
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConformational control of the bacterial Clp protease by natural product antibiotics
I. T.
Malik
 and
H.
Brötz-Oesterhelt
*
and
H.
Brötz-Oesterhelt
*
Department of Microbial Bioactive Compounds, Interfaculty Institute of Microbiology and Infection Medicine, University of Tuebingen, Germany. E-mail: heike.broetz-oesterhelt@uni-tuebingen.de
First published on 4th April 2017
Abstract
Covering: up to 2017
The bacterial Clp protease is a highly conserved and structurally versatile machine. It has gained a lot of recognition during the last decade as a novel antibacterial drug target with an unprecedented mechanism of action. Due to its complexity, there are distinct means of interfering with its natural functions and several compounds targeting this machine have been identified. In this review, we summarize the current state of knowledge about natural products deregulating Clp proteolysis, a crucial and delicate process within the cell. Among those, acyldepsipeptide antibiotics of the ADEP class (ADEPs) are characterized best. The molecular mechanism of ADEP-mediated deregulation sheds light on the inner workings of the Clp protease.
 I. T. Malik | Imran T. Malik received his diploma in Biology from Freiburg University in 2012. Currently, he is pursuing his Ph.D. in Microbiology at the Eberhard Karls University of Tübingen. His main field of interest is the biochemical and structural characterization of large proteolytic machineries by means of enzymatic assays and fluorescence spectroscopy. |
 H. Brötz-Oesterhelt | Heike Brötz-Oesterhelt is a professor for Microbiology at the University of Tuebingen. For more than 20 years, she has worked in the field of antibacterial drug discovery in the academic as well as in the pharmaceutical industry setting. She is particularly interested in molecular mode of action studies of new antibiotic lead structures. |
1 Introduction
Bacterial resistances towards antibiotics pose a huge problem for the treatment of infectious diseases.1,2 Until recently, antibiotic development largely relied on synthetic modifications of established antibiotic classes to overcome resistances by compound derivatization. However, this source is running dry and new antibiotic classes with unprecedented core structures are urgently needed to overcome the plethora of resistance mechanisms spreading through the bacterial population. Antibacterial agents with unrelated chemical scaffolds often act by novel mechanisms of bacterial growth inhibition and are less affected by widespread resistance traits. New means of killing multi-drug resistant bacteria must be found.Microbial natural products are a privileged source of antibacterial lead structures. Being produced by microorganisms themselves and optimized through co-evolution with bacterial competitors for billions of years, they often surpass synthetic comparators with regard to cell entry and complex target interactions.3,4 Most antibiotics in therapeutic use to date inhibit essential functions in DNA, RNA, protein or cell wall syntheses, whereas daptomycin and polymyxins interfere with membrane integrity, but rarely is a completely unrelated mode of action described for a novel antibacterial agent with good tolerance and promising efficacy in infection models.
During the last decade, a bacterial protease has emerged as an unprecedented antibacterial target in the course of mode of action studies on acyldepsipeptide antibiotics (ADEPs). Isolated from the fermentation broth of Streptomyces hawaiiensis, the natural products A54556 A and B (factor A and B) showed good antibiotic activity in vitro without mechanism-based cross-resistance to other known antibiotics.5,6 Identification of the resistance-mediating mutation within an ADEP-resistant Escherichia coli mutant and affinity chromatography with an immobilized ADEP congener led to ClpP as the direct target.6 Medicinal chemistry campaigns established the structure–activity relationship and yielded a number of derivatives with enhanced in vitro potency and stability.7–10 Furthermore, ADEP treatment proved successful in lethal bacterial infections in rodents, including deep-seated biofilm infections, and, in combination with e.g. rifampicin, eradicated persister cells of methicillin-resistant Staphylococcus aureus.6,7,11
ClpP is a serine peptidase with active sites shielded within its barrel shaped proteolytic chamber. It can only degrade peptides on its own but is capable of protein degradation when partnering with a cognate Clp/Hsp100 chaperone (Clp-ATPase). As ClpP can act in conjunction with several Clp-ATPases, the resulting proteolytic complexes differ in their respective Hsp100 module. However, as they uniquely contain ClpP as the proteolytic core, we will refer to the complex consisting of ClpP and any corresponding Clp-ATPase as the “Clp protease”. The Clp protease system has a multitude of functions in bacteria, including protein quality control and homeostasis, stress management, virulence factor expression, and regulation of cell differentiation programmes.12–14 Its function is also essential for viability in actinobacteria including Mycobacterium tuberculosis.15
Following the first reports on ADEP, other natural products were recently discovered to modulate, i.e. inhibit or activate, either the proteolytic core ClpP or its Hsp100 partners. In this review, while briefly touching on the promising biological activities of these compounds, we will focus on the intriguing mechanistic interaction between the Clp protease system and its natural product modulators. Among those, ADEP is best understood. The interplay between ClpP and ADEP stands representative for a new principle of killing bacteria by targeting and deregulating a protease system. Furthermore, ADEPs are instrumental in understanding the molecular operation mode of the complex Clp protease machinery.
2 ClpP structure and function
2.1 ClpP and AAA+ chaperones regulate protein homeostasis, stress response, cellular differentiation, and virulence
The Clp protease system is widely conserved within the bacterial domain and is functionally organized in two separate compartments. The Clp/Hsp100 enzymes of the AAA+ super family of chaperones (Clp-ATPases) select substrates for degradation, unfold and thread them into the proteolytically active ClpP in an ATP-dependent fashion. There is a number of different Clp-ATPases that associate with ClpP for protein degradation, e.g. ClpX and ClpA in E. coli, ClpX and ClpC in Staphylococcus aureus, ClpX and ClpC1 in Mycobacterium tuberculosis, to name a few. Each shows distinct substrate specificities and performs different cellular functions, albeit there are some redundancies. In the beginning, the characterization of the Clp protease took place primarily in non-pathogenic E. coli and Bacillus subtilis strains. In E. coli, loss-of-function of ClpP or Clp-ATPases causes only a mild phenotype, as here, the Clp system shares functions in protein homoeostasis with the Lon protease.16,17 In B. subtilis, clpP deletion prevents motility, sporulation and genetic competence.18 Heat tolerance and stationary phase survival are also reduced and accumulation of the stress regulator Spx in a clpP or clpX mutant is toxic and impairs growth.18,19 Trapping experiments with a proteolytically inactive ClpP variant revealed protein substrates with important functions in global stress management, cell division, global transcription regulation, DNA damage repair, and protein synthesis.13,20 Meanwhile, the Clp protease is validated as a drug target in pathogenic organisms, as recently reviewed.21–23 For instance, in S. aureus, the Clp protease is responsible for stress tolerance and involved in virulence regulation.24,25clpP and clpX deletions were shown to attenuate S. aureus in a murine skin abscess model and a Staphylococcus epidermidis clpP deletion mutant proved less virulent in a catheter infection in rats.26,27 A synthetic β-lactone acting as a covalent suicide inhibitor of ClpP was effective in treating staphylococcal skin abscesses in mice, demonstrating druggability of ClpP's catalytic triad.28clpP deletions further prevented Streptococcus pneumoniae from colonizing the nasopharynx and infecting lungs of mice, and reduced survival of Listeria monocytogenes within macrophages.29,30 While the Clp-ATPases and the ClpP peptidase work together in general degradation of misfolded and aggregated proteins as well as in directed regulatory proteolysis, Clp-ATPases also possess chaperone activity independently of the peptidase.31,32 They can actively induce structural changes within their substrates altering their biological activity. For instance, one of the first characterized substrates of E. coli ClpX was the Mu transposase.33 ClpX alters its conformation and thereby initiates the transition from recombination to Mu phage replication without the need of a partner peptidase.33,34 Secondly, expression of spa, the gene encoding protein A in S. aureus, is nearly abolished in a clpX but not a clpP mutant.26 Protein A is a virulence factor expressed in the early growth phase and hampers detection by the host immune system. It is under negative regulatory control of the accessory gene regulator (agr) quorum sensing system, which is activated in the late exponential phase of growth and down-regulates protein A expression by post-transcriptional inhibition.35,36 The repression of spa in a clpX mutant works independently of agr and counteracts the normally occurring derepression in an agr negative strain.24 ClpC in S. aureus plays an important role in acetate catabolism and has been further characterized as a global regulator in late growth phase carbon metabolism.37,38 In M. tuberculosis, the entire Clp protease system including the chaperones ClpX, ClpC1 and ClpB as well as the two ClpP paralogs, ClpP1 and ClpP2, is essential for growth.15,39–42 Compounds targeting the ClpC1 ATPase in mycobacteria display potent antibacterial activity. In Gram-negatives, the Clp protease regulates expression of the type III secretion system and a lack of functional ClpX and ClpP results in severely attenuated or abolished virulence.43–45 Reviewed examples and similar reports established the Clp protease as a promising novel drug target.2.2 ClpP forms the proteolytic core of a compartmentalized protease
Fourteen ClpP protomers arrange themselves to form a tetradecameric barrel-shaped complex in a stack of two heptameric rings (Fig. 1a). The catalytic residues are located within the sequestered space of the barrel. Seven hydrophobic pockets on both sides of the barrel serve as anchors for partner Clp-ATPases during translocation of substrates into the proteolytic chamber of ClpP (Fig. 2).46,47 Several crystal structures of ClpP from different species have revealed distinct barrel conformations, namely compressed, compact and extended state.48–52 Based on molecular dynamics (MD) simulation experiments, the compact conformation has been speculated to represent a stable intermediate state between extended and compressed, showing a local energy minimum during transitions between extended and compressed conformations.48 Here, we focus only on the two end-points of this transition (Fig. 1a). The compressed conformation, about 80 Å in height, is deemed to be inactive because of the arrangement of the active site residues with an increased distance between the serine and the histidine side chain, whereas crystal structures of extended conformations, where the barrel is elongated by approximately 10 Å along the rotational axis, show the active site residues in reduced distance (Fig. 1a).49,53–55 In the course of the nucleophilic attack of the active site serine on the electron deficient carbonyl carbon of the peptide bond, the proton of the serine hydroxyl group is abstracted by the histidine imidazole and the positive charge thereby generated at the histidine imidazole is stabilized by the carboxyl function of the aspartate (Fig. 1b). The resulting acyl–ester intermediate then undergoes hydrolysis and the serine side chain is regenerated to undergo the next cycle of catalysis. In the catalytically competent conformation, these three side chains are in the correct distance to form hydrogen bonds (see green dotted lines in Fig. 1b), which strongly enhance nucleophilicity of the serine. This competent conformation has so far only been observed in crystal structures that captured ClpP in the extended conformation, but never in crystals containing compressed conformations. Notably, other key structural elements show considerable shifts between the two conformations in X-ray crystallography structures. These shifts correlate with predicted residue flexibility from MD simulation experiments.55 The most flexible domains of a ClpP protomer are the N-terminal loop and the α5-helix. The latter is associated with the ring–ring interface in the equatorial plane and is also known as the handle region.50 The handle of each ClpP subunit adopts a straight orientation in the extended conformation while it is kinked in the compressed structure (Fig. 1a). The physiological significance of these conformations and their regulation are still subject to investigation.49,51 A crystal structure of ClpP with an inhibitor covalently bound to the active site displayed a slightly more compacted conformation, corroborating the idea of a functional link between conformation and catalysis.56 One model proposes an exit route for peptide products via transient equatorial pore openings in the compressed situation with the compression motion being part of a natural cycle of ClpP dynamics.57–61 Introducing covalent cross-links between handle regions of neighbouring ClpP subunits resulted in decreased handle flexibility and displayed increased substrate retention times within the ClpP lumen.59 This finding strongly supports the idea of an involvement of the handle region in product release. Nonetheless, product release via the axial pores or a combination of both cannot be ruled out, yet. While most compressed crystal structures of ClpP show a disordered tip of the α5-helix, two structures from S. aureus reveal a kinked orientation.49,55 In this state, the handle is stabilized by hydrogen bonds within its own subunit.48 In the extended state, a straight α5-helix is involved in a network of hydrogen bonds stabilizing the ring–ring connection between the two heptamers.55 This network of hydrogen bonds connects the tip of the extended helix of one ClpP subunit (Fig. 1a, green helix) to amino acid residues in the α6-helix of the opposing ClpP subunit (Fig. 1a, red helix). Mutation studies of these amino acid residues termed “oligomerization sensors” showed defects in oligomeric state formation and catalytic capabilities, stressing the importance of the handle to make contact to the opposing ring.51 Interestingly, MD simulations indicate a tendency towards the compressed state in the absence of the hydrogen bond network due to favourable thermodynamics.48,55 Therefore, it can be assumed that control over the conformational state is part of a protection system in the cell that requires the proteolytic extended form to be actively promoted. | ||
| Fig. 1 The proteolytic core of the Clp protease, ClpP. (a) Crystal structures of ClpP from S. aureus. Overview of the ClpP architecture both in active extended (PDB code: 3V5E) and in inactive compressed conformation (pdp code: 3qwd). Two opposing subunits are highlighted in colour. The α5 and α6 helices responsible for the ring–ring connection are coloured in green and red, respectively. The top and inside views are depicted in a surface fill model representation. The inside view reveals the catalytic sites within the barrel and the orientations of the residue side chains. The histidine side chain imidazole is rotated away from the hydrolytic serine hydroxyl function in the inactive arrangement and cannot form a hydrogen bond. The hydrogen bond network required to stabilize the reaction intermediate cannot be formed. (b) Catalytic mechanism of the model serine protease ClpP. Key interactions within the catalytic triad during peptide bond cleavage are indicated (reaction details are given in the text). | ||
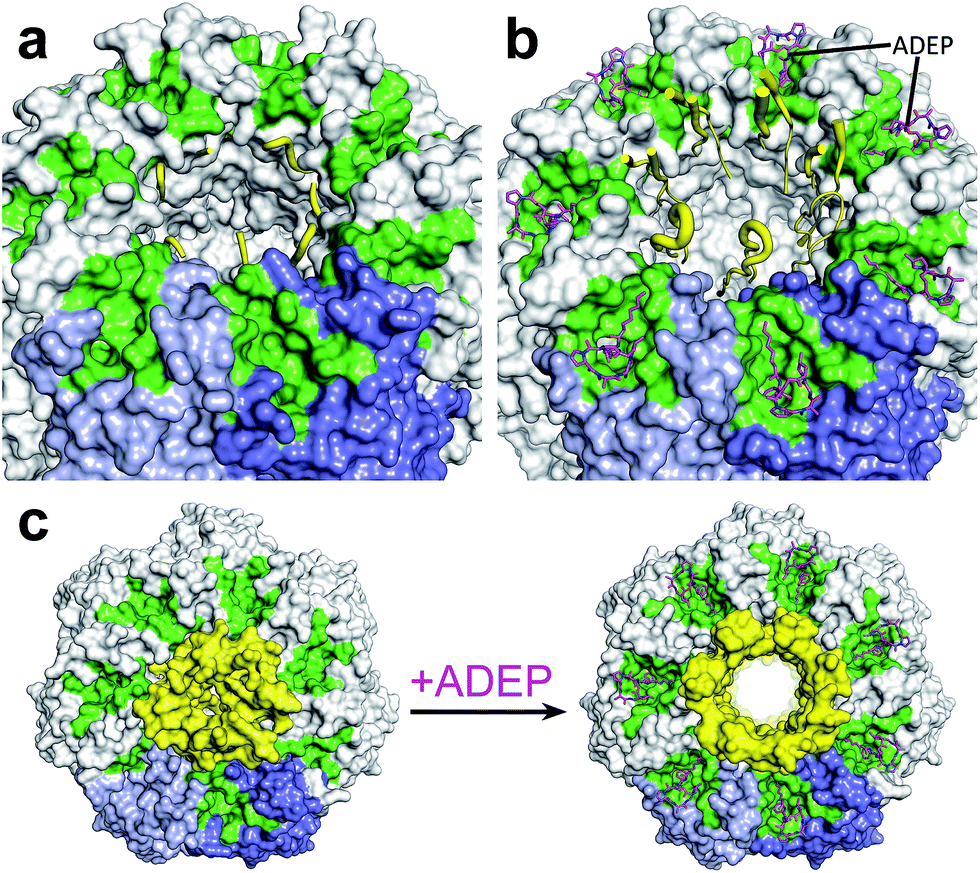 | ||
| Fig. 2 View on the apical surface of a ClpP tetradecamer from E. coli. (a) “down”-conformation of the N-terminus of ClpP (PDB code: 1YG6): two neighbouring ClpP subunits are coloured in shades of blue, the hydrophobic pockets, each spanning two ClpP subunits, are highlighted in green. The N-terminal loops (yellow) surrounding the axial pore are disordered and close the channel by serving as a hydrophobic plug. (b) “open gate”-conformation of the N-terminus of the ClpP/ADEP-complex (PDB code: 3MT6). In the representation of the N-terminal loops, strength of the ribbon correlates with flexibility. Three loops are completely resolved and show a β-hairpin structure that points upwards. Flexibility of the N-terminal loops increases towards the tip region, indicated by unresolved structures in the four other loops. In contrast to the “up”-conformation reported for apo-ClpP, this structure has an increased pore diameter of 20 Å. (c) Top view of the structured axial channel in the apo form of ClpP in the “up”-conformation (left) and the widened pore of ClpP in complex with ADEP (right). | ||
2.3 The apical side of the ClpP barrel harbours interaction sites for the cognate Clp-ATPases
In the absence of a cognate Clp-ATPase, ClpP can only degrade small peptides.62,63 The axial channels are the only opening into the catalytic chamber of ClpP.64,65 These channels are bordered by the respective N-termini of the ClpP subunits (Fig. 2). A crystal structure of E. coli ClpP in the apo form displayed an “up”-conformation of the N-terminus at one apical side and a “down”-conformation at the opposite side suggesting a gating functionality of the N-terminus for substrate entry.66 Although the N-terminal residues of the “down”-conformation are unresolved, it has been proposed that in this conformation, clustering of hydrophobic residues within the axial channel serves as a hydrophobic plug and presents the closed gate of ClpP.67 Interestingly, the observation that only six out of seven N-terminal loops were in the “up”-conformation led the authors to speculate about a pseudo-6-fold symmetry matching the 6-fold symmetry of the partner Clp-ATPase.66 Cryo-EM studies with a ClpP tetradecamer bound to a ClpA hexamer at one apical side only, showed an open channel with a diameter of 12 Å at the apical side facing ClpA, forming a continuous channel with the Clp-ATPase.68 The N-terminal domain of ClpP on the ClpA-free side was blocked. Whether the “up”-conformation observed in the E. coli ClpP crystal (i.e. the state with upraised N-termini yet narrow pore diameter) resembles the open gate in the natural context when bound to Clp-ATPases, is still controversially discussed. Alexopoulos et al. argue that this “up”-conformation might not be identical to the Clp-ATPase-bound open conformation in the substrate feeding process.69 For more details on N-terminal gating, refer to ref. 69. Structural investigations with the help of ADEP activators, which mimic binding of Clp-ATPases by employing the same binding pocket, have been instrumental in addressing the question of how these conformations are related to pore gating (compare Section 3.2).53,69,70 Furthermore, is was suggested that the “up”-conformation stabilizes the intermediate substrate-bound form of ClpP while the “down”-conformation facilitates substrate hydrolysis, thus directly involving the N-terminus in the catalytic cycle of ClpP.712.4 Proteolysis by ClpP is tightly regulated
Substrates tagged with a degradation signal like the C-terminal ssrA-tag for incomplete translation of nascent polypeptide chains are recognized and bound by ClpX.72,73 Subsequently, ClpX assembles into hexamers and makes contact with ClpP by binding to the hydrophobic pockets via loops containing highly conserved (L/I/V)-GF tripeptide motifs necessary for association with ClpP and by binding the N-terminal stem loop of ClpP via its pore-2-loops.46,47,74 ATP binding and hydrolysis by ClpX then provides the energy to mechanically unfold and translocate linearized protein into ClpP.75–78 Within the proteolytic chamber, substrate is cleaved into small peptide fragments of around 6–8 residues (Fig. 3).79,80 Thus, substrate specificity is not defined by the amino acid sequence rather than by Clp-ATPases that recognize specific degradation signals and interact with certain adaptor proteins.20,73,81–83 Exposure to the active site residues is sufficient for cleavage with no strict specificity, albeit a preference for certain amino acids at the P1 position exists.84,85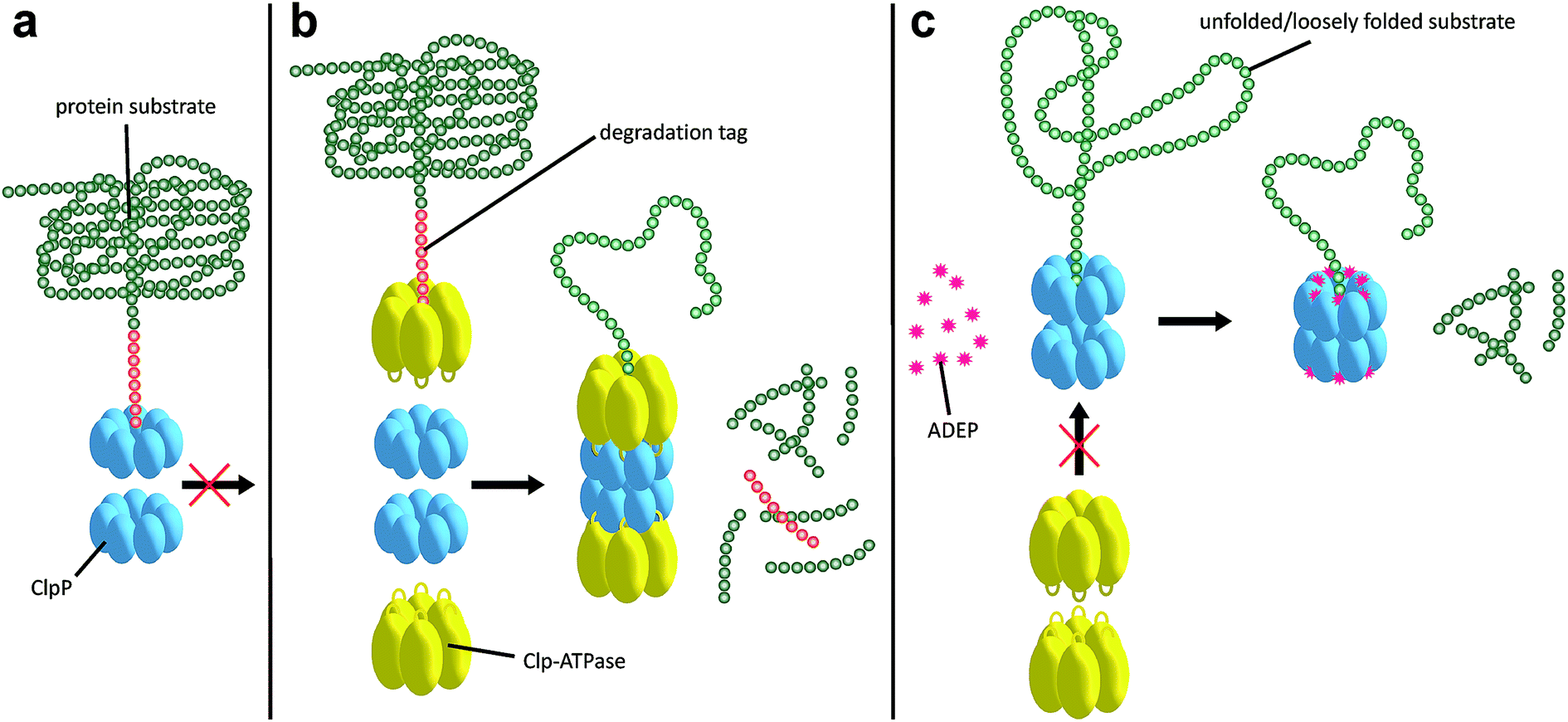 | ||
| Fig. 3 Operation mode of the Clp protease system and deregulation by ADEP. (a) In the dormant, inactive state the ClpP barrel is either not assembled or, if already assembled, the axial pores are closed. Proteins are not degraded by ClpP when on its own. (b) In the natural context, substrates are recognized by the Clp-ATPase via specific degradation tags and with the aid of adaptor proteins. The Clp-ATPase docks to the “hydrophobic pockets” of ClpP via surface loops presenting a conserved tripeptide signal, thereby initiating assembly of the ClpP tetradecamer in a conformation competent for catalysis. Furthermore, the Clp-ATPase actively unfolds the protein substrate using ATP hydrolysis and threads it into the entry pores of the catalytic chamber. (c) ADEP binding to the “hydrophobic pockets” of ClpP also assembles the ClpP barrel in a catalytically competent state. By steric hindrance, ADEP efficiently prevents the interaction of ClpP with the Clp-ATPases. As one consequence, none of the natural protein substrates can be degraded any more. As a second consequence, ClpP pores open and some protein substrates and nascent polypeptide chains necessary for bacterial growth and survival are now degraded (dual mechanism). Degradation tags are not required for ADEP-mediated protein degradation. | ||
The Clp protease is a paradigm of self-compartmentalized proteases and shares typical architectural features with other compartmentalized proteases like HslUV or the 26S proteasome. The term self-compartmentalized expresses that the active sites reside within a proteolytic chamber (“compartment”), which is shielded from the cytoplasm and inaccessible to potential protein substrates (Fig. 3a). Cleavage is performed in this sequestered space only after active unfolding and translocation of substrate through the narrow axial pores by Clp-ATPases (Fig. 3b). These in turn select substrates by either decisive degradation signals, through contacts mediated by specific adaptor proteins, or a combination of both. Furthermore, binding of Clp-ATPases to ClpP initiates structural reorganizations within ClpP that render substrate cleavage possible.68,86 Findings derived from the interaction of ClpP with ADEPs strongly contributed to our understanding of these reorganizations (see below).53,67,69,70,87 In the case of B. subtilis ClpP, protomers do not assemble to a barrel in vitro unless either ClpX or ClpC is active to bind and deliver substrate.88 Architectural restrictions are common among these proteolytic machines; they prohibit uncontrolled substrate processing and serve as safeguards against potentially harmful self-digest.89
3 Deregulation of the Clp protease by natural products
3.1 Uncontrolled ClpP activity is bactericidal
ADEP mimics binding of Clp-ATPases to the hydrophobic pockets. This event inhibits association of ClpP with Clp-ATPases, and thereby abolishes all natural functions of the Clp protease that require Clp-ATPase mediated degradation (Fig. 3c).90,91 The affinity of ADEP for ClpP with a KD of approximately 2 μM is much stronger compared to the Clp-ATPases, as a single ADEP molecule is sufficient for displacing a full ClpX hexamer.91 Clp protease function is essential for stress regulation and virulence in firmicutes. But, disturbing the ClpP–Clp-ATPase interaction under non-stressing in vitro conditions alone does not result in cell death as exemplified by clpP and clpX deletion mutants.12,21,22 Likewise covalent β-lactone inhibitors of ClpP lead to decreased virulence factor excretion in S. aureus but show no growth inhibition in in vitro assays for antibacterial activity.92The ADEP mode of killing in firmicutes is uncontrolled proteolysis by the ClpP/ADEP complex, i.e. proteins that are not tagged for degradation or recognized by adaptor proteins are still unspecifically targeted by ClpP. ADEP binding circumvents the above mentioned safeguards and initiates structural shifts that enable ClpP to degrade the loosely folded model substrate casein and nascent polypeptide chains in an unregulated fashion.2,37,70 The overactivated ClpP causes degradation of bacterial cell division protein FtsZ, as demonstrated in B. subtilis, S. aureus, and Wolbachia sp., resulting in cell division inhibition and eventually cell death.93,94 Furthermore, Conlon et al. performed proteomic analysis of non-replicating methicillin-resistant S. aureus after long-term exposure to ADEP and identified decreased abundance in 417 proteins compared to a non-treated control.11
Besides acyldepsipeptide antibiotics, a number of compounds have been described to target the Clp protease (Fig. 4). A non-peptide-based natural product activator of ClpP, sclerotiamide, has recently been identified in a screening for β-casein degradation.95 As of yet, there is no available data on neither the mechanism of ClpP binding nor antibacterial activity of this compound. In comparison to the natural product ADEP1, casein degradation was slow and rather high concentrations of sclerotiamide were required. Furthermore, sclerotiamide activity was restricted to ClpP from E. coli and ClpP from B. subtilis could not be activated. So far, ADEP is the only natural product activator that is confirmed to target ClpP from a wide variety of organisms.
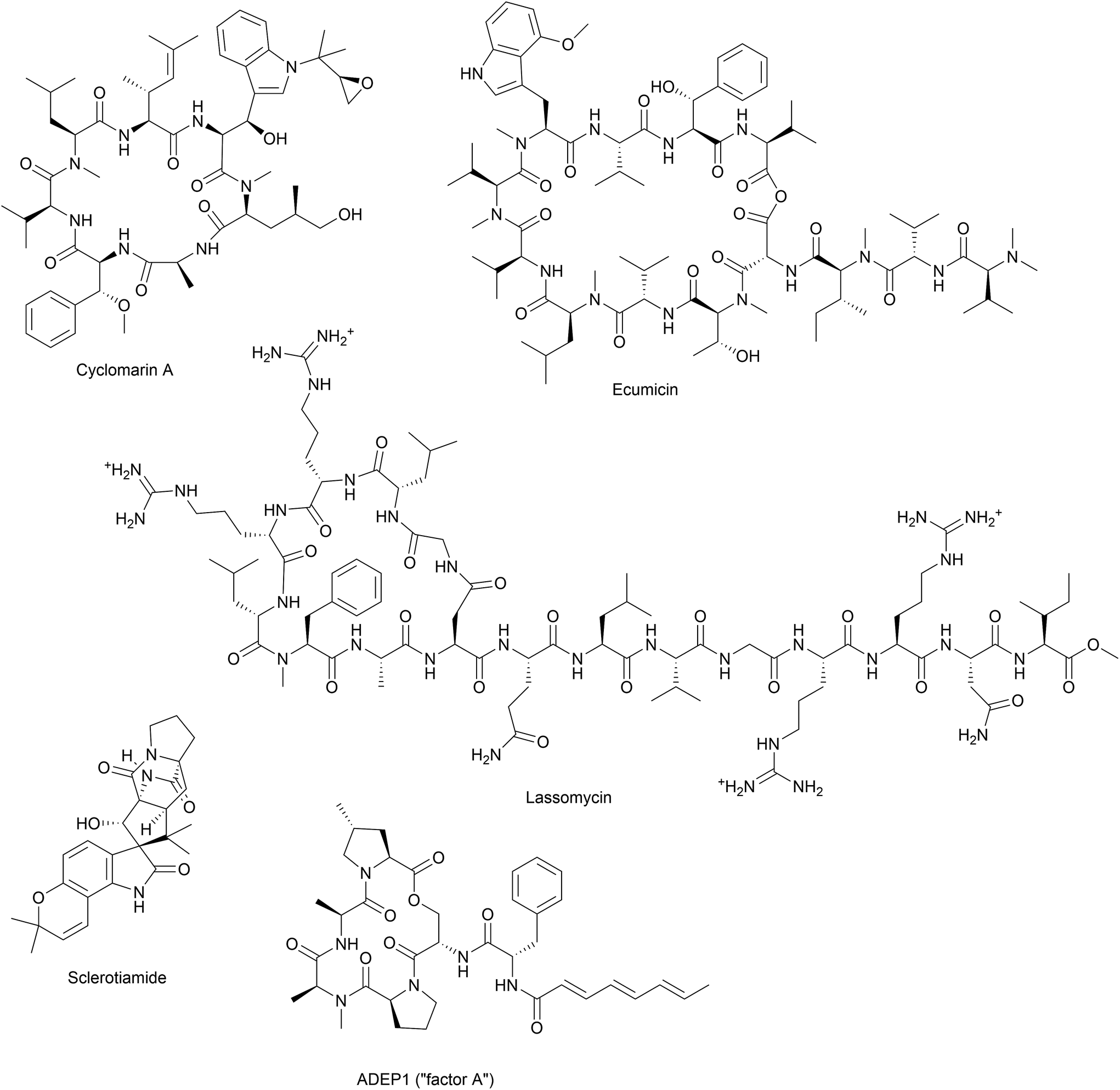 | ||
| Fig. 4 Natural product modulators of the Clp protease. | ||
3.2 ClpP in complex with ADEP adopts an “open-gate”-conformation
The ClpP/ADEP complex adopts a proteolytically active conformation strongly resembling the extended form of apo-ClpP, yet distinct, because of an increased diameter of the axial pores (Fig. 2).53,70 The crystal structure of E. coli ClpP in complex with ADEPs shows the N-terminal loop pointing upwards, similar to the “up”-conformation of apo ClpP, but with a widened axial pore of 20 Å in diameter, henceforth referred to as the “open gate”-conformation (Fig. 2b).70 It is not yet known, if such an increased diameter which was not observed in the cryo-EM structure of the E. coli ClpP/ClpA complex (‘ClpAP’) might present a special case exclusive to the ClpP/ADEP complex or if it occurs in a similar form also during the ClpP/Clp-ATPase interaction. In contrast to the structured terminal β-hairpins of the E. coli ClpP/ADEP crystals, the axial pores seemed unstructured in ClpP crystals from B. subtilis in complex with ADEP. This difference was attributed to tight packing of the B. subtilis ClpP/ADEP complexes in the crystals, while the E. coli crystals showed less tight packing in the N-terminal region. Thus, it was proposed that the N-terminal loops of ClpP/ADEP indeed form a structured channel (Fig. 2b).69,96 Bound ADEP molecules provide additional hydrophobic anchor points for the N-terminal tail of ClpP.67 The whole ClpP/ADEP complex displays reduced structural flexibility, as demonstrated in hydrogen/deuterium exchange experiments.67 In this study, ClpP was incubated in a deuterated solution both in absence and in presence of ADEP1. Hydrogen/deuterium exchange as a measure of flexibility occurred to a lesser extent in the presence of ADEP1, especially in the equatorial plane where the α5-helices are located.67 This finding underlines the allosteric nature of the ClpP/ADEP interaction. Structural dynamics in regions of the ClpP macromolecule distant from the ADEP binding site are affected. For some years, pore gating has been considered to be the only structural determinant of ADEP-mediated activation. However, recent data reveal additional activating rearrangements (see below).3.3 Disruption of the functional ClpP–Clp-ATPase interaction kills mycobacteria
Other natural compounds that specifically target the mycobacterial ClpC1 chaperone are lassomycin, ecumicin and a recently reported rufomycin analogue.106–108
The effort to find new antitubercular compounds also led to the discovery of RUF-I, an analogue of the natural product rufomycin, with so far undisclosed structure.108 RUF-I was also reported to target ClpC1, but so far, little information is available on this compound.108 An initial study showed no cross-resistance between ecumicin and rufomycin and resistant clones generated by exposure to either ecumicin or rufomycin, showed distinct single point mutations in clpC1, implicating different binding modes.
4 Molecular interaction between ADEP and ClpP
4.1 ADEP structure–activity-relationship (SAR)
 | ||
| Fig. 5 Natural product ADEP1 and synthetic derivatives. Important structural elements of ADEP1 are colour coded. Red: aliphatic side chain; green: phenylalanine linker (bisfluorinated for increased potency in the synthetic congeners); blue: methylproline; purple: N-methylalanine (modified to a pipecolate moiety for increased potency in ADEP4). | ||
4.2 ADEP – biological activity
Synthetic ADEP congeners, including ADEP4, B315 and compound 26 (Fig. 4) have MIC values in the low nanomolar range against a broad panel of Gram-positive pathogens.6,9,10 ADEP4 cured mice with lethal systemic S. aureus infections where ADEP1 failed due to limited antibacterial activity, poor chemical stability and high metabolic clearance.6,7 ADEP4 also outmatched marketed linezolid in murine lethal systemic infections caused by S. aureus, S. pneumoniae or E. faecalis and B315 was more effective than vancomycin in reducing the bacterial load of S. aureus in livers and kidneys of mice.6,116 Furthermore, ADEP4 showed exceptional activity against persister cells of S. aureus. In side-by-side experiments with stationary cells, where ciprofloxacin, linezolid, rifampicin and vancomycin were literally inactive, ADEP4 reduced the number of colony forming units of S. aureus by 4 log units.11 When ADEP4 was combined with either ciprofloxacin, or linezolid, or rifampicin, bacteria were eradicated to the level of detection.11 A combination treatment of ADEP4 and rifampicin also eradicated S. aureus from a biofilm that had developed during a deep-seated thigh infection in neutropenic mice.11 ADEP was also effective in killing a persisting Enterococcus faecium strain isolated from a neutropenic patient.117 ADEP4 at 0.2 μM eradicated this clinical isolate even in a preformed biofilm, whereas vancomycin and daptomycin failed at 256 and 50 μg ml–1, respectively.117 The antibacterial potency of ADEPs proves the potential of ClpP's hydrophobic pocket as a druggable target site, where bactericidal and anti-persister activity can be achieved covering a broad spectrum of bacteria. ClpP mutations, which were observed in firmicutes during ADEP treatment under moderate growth conditions in vitro, should play less of a role under the stressed conditions of the infection process, where ClpP is essential for virulence and fitness.6,24,25,115 Nonetheless, combination therapy is probably the therapeutic application strategy for ADEP, also considering the observed synergy against persisters.Specificity of ADEPs for procaryotes is high and eukaryotic cells are not affected up to the micromolar concentration range.6,118 Recently, a first organ histology study was published.116 Histological analyses of liver and kidney sections from healthy mice treated with 50 mg kg−1 B315 did not indicate any tissue toxicity, whereas vancomycin, which had been run in parallel, showed pronounced kidney toxicity as obvious from vacuolization of kidney cells and protein accumulation. Despite these promising preliminary studies on in vivo efficacy, the current leads require further improvement, e.g. with regard to solubility (for intravenous application), metabolism, and chemical stability. When considering combination therapy, pharmacokinetic and pharmacodynamics parameters of the partners must also be compatible and drug–drug interaction must be avoided. Efforts concerning pharmacological optimization are underway.
4.3 Interaction of ADEP with the hydrophobic pocket of ClpP
The ADEP binding pocket ranges over two neighboring ClpP subunits. In accordance with the minimal structural requirement for ADEP to take effect, the aliphatic side chain and the benzene ring of the phenylalanine are buried deeply within the hydrophobic pocket of ClpP (Fig. 6b). The relevance of this part of the molecule is represented by the number of its hydrophobic interactions (Fig. 6a).53 As each ADEP molecule establishes direct contacts with two neighboring ClpP subunits within the same ring, the stabilizing effects within the heptameric ring are obvious.53,90 When studying the surface model of B. subtilis ClpP with bound ADEP1, the structural confinements of the hydrophobic side-chain in terms of length as well as sterical limitations for substituents at the benzene ring become apparent. Modelling of a superimposition of the Helicobacter pylori ClpX LGF-loop extracted from a HpClpX crystal structure with an ADEP structure from an ADEP-bound E. coli ClpP crystal, suggests that the ADEP phenyl moiety and aliphatic side chain mimic binding of the LGF motif (corresponding consensus sequence in E. coli: IGF).70,119 Both the N-acylphenylalanine moiety of ADEP1 and the LGF motif overlap closely (Fig. 7c). The LGF leucine side chain extends into the channel that is otherwise occupied by the ADEP aliphatic side chain. The phenylalanine moiety of ADEP1 closely overlaps with the LGF phenylalanine. This is especially remarkable considering that the N-acylphenylalanine moiety, which closely resembles the (L/I)-GF-motif of ClpX, is also the minimal structural requirement for ADEP activity (see Section 4.1). Modifications in solvent-exposed regions of the macrocycle are promising for further compound optimization in terms of physiochemistry and ADME, whereas the potential for improvement of the N-acylphenylalanine part of ADEP seems limited due to the strict SAR and space limitations of the binding pocket. The ADEP binding pocket presents a hot spot for ClpP modulation and serves as a model target for protein–protein-interaction modulators. Efforts to screen for potential alternative ClpP binders have been undertaken with first positive results.120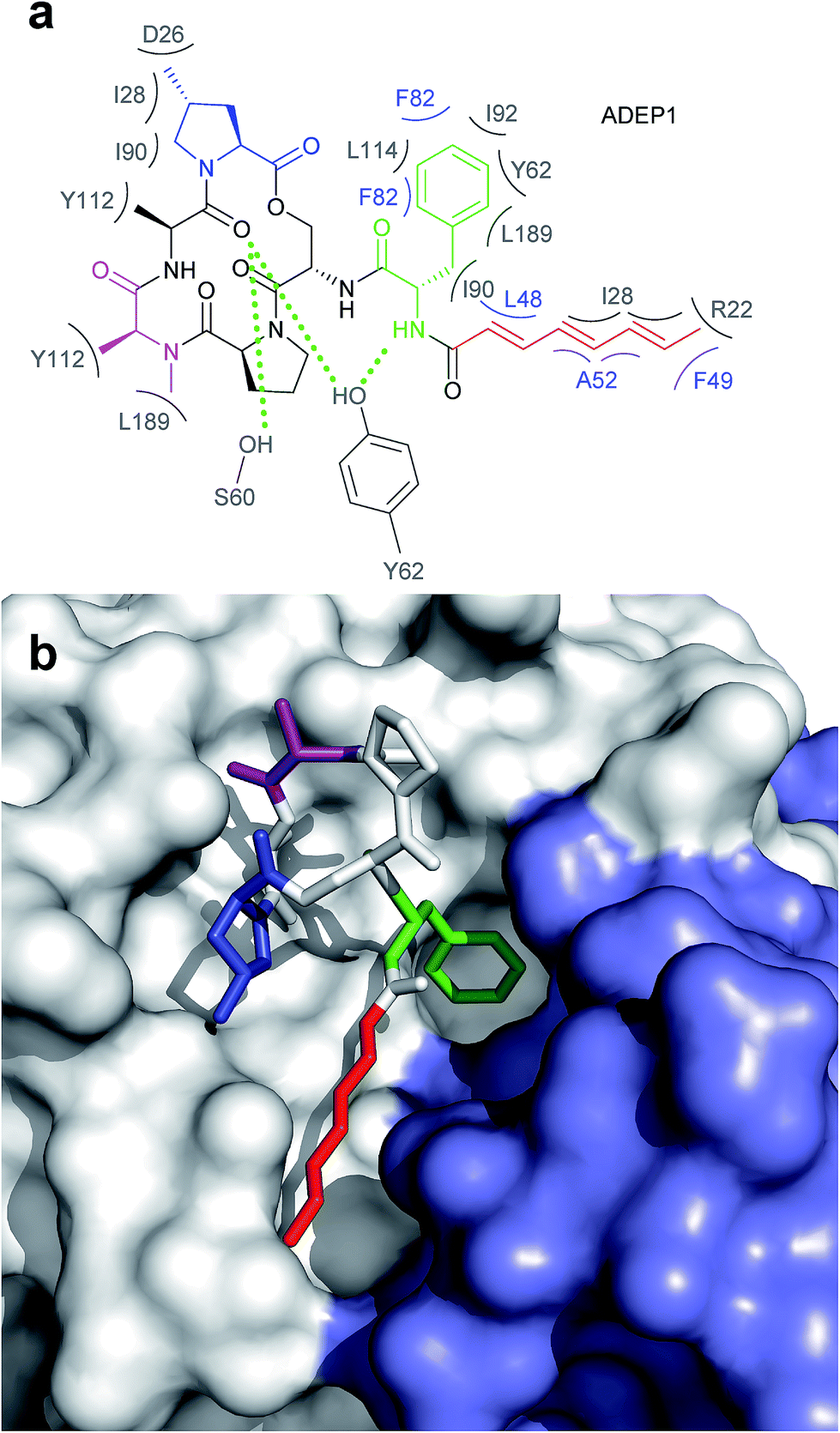 | ||
| Fig. 6 Interaction between ADEP1 and B. subtilis ClpP. (a) Structure of ADEP1 and ClpP amino acid residues involved in binding. Two neighbouring ClpP subunits contribute to ADEP binding with their respective amino acid residues highlighted in grey and blue. Residues coloured in grey belong to the white ClpP subunit in Fig. 3b, blue residues originate from the blue ClpP subunit in Fig. 3b. Hydrophobic interactions are denoted by brackets, hydrogen bonds by green dotted lines. (b) Surface fill model of the B. subtilis ClpP crystal structure complexed with ADEP1. Two adjacent ClpP monomers are depicted in white and blue, respectively. The N-acylphenylalanine moiety inserts deeply into the hydrophobic pocket and comprises the green (phenylalanine) and red (aliphatic side chain) functional groups. The macrocycle backbone is more solvent-exposed. It contains the N-methylalanine (purple) and the methylproline (blue) moieties beneficial for activity. | ||
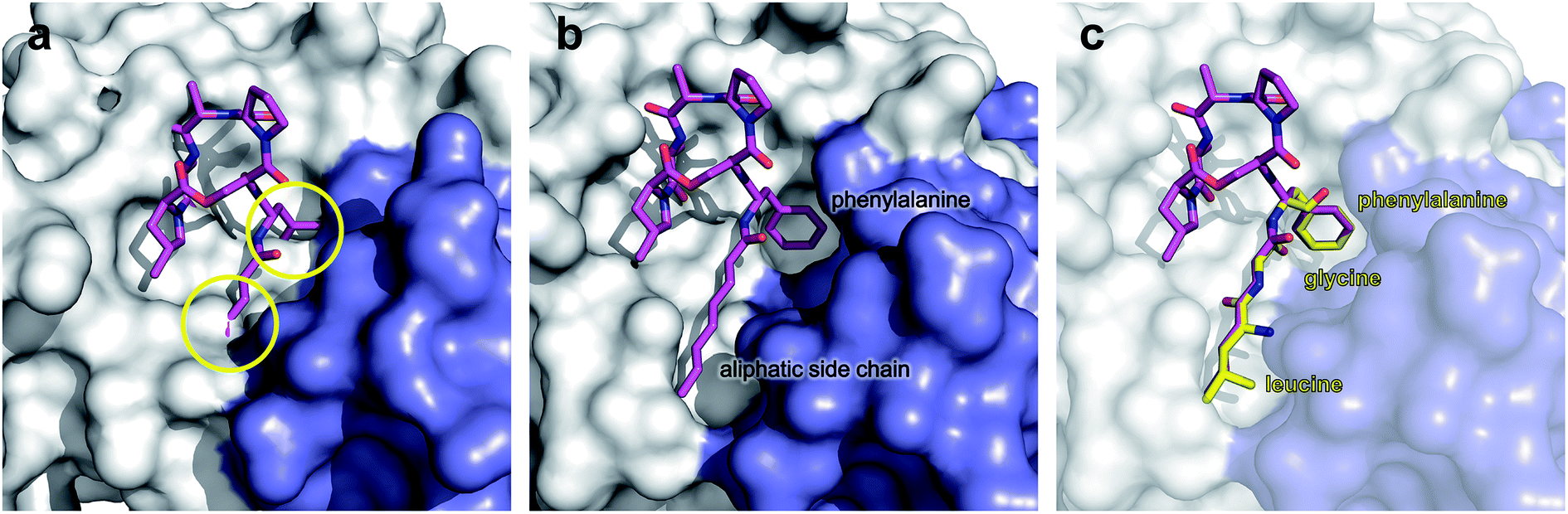 | ||
| Fig. 7 Surface fill model of the B. subtilis ClpP crystal structure in complex with ADEP1. (a) Modelling of an ADEP1 molecule into the ADEP binding pocket of ClpP in the compressed conformation (PDB code: 3TT6) predicts sterical clashes (circles). This is also the case with other ADEP derivatives as well as different compressed ClpP structures. (b) Close-up view of an ADEP1 molecule in the hydrophobic pocket of ClpP in its extended state (PDB code: 3KTI). (c) Superimposition of the LGF motif from of Helicobacter pylori ClpX (PDB code: 1UM8) with the N-acylphenylalanine moiety of ADEP1. The leucine and glycine residues overlap closely with the aliphatic side chain of ADEP1. | ||
Recently, discovery of a gain-of-function ClpP mutant of S. aureus provided new insight into the architectural properties of the ADEP binding site.121 The Tyr62 residue of B. subtilis ClpP had already been shown to form two hydrogen bonds and also hydrophobic interactions with ADEP1 (Fig. 6a).53 The H-bonds are formed between the tyrosine residue and the macrocycle backbone as well as the proximal part of the aliphatic chain. Hydrophobic interactions with the benzene ring of the ADEP phenylalanine residue further anchor the molecule. Upon closer look, Ni et al. found that said tyrosine residue was rotated by approximately 90° in the ADEP-bound structures of BsClpP, EcClpP, and MtClpP1P2/agonist when compared to the apo forms of the respective ClpPs.53,70,87,121 They argue that the energy barrier for adopting this rotated conformation is probably too high in the apo form of ClpP. Mutating Tyr63 in S. aureus, the corresponding tyrosine residue according to SaClpP nomenclature, to an alanine, however, led to a rotated peptide backbone even in the absence of activator and bestowed β-casein and even FtsZ degradation capability onto ClpP alone. This marked an important step in the elucidation of the ADEP pocket organization and possibly revealed the key switch for turning on the protease.
4.4 ADEP exerts conformational control over the entire ClpP barrel
ADEPs provide an elegant means for investigating the implications of “filling” the hydrophobic binding pocket of ClpP (Fig. 7). N-Terminal gate closure serves as a safeguard against uncontrolled digestion of proteins. A gated pore mechanism has long been presumed to be the critical factor in ClpP control. The available data however reveal a more global regulatory principle that includes additional safety measures.Cumulative evidence indicates that the conformational control of ClpP by ADEP reaches beyond the N-terminal region lining the central entrance pore. Thermal shift assays revealed that ADEP binding increases the overall folding stability of the S. aureus ClpP complex.91 In contrast to wild type ClpP from S. aureus, the catalytic site mutant D172N showed no residual peptidase activity in in vitro degradation assays but could be successfully activated by addition of either ADEPs or ClpX to degrade full-length protein and small peptides. Small angle X-ray scattering analysis showed a more compacted conformation for the D172N mutant with no difference in oligomeric state. ADEPs promote the extended conformation in this mutant that is otherwise only observed in a compacted conformation.91 Hydrogen–deuterium exchange experiments using E. coli ClpP showed a remarkable increase in rigidity of the handle region after addition of ADEP.67 The concept that “filling” the hydrophobic pocket stabilizes the handle in an extended conformation and the catalytic site in a competent conformation is in line with the crystallographic data.53,70 This is remarkable insofar as the hydrophobic pocket is connected via intramolecular relays not just to the N-terminal region but also the more distant handle region. Recent insights into the workings of M. tuberculosis ClpP are particularly interesting in this respect. M. tuberculosis encodes two ClpP paralogs on a single operon, namely clpP1 and clpP2.122 In the presence of an agonist peptide, ClpP1 and ClpP2 arrange into heterotetradecameric ClpP1P2 complexes in vitro, composed of two homoheptameric ClpP1 and ClpP2 rings, respectively.123,124 This heterotetradecamer interacts asymmetrically with cognate Clp-ATPases ClpX and ClpC1 which bind only to ClpP2.125 In accordance, X-ray crystallographic structural data of the ClpP1P2/ADEP complex show ADEP binding only at one ring, ClpP2.87 Remarkably, pore opening was still triggered at the ClpP1 ring.87 Thus, the conformation rearrangement that ADEP binding sets in motion must propagate from the hydrophobic pockets of ClpP2 via the ring–ring interface to the N-termini of ClpP1 over a distance of approximately 90 Å. This finding implies a long-distance relay within the complex that extends beyond single subunits.
To determine whether ADEP binding also effects the catalytic centers of ClpP, known inhibitors have been deployed in combination with ADEP treatment. β-Lactones are covalent inhibitors of the ClpP peptidase (suicide inhibitors).126,127 Instead of a peptide bond, the active site serine attacks the carbonyl atom of the β-lactone ring. This event opens the ring to establish an acyl–ester linkage with the inhibitor which is much more stable than the acyl–ester intermediate in polypeptide degradation.127 The following hydrolysis reaction regenerating the active site serine is substantially slowed (see catalysis mechanism of serine proteases in Fig. 1b). When ClpP was exposed to ADEP and β-lactone in combination, the catalytic efficiency of the first step, i.e. β-lactone binding, was two-fold accelerated for all β-lactone inhibitors employed despite varying side-chains.91 Even more striking was the stimulating effect of ADEP on the β-lactone hydrolysis reaction. Hydrolysis rates of the bound inhibitors were stimulated from two-fold to approximately 20-fold depending on the β-lactone structure tested.91 Notably, ADEPs affect the hydrolysis activity independently of the open state of the axial pores and stimulate catalysis as allosteric activators.
In conclusion, the hydrophobic pocket serves as a sort of “master switch” to turn the whole complex into an “on”-state. Turning the switch leads to the formation of an active extended conformation of ClpP, accelerated substrate turnover by the catalytic sites, widened axial pores, and an overall more stable and structurally less flexible machine. Taking a closer look at the geometry of the ADEP binding pocket provides a clue as to how ADEPs may achieve this transition. Modeling of ADEP1 (as well as ADEP derivatives) into the binding pocket of the inactive compressed ClpP structure from B. subtilis predicts a sterical clash of the aliphatic side chain (Fig. 7a). Thus, ADEPs can be regarded as sterical locks fixing ClpP in the active extended form and restricting transition to the inactive compressed form. This idea is further supported by the complete lack of compressed structures that have been co-crystallized with ADEP. It has been established that ClpP is a highly dynamic macromolecule.52 We propose a model in which ClpP resides in an equilibrium between an active and inactive state and transiently adopts all of the above mentioned states. However, ADEP binding strongly shifts this equilibrium towards the active state via conformational control (Fig. 8). ClpP functions like a clockwork where each cogwheel is tightly interconnected. Mutation of a single amino acid residue can alter the structural organization of the whole macromolecule, for instance the active site mutation D172N, which results in a more compacted structure.91 Mutation of the Y63 to alanine enables an otherwise tightly controlled peptidase to degrade a folded protein substrate.121 Therefore, structural shifts at one position are accompanied by a cascade of structural rearrangements that span the whole macromolecule. This structural interdependency is most vividly demonstrated by the observation that ADEP binding to the ClpP2 ring leads to pore opening in the opposing ClpP1 ring of the mycobacterial ClpP1P2 complex.87 According to this model, ADEP binding is the equivalent to blocking one cogwheel with an iron rod and, as a consequence, bringing the whole clockwork to a halt.
 | ||
| Fig. 8 Current model of ClpP conformational control by ADEPs. ClpP resides in a natural equilibrium between the inactive compressed and the active extended states. None of these states are proteolytically competent because the digestion chamber is inaccessible for proteins. ADEP binding shifts this equilibrium towards the active extended state, opens the axial entry pores and, in addition, enhances catalytic efficiency. | ||
4.5 Comparison between ADEP-mediated and Clp-ATPase-mediated activation of ClpP
To this day, no co-crystallization of ClpP with any partner Clp-ATPase has been achieved. ADEPs have served as a tool to investigate the interaction between ClpP and its cognate unfoldases. Some of the findings gained by investigating the interaction between ADEP and ClpP have been transferred to Clp-ATPase/ClpP interaction. But, there are differences. Clp-ATPases actively unfold and thread the unfolded protein strand into the catalytic space of ClpP. In cryo-EM structures with Clp-ATPases, the axial gate of ClpP appeared smaller than in the “open-gate”-conformation of the ClpP/ADEP crystal structures.68,69 The α5-helices of ClpP can adopt an unstructured conformation without destroying the oligomeric state and several studies suggest ClpP dynamics to be crucial in the interaction with Clp-ATPases.58 While this motion of ClpP has been suggested to be part of a natural cycle required for catalysis and product release, dynamics of ADEP-mediated activity might be different.In hydrogen exchange experiments, ADEP binding has been shown to induce a rigidification of the α5-helices in the equatorial plane.67 As ADEPs binds to the hydrophobic pockets of ClpP with high affinity, it can be assumed that many, if not all, hydrophobic pockets are occupied by ADEP molecules at the same time. Clp-ATPase binding is comparatively weak and one Clp-ATPase hexamer can never fill more than six hydrophobic pockets at once. In accordance with the notion that “filling” the hydrophobic pocket enforces the active extended state, weak and partial binding by Clp-ATPases might have evolved to ensure ClpP dynamics important for natural functions. Here, we propose that the tight ADEP binding event shifts the dynamic equilibrium of ClpP more strongly towards the active extended conformation than binding of Clp-ATPases does. In that case, equatorial pore opening would occur to a smaller extent due to predominantly extended handles. The widened axial pore of the ClpP/ADEP complex then could not only allow for substrate entry but also, at least partially, accommodate product exit, thus altering the presumed operation mode of the structurally versatile ClpP.
5 Outlook
The bacterial Clp protease is a novel potential drug target that had not been included in the large antibacterial screening campaigns of pharma companies in the 1990s/2000s due to its inessentiality for growth of most pathogens under moderate conditions and due to the fact that mechanism-wise only inhibition but not activation and deregulation of a target were envisioned. It took the study of ADEP as a natural product to understand the elaborate mechanism by which a protease machine can be taken out of its natural context and misdirected to kill multi-resistant and persisting bacterial pathogens. Bacterial Clp protease activity can be deregulated in a multi-facetted way with two different major consequences. Abrogating the interaction or productive enzymatic cycle between the proteolytic core ClpP and its cognate Clp-ATPases prevents all natural functions of the protease in general and regulated proteolysis. This consequence is achieved by known modulators blocking either the hydrophobic pocket of ClpP (ADEP) or by binding to the N-terminus of the mycobacterial Clp-ATPase ClpC1 (lassomycin, ecumicin, and potentially cyclomarin A). In addition, extensive conformational control of ClpP by ADEP leads to unregulated proteolysis and the degradation of vital bacterial proteins. Notably, it is the latter aspect by which ClpP as a non-essential target in firmicutes leads to bactericidal activity. Since the first report of ClpP as the target of ADEP about ten years ago, four additional and structurally unrelated natural product classes (Fig. 4) have been discovered to act on the Clp protease by either binding to ClpP or the cognate Clp-ATPase partner. It is likely that further natural product modulators of the Clp system remain to be found.6 Acknowledgements
The research group of H. B.-O. acknowledges financial support of the German Research Foundation (SFB 766).7 References
- WHO, 2014, ISBN: 978 92 4 156474 8.
- J. O'Neill, AMR Review, 2014, Antimicrobial Resistance: Tackling a Crisis for the Future Health and Wealth of Nations Search PubMed.
- T. A. Wencewicz, Bioorg. Med. Chem., 2016, 24, 6227–6252 CrossRef CAS PubMed.
- L. L. Silver, Future Microbiol., 2015, 10, 1711–1718 CrossRef CAS PubMed.
- K. H. Michel and R. E. Kastner, US Pat., 4492650, 1985.
- H. Brötz-Oesterhelt, D. Beyer, H.-P. Kroll, R. Endermann, C. Ladel, W. Schroeder, B. Hinzen, S. Raddatz, H. Paulsen, K. Henninger, J. E. Bandow, H.-G. Sahl and H. Labischinski, Nat. Med., 2005, 11, 1082–1087 CrossRef PubMed.
- B. Hinzen, S. Raddatz, H. Paulsen, T. Lampe, A. Schumacher, D. Häbich, V. Hellwig, J. Benet-Buchholz, R. Endermann, H. Labischinski and H. Brötz-Oesterhelt, ChemMedChem, 2006, 1, 689–693 CrossRef CAS PubMed.
- A. M. Socha, N. Y. Tan, K. L. Laplante and J. K. Sello, Bioorg. Med. Chem., 2010, 18, 7193–7202 CrossRef CAS PubMed.
- D. W. Carney, K. R. Schmitz, J. V. Truong, R. T. Sauer and J. K. Sello, J. Am. Chem. Soc., 2014, 136, 1922–1929 CrossRef CAS PubMed.
- J. D. Goodreid, J. Janetzko, J. P. Santa Maria, K. S. Wong, E. Leung, B. T. Eger, S. Bryson, E. F. Pai, S. D. Gray-Owen, S. Walker, W. A. Houry and R. A. Batey, J. Med. Chem., 2016, 59, 624–646 CrossRef CAS PubMed.
- B. P. Conlon, E. S. Nakayasu, L. E. Fleck, M. D. LaFleur, V. M. Isabella, K. Coleman, S. N. Leonard, R. D. Smith, J. N. Adkins and K. Lewis, Nature, 2013, 503, 365–370 CrossRef CAS PubMed.
- D. Frees, U. Gerth and H. Ingmer, Int. J. Med. Microbiol., 2014, 304, 142–149 CrossRef CAS PubMed.
- J. Feng, S. Michalik, A. N. Varming, J. H. Andersen, D. Albrecht, L. Jelsbak, S. Krieger, K. Ohlsen, M. Hecker, U. Gerth, H. Ingmer and D. Frees, J. Proteome Res., 2013, 12, 547–558 CrossRef CAS PubMed.
- S. Runde, N. Molière, A. Heinz, E. Maisonneuve, A. Janczikowski, A. K. W. Elsholz, U. Gerth, M. Hecker and K. Turgay, Mol. Microbiol., 2014, 91, 1036–1052 CrossRef CAS PubMed.
- C. M. Sassetti, D. H. Boyd and E. J. Rubin, Mol. Microbiol., 2003, 48, 77–84 CrossRef CAS PubMed.
- W. F. Wu, Y. Zhou and S. Gottesman, J. Bacteriol., 1999, 181, 3681–3687 CAS.
- C. K. Smith, T. A. Baker and R. T. Sauer, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6678–6682 CrossRef CAS.
- T. Msadek, V. Dartois, F. Kunst, M. L. Herbaud, F. Denizot and G. Rapoport, Mol. Microbiol., 1998, 27, 899–914 CrossRef CAS PubMed.
- S. Nakano, G. Zheng, M. M. Nakano and P. Zuber, J. Bacteriol., 2002, 184, 3664–3670 CrossRef CAS PubMed.
- J. M. Flynn, S. B. Neher, Y. I. Kim, R. T. Sauer and T. A. Baker, Mol. Cell, 2003, 11, 671–683 CrossRef CAS PubMed.
- P. Sass and H. Brötz-Oesterhelt, Curr. Opin. Microbiol., 2013, 16, 522–530 CrossRef CAS PubMed.
- H. Brötz-Oesterhelt and P. Sass, Int. J. Med. Microbiol., 2014, 304, 23–30 CrossRef PubMed.
- E. Culp and G. D. Wright, J. Antibiot., 2016 DOI:10.1038/ja.2016.138.
- D. Frees, K. Sørensen and H. Ingmer, Infect. Immun., 2005, 73, 8100–8108 CrossRef CAS PubMed.
- D. Frees, J. H. Andersen, L. Hemmingsen, K. Koskenniemi, K. T. Bæk, M. K. Muhammed, D. D. Gudeta, T. A. Nyman, A. Sukura, P. Varmanen and K. Savijoki, J. Proteome Res., 2012, 11, 95–108 CrossRef CAS PubMed.
- D. Frees, S. N. A. Qazi, P. J. Hill and H. Ingmer, Mol. Microbiol., 2003, 48, 1565–1578 CrossRef CAS PubMed.
- C. Wang, M. Li, D. Dong, J. Wang, J. Ren, M. Otto and Q. Gao, Microbes Infect., 2007, 9, 1376–1383 CrossRef CAS PubMed.
- F. Weinandy, K. Lorenz-Baath, V. S. Korotkov, T. Böttcher, S. Sethi, T. Chakraborty and S. A. Sieber, ChemMedChem, 2014, 9, 710–713 CrossRef CAS PubMed.
- H. Y. Kwon, A. D. Ogunniyi, M. H. Choi, S. N. Pyo, D. K. Rhee and J. C. Paton, Infect. Immun., 2004, 72, 5646–5653 CrossRef CAS PubMed.
- O. Gaillot, E. Pellegrini, S. Bregenholt, S. Nair and P. Berche, Mol. Microbiol., 2000, 35, 1286–1294 CrossRef CAS PubMed.
- H. P. Feng and L. M. Gierasch, Curr. Biol., 1998, 8, R464–R467 CrossRef CAS PubMed.
- J. Porankiewlcz, J. Wang and A. K. Clarke, Mol. Microbiol., 1999, 32, 449–458 CrossRef.
- I. Levchenko, L. Luo and T. A. Baker, Genes Dev., 1995, 9, 2399–2408 CrossRef CAS PubMed.
- R. Kruklitis, D. J. Welty and H. Nakai, EMBO J., 1995, 9, 935–944 Search PubMed.
- B. Felden, F. Vandenesch, P. Bouloc and P. Romby, PLoS Pathog., 2011, 7, e1002006 CAS.
- R. Singh and P. Ray, Future Microbiol., 2014, 9, 669–681 CrossRef CAS PubMed.
- I. Chatterjee, P. Becker, M. Grundmeier, M. Bischoff, G. A. Somerville, G. Peters, B. Sinha, N. Harraghy, R. A. Proctor and M. Herrmann, J. Bacteriol., 2005, 187, 4488–4496 CrossRef CAS PubMed.
- I. Chatterjee, S. Schmitt, C. F. Batzilla, S. Engelmann, A. Keller, M. W. Ring, R. Kautenburger, W. Ziebuhr, M. Hecker, K. T. Preissner, M. Bischoff, R. A. Proctor, H. P. Beck, H. P. Lenhof, G. A. Somerville and M. Herrmann, Proteomics, 2009, 9, 1152–1176 CrossRef CAS PubMed.
- J. E. Griffin, J. D. Gawronski, M. A. DeJesus, T. R. Ioerger, B. J. Akerley and C. M. Sassetti, PLoS Pathog., 2011, 7, e1002251 CAS.
- J. Ollinger, T. O'malley, E. A. Kesicki, J. Odingo and T. Parish, J. Bacteriol., 2012, 194, 663–668 CrossRef CAS PubMed.
- R. M. Raju, M. Unnikrishnan, D. H. F. Rubin, V. Krishnamoorthy, O. Kandror, T. N. Akopian, A. L. Goldberg and E. J. Rubin, PLoS Pathog., 2012, 8, e1002511 CAS.
- D. M. Roberts, Y. Personne, J. Ollinger and T. Parish, Future Microbiol., 2013, 8, 621–631 CrossRef CAS PubMed.
- M. W. Jackson, E. Silva-Herzog and G. V. Plano, Mol. Microbiol., 2004, 54, 1364–1378 CrossRef CAS PubMed.
- C. Webb, M. Moreno, M. Wilmes-Riesenberg, R. Curtiss and J. W. Foster, Mol. Microbiol., 1999, 34, 112–123 CrossRef CAS PubMed.
- R. Hengge-Aronis, Microbiol. Mol. Biol. Rev., 2002, 66, 373–395 CrossRef CAS PubMed.
- S. A. Joshi, G. L. Hersch, T. A. Baker and R. T. Sauer, Nat. Struct. Mol. Biol., 2004, 11, 404–411 CAS.
- Y. I. Kim, I. Levchenko, K. Fraczkowska, R. V. Woodruff, R. T. Sauer and T. A. Baker, Nat. Struct. Biol., 2001, 8, 230–233 CrossRef CAS PubMed.
- F. Ye, J. Zhang, H. Liu, R. Hilgenfeld, R. Zhang, X. Kong, L. Li, J. Lu, X. Zhang, D. Li, H. Jiang, C. G. Yang and C. Luo, J. Biol. Chem., 2013, 288, 17643–17653 CrossRef CAS PubMed.
- S. R. Geiger, T. Böttcher, S. A. Sieber and P. Cramer, Angew. Chem., Int. Ed., 2011, 50, 5749–5752 CrossRef CAS PubMed.
- J. Wang, J. A. Hartling and J. M. Flanagan, Cell, 1997, 91, 447–456 CrossRef CAS PubMed.
- M. Gersch, A. List, M. Groll and S. A. Sieber, J. Biol. Chem., 2012, 287, 9484–9494 CrossRef CAS PubMed.
- K. Liu, A. Ologbenla and W. A. Houry, Crit. Rev. Biochem. Mol. Biol., 2014, 49, 400–412 CrossRef CAS PubMed.
- B.-G. Lee, E. Y. Park, K.-E. Lee, H. Jeon, K. H. Sung, H. Paulsen, H. Rübsamen-Schaeff, H. Brötz-Oesterhelt and H. K. Song, Nat. Struct. Mol. Biol., 2010, 17, 471–478 CAS.
- B.-G. Lee, M. K. Kim and H. K. Song, Mol. Cells, 2011, 32, 589–595 CrossRef CAS PubMed.
- J. Zhang, F. Ye, L. Lan, H. Jiang, C. Luo and C. G. Yang, J. Biol. Chem., 2011, 286, 37590–37601 CrossRef CAS PubMed.
- A. Szyk and M. R. Maurizi, J. Struct. Biol., 2006, 156, 165–174 CrossRef CAS PubMed.
- M. R. Maurizi, S. K. Singh, M. W. Thompson, M. Kessel and A. Ginsburg, Biochemistry, 1998, 37, 7778–7786 CrossRef CAS PubMed.
- A. Gribun, M. S. Kimber, R. Ching, R. Sprangers, K. M. Fiebig and W. A. Houry, J. Biol. Chem., 2005, 280, 16185–16196 CrossRef CAS PubMed.
- R. Sprangers, A. Gribun, P. M. Hwang, W. A. Houry and L. E. Kay, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16678–16683 CrossRef CAS PubMed.
- Ž. Maglica, K. Kolygo and E. Weber-Ban, Structure, 2009, 17, 508–516 CrossRef PubMed.
- M. S. Kimber, A. Y. H. Yu, M. Borg, E. Leung, H. S. Chan and W. A. Houry, Structure, 2010, 18, 798–808 CrossRef CAS PubMed.
- K. M. Woo, W. J. Chung, D. B. Ha, A. L. Goldberg and C. H. Chung, J. Biol. Chem., 1989, 264, 2088–2091 CAS.
- M. W. Thompson, S. K. Singh and M. R. Maurizi, J. Biol. Chem., 1994, 269, 18209–18215 CAS.
- J. Ortega, S. K. Singh, T. Ishikawa, M. R. Maurizi and A. C. Steven, Mol. Cell, 2000, 6, 1515–1521 CrossRef CAS PubMed.
- T. Ishikawa, F. Beuron, M. Kessel, S. Wickner, M. R. Maurizi and A. C. Steven, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 4328–4333 CrossRef CAS PubMed.
- M. C. Bewley, V. Graziano, K. Griffin and J. M. Flanagan, J. Struct. Biol., 2006, 153, 113–128 CrossRef CAS PubMed.
- M. A. Sowole, J. A. Alexopoulos, Y. Q. Cheng, J. Ortega and L. Konermann, J. Mol. Biol., 2013, 425, 4508–4519 CrossRef CAS PubMed.
- G. Effantin, M. R. Maurizi and A. C. Steven, J. Biol. Chem., 2010, 285, 14834–14840 CrossRef CAS PubMed.
- J. A. Alexopoulos, A. Guarné and J. Ortega, J. Struct. Biol., 2012, 179, 202–210 CrossRef CAS PubMed.
- D. H. S. Li, Y. S. Chung, M. Gloyd, E. Joseph, R. Ghirlando, G. D. Wright, Y. Q. Cheng, M. R. Maurizi, A. Guarné and J. Ortega, Chem. Biol., 2010, 17, 959–969 CrossRef CAS PubMed.
- L. D. Jennings, J. Bohon, M. R. Chance and S. Licht, Biochemistry, 2008, 47, 11031–11040 CrossRef CAS PubMed.
- K. C. Keiler, P. R. Waller and R. T. Sauer, Science, 1996, 271, 990–993 CAS.
- S. Gottesman, E. Roche, Y. Zhou and R. T. Sauer, Genes Dev., 1998, 12, 1338–1347 CrossRef CAS PubMed.
- A. Martin, T. A. Baker and R. T. Sauer, Mol. Cell, 2007, 27, 41–52 CrossRef CAS PubMed.
- J. A. Kenniston, T. A. Baker, J. M. Fernandez and R. T. Sauer, Cell, 2003, 114, 511–520 CrossRef CAS PubMed.
- B. M. Stinson, V. Baytshtok, K. R. Schmitz, T. A. Baker and R. T. Sauer, Nat. Struct. Mol. Biol., 2015, 22, 411–416 CAS.
- A. Martin, T. A. Baker and R. T. Sauer, Nat. Struct. Mol. Biol., 2008, 15, 139–145 CAS.
- A. Martin, T. A. Baker and R. T. Sauer, Nature, 2005, 437, 1115–1120 CrossRef CAS PubMed.
- K. H. Choi and S. Licht, Biochemistry, 2005, 44, 13921–13931 CrossRef CAS PubMed.
- L. D. Jennings, D. S. Lun, M. Médard and S. Licht, Biochemistry, 2008, 47, 11536–11546 CrossRef CAS PubMed.
- I. Levchenko, M. Seidel, R. T. Sauer and T. A. Baker, Science, 2000, 289, 2354–2356 CrossRef CAS PubMed.
- J. M. Flynn, I. Levchenko, M. Seidel, S. H. Wickner, R. T. Sauer and T. A. Baker, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10584–10589 CrossRef CAS PubMed.
- J. Kirstein, N. Molière, D. A. Dougan and K. Turgay, Nat. Rev. Microbiol., 2009, 7, 589–599 CrossRef CAS PubMed.
- M. W. Thompson and M. R. Maurizi, J. Biol. Chem., 1994, 269, 18201–18208 CAS.
- T. A. Baker and R. T. Sauer, Biochim. Biophys. Acta, Mol. Cell Res., 2012, 1823, 15–28 CrossRef CAS PubMed.
- S. G. Kang, M. N. Dimitrova, J. Ortega, A. Ginsburg and M. R. Maurizi, J. Biol. Chem., 2005, 280, 35424–35432 CrossRef CAS PubMed.
- K. R. Schmitz, D. W. Carney, J. K. Sello and R. T. Sauer, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E4587–E4595 CrossRef CAS PubMed.
- J. Kirstein, T. Schlothauer, D. A. Dougan, H. Lilie, G. Tischendorf, A. Mogk, B. Bukau and K. Turgay, EMBO J., 2006, 25, 1481–1491 CrossRef CAS PubMed.
- T. A. Baker and R. T. Sauer, Trends Biochem. Sci., 2006, 31, 647–653 CrossRef CAS PubMed.
- J. Kirstein, A. Hoffmann, H. Lilie, R. Schmidt, H. Rübsamen-Waigmann, H. Brötz-Oesterhelt, A. Mogk and K. Turgay, EMBO Mol. Med., 2009, 1, 37–49 CrossRef CAS PubMed.
- M. Gersch, K. Famulla, M. Dahmen, C. Göbl, I. Malik, K. Richter, V. S. Korotkov, P. Sass, H. Rübsamen-Schaeff, T. Madl, H. Brötz-Oesterhelt and S. A. Sieber, Nat. Commun., 2015, 6, 6320 CrossRef CAS PubMed.
- T. Böttcher and S. A. Sieber, ChemBioChem, 2009, 10, 663–666 CrossRef PubMed.
- P. Sass, M. Josten, K. Famulla, G. Schiffer, H.-G. Sahl, L. Hamoen and H. Brötz-Oesterhelt, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 17474–17479 CrossRef CAS PubMed.
- A. Schiefer, J. Vollmer, C. Lämmer, S. Specht, C. Lentz, H. Ruebsamen-Schaeff, H. Brötz-Oesterhelt, A. Hoerauf and K. Pfarr, J. Antimicrob. Chemother., 2013, 68, 1790–1800 CrossRef CAS PubMed.
- N. P. Lavey, J. A. Coker, E. A. Ruben and A. S. Duerfeldt, J. Nat. Prod., 2016, 79, 1193–1197 CrossRef CAS PubMed.
- J. Alexopoulos, B. Ahsan, L. Homchaudhuri, N. Husain, Y. Q. Cheng and J. Ortega, Mol. Microbiol., 2013, 90, 167–180 CAS.
- M. K. Renner, Y. C. Shen, X. C. Cheng, P. R. Jensen, W. Frankmoelle, C. A. Kauffman, W. Fenical, E. Lobkovsky and J. Clardy, J. Am. Chem. Soc., 1999, 121, 11273–11276 CrossRef CAS.
- E. K. Schmitt, M. Riwanto, V. Sambandamurthy, S. Roggo, C. Miault, C. Zwingelstein, P. Krastel, C. Noble, D. Beer, S. P. S. Rao, M. Au, P. Niyomrattanakit, V. Lim, J. Zheng, D. Jeffery, K. Pethe and L. R. Camacho, Angew. Chem., Int. Ed., 2011, 50, 5889–5891 CrossRef CAS PubMed.
- D. Vasudevan, S. P. S. Rao and C. G. Noble, J. Biol. Chem., 2013, 288, 30883–30891 CrossRef CAS PubMed.
- K. Turgay, L. W. Hamoen, G. Venema and D. Dubnau, Genes Dev., 1997, 11, 119–128 CrossRef CAS PubMed.
- T. Schlothauer, A. Mogk, D. A. Dougan, B. Bukau and K. Turgay, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 2306–2311 CrossRef CAS PubMed.
- D. B. Trentini, M. J. Suskiewicz, A. Heuck, R. Kurzbauer, L. Deszcz, K. Mechtler and T. Clausen, Nature, 2016, 539, 48–53 CrossRef CAS PubMed.
- S. Barik, K. Sureka, P. Mukherjee, J. Basu and M. Kundu, Mol. Microbiol., 2010, 75, 592–606 CrossRef CAS PubMed.
- F. Wang, Z. Mei, Y. Qi, C. Yan, Q. Hu, J. Wang and Y. Shi, Nature, 2011, 471, 331–335 CrossRef CAS PubMed.
- E. U. Weber-Ban, B. G. Reid, A. D. Miranker and A. L. Horwich, Nature, 1999, 401, 90–93 CrossRef CAS PubMed.
- E. Gavrish, C. S. Sit, S. Cao, O. Kandror, A. Spoering, A. Peoples, L. Ling, A. Fetterman, D. Hughes, A. Bissell, H. Torrey, T. Akopian, A. Mueller, S. Epstein, A. Goldberg, J. Clardy and K. Lewis, Chem. Biol., 2014, 21, 509–518 CrossRef CAS PubMed.
- W. Gao, J. Y. Kim, S. N. Chen, S. H. Cho, J. Choi, B. U. Jaki, Y. Y. Jin, D. C. Lankin, J. E. Lee, S. Y. Lee, J. B. McAlpine, J. G. Napolitano, S. G. Franzblau, J.-W. Suh and G. F. Pauli, Org. Lett., 2014, 16, 6044–6047 CrossRef CAS PubMed.
- M. Choules, Y. Yu, S. H. Cho, J. Anderson, W. Gao, L. Klein, D. C. Lankin, J. Y. Kim, J. Cheng, S. H. Yang, H. Lee, J.-W. Suh, S. G. Franzblau and G. F. Pauli, Planta Med., 2015, 81, CL2 CrossRef PubMed.
- W. Gao, J. G. Napolitano, J. Y. Kim, I.-A. Lee, J. E. Lee, J. Choi, M. F. Rodríguez-Brasco, B. U. Jaki, S. Cho, J. B. McAlpine, G. F. Pauli, J. Kim, J.-W. Suh and S. G. Franzblau, Planta Med., 2012, 78, PJ134 Search PubMed.
- W. Gao, J. Y. Kim, J. R. Anderson, T. Akopian, S. Hong, Y. Y. Jin, O. Kandror, J. W. Kim, I.-A. Lee, S. Y. Lee, J. B. McAlpine, S. Mulugeta, S. Sunoqrot, Y. Wang, S. H. Yang, T.-M. Yoon, A. L. Goldberg, G. F. Pauli, J.-W. Suh, S. G. Franzblau and S. Cho, Antimicrob. Agents Chemother., 2015, 59, 880–889 CrossRef PubMed.
- K. Famulla, P. Sass, I. Malik, T. Akopian, O. Kandror, M. Alber, B. Hinzen, H. Ruebsamen-Schaeff, R. Kalscheuer, A. L. Goldberg and H. Brötz-Oesterhelt, Mol. Microbiol., 2016, 101, 194–209 CrossRef CAS PubMed.
- T. Akopian, O. Kandror, R. M. Raju, M. Unnikrishnan, E. J. Rubin and A. L. Goldberg, EMBO J., 2012, 31, 1529–1541 CrossRef CAS PubMed.
- K. R. Schmitz and R. T. Sauer, Mol. Microbiol., 2014, 93, 617–628 CrossRef CAS PubMed.
- R. M. Raju, M. P. Jedrychowski, J.-R. Wei, J. T. Pinkham, A. S. Park, K. O'Brien, G. Rehren, D. Schnappinger, S. P. Gygi and E. J. Rubin, PLoS Pathog., 2014, 10, e1003994 Search PubMed.
- D. W. Carney, C. L. Compton, K. R. Schmitz, J. P. Stevens, R. T. Sauer and J. K. Sello, ChemBioChem, 2014, 15, 2216–2220 CrossRef CAS PubMed.
- M. Arvanitis, G. Li, D. D. Li, D. Cotnoir, L. Ganley-Leal, D. W. Carney, J. K. Sello and E. Mylonakis, PLoS One, 2016, 11, e0153912 Search PubMed.
- E. S. Honsa, V. S. Cooper, M. N. Mhaissen, M. Frank, J. Shaker, A. Iverson, J. Rubnitz, R. T. Hayden, R. E. Lee, C. O. Rock, E. I. Tuomanen, J. Wolf and J. W. Rosch, mBio, 2017, 8, e02124-16 CrossRef PubMed.
- S. Xu, P. Guo, Y. Gao, Q. Shi, D. He, Y. Gao and H. Zhang, Biochem. Biophys. Res. Commun., 2013, 438, 468–472 CrossRef CAS PubMed.
- D. Y. Kim and K. K. Kim, J. Biol. Chem., 2003, 278, 50664–50670 CrossRef CAS PubMed.
- E. Leung, A. Datti, M. Cossette, J. Goodreid, S. E. McCaw, M. Mah, A. Nakhamchik, K. Ogata, M. El Bakkouri, Y. Q. Cheng, S. J. Wodak, B. T. Eger, E. F. Pai, J. Liu, S. Gray-Owen, R. A. Batey and W. A. Houry, Chem. Biol., 2011, 18, 1167–1178 CrossRef CAS PubMed.
- T. Ni, F. Ye, X. Liu, J. Zhang, H. Liu, J. Li, Y. Zhang, Y. Sun, M. Wang, C. Luo, H. Jiang, L. Lan, J. Gan, A. Zhang, H. Zhou and C. G. Yang, ACS Chem. Biol., 2016, 11, 1964–1972 CrossRef CAS PubMed.
- S. T. Cole, R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S. V. Gordon, K. Eiglmeier, S. Gas, C. E. Barry, F. Tekaia, K. Badcock, D. Basham, D. Brown, T. Chillingworth, R. Connor, R. Davies, K. Devlin, T. Feltwell, S. Gentles, N. Hamlin, S. Holroyd, T. Hornsby, K. Jagels, A. Krogh, J. McLean, S. Moule, L. Murphy, K. Oliver, J. Osborne, M. A. Quail, M.-A. Rajandream, J. Rogers, S. Rutter, K. Seeger, J. Skelton, R. Squares, S. Squares, J. E. Sulston, K. Taylor, S. Whitehead and B. G. Barrell, Nature, 1998, 393, 537–544 CrossRef CAS PubMed.
- H. Ingvarsson, M. J. Maté, M. Högbom, D. Portnoï, N. Benaroudj, P. M. Alzari, M. Ortiz-Lombardía and T. Unge, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2007, 63, 249–259 CrossRef CAS PubMed.
- N. Benaroudj, B. Raynal, M. Miot and M. Ortiz-Lombardia, BMC Biochem., 2011, 12, 61 CrossRef CAS PubMed.
- J. Leodolter, J. Warweg and E. Weber-Ban, PLoS One, 2015, 10, e0125345 Search PubMed.
- T. Böttcher and S. A. Sieber, Angew. Chem., Int. Ed., 2008, 47, 4600–4603 CrossRef PubMed.
- M. Gersch, F. Gut, V. S. Korotkov, J. Lehmann, T. Böttcher, M. Rusch, C. Hedberg, H. Waldmann, G. Klebe and S. A. Sieber, Angew. Chem., Int. Ed., 2013, 52, 3009–3014 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |