
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of graphene oxide supports on solution-phase catalysis of thiolate-protected palladium nanoparticles in water†
Vivian
Chen
,
Hanqing
Pan
,
Roxanne
Jacobs
,
Shahab
Derakhshan
and
Young-Seok
Shon
 *
*
Department of Chemistry and Biochemistry, California State University, Long Beach, 1250 Bellflower Blvd., Long Beach, California 90840-9507, USA. E-mail: ys.shon@csulb.edu
First published on 11th November 2016
Abstract
The influence of graphene oxide supports and thiolate surface ligands on the catalytic activity of colloidal Pd nanoparticles for alkyne hydrogenation in water is investigated. The studies show that unsupported, water-soluble thiolate-capped Pd nanoparticle catalysts favor semi-hydrogenation over full-hydrogenation of dimethyl acetylene dicarboxylate (DMAD) under atmospheric pressure and at room temperature. Pd nanoparticles supported on graphene oxide exhibit a similar activity for the hydrogenation of DMAD, but they show an improved long-term colloidal stability in aqueous solution after multiple catalytic cycles. After the heat treatment of Pd nanoparticles supported on graphene oxide at 300 °C, these heated hybrids exhibit an enhanced catalytic activity towards full-hydrogenation. Overall, the studies suggest some influences of graphene oxide supports on the stability, and thiolate surface ligands on the activity and selectivity of Pd nanoparticle catalysts.
Introduction
Due to the high surface area-to-mass ratio and size/shape dependent activity,1–3 metal nanoparticles have gained more interest for practical applications as efficient catalysts that offer the combination of activity, selectivity, and recyclability.4–6 In particular, palladium nanoparticles (PdNPs) have been frequently used as catalysts in organic reactions including hydrogenation and cross-coupling reactions.7,8 Since small PdNPs are thermodynamically unstable and tend to aggregate or experience core dissolution during catalytic reactions, an increase in stability of PdNPs is necessary for improved recovery and recycling by using either solid supports or more effective stabilizing ligands.7–11Previous studies have shown that the performance of nanoparticle catalysts is influenced by the presence of various surface ligand stabilizers, such as polymers,9 dendrimers,11 and small organic ligands including alkylammoniums, thioethers, isocyanides and amines.7 Both inorganic (sulfur, selenium, barium sulfate, calcium carbonate or lead acetate) and organic (nitrogen heterocycles like pyridine and quinoline) reagents are also known as surface-bound poisons that control the activity of Pd catalysts.12,13 Lindlar's catalyst is an excellent example of poisoned heterogeneous Pd catalysts useful for selective hydrogenation of alkynes to alkenes.12 Recent studies have shown that thiols can be utilized to deactivate the surface of supported Pd catalysts providing higher selectivity for certain organic reactions.14–16 Previous work by our group also proved that alkanethiolate-capped PdNPs generated from sodium S-alkylthiosulfate is an excellent catalyst for regio- and chemo-selective organic reactions including the hydrogenation and/or isomerization of various alkenes, dienes, alkynols, and allylic alcohols.17–20
Controls over the structure and functionality of thiolate ligands of these PdNPs also provided the ability to create either homogeneously soluble or heterogeneous catalytic systems in different solvents.21,22 For example, the presence of a terminal carboxylate group in the thiolate ligand allows for the formation of homogeneously soluble aqueous systems, while that of simple alkanethiolate ligands led to heterogeneous systems in water.22 This enabled PdNPs capped with water-soluble thiolate ligands to be useful in colloidal catalytic reactions in aqueous environments as nanoparticle-based green catalysts.
In consideration of the relatively low colloidal stability of nanoparticles in the solution phase, the attachment of PdNPs to water-dispersible support materials would be an ideal strategy for enhancing the utility of nanoparticle catalysts as support-stabilized aqueous catalytic systems.23–25 Graphene oxide (GO) would be a perfect candidate as a support material for colloidal catalysts considering its characteristics of forming a highly stable aqueous dispersion and possessing a large flat area-to-mass ratio. Therefore, the catalytic PdNPs could possibly avoid extensive aggregation in the solution phase after the immobilization on GO.26,27 In addition, the presence of hydrophobic groups in GO surfaces may facilitate the adsorption of substrates and accelerate the catalytic reactions for hydrophobic organic compounds.
Previously, others have shown that metal nanoparticles anchored on graphene sheets can exhibit enhanced catalytic, magnetic, electrical and optical activities.28–34 Most other attempts to develop hybrid GO materials were aimed at maximizing their potential for device applications such as batteries, supercapacitors, fuel cells, photovoltaic devices, sensors and surface enhanced Raman scattering (SERS).35–37 This paper specifically focuses on understanding the effects of GO supports and surface thiolate ligands on controlling the activity and selectivity of water-soluble PdNP catalysts for a model hydrophobic alkyne hydrogenation reaction.
Experimental
Materials and methods
The following materials were purchased from the indicated suppliers and used as received: potassium tetrachloropalladate(II) (K2PdCl4), tetraoctylammonium bromide (TOAB), sodium borohydride (NaBH4) and 6-bromohexanoic acid were purchased from ACROS. Dimethyl acetylene dicarboxylate (DMAD) and graphene oxide (GO) [4 mg mL−1 in H2O] were purchased from Sigma Aldrich. D2O was purchased from Cambridge Isotope Laboratories. Sodium thiosulfate (Na2S2O3·5H2O), toluene, acetone, acetonitrile, methanol, ethanol, chloroform and tetrahydrofuran (THF) were obtained from Fisher Scientific. Spectra/Por cellulose ester (CE) dialysis membranes (MW = 8000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Daltons) were purchased from Spectrum Laboratories, Inc. Water was purified using a Barnstead NANOpure Diamond ion exchange resin purification unit.
000 Daltons) were purchased from Spectrum Laboratories, Inc. Water was purified using a Barnstead NANOpure Diamond ion exchange resin purification unit.
Preparations
Catalysis
Catalysis experiments were performed by dissolving 4.8 mol% (based on Pd/substrate) of PdNPs, unheated PdNP/GO or heated PdNP/GO hybrid catalysts in 2 mL D2O in a 35 mL reaction flask. The amount of Pd remained constant for all catalysts. The flask was sealed and purged with H2 gas for 15 min with the pressure being monitored using a manometer to maintain a flow rate of around 3.4 psi. Once purging was complete, 90 μL (0.73 mmol) of dimethyl acetylene dicarboxylate (DMAD) was then injected into the reaction mixture. The reaction was stirred under atmospheric pressure and at room temperature. 1H NMR spectra of the solutions were obtained after 3, 12 or 24 hours.Recycling of catalysts
Recycling experiments were performed following the catalysis procedure. After the 1H NMR data were analysed, the PdNP, PdNP/GO or heated PdNP/GO catalysts in the NMR tubes were returned to the reaction flask. The flask was then washed with methanol followed by centrifugation of the nanoparticles. The centrifugation was repeated twice more with washings of methanol. The catalysts were then dried in a vacuum before the next cycle.Characterizations
1H NMR spectra were recorded on Bruker Fourier or Advances II FT-NMR spectrometers operating at either 300 or 400 MHz, respectively. PdNPs were dissolved in D2O, which was internally referenced at δ 4.81 ppm. UV-vis measurements of the NP solutions were carried out using a UV-2450 Shimadzu UV-vis spectrophotometer using quartz cells. The spectra were recorded from 4500 to 450 cm−1. Infrared spectra were recorded using a Perkin Elmer Spectrum 100 FT-IR spectrometer. The spectra were recorded from 800 to 200 cm−1. Transmission electron microscopy (TEM) images were taken using a JEOL 1200 EX II electron microscope operating at 90 or 80 KeV. Samples dissolved in nanopure water were cast onto carbon-coated copper mesh grids, and dried for at least 24 h before analysis. PdNP core sizes were analysed with Scion Image Beta Release 2.0. Powder X-ray diffraction (XRD) data were collected using a PANalytical X'Pert Pro MPD diffractometer, equipped with a linear X'Celerator detector, with monochromatic Cu-Kα1 radiation. The data were collected at room temperature in the range 17° < 2Theta < 100° with ≈0.008° intervals. The pattern match process was performed using the HighScore Plus software.Results and discussion
Spectroscopic and microscopic characterization of Pd nanoparticles and Pd nanoparticle–graphene oxide hybrids
Water-soluble ω-carboxylate-1-hexanethiolate-capped PdNPs were synthesized by utilizing sodium ω-carboxyl-S-hexanethiosulfate as a ligand precursor in a biphasic synthetic system described in our previous report.22 Briefly, the synthesis began with the phase transfer of PdCl42− to an organic layer by TOAB (Scheme 1). After the completion of the phase transfer, the ω-carboxyl-S-hexanethiosulfate salt was added along with additional TOAB. The addition of NaBH4 resulted in the formation of PdNPs in the organic phase, which quickly transferred into the aqueous layer. FT-IR and UV-vis spectra and TGA results of isolated PdNPs were identical with the previous results and confirmed the formation of thiolate-capped PdNPs (Fig. S1 and S2, ESI†). The TEM image and histogram of PdNPs showed that the nanoparticles were approximately 1.9 ± 0.9 nm in diameter without any evidence of aggregation and their cores were spherical and relatively monodispersed (Fig. 1(a)).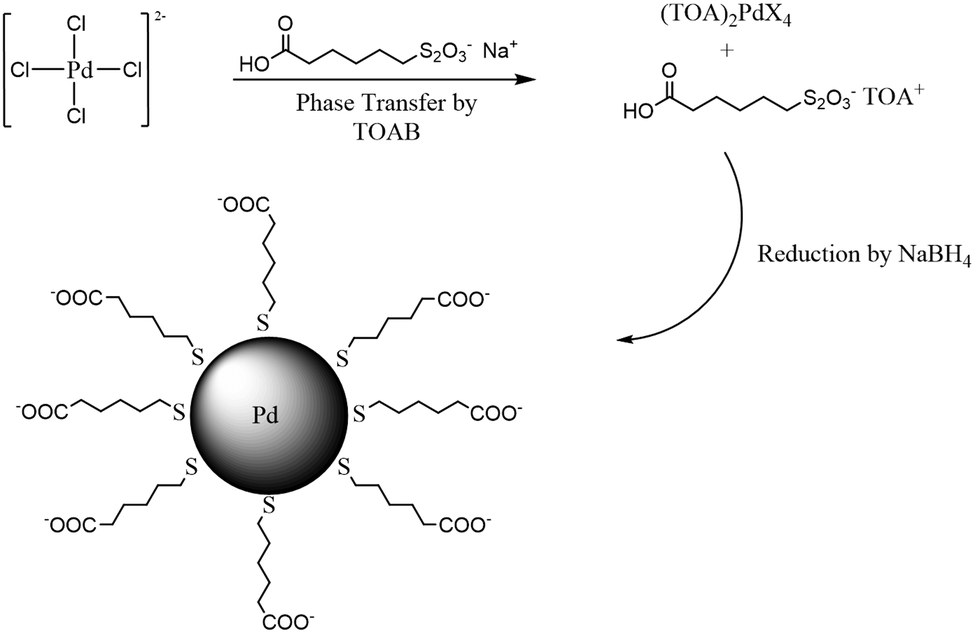 | ||
| Scheme 1 Synthetic scheme for ω-carboxylate-1-hexanethiolate-capped palladium nanoparticles (PdNPs). | ||
 | ||
| Fig. 1 TEM images and histograms of (a) PdNPs and (b) PdNP/GO hybrids. Scale bars are 20 nm. | ||
PdNPs were self-assembled onto the surface of GO via interactions between the COO−/COOH/OH groups of GO and the COO−/COOH groups of PdNPs.39–41 The UV-vis spectrum of the PdNP/GO hybrids shown in Fig. S3(a) is consistent with the spectra of other palladium nanoparticles with similar core sizes, which show no absorption or plasmon bands with only an exponential decay with increasing wavelength from 300 nm to 800 nm.19 The IR spectrum of PdNP/GO shown in Fig. S3(b) showed a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch at 1725 cm−1, a sp3 C–H stretch at 2938–2878 cm−1 and a broad O–H stretch at 3300–2800 cm−1, corresponding to the IR characteristics of ω-carboxylate-1-hexanethiolate ligands and GO. Fig. 1(b) shows the TEM image and histogram of PdNPs after the immobilization on GO, indicating the retention of the core size and dispersity of the assembled nanoparticles (2.0 ± 0.8 nm) compared to the PdNPs before the adsorption (1.9 ± 0.9 nm).
O stretch at 1725 cm−1, a sp3 C–H stretch at 2938–2878 cm−1 and a broad O–H stretch at 3300–2800 cm−1, corresponding to the IR characteristics of ω-carboxylate-1-hexanethiolate ligands and GO. Fig. 1(b) shows the TEM image and histogram of PdNPs after the immobilization on GO, indicating the retention of the core size and dispersity of the assembled nanoparticles (2.0 ± 0.8 nm) compared to the PdNPs before the adsorption (1.9 ± 0.9 nm).
Heat treatments of palladium nanoparticle–graphene oxide hybrids
Heat treatments of PdNP/GO at a temperature ranging from 50 to 400 °C in air were performed to understand the chemical and morphological transformations of PdNPs on GO. The IR and UV-vis spectra of PdNP/GO heated at 50–400 °C are shown in Fig. S4 (ESI†). The IR spectra confirm the near-absence of hexanethiolate ligands after heat treatment at a temperature of 300 °C. The UV-vis spectra showed a steady decrease in the intensity of the exponential curves, indicating a gradual loss in the solubility of PdNP/GO with heat treatments at higher temperatures. TEM images showed that the extensive coarsening process started to take place for PdNP/GO when heated at a temperature of 300 °C and above (Fig. 2). Based on the IR results, however, this treatment was necessary for the removal of most thiolate ligands from the nanoparticles. Table S1 (ESI†) summarizes the transitions of the average core size of the heated PdNP/GO. The average core size of the heated PdNP/GO at 300 °C was 7.1 ± 2.9 nm. However, the TEM image in Fig. 2 shows the presence of many small particles on GO after heat treatments at this temperature. These results, therefore, confirm the efficiency of GO in preventing PdNPs from extensive aggregation and coarsening even after the removal of the surface thiolate ligands at 300 °C. The heated PdNP/GO catalysts at 300 °C were no longer dispersed in water due to the loss of the solubilizing ligands of the nanoparticles and the carboxyl groups of GO.39 The powder XRD pattern of the heated Pd/GO at 300 °C clearly indicates the presence of Pd oxide in the hybrid after the treatment (Fig. S5, ESI†). The lack of stabilizing surface ligands on the Pd surface should be the main reason for this facile oxidation of the exposed Pd surface under an atmospheric environment with a high temperature. In comparison, a well-defined XRD pattern could not be obtained from the PdNPs and PdNP/GO samples due to the lack of crystalline structures related to the presence of surface ligands and GO supports.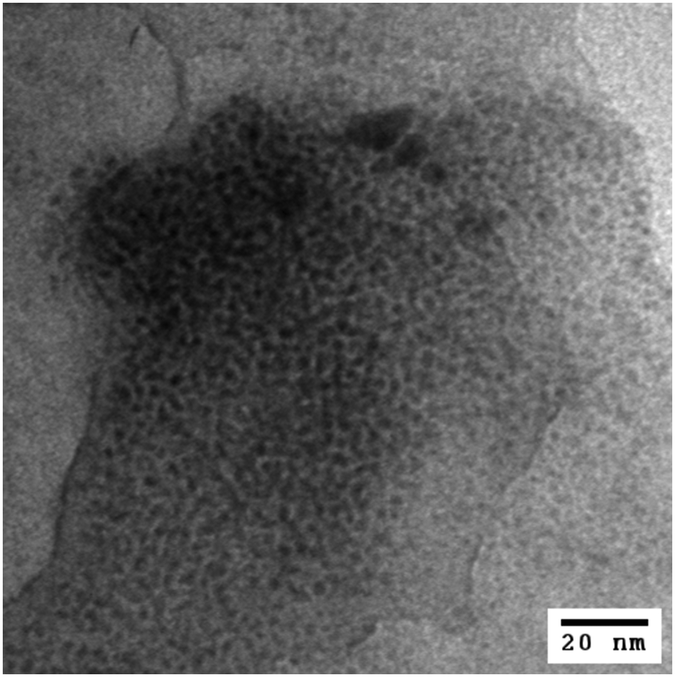 | ||
| Fig. 2 TEM image of PdNP/GO heated at 300 °C. Scale bar is 20 nm. | ||
Catalysis studies comparing PdNPs, PdNP/GO and heated PdNP/GO
The activity and selectivity of PdNPs, PdNP/GO, and heated PdNP/GO (300 °C) were analysed using dimethyl acetylene dicarboxylate (DMAD) as the model alkyne substrate in a reaction involving 4.8 mol% (based on the Pd:DMAD mole ratio) of the catalysts. Reactions were run using a balloon as a constant source of H2 gas, in an attempt to push the reactions to completion and to keep the system at equilibrium for the catalysts. The reactions were monitored using 1H NMR spectroscopy (see Fig. S6, ESI,† for examples). DMAD shows one distinct chemical shift at δ 3.86 ppm (–OC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3). The semi-hydrogenation product has two distinct hydrogens at δ 3.80 ppm (–OC3) and δ 6.50 ppm (CC–). The fully hydrogenated product also has two distinct hydrogens at δ 3.70 ppm (–CO2C3) and δ 2.69 ppm (CH2–C2). By comparing the integrations of the methoxy group (–OC3: δ 3.70–3.86 ppm) of DMAD, the reaction progress could be easily monitored. 1H NMR results were taken after 3 h, 12 h, and 24 h in order to monitor the progress of the reactions.
3). The semi-hydrogenation product has two distinct hydrogens at δ 3.80 ppm (–OC3) and δ 6.50 ppm (CC–). The fully hydrogenated product also has two distinct hydrogens at δ 3.70 ppm (–CO2C3) and δ 2.69 ppm (CH2–C2). By comparing the integrations of the methoxy group (–OC3: δ 3.70–3.86 ppm) of DMAD, the reaction progress could be easily monitored. 1H NMR results were taken after 3 h, 12 h, and 24 h in order to monitor the progress of the reactions.
The kinetic plots of the catalytic hydrogenation of DMAD by three catalysts revealed that PdNP and PdNP/GO catalysts are more selective towards the semi-hydrogenation product (Fig. 3). The presence of GO seemed to have no significant impact over the catalytic activity and selectivity of the PdNPs. The increase in the kinetics of substrate delivery by the spill-over, which has been observed by others for catalysts on graphene substrates, was absent for the colloidal PdNP/GO catalysis.40,41 For PdNP catalysts, most DMAD was hydrogenated to maleic ester, the semi-hydrogenated product in a cis-form (>99% selectivity), in less than 3 h of reaction time. However, the initial rate of reaction using PdNP/GO catalysts was slightly slower despite the fact that the reaction resulted in products with similar selectivities (as shown in Table S2, ESI,† which summarizes the turn-over-frequency (TOF) for semi- and fully hydrogenated products in the hydrogenation of DMAD). Overall, the catalytic activity and selectivity of PdNPs and PdNP/GO were quite comparable to those of other supported Pd catalysts with surface modifiers, which exhibited typical conversion yields of 93–99% with selectivities ranging from 88 to 96% for semi-hydrogenation products at room temperature and under atmospheric pressure.11,22
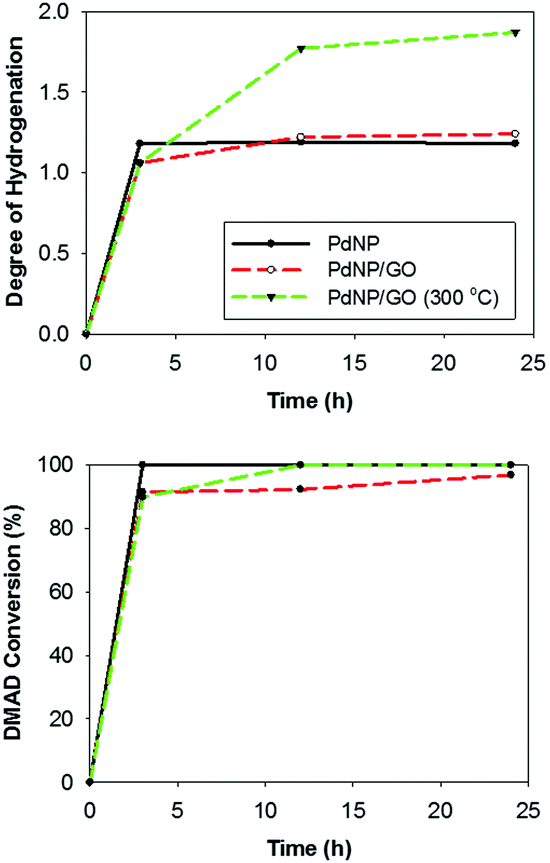 | ||
| Fig. 3 Kinetic plot of the hydrogenation of DMAD using the PdNP, PdNP/GO, and heated PdNP/GO (300 °C) catalysts. The complete hydrogenation of alkyne to alkene requires the degree of hydrogenation to be 2. | ||
The catalytic activity of heated PdNP/GO at 300 °C, which contains an extensive amount of Pd oxide on the surface, was compared to that of the unheated PdNP/GO catalyst. The heated PdNP/GO catalysts mainly produced the fully hydrogenated product (∼87%), indicating their high catalytic activity and the availability of active metal surfaces capable of further hydrogenation. The powder XRD pattern of heated PdNP/GO after 1 catalytic cycle showed no signals corresponding to palladium oxide, which can be understood based on the reduction of PdO during the catalytic reaction in the presence of excess H2 gas (Fig. S7, ESI†). This also explains why the heated PdNP/GO could exhibit enhanced catalytic activity, despite the initial formation of PdO on the surface. The results confirm that the strategy of using GO as a colloidal support for ligand-capped nanoparticle catalysts can be applied for the calcination of thiolate ligands from PdNP catalysts. The initial rate of the reaction was, however, not improved compared to the rate of those in the presence of PdNPs and PdNP/GO. This is likely due to the delay time required for the initial reduction of Pd(II) to Pd(0) and the heated PdNP/GO being a heterogeneous system with low dispersity.
Since GO has shown some activity as carbocatalysts for reactions such as nitrobenzene hydrogenation, it was tested for DMAD hydrogenation.42 The conclusion from these tests was that the effect of direct GO participation in the hydrogenation of DMAD was less than 5% over a 24 h period. The PdNP/GO catalysts heated at 150 °C were also tested for the DMAD hydrogenation, which resulted in poor conversion yields for both semi- and full-hydrogenation products. The results suggest that annealing at 150 °C causes the rearrangement of thiolate ligands on the surface rather than the removal from the surface, decreasing the overall activity of the PdNPs.
Recyclability and colloidal stability of PdNPs, PdNP/GO and heated PdNP/GO
The recyclability of the catalysts was tested in order to determine the reusability and stability of PdNPs with and without GO.43,44 Results obtained from the reactions after the third cycle of catalyst recycling were compared to the averages of the 24 h reaction results using fresh catalysts to determine the catalysts' effectiveness after multiple cycles (Fig. 4). The recycling of the PdNPs and the PdNP/GO hybrid catalysts was successful with both catalysts retaining their catalytic properties after three uses. It was found that the overall rate of hydrogenation dropped slightly (Table S2, ESI†), which was likely due to a small loss of the catalysts during catalyst purification and product isolation. The reactions were still all completed in less than 24 h reaction time. This showed that both the PdNPs and the PdNP/GO hybrid catalysts retained their properties as selective hydrogenation catalysts for alkyne. The heated PdNP/GO experienced, however, a dramatic decrease in catalytic activity after multiple uses, producing more semi-hydrogenation products than full-hydrogenation products after the third cycle. The permanent adsorption of the substrate or intermediates to the bare surface of the heated PdNP/GO was likely the reason for the decrease in the number of available active sites and its overall activity as a consequence. A similar deactivation of metal catalysts has been observed many times by others.45,46 This adsorption problem has been confirmed by the successful regeneration of heated PdNP/GO, which required reheating the hybrid at 300 °C for 1 hour. These regenerated, heated PdNP/GO catalysts yielded the fully hydrogenated product almost quantitatively, indicating that their catalytic activity was regained after the second heat treatment. The powder XRD pattern of the thrice cycled PdNP/GO did not show any evidence for the presence of palladium oxide.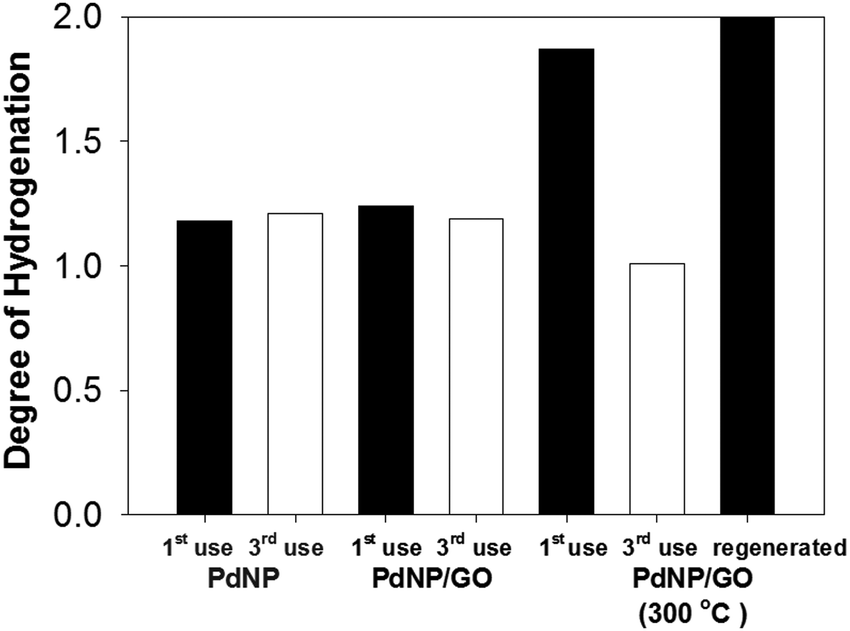 | ||
| Fig. 4 Results from the 3rd recycling reactions for all three catalysts compared to the average of catalytic reactions using fresh catalysts (1st use). The catalysis result from the reaction using the regenerated PdNP/GO at 300 °C is also included in the figure. The complete hydrogenation of alkyne to alkene requires the degree of hydrogenation to be 2. | ||
It was found that PdNP catalysts occasionally lost their colloidal stability in water after the catalytic reaction and formed precipitates, indicating possible particle agglomeration through ligand interactions (Fig. 5(a)). In comparison, the recycling studies of PdNP/GO catalysts showed that the presence of GO as a support for PdNPs could enhance the colloidal stability of PdNPs and allow the catalysts to maintain their dispersity in water after multiple catalyst recycling (Fig. 5(b)). The heated PdNP/GO catalysts meanwhile were already in heterogeneous conditions with low dispersity even before the catalytic reaction (Fig. 5(c)).
 | ||
| Fig. 5 Reaction systems before and after 24 hour DMAD hydrogenation reactions with (a) PdNPs, (b) PdNP/GO, and (c) PdNP/GO heated at 300 °C. | ||
A change in pH of the reaction solution before and after the reaction was monitored and its role on the colloidal stability of the catalyst was investigated. For PdNPs, the pH of the solutions before the reaction was around neutral (pH = ∼7.8), but during the course of the reaction with DMAD, the pH decreased by more than a couple of units. The decrease in pH for the PdNP solution would cause some protonation of the carboxylate ligands, potentially affecting the solubility of the PdNPs. In comparison, the initial pH of both the heated and unheated PdNP/GO catalysts was already below neutral (pH = 5–6). This was caused by the addition of graphene oxide solution, which itself has a pH of 2–3. For both the unheated and heated PdNP/GO catalysts, the pH of the solution increased slightly with the progression of the reaction. With no additional decrease in the pH of the solution after the catalytic reaction, PdNP/GO catalysts could maintain their dispersity in the aqueous environment, indicating the positive influence of GO on the colloidal stability of PdNPs.
UV-vis spectra of crude reaction mixtures were recorded in order to observe the integrity of the catalysts and the formation of any side product.47 The UV-vis spectra showed a very small peak at around 400 nm for both the PdNP and PdNP/GO catalysis samples, indicating the formation of a small amount of Pd(II) species. This suggested that the small leaching of Pd atoms could not be completely eliminated in water using either thiolate ligand protection or the GO supporting strategy. However, the presence of a small amount of leached Pd atoms clearly did not have any noticeable influence on the catalytic activity and selectivity of both catalytic systems. TEM images were also obtained for the recycled catalysts to determine whether recycling caused any morphological changes to the PdNP or PdNP/GO catalysts. TEM images of both PdNP and PdNP/GO catalysts after the reaction showed the formation of some small aggregates among many small nanoparticles, but no significant shape changes (Fig. S8, ESI†). In addition, the TEM image of PdNP/GO clearly showed the presence of many small nanoparticles still deposited on the surface of GO, indicating the relatively strong interactions between GO and the PdNPs.
Conclusions
This article described the synthesis of water-soluble palladium nanoparticles capable of catalysing selective hydrogenation of alkyne under mild conditions (atmospheric pressure and room temperature). The palladium nanoparticles supported on graphene oxide nanosheets were prepared using the strategy of pre-formed nanoparticle self-assembly. The annealing studies at different temperatures revealed that most thiolate ligands could be removed from the palladium nanoparticle–graphene oxide hybrids at 300 °C without extensive coalescence of particles. By comparing the activity and selectivity of three different catalysts, it was concluded that the catalytic properties of palladium nanoparticles and palladium nanoparticle–graphene oxide hybrids were similar. However, with the addition of the graphene oxide support, the palladium nanoparticle–graphene oxide hybrids could exhibit better colloidal stability during the catalytic reaction. When the surface ligands were removed from the palladium nanoparticle–graphene oxide hybrids via heat treatment, the activity of the hybrid catalysts was increased producing the fully hydrogenated products in high yields.Acknowledgements
This study was supported by the National Institute of General Medical Science (#SC3GM089562) and the Undergraduate Education Grant Program of the W. M. Keck Foundation. NMR instrumentation was provided for by the National Science Foundation (MRI CHE-1337559).Notes and references
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS PubMed.
- D. Astruc, Inorg. Chem., 2007, 46, 1884–1894 CrossRef CAS PubMed.
- S. Bhattacharjee, D. M. Dotzauer and M. L. Bruening, J. Am. Chem. Soc., 2009, 131, 3601–3610 CrossRef CAS PubMed.
- M. Crespo-Quesada, F. Cardenas-Lizana, A.-L. Dessimoz and L. Kiwi-Minsker, ACS Catal., 2012, 2, 1773–1786 CrossRef CAS.
- L. L. Chng, N. Erathodiyil and J. Y. Ying, Acc. Chem. Res., 2013, 46, 1825–1837 CrossRef CAS PubMed.
- H. Cong and J. A. Porco, ACS Catal., 2012, 2, 65–70 CrossRef CAS PubMed.
- D. J. Gavia and Y.-S. Shon, ChemCatChem, 2015, 7, 892–900 CrossRef CAS PubMed.
- C. Deraedt and D. Astruc, Acc. Chem. Res., 2014, 47, 494–503 CrossRef CAS PubMed.
- P. Wagener, A. Schwenke and S. Barcikowski, Langmuir, 2012, 28, 6132–6140 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Acc. Chem. Res., 2012, 45, 1657–1666 CrossRef CAS PubMed.
- V. S. Myers, M. G. Weir, E. V. Carino, D. F. Yancey, S. Pande and R. M. Crooks, Chem. Sci., 2011, 2, 1632–1646 RSC.
- H. Lindlar, Helv. Chim. Acta, 1952, 35, 446 CrossRef CAS.
- L.-Y. Gan, Y.-X. Zhang and Y.-J. Zhao, J. Phys. Chem. C, 2009, 114, 996–1003 Search PubMed.
- S. T. Marshall, M. O’Brien, B. Oetter, A. Corpuz, R. M. Richards, D. K. Schwarts and J. W. Medlin, Nat. Mater., 2010, 9, 853–858 CrossRef CAS PubMed.
- S. H. Pang, C.-H. Lien and J. W. Medlin, Catal. Sci. Technol., 2016, 6, 2413–2418 CAS.
- B. P. S. Chauhan, J. S. Rathore and T. Bandoo, J. Am. Chem. Soc., 2004, 126, 8493 CrossRef CAS PubMed.
- J. S. Zhu and Y.-S. Shon, Nanoscale, 2015, 7, 17786–17790 RSC.
- D. J. Gavia, J. Koeppen, E. Sadeghmoghaddam and Y.-S. Shon, RSC Adv., 2013, 3, 13642–13645 RSC.
- D. J. Gavia and Y.-S. Shon, Langmuir, 2012, 28, 14502–14508 CrossRef CAS PubMed.
- E. Sadeghmoghaddam, K. Gaïeb and Y.-S. Shon, Appl. Catal., A, 2011, 405, 137–141 CrossRef CAS.
- E. Sadeghmoghaddam, H. Gu and Y.-S. Shon, ACS Catal., 2012, 2, 1838–1845 CrossRef CAS PubMed.
- D. J. Gavia, M. S. Maung and Y.-S. Shon, ACS Appl. Mater. Interfaces, 2013, 5, 12432–12440 CAS.
- W. Long, N. A. Brunelli, S. A. Didas, E. W. Ping and C. W. Jones, ACS Catal., 2013, 3, 1700–1708 CrossRef CAS.
- Y. Yabe, Y. Sawama, Y. Monguchi and H. Sajiki, Catal. Sci. Technol., 2014, 4, 260 CAS.
- C. Tan, X. Huang and H. Zhang, Mater. Today, 2013, 16, 29–36 CrossRef CAS.
- Z. Zhang, T. Sun, C. Chen, F. Xiao, Z. Gong and S. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 21035–21040 CAS.
- S. Sabater, J. Mata and E. Peris, ACS Catal., 2014, 4, 2038–2047 CrossRef CAS.
- S. Sun and P. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 3481–3486 CAS.
- G. M. Scheuermann, L. Rumi, P. Steurer, W. Bannwarth and R. Mülhaupt, J. Am. Chem. Soc., 2009, 131, 8262–8270 CrossRef CAS PubMed.
- H. Ismaili, D. Geng, A. X. Sun, T. T. Kantzas and M. S. Workentin, Langmuir, 2011, 27, 13261–13268 CrossRef CAS PubMed.
- Y. Gao, X. Chen, J. Zhang, H. Asakura, T. Tanaka, K. Teramura, D. Ma and N. Yan, Adv. Mater., 2015, 27, 4688–4694 CrossRef CAS PubMed.
- Y. Wang, S. De and N. Yan, Chem. Commun., 2016, 52, 6210–6224 RSC.
- Z. Zhang, Y. Dong, L. Wang and S. Wang, Chem. Commun., 2015, 51, 8357–8360 RSC.
- T. Sun, Z. Zhang, J. Xiao, C. Chen, F. Xiao, S. Wang and Y. Liu, Sci. Rep., 2013, 3, 2527 Search PubMed.
- J. Pyun, Angew. Chem., Int. Ed., 2011, 50, 46–48 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, M. Alvaro, P. Concepcion, V. Fornes and H. Garcia, Chem. Commun., 2012, 48, 5443–5445 RSC.
- Y. Gao, D. Ma, C. Wang, J. Guan and X. Bao, Chem. Commun., 2011, 47, 2432–2434 RSC.
- H. Pan, S. Low, N. Weerasuriya and Y.-S. Shon, ACS Appl. Mater. Interfaces, 2015, 7, 3406–3413 CAS.
- A. F. Zedan, S. Moussa, J. Terner, G. Atkinson and M. S. El-Shall, ACS Nano, 2013, 7, 627–636 CrossRef CAS PubMed.
- C. Xu, D. Yang, L. Mei, B. Lu, L. Chen, Q. Li, H. Zhu and T. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 2715–2724 CAS.
- S. Pevzner, I. Pri-Bar, I. Lutzky, E. Ben-Yehuda, E. Ruse and O. Regev, J. Phys. Chem. C, 2014, 118, 27164–27169 CAS.
- J. Huang, L. Zhang, B. Chen, N. Ji, F. Chen, Y. Zhang and Z. Zhang, Nanoscale, 2010, 2, 2733–2738 RSC.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Rev., 2014, 114, 6179–6212 CrossRef CAS PubMed.
- Z. Niu and Y. Li, Chem. Mater., 2014, 26, 72–83 CrossRef CAS.
- V. P. Ananikov and I. P. Beletskaya, Organometallics, 2012, 31, 1595–1604 CrossRef CAS.
- A. M. Buchbinder, N. A. Ray, J. Lu, R. P. Van Duyne, P. C. Stair, E. Weitz and F. M. Geiger, J. Am. Chem. Soc., 2011, 133, 17816 CrossRef CAS PubMed.
- L. Huang, T. P. Ang, Z. Wang, J. Tan, J. Chen and P. K. Wong, Inorg. Chem., 2011, 50, 2094 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR results of sodium ω-carboxyl-S-hexanethiosulfate. UV-vis, FT-IR, and TGA results of PdNP. UV-vis, FT-IR, and TEM results of PdNP/GO and heated PdNP/GO. Powder XRD patterns of heated PdNP/GO and recycled PdNP/GO. 1H NMR example of catalysis studies. TEM images of recycled PdNP and PdNP/GO. See DOI: 10.1039/c6nj02898e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |