- C. Salvador-Morales, E. Flahaut, E. Sim, J. Sloan, M. L. Green and R. B. Sim, Mol. Immunol., 2006, 43, 193–201 CrossRef CAS PubMed.
- C. Kemper, L. M. Mitchell, L. Zhang and D. E. Hourcade, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9023–9028 CrossRef CAS PubMed.
- W. Xu, S. P. Berger, L. A. Trouw, H. C. de Boer, N. Schlagwein, C. Mutsaers, M. R. Daha and C. van Kooten, J. Immunol., 2008, 180, 7613–7621 CrossRef CAS.
- L. Kouser, B. Paudyal, A. Kaur, E. Flauhat, H. A. Khan, C. M. Stover, R. B. Sim and U. Kishore, Front. Immunol., 2016 Search PubMed , submitted.
- I. Hamad, A. Christy Hunter, K. J. Rutt, Z. Liu, H. Dai and S. Moein Moghimi, Mol. Immunol., 2008, 45, 3797–3803 CrossRef CAS PubMed.
- K. M. Pondman, L. Pednekar, B. Paudyal, A. G. Tsolaki, L. Kouser, H. A. Khan, M. H. Shamji, B. Ten Haken, G. Stenbeck, R. B. Sim and U. Kishore, Nanomedicine, 2015, 11, 2109–2118 CrossRef CAS PubMed.
- J. K. Gbadamosi, A. C. Hunter and S. M. Moghimi, FEBS Lett., 2002, 532, 338–344 CrossRef CAS PubMed.
- A. J. Andersen, J. T. Robinson, H. Dai, A. C. Hunter, T. L. Andresen and S. M. Moghimi, ACS Nano, 2013, 7, 1108–1119 CrossRef CAS PubMed.
- A. J. Andersen, B. Windschiegl, S. Ilbasmis-Tamer, I. T. Degim, A. C. Hunter, T. L. Andresen and S. M. Moghimi, Nanomedicine, 2013, 9, 469–473 CrossRef CAS PubMed.
- J. Hellwage, T. Meri, T. Heikkila, A. Alitalo, J. Panelius, P. Lahdenne, I. J. Seppala and S. Meri, J. Biol. Chem., 2001, 276, 8427–8435 CrossRef CAS PubMed.
- R. Gref, M. Luck, P. Quellec, M. Marchand, E. Dellacherie, S. Harnisch, T. Blunk and R. H. Muller, Colloids Surf., B, 2000, 18, 301–313 CrossRef CAS PubMed.
- C. A. Ogden, A. deCathelineau, P. R. Hoffmann, D. Bratton, B. Ghebrehiwet, V. A. Fadok and P. M. Henson, J. Exp. Med., 2001, 194, 781–795 CrossRef CAS PubMed.
- J. Meng, X. Li, C. Wang, H. Guo, J. Liu and H. Xu, ACS Appl. Mater. Interfaces, 2015, 7, 3180–3188 CAS.
- S. Hussain, J. A. Vanoirbeek and P. H. Hoet, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2012, 4, 169–183 CrossRef CAS PubMed.
- L. Tabet, C. Bussy, A. Setyan, A. Simon-Deckers, M. J. Rossi, J. Boczkowski and S. Lanone, Part. Fibre Toxicol., 2011, 8, 3 CrossRef CAS PubMed.
- K. Kostarelos, L. Lacerda, G. Pastorin, W. Wu, S. Wieckowski, J. Luangsivilay, S. Godefroy, D. Pantarotto, J. Briand, S. Muller, M. Prato and A. Bianco, Nat. Nanotechnol., 2007, 2, 108–113 CrossRef CAS PubMed.
- J. Palomaki, E. Valimaki, J. Sund, M. Vippola, P. A. Clausen, K. A. Jensen, K. Savolainen, S. Matikainen and H. Alenius, ACS Nano, 2011, 5, 6861–6870 CrossRef CAS PubMed.
- G. Laverny, A. Casset, A. Purohit, E. Schaeffer, C. Spiegelhalter, F. de Blay and F. Pons, Toxicol. Lett., 2013, 217, 91–101 CrossRef CAS PubMed.
- N. W. Kam, Z. Liu and H. Dai, Angew. Chem., Int. Ed., 2006, 45, 577–581 CrossRef CAS PubMed.
- B. Kang, D. C. Yu, S. Q. Chang, D. Chen, Y. D. Dai and Y. Ding, Nanotechnology, 2008, 19, 375103 CrossRef PubMed , epub 2008 Aug 1.
- Q. Mu, D. L. Broughton and B. Yan, Nano Lett., 2009, 9, 4370–4375 CrossRef CAS PubMed.
- H. Jin, D. A. Heller and M. S. Strano, Nano Lett., 2008, 8, 1577–1585 CrossRef PubMed.
- F. A. Murphy, A. Schinwald, C. A. Poland and K. Donaldson, Part. Fibre Toxicol., 2012, 9, 8 CrossRef CAS PubMed.
- D. Liu, L. Wang, Z. Wang and A. Cuschieri, Nanoscale Res. Lett., 2012, 7, 361 CrossRef PubMed.
- D. M. Brown, I. A. Kinloch, U. Bangert, A. H. Windle, D. M. Walter, G. S. Walker, C. A. Scotchford, K. Donaldson and V. Stone, Carbon, 2007, 45, 1743 CrossRef CAS.
- C. P. Chio, S. C. Chen, K. C. Chiang, W. C. Chou and C. M. Liao, Sci. Total Environ., 2007, 387, 113–127 CrossRef CAS PubMed.
- E. Meunier, A. Coste, D. Olagnier, H. Authier, L. Lefevre, C. Dardenne, J. Bernad, M. Beraud, E. Flahaut and B. Pipy, Nanomedicine, 2012, 8, 987–995 CrossRef CAS PubMed.
- E. Koike, H. Takano, K. Inoue, R. Yanagisawa and T. Kobayashi, Chemosphere, 2008, 73, 371–376 CrossRef CAS PubMed.
- M. Yang, K. Flavin, I. Kopf, G. Radics, C. H. Hearnden, G. J. McManus, B. Moran, A. Villalta-Cerdas, L. A. Echegoyen, S. Giordani and E. C. Lavelle, Small, 2013, 9, 4194–4206 CrossRef CAS PubMed.
- V. C. Sanchez, J. R. Pietruska, N. R. Miselis, R. H. Hurt and A. B. Kane, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2009, 1, 511–529 CrossRef CAS PubMed.
- C. W. Lam, J. T. James, R. McCluskey and R. L. Hunter, Toxicol. Sci., 2004, 77, 126–134 CrossRef CAS PubMed.
- D. B. Warheit, B. R. Laurence, K. L. Reed, D. H. Roach, G. A. Reynolds and T. R. Webb, Toxicol. Sci., 2004, 77, 117–125 CrossRef CAS PubMed.
- J. B. Mangum, E. A. Turpin, A. Antao-Menezes, M. F. Cesta, E. Bermudez and J. C. Bonner, Part. Fibre Toxicol., 2006, 3, 15 CrossRef PubMed.
- A. A. Shvedova, E. R. Kisin, R. Mercer, A. R. Murray, V. J. Johnson, A. I. Potapovich, Y. Y. Tyurina, O. Gorelik, S. Arepalli, D. Schwegler-Berry, A. F. Hubbs, J. Antonini, D. E. Evans, B. K. Ku, D. Ramsey, A. Maynard, V. E. Kagan, V. Castranova and P. Baron, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2005, 289, L698–L708 CrossRef CAS PubMed.
- A. R. Murray, E. R. Kisin, A. V. Tkach, N. Yanamala, R. Mercer, S. H. Young, B. Fadeel, V. E. Kagan and A. A. Shvedova, Part. Fibre Toxicol., 2012, 9, 10 CrossRef CAS PubMed.
- A. A. Shvedova, J. P. Fabisiak, E. R. Kisin, A. R. Murray, J. R. Roberts, Y. Y. Tyurina, J. M. Antonini, W. H. Feng, C. Kommineni, J. Reynolds, A. Barchowsky, V. Castranova and V. E. Kagan, Am. J. Respir. Cell Mol. Biol., 2008, 38, 579–590 CrossRef CAS PubMed.
- A. A. Shvedova, E. Kisin, A. R. Murray, V. J. Johnson, O. Gorelik, S. Arepalli, A. F. Hubbs, R. R. Mercer, P. Keohavong, N. Sussman, J. Jin, J. Yin, S. Stone, B. T. Chen, G. Deye, A. Maynard, V. Castranova, P. A. Baron and V. E. Kagan, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2008, 295, L552–L565 CrossRef CAS PubMed.
- A. A. Shvedova, E. R. Kisin, A. R. Murray, C. Kommineni, V. Castranova, B. Fadeel and V. E. Kagan, Toxicol. Appl. Pharmacol., 2008, 231, 235–240 CrossRef CAS PubMed.
- D. W. Porter, A. F. Hubbs, R. R. Mercer, N. Wu, M. G. Wolfarth, K. Sriram, S. Leonard, L. Battelli, D. Schwegler-Berry, S. Friend, M. Andrew, B. T. Chen, S. Tsuruoka, M. Endo and V. Castranova, Toxicology, 2010, 269, 136–147 CrossRef CAS PubMed.
- U. Kishore, T. J. Greenhough, P. Waters, A. K. Shrive, R. Ghai, M. F. Kamran, A. L. Bernal, K. B. Reid, T. Madan and T. Chakraborty, Mol. Immunol., 2006, 43, 1293–1315 CrossRef CAS PubMed.
- C. Salvador-Morales, Z. Khan, J. Zamory, V. Tran, A. Cedeno, J. Umanzor-Alvarez, U. Kishore and R. B. Sim, J. Adv. Microsc. Res., 2013, 8, 93–99 CrossRef CAS.
- M. Marchetti, M. S. Shaffer, M. Zambianchi, S. Chen, F. Superti, S. Schwander, A. Gow, J. J. Zhang, K. F. Chung, M. P. Ryan, A. E. Porter and T. D. Tetley, Philos. Trans. R. Soc., B, 2015, 370, 20140038 CrossRef PubMed.
- A. A. Kapralov, W. H. Feng, A. A. Amoscato, N. Yanamala, K. Balasubramanian, D. E. Winnica, E. R. Kisin, G. P. Kotchey, P. Gou, L. J. Sparvero, P. Ray, R. K. Mallampalli, J. Klein-Seetharaman, B. Fadeel, A. Star, A. A. Shvedova and V. E. Kagan, ACS Nano, 2012, 6, 4147–4156 CrossRef CAS PubMed.
- M. Gasser, P. Wick, M. J. Clift, F. Blank, L. Diener, B. Yan, P. Gehr, H. F. Krug and B. Rothen-Rutishauser, Part. Fibre Toxicol., 2012, 9, 17-8977-9-17 CrossRef PubMed.
- K. M. Pondman, B. Paudyal, R. B. Sim, A. Kaur, L. Kouser, A. G. Tsolaki, A. L. Jones, C. Salvador-Morales, A. H. Khan, B. Ten Haken, G. Stenbeck and U. Kishore, Nanoscale, 2017, 9, 1097–1109 RSC.
- R. Li, X. Wang, Z. Ji, B. Sun, H. Zhang, C. H. Chang, S. Lin, H. Meng, Y. P. Liao, M. Wang, Z. Li, A. A. Hwang, T. B. Song, R. Xu, Y. Yang, J. I. Zink, A. E. Nel and T. Xia, ACS Nano, 2013, 7, 2352–2368 CrossRef CAS PubMed.
- S. Jain, V. S. Thakare, M. Das, C. Godugu, A. K. Jain, R. Mathur, K. Chuttani and A. K. Mishra, Chem. Res. Toxicol., 2011, 24, 2028–2039 CrossRef CAS PubMed.
- H. Nagai, Y. Okazaki, S. H. Chew, N. Misawa, Y. Yamashita, S. Akatsuka, T. Ishihara, K. Yamashita, Y. Yoshikawa, H. Yasui, L. Jiang, H. Ohara, T. Takahashi, G. Ichihara, K. Kostarelos, Y. Miyata, H. Shinohara and S. Toyokuni, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, E1330–E1338 CrossRef PubMed.
- B. Kang, S. Chang, Y. Dai, D. Yu and D. Chen, Small, 2010, 6, 2362–2366 CrossRef CAS PubMed.
- H. Haniu, N. Saito, Y. Matsuda, Y. A. Kim, K. C. Park, T. Tsukahara, Y. Usui, K. Aoki, M. Shimizu, N. Ogihara, K. Hara, S. Takanashi, M. Okamoto, N. Ishigaki, K. Nakamura and H. Kato, Int. J. Nanomed., 2011, 6, 3295–3307 CrossRef CAS PubMed.
- Y. Zhang, Y. Xu, Z. Li, T. Chen, S. M. Lantz, P. C. Howard, M. G. Paule, W. Slikker Jr, F. Watanabe, T. Mustafa, A. S. Biris and S. F. Ali, ACS Nano, 2011, 5, 7020–7033 CrossRef CAS PubMed.
- M. H. Cato, F. D'Annibale, D. M. Mills, F. Cerignoli, M. I. Dawson, E. Bergamaschi, N. Bottini, A. Magrini, A. Bergamaschi, N. Rosato, R. C. Rickert, T. Mustelin and M. Bottini, J. Nanosci. Nanotechnol., 2008, 8, 2259–2269 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |